 Ikuo Shoji*
Ikuo Shoji* Lin Deng and Hak Hotta
Lin Deng and Hak Hotta
- Division of Microbiology, Center for Infectious Diseases, Kobe University Graduate School of Medicine, Kobe, Japan
Hepatitis C virus (HCV) infection causes not only intrahepatic diseases but also extrahepatic manifestations, including metabolic disorders. Chronic HCV infection is often associated with type 2 diabetes. However, the precise mechanism underlying this association is still unclear. Glucose is transported into hepatocytes via glucose transporter 2 (GLUT2). Hepatocytes play a crucial role in maintaining plasma glucose homeostasis via the gluconeogenic and glycolytic pathways. We have been investigating the molecular mechanism of HCV-related type 2 diabetes using HCV RNA replicon cells and HCV J6/JFH1 system. We found that HCV replication down-regulates cell surface expression of GLUT2 at the transcriptional level. We also found that HCV infection promotes hepatic gluconeogenesis in HCV J6/JFH1-infected Huh-7.5 cells. HCV infection transcriptionally up-regulated the genes for phosphoenolpyruvate carboxykinase (PEPCK) and glucose 6-phosphatase (G6Pase), the rate-limiting enzymes for hepatic gluconeogenesis. Gene expression of PEPCK and G6Pase was regulated by the transcription factor forkhead box O1 (FoxO1) in HCV-infected cells. Phosphorylation of FoxO1 at Ser319 was markedly diminished in HCV-infected cells, resulting in increased nuclear accumulation of FoxO1. HCV NS5A protein was directly linked with the FoxO1-dependent increased gluconeogenesis. This paper will discuss the current model of HCV-induced glucose metabolic disorders.
Introduction
Hepatitis C virus (HCV) is a positive-sense, single stranded RNA virus that belongs to the genus Hepacivirus of the family Flaviviridae. The approximately 9.6-kb HCV genome encodes a unique open reading frame that is translated into a polyprotein of about 3,000 amino acids, which is cleaved by cellular signalases and viral proteases to generate at least 10 viral proteins, such as core, envelope 1 (E1) and E2, p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B (Choo et al., 1991; Lemon et al., 2007).
Hepatitis C virus is the main cause of chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma. More than 170 million people worldwide are chronically infected with HCV (Poynard et al., 2003). Persistent HCV infection causes not only liver diseases but also extrahepatic manifestations. It is well established that HCV perturbs the glucose metabolism, leading to insulin resistance and type 2 diabetes in predisposed individuals. Several epidemiological, clinical, and experimental data suggested that HCV infection serves as an additional risk factor for the development of diabetes (Mason et al., 1999; Negro and Alaei, 2009; Negro, 2011). HCV-related glucose metabolic changes and insulin resistance and diabetes have significant clinical consequences, such as accelerated fibrogenesis, increased incidence of hepatocellular carcinoma, and reduced virological response to interferon (IFN)-α-based therapy (Negro, 2011). Therefore, it is very important to clarify the molecular mechanism of HCV-related diabetes. However, the precise mechanisms are poorly understood.
Experimental data suggest a direct interference of HCV with the insulin signaling pathway. Transgenic mice expressing HCV core gene exhibit insulin resistance (Shintani et al., 2004; Koike, 2007). In this transgenic mice model, both tyrosine phosphorylation of the insulin receptor substrate (IRS)-1 and IRS-2 are decreased. These decreases are recovered when the proteasome activator PA28γ is deleted, suggesting that the HCV core protein suppresses insulin signaling through a PA28γ-dependent pathway (Miyamoto et al., 2007). Several other reports also showed a link of the HCV core protein with insulin resistance (Kawaguchi et al., 2004; Pazienza et al., 2007).
