 Kohji Moriishi1* and Yashiharu Matsuura2
Kohji Moriishi1* and Yashiharu Matsuura2
- 1 Department of Microbiology, Faculty of Medicine, University of Yamanashi, Chuo-shi, Yamanashi, Japan
- 2 Department of Molecular Virology, Research Institute for Microbial Diseases, Osaka University, Suita-shi, Osaka, Japan
Hepatitis C virus (HCV), which is a major causative agent of blood-borne hepatitis, has chronically infected about 170 million individuals worldwide and leads to chronic infection, resulting in development of steatosis, cirrhosis, and eventually hepatocellular carcinoma. Hepatocellular carcinoma associated with HCV infection is not only caused by chronic inflammation, but also by the biological activity of HCV proteins. HCV core protein is known as a main component of the viral nucleocapsid. It cooperates with host factors and possesses biological activity causing lipid alteration, oxidative stress, and progression of cell growth, while other viral proteins also interact with host proteins including molecular chaperones, membrane-anchoring proteins, and enzymes associated with lipid metabolism to maintain the efficiency of viral replication and production. HCV core protein is localized on the surface of lipid droplets in infected cells. However, the role of lipid droplets in HCV infection has not yet been elucidated. Several groups recently reported that other viral proteins also support viral infection by regulation of lipid droplets and core localization in infected cells. Furthermore, lipid components are required for modification of host factors and the intracellular membrane to maintain or up-regulate viral replication. In this review, we summarize the current status of knowledge regarding the exploitation of lipid components by viral and host proteins in HCV infection.
Introduction
Hepatitis C virus (HCV) is a major causative agent of chronic liver disease including steatosis, cirrhosis, and hepatocellular carcinoma. Epidemiological studies indicate that HCV is also associated with extrahepatic manifestations including type 2 diabetes mellitus, B-cell non-Hodgkin lymphoma, mixed cryoglobulinemia, and Sjögren’s syndrome (Jacobson et al., 2010). It has been estimated that there are 170 million patients worldwide, of whom most are infected with HCV. Combination therapy with pegylated interferon (PEG-IFN) and ribavirin has been the standard treatment but it fails to cure ∼50% of treated patients (Soriano et al., 2009).
Hepatitis C virus belongs to the genus Hepacivirus of the family Flaviviridae. The viral genome of HCV is characterized by a single positive strand RNA with a nucleotide length of 9.6 kb and it encodes a single polypeptide (Figure 1). This polyprotein is cleaved by host and viral proteases into structural and non-structural proteins (Harada et al., 1991; Hijikata et al., 1991; Grakoui et al., 1993a,b). Structural proteins, including the core protein and two envelope proteins, and the viroporin p7 are located within one-third of the N-terminal, while the remaining viral proteins are classified as non-structural proteins which form a replication complex with host factors (Grakoui et al., 1993c). HCV core protein is cleaved by signal peptide cleavage and then released from E1 (Santolini et al., 1994). After cleavage by signal peptidase (SP), the C-terminal transmembrane region of the core protein is further cleaved by signal peptide peptidase (SPP; Hussy et al., 1996; McLauchlan et al., 2002). The nucleocapsid, composed of matured core proteins and the viral genome, is surrounded by an envelope composed of host lipids and viral envelope proteins (Wakita et al., 2005). The life cycle of HCV is shown in Figure 2. The viral envelope proteins play a role in the binding to host receptors and membrane fusion for uncoating. Recently, several groups reported that the viral particle binds to a very low-density lipoprotein (VLDL), including