Hepatocytes play a crucial role in maintaining plasma glucose homeostasis by adjusting the balance between hepatic glucose production and utilization via the gluconeogenic and glycolytic pathways, respectively. Gluconeogenesis is mainly regulated at the transcriptional level of the glucose 6-phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEPCK) genes, whereas glycolysis is mainly regulated by glucokinase (GK). Gluconeogenesis and glycolysis are coordinated so that one pathway is highly active within a cell while the other is relatively inactive. It is well known that increased hepatic glucose production via gluconeogenesis is a major feature of type 2 diabetes (Clore et al., 2000).
To identify a novel mechanism of HCV-related diabetes, we have been investigating the effects of HCV on glucose production in hepatocytes using HCV RNA replicon cells (Lohmann et al., 1999) and HCV J6/JFH1 cell culture system (Lindenbach et al., 2005; Wakita et al., 2005; Bungyoku et al., 2009). We previously reported that HCV replication suppresses cellular glucose uptake through down-regulation of cell surface expression of glucose transporter 2 (GLUT2; Kasai et al., 2009). Furthermore, we recently reported that HCV promotes hepatic gluconeogenesis via an NS5A-mediated, forkhead box O1 (FoxO1)-dependent pathway, resulting in increased cellular glucose production in hepatocytes (Deng et al., 2011). This paper discusses our current model for HCV-induced glucose metabolic disorders.
HCV Replication Down-Regulates Cell Surface Expression of GLUT2
The uptake of glucose into cells is conducted by the facilitative glucose carrier, glucose transporters (GLUTs). GLUTs are integral membrane proteins that contain 12 membrane-spanning helices. To date, a total of 14 isoforms have been identified in the GLUT family (Wu and Freeze, 2002; Macheda et al., 2005; Godoy et al., 2006). Glucose is transported into hepatocytes by GLUT2. We previously reported that HCV J6/JFH1 infection suppresses hepatocytic glucose uptake through down-regulation of surface expression of GLUT2 in human hepatoma cell line, Huh-7.5 cells (Kasai et al., 2009). We also demonstrated that GLUT2 expression in hepatocytes of the liver tissues from HCV-infected patients was significantly lower than in those from patients without HCV infection. Our data suggest that HCV infection down-regulates GLUT2 expression at transcriptional level. We are currently analyzing transcriptional control of human GLUT2 promoter in HCV replicon cells as well as in HCV J6/JFH1-infected cells.
HCV Infection Promotes Hepatic Gluconeogenesis
Then we analyzed hepatic glucose production and expression of transcription factors using HCV replicon cells and HCVcc system in order to clarify a role of HCV infection in glucose metabolic changes. Hepatic glucose production is usually regulated by gluconeogenesis and glycolysis. Therefore, we examined whether HCV infection induces gluconeogenesis or glycolysis. We found that the PEPCK and G6Pase genes were transcriptionally up-regulated in J6/JFH1-infected cells (Figure 1). On the other hand, the GK gene was transcriptionally down-regulated in HCV-infected cells. We obtained similar data in HCV replicon cells (both in subgenomic replicon cells and full-genomic replicon cells). When HCV replication was suppressed by IFN treatment, the up-regulation of PEPCK and G6Pase gene expression as well as the down-regulation of GK gene expression were canceled. From these results, HCV infection selectively up-regulates PEPCK and G6Pase genes, whereas HCV infection down-regulates GK gene (Deng et al., 2011).
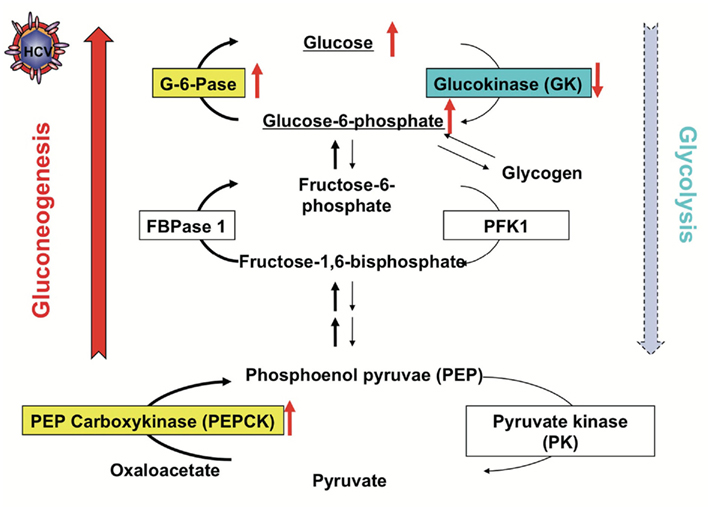
Figure 1. Regulation of Gluconeogenesis and Glycolysis in the HCV-infected cells. HCV infection promotes gluconeogenesis via transcriptional up-regulation of the genes for PEPCK and G6Pase, the rate-limiting enzymes for hepatic gluconeogenesis, and transcriptional down-regulation of the gene for GK, the rate-limiting enzyme for hepatic glycolysis.
Both HCV replicon cells and HCV-infected cells produced greater amounts of glucose than the control cells. IFN treatment canceled the enhanced glucose production in HCV replicon cells as well as in HCV-infected cells. G6P is an important precursor molecule that is converted to glucose in the gluconeogenesis pathway (Figure 1). Our metabolite analysis showed that a significantly higher level of G6P was accumulated in HCV-infected cells than in the control cells, suggesting that HCV indeed promotes hepatic gluconeogenesis to cause hyperglycemia. There is a trend toward an increase in gluconeogenesis in HCV-infected cells (Figure 1).
HCV Suppresses FoxO1 Phosphorylation at Ser319, Leading to the Nuclear Accumulation of FoxO1
It has been reported that G6Pase, PEPCK, and GK are regulated by certain transcription factors, including FoxO1 (Hirota et al., 2008), hepatic nuclear factor 4α (HNF-4α; Hirota et al., 2008), Krüppel-like factor 15 (KLF15; Takashima et al., 2010), and cyclic AMP (cAMP) response element binding protein (CREB; Rozance et al., 2008). While we were analyzing these factors in both HCV replicon cells and HCV J6/JFH1-infected cells, we found the involvement of the FoxO1 in the transcriptional activation of G6Pase and PEPCK (Deng et al., 2011). It is known that the FoxO1 enhances gluconeogenesis through the transcriptional activation of various genes, including G6Pase and PEPCK (Gross et al., 2008). The function of FoxO1 is regulated by post-translational modifications, including phosphorylation, ubiquitylation, and acetylation (Tzivion et al., 2011). The phosphorylated form of FoxO1 is exported from the nucleus to the cytosol, resulting in loss of its transcriptional activity (Figure 2). Phosphorylation status of FoxO1 at Ser319 is critical for FoxO1 nuclear exclusion (Zhao et al., 2004). Although the total amounts of FoxO1 protein were unchanged, FoxO1 phosphorylation at Ser319 was markedly suppressed in HCV-infected cells compared to that in the mock-infected cells. It is known that the FoxO1 is phosphorylated by the protein kinase Akt and is exported from the nucleus to the cytosol, resulting in loss of its transcriptional activity (Tzivion et al., 2011). The majority of FoxO1 was accumulated in the nuclear fraction in HCV-infected cells, whereas in control cells FoxO1 was distributed in both the nuclear and cytoplasmic fractions. Akt phosphorylation was enhanced in HCV-infected cells, although the protein levels of total Akt protein were comparable, which is consistent with the report by Burdette et al. (2010). Our findings suggest an interesting scenario in which the HCV-mediated suppression in FoxO1 phosphorylation is caused by an unknown mechanism independent of Akt activity.
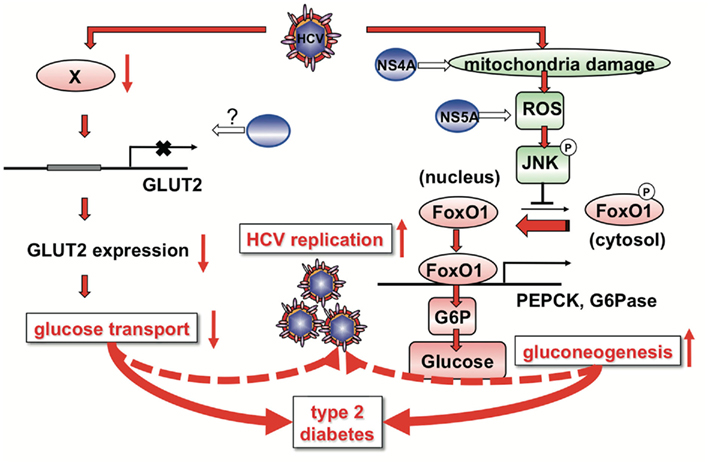
Figure 2. A proposed mechanism of HCV-induced glucose metabolic disorders. HCV infection down-regulates cell surface expression of GLUT2 in hepatocytes at the transcriptional level. HCV down-regulates a transcription factor involved in GLUT2 gene expression through an unknown mechanism. HCV infection induces mitochondria damage and ROS production, leading to JNK activation. HCV NS4A protein is involved in mitochondrial damage. HCV NS5A protein is involved in ROS production. HCV-induced ROS production causes JNK activation, resulting in the decreased phosphorylation and nuclear accumulation of FoxO1. Nuclear accumulation of FoxO1 up-regulates gene expression of PEPCK and G6Pase, leading eventually to increased glucose production by gluconeogenesis. High glucose levels in the hepatocytes may confer an advantage in efficient replication of HCV.
HCV-Induced JNK Activation is Involved in the Suppression of FoxO1 Phosphorylation
It is known that the stress-sensitive serine/threonine kinase JNK regulates FoxO at multiple levels (van der Horst and Burgering, 2007; Karpac and Jasper, 2009). We demonstrated that HCV infection induces phosphorylation and activation of JNK in a time-dependent manner, which is similar to that observed for the suppression of FoxO1 phosphorylation. As a result, c-Jun, a key substrate for JNK, got phosphorylated and activated in HCV-infected cells. The JNK inhibitor SP600125 clearly prevented the phosphorylation of c-Jun, and concomitantly recovered the suppression of FoxO1 phosphorylation in HCV-infected cells, suggesting that HCV activates the JNK/c-Jun signaling pathway, resulting in the nuclear accumulation of FoxO1 by reducing its phosphorylation status. The detailed mechanisms of HCV-induced suppression of FoxO1 phosphorylation via the JNK/c-Jun signaling pathway remain to be explored. There are at least two possibilities. The JNK/c-Jun signaling pathway (1) suppresses a protein kinase, or (2) activates a protein phosphatase to reduce phosphorylation of FoxO1.
HCV-Induced Mitochondrial Reactive Oxygen Species Production is Involved in Increased Glucose Production Through JNK Activation
Hepatitis C virus infection increases mitochondrial reactive oxygen species (ROS) production (Deng et al., 2008). N-acetyl cysteine (NAC; a general antioxidant) clearly prevented the phosphorylation of JNK, and concomitantly canceled the suppression of FoxO1 phosphorylation in HCV-infected cells, suggesting that HCV-induced ROS production is involved in the JNK activation. There was no significant difference in HCV RNA replication or infectious virus release between SP600125- or NAC-treated HCV-infected cells and non-treated HCV-infected cells. These results suggest that ROS-mediated JNK activation plays a key role in the suppression of FoxO1 phosphorylation, nuclear accumulation of FoxO1, and enhancement of glucose production in HCV-infected cells (Deng et al., 2011).
HCV NS5A is Involved in the Enhancement of Glucose Production
Then we sought to determine which HCV protein(s) is involved in the enhancement of glucose production. Transient expression of NS5A protein in Huh-7.5 cells significantly promoted the gene expression levels of G6Pase and PEPCK determined by real time quantitative RT-PCR. Promoter assay revealed that the level of PEPCK promoter activity was significantly higher in NS5A-expressing cells than in the control cells. Our results suggest that NS5A activate both the PEPCK promoter and the G6Pase promoter, leading to an increase in glucose production (Deng et al., 2011). The study by Banerjee et al. (2010) suggests that the HCV core protein modulates FoxO1 and FoxA2 activation and affects insulin-induced metabolic gene regulation in human hepatocytes. Our results, however, suggest that the HCV core protein is not significantly involved in the increased gluconeogenesis (Deng et al., 2011). The difference between these two studies needs to be explored.
There were previous reports suggesting that ROS production is induced in NS5A-expressing cells (Dionisio et al., 2009) or in hepatocytes of NS5A transgenic mice (Wang et al., 2009). We therefore sought to determine whether NS5A contributes to increased hepatic gluconeogenesis through the induction of ROS production. NS5A-expressing cells displayed a much stronger signal of ROS than in control cells. NS5A-expressing cells promoted phosphorylation level at Ser63 of c-Jun and suppressed FoxO1 phosphorylation at Ser319, suggesting that NS5A mediates JNK/c-Jun activation and FoxO1 phosphorylation suppression. These results suggest that NS5A play a role in the HCV-induced enhancement of hepatic gluconeogenesis through JNK/c-Jun activation and FoxO1 phosphorylation suppression.
Conclusion and Future Perspectives
Taken together, we propose a model of HCV-induced glucose metabolic disorders as shown in Figure 2. HCV infection down-regulates cell surface expression of GLUT2 in hepatocytes at the transcriptional level. HCV down-regulates a transcription factor involved in GLUT2 gene expression through an unknown mechanism. As GLUT2 is a facilitative GLUT, it ensures large bidirectional fluxes of glucose in and out the cell due to its low affinity and high capacity (Leturque et al., 2009). Down-regulated cell surface expression of GLUT2 results in disruption of bidirectional transport of glucose in hepatocytes. Even in the fasting state, down-regulation of GLUT2 may result in low glucose uptake of hepatocytes, causing hyperglycemia. In the fed state, glucose secretion from hepatocytes may be suppressed due to low level cell surface expression of GLUT2, as GLUT2 is a bidirectional transporter.
Hepatitis C virus infection induces mitochondria damage and ROS production, leading to JNK activation. HCV NS4A protein is involved in mitochondrial damage (Nomura-Takigawa et al., 2006). HCV NS5A protein is involved in ROS production (Dionisio et al., 2009; Wang et al., 2009; Deng et al., 2011). HCV-induced ROS production causes JNK activation, which results in the decreased phosphorylation and nuclear accumulation of FoxO1 by an unidentified mechanism. Nuclear accumulation of FoxO1 up-regulates gene expression of PEPCK and G6Pase, leading eventually to increased glucose production by gluconeogenesis (Deng et al., 2011).
These two pathways, HCV-induced down-regulation of GLUT2 expression and up-regulation of gluconeogenesis, may contribute to development of type 2 diabetes in HCV-infected patients at least to some extent. HCV-induced down-regulation of GLUT2 expression and up-regulation of gluconeogenesis may result in high concentration of glucose in HCV-infected hepatocytes. As suggested in a recent study, low glucose concentration in the hepatocytes inhibits HCV replication (Nakashima et al., 2011). Therefore, high glucose levels in the hepatocytes may confer an advantage in efficient replication of HCV.
Our understanding of HCV-induced glucose metabolic disorders will require much more work to fully unfold this pathway. Further investigation including the mechanism of HCV-induced GLUT2 downregualtion, JNK-mediated decreased phosphorylation of FoxO1, and the possible effect(s) of the dysregulation of hepatic gluconeogenesis on the HCV life cycle and host cells are currently under way.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to all of their co-workers who contributed to the studies cited here. This work was supported in part by grants-in-aid for Research on Hepatitis from the Ministry of Health, Labor and Welfare, Japan, and the Japan Initiative for Global Research Network on Infectious Diseases (J-GRID) program of Ministry of Education, Culture, Sports, Science and Technology, Japan. This study was also carried out as part of the Global Center of Excellence program of Kobe University Graduate School of Medicine, and the Science and Technology Research Partnership for Sustainable Development (SATREPS) program of Japan Science and Technology Agency (JST) and Japan International Cooperation Agency (JICA).
References
Banerjee, A., Meyer, K., Mazumdar, B., Ray, R. B., and Ray, R. (2010). Hepatitis C virus differentially modulates activation of forkhead transcription factors and insulin-induced metabolic gene expression. J. Virol. 84, 5936–5946.
Bungyoku, Y., Shoji, I., Makine, T., Adachi, T., Hayashida, K., Nagano-Fujii, M., Ide, Y. H., Deng, L., and Hotta, H. (2009). Efficient production of infectious hepatitis C virus with adaptive mutations in cultured hepatoma cells. J. Gen. Virol. 90, 1681–1691.
Burdette, D., Olivarez, M., and Waris, G. (2010). Activation of transcription factor Nrf2 by hepatitis C virus induces the cell-survival pathway. J. Gen. Virol. 91, 681–690.
Choo, Q. L., Richman, K. H., Han, J. H., Berger, K., Lee, C., Dong, C., Gallegos, C., Coit, D., Medina-Selby, R., Barr, P. J., Weiner, A. J., Bredley, D. W., Kuo, G., and Houghton, M. (1991). Genetic organization and diversity of the hepatitis C virus. Proc. Natl. Acad. Sci. U.S.A. 88, 2451–2455.
Clore, J. N., Stillman, J., and Sugerman, H. (2000). Glucose-6-phosphatase flux in vitro is increased in type 2 diabetes. Diabetes 49, 969–974.
Deng, L., Adachi, T., Kitayama, K., Bungyoku, Y., Kitazawa, S., Ishido, S., Shoji, I., and Hotta, H. (2008). Hepatitis C virus infection induces apoptosis through a Bax-triggered, mitochondrion-mediated, caspase 3-dependent pathway. J. Virol. 82, 10375–10385.
Deng, L., Shoji, I., Ogawa, W., Kaneda, S., Soga, T., Jiang, D. P., Ide, Y. H., and Hotta, H. (2011). Hepatitis C virus infection promotes hepatic gluconeogenesis through an NS5A-mediated, FoxO1-dependent pathway. J. Virol. 85, 8556–8568.
Dionisio, N., Garcia-Mediavilla, M. V., Sanchez-Campos, S., Majano, P. L., Benedicto, I., Rosado, J. A., Salido, G. M., and Gonzalez-Gallego, J. (2009). Hepatitis C virus NS5A and core proteins induce oxidative stress-mediated calcium signalling alterations in hepatocytes. J. Hepatol. 50, 872–882.
Godoy, A., Ulloa, V., Rodriguez, F., Reinicke, K., Yanez, A. J., Garcia Mde, L., Medina, R. A., Carrasco, M., Barberis, S., Castro, T., Martinez, F., Koch, X., Vera, J. C., Poblete, M. T., Figueroa, C. D., Peruzzo, B., Perez, F., and Nualart, F. (2006). Differential subcellular distribution of glucose transporters GLUT1-6 and GLUT9 in human cancer: ultrastructural localization of GLUT1 and GLUT5 in breast tumor tissues. J. Cell. Physiol. 207, 614–627.
Gross, D. N., van den Heuvel, A. P., and Birnbaum, M. J. (2008). The role of FoxO in the regulation of metabolism. Oncogene 27, 2320–2336.
Hirota, K., Sakamaki, J., Ishida, J., Shimamoto, Y., Nishihara, S., Kodama, N., Ohta, K., Yamamoto, M., Tanimoto, K., and Fukamizu, A. (2008). A combination of HNF-4 and Foxo1 is required for reciprocal transcriptional regulation of glucokinase and glucose-6-phosphatase genes in response to fasting and feeding. J. Biol. Chem. 283, 32432–32441.
Karpac, J., and Jasper, H. (2009). Insulin and JNK: optimizing metabolic homeostasis and lifespan. Trends Endocrinol. Metab. 20, 100–106.
Kasai, D., Adachi, T., Deng, L., Nagano-Fujii, M., Sada, K., Ikeda, M., Kato, N., Ide, Y. H., Shoji, I., and Hotta, H. (2009). HCV replication suppresses cellular glucose uptake through down-regulation of cell surface expression of glucose transporters. J. Hepatol. 50, 883–894.
Kawaguchi, T., Yoshida, T., Harada, M., Hisamoto, T., Nagao, Y., Ide, T., Taniguchi, E., Kumemura, H., Hanada, S., Maeyama, M., Baba, S., Koga, H., Kumashiro, R., Ueno, T., Ogata, H., Yoshimura, A., and Sata, M. (2004). Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am. J. Pathol. 165, 1499–1508.
Koike, K. (2007). Hepatitis C virus contributes to hepatocarcinogenesis by modulating metabolic and intracellular signaling pathways. J. Gastroenterol. Hepatol. 22(Suppl. 1), S108–S111.
Lemon, S. M., Walker, C., Alter, M. J., and Yi, M. (2007). “Hepatitis C virus,” in Fields’ Virology, 5th Edn, eds B. N. Fields, D. M. Knipe, and P. M. Howley (Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams and Wilkins), 1291–1304.
Leturque, A., Brot-Laroche, E., and Le Gall, M. (2009). GLUT2 mutations, translocation, and receptor function in diet sugar managing. Am. J. Physiol. Endocrinol. Metab. 296, E985–E992.
Lindenbach, B. D., Evans, M. J., Syder, A. J., Wolk, B., Tellinghuisen, T. L., Liu, C. C., Maruyama, T., Hynes, R. O., Burton, D. R., McKeating, J. A., and Rice, C. M. (2005). Complete replication of hepatitis C virus in cell culture. Science 309, 623–626.
Lohmann, V., Korner, F., Koch, J., Herian, U., Theilmann, L., and Bartenschlager, R. (1999). Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285, 110–113.
Macheda, M. L., Rogers, S., and Best, J. D. (2005). Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 202, 654–662.
Mason, A. L., Lau, J. Y., Hoang, N., Qian, K., Alexander, G. J., Xu, L., Guo, L., Jacob, S., Regenstein, F. G., Zimmerman, R., Everhart, J. E., Wasserfall, C., Maclaren, N. K., and Perrillo, R. P. (1999). Association of diabetes mellitus and chronic hepatitis C virus infection. Hepatology 29, 328–333.
Miyamoto, H., Moriishi, K., Moriya, K., Murata, S., Tanaka, K., Suzuki, T., Miyamura, T., Koike, K., and Matsuura, Y. (2007). Involvement of the PA28gamma-dependent pathway in insulin resistance induced by hepatitis C virus core protein. J. Virol. 81, 1727–1735.
Nakashima, K., Takeuchi, K., Chihara, K., Hotta, H., and Sada, K. (2011). Inhibition of hepatitis C virus replication through adenosine monophosphate-activated protein kinase-dependent and -independent pathways. Microbiol. Immunol. 55, 774–782.
Negro, F. (2011). Mechanisms of hepatitis C virus-related insulin resistance. Clin. Res. Hepatol. Gastroenterol. 35, 358–363.
Negro, F., and Alaei, M. (2009). Hepatitis C virus and type 2 diabetes. World J. Gastroenterol. 15, 1537–1547.
Nomura-Takigawa, Y., Nagano-Fujii, M., Deng, L., Kitazawa, S., Ishido, S., Sada, K., and Hotta, H. (2006). Non-structural protein 4A of Hepatitis C virus accumulates on mitochondria and renders the cells prone to undergoing mitochondria-mediated apoptosis. J. Gen. Virol. 87, 1935–1945.
Pazienza, V., Clement, S., Pugnale, P., Conzelman, S., Foti, M., Mangia, A., and Negro, F. (2007). The hepatitis C virus core protein of genotypes 3a and 1b downregulates insulin receptor substrate 1 through genotype-specific mechanisms. Hepatology 45, 1164–1171.
Poynard, T., Yuen, M. F., Ratziu, V., and Lai, C. L. (2003). Viral hepatitis C. Lancet 362, 2095–2100.
Rozance, P. J., Limesand, S. W., Barry, J. S., Brown, L. D., Thorn, S. R., LoTurco, D., Regnault, T. R., Friedman, J. E., and Hay, W. W. Jr. (2008). Chronic late-gestation hypoglycemia upregulates hepatic PEPCK associated with increased PGC1alpha mRNA and phosphorylated CREB in fetal sheep. Am. J. Physiol. Endocrinol. Metab. 294, E365–E370.
Shintani, Y., Fujie, H., Miyoshi, H., Tsutsumi, T., Tsukamoto, K., Kimura, S., Moriya, K., and Koike, K. (2004). Hepatitis C virus infection and diabetes: direct involvement of the virus in the development of insulin resistance. Gastroenterology 126, 840–848.
Takashima, M., Ogawa, W., Hayashi, K., Inoue, H., Kinoshita, S., Okamoto, Y., Sakaue, H., Wataoka, Y., Emi, A., Senga, Y., Matsuki, Y., Watanabe, E., Hiramatsu, R., and Kasuga, M. (2010). Role of KLF15 in regulation of hepatic gluconeogenesis and metformin action. Diabetes 59, 1608–1615.
Tzivion, G., Dobson, M., and Ramakrishnan, G. (2011). FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta 1813, 1938–1945.
van der Horst, A., and Burgering, B. M. (2007). Stressing the role of FoxO proteins in lifespan and disease. Nat. Rev. Mol. Cell Biol. 8, 440–450.
Wakita, T., Pietschmann, T., Kato, T., Date, T., Miyamoto, M., Zhao, Z., Murthy, K., Habermann, A., Krausslich, H. G., Mizokami, M., Bartenschlager, R., and Liang, T. J. (2005). Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11, 791–796.
Wang, A. G., Lee, D. S., Moon, H. B., Kim, J. M., Cho, K. H., Choi, S. H., Ha, H. L., Han, Y. H., Kim, D. G., Hwang, S. B., and Yu, D. Y. (2009). Non-structural 5A protein of hepatitis C virus induces a range of liver pathology in transgenic mice. J. Pathol. 219, 253–262.
Wu, X., and Freeze, H. H. (2002). GLUT14, a duplicon of GLUT3, is specifically expressed in testis as alternative splice forms. Genomics 80, 553–557.
Keywords: HCV, diabetes, gluconeogenesis, GLUT2, FoxO1, JNK, NS5A
Citation: Shoji I Deng L and Hotta H (2012) Molecular mechanism of hepatitis C virus-induced glucose metabolic disorders. Front. Microbio. 2:278. doi: 10.3389/fmicb.2011.00278
Received: 01 December 2011; Accepted: 25 December 2011; Published online: 10 January 2012.
Edited by:
Yasuko Yokota, National Institute of Infectious Diseases, JapanReviewed by:
Koji Ishii, National Institute of Infectious Diseases, JapanKohji Moriishi, University of Yamanashi, Japan
Copyright: © 2012 Shoji, Deng and Hotta. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Ikuo Shoji, Division of Microbiology, Center for Infectious Diseases, Kobe University Graduate School of Medicine, 7-5-1 Kusunoki-cho, Chuo-ku, Kobe, Hyogo 650-0017, Japan. e-mail:aXNob2ppQG1lZC5rb2JlLXUuYWMuanA=