apolipoprotein E (apoE), which is required for the binding step (Andre et al., 2002; Nielsen et al., 2006; Chang et al., 2007; Benga et al., 2010) as described below. The virus infects hepatocytes via entry factors known as receptors and co-receptors. The viral particle complex composed of the enveloped nucleocapsid and VLDL including apoE (Merz et al., 2011), is reported to bind to heparin sulfate (HS; Barth et al., 2003) and the low-density lipoprotein (LDL) receptor (LDLR; Agnello et al., 1999), although Albecka et al. (2012) recently reported that LDLR is required for optimal replication of the HCV genome rather than entry of the infectious viral particle. Other host factors may be involved in apoE-mediated entry. The HCV viral particle is transferred to the scavenger receptor class B type I (SR-BI; Scarselli et al., 2002; Bartosch et al., 2003) and CD81 (Pileri et al., 1998) through E2 binding and then enters cells with claudin-1 (CLDN1; Evans et al., 2007) and occludin (OCLN; Ploss et al., 2009) by endocytosis. The Niemann–Pick C1-like 1 cholesterol absorption receptor has recently been reported to be an HCV cell entry factor that is involved in the entry step between post-binding and pre-fusion (Sainz et al., 2012). The viral envelope fuses with the host plasma membrane in an endosome under a low pH condition (Takikawa et al., 2000; Hsu et al., 2003; Blanchard et al., 2006; Codran et al., 2006; Meertens et al., 2006; Tscherne et al., 2006). The capsid protein and viral genome are expected to be released into the cytoplasm of infected cells. The viral replication, assembly, and budding are summarized in Figure 3 on the basis of current information. The viral genome is translated dependent on own internal ribosome entry site (Tsukiyama-Kohara et al., 1992) and transcribed by the translated and processed NS3 to NS5B (Lohmann et al., 1999). The viral protein NS4B induces a convoluted membrane structure (termed a membranous web) with host lipid components and proteins, in which the viral replication is carried out (Egger et al., 2002; Gosert et al., 2005; Ferraris et al., 2010). The newly synthesized viral positive stranded RNA genome is released from the membranous web and passes to the core protein via NS5A (Masaki et al., 2008). The core protein is translocated on the surface of the lipid droplet or endoplasmic reticulum (ER) membrane for efficient formation of viral particles, and then encloses the synthesized viral genome to form a capsid near the membranous web (Miyanari et al., 2007; Boson et al., 2011). The capsids are enclosed by an endoplasmic membrane containing the viral envelope proteins E1 and E2 and are then released into ER lumen side, since intracellular envelope proteins are categorized as high-mannose type glycoproteins and the viral particle composed of core proteins and envelope proteins egresses into the lumen side of the intracellular compartment associated with lipid droplets (Miyanari et al., 2007; Vieyres et al., 2010). The viral particle is secreted through a host secretion pathway, although the mechanism by which HCV particles are secreted in infected cells remains poorly understood.
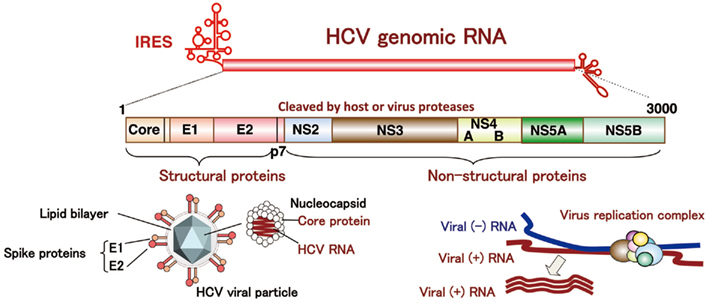
Figure 1. Structure of HCV. HCV RNA encodes a polyprotein composed of about 3,000 amino acids. The core protein, and two envelope proteins are classified as structural protein, while NS2, NS3, NS4A, NS4B, NS5A, and NS5B are non-structural proteins.
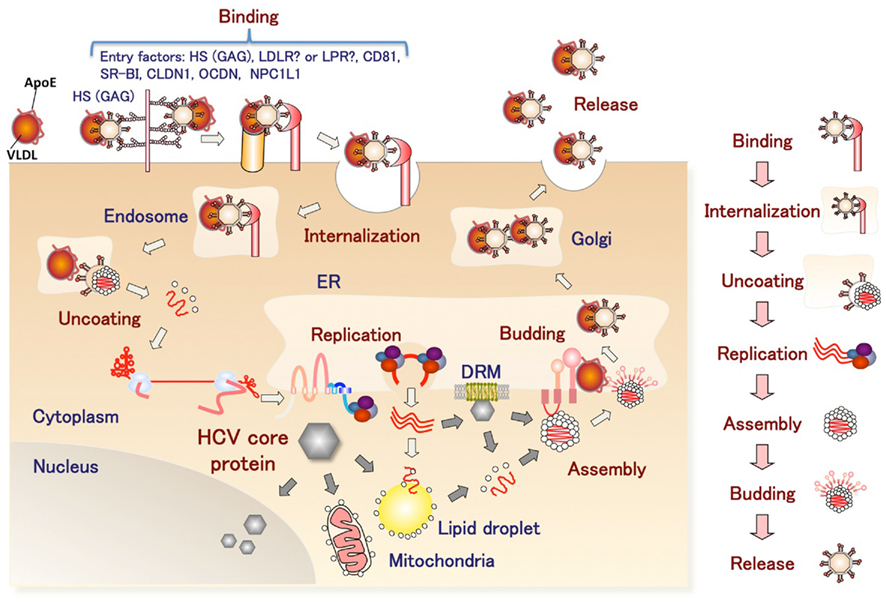
Figure 2. The infection cycle of HCV. The HCV complex with VLDL binds to entry factors on the surface of hepatocytes and then enters cells by endocytosis. After uncoating, viral replication is carried out in the convoluted membrane structure called the membranous web. The viral nucleocapsid egresses into the lumen side of the ER and binds to VLDL. The HCV complex with VLDL is released from the infected cells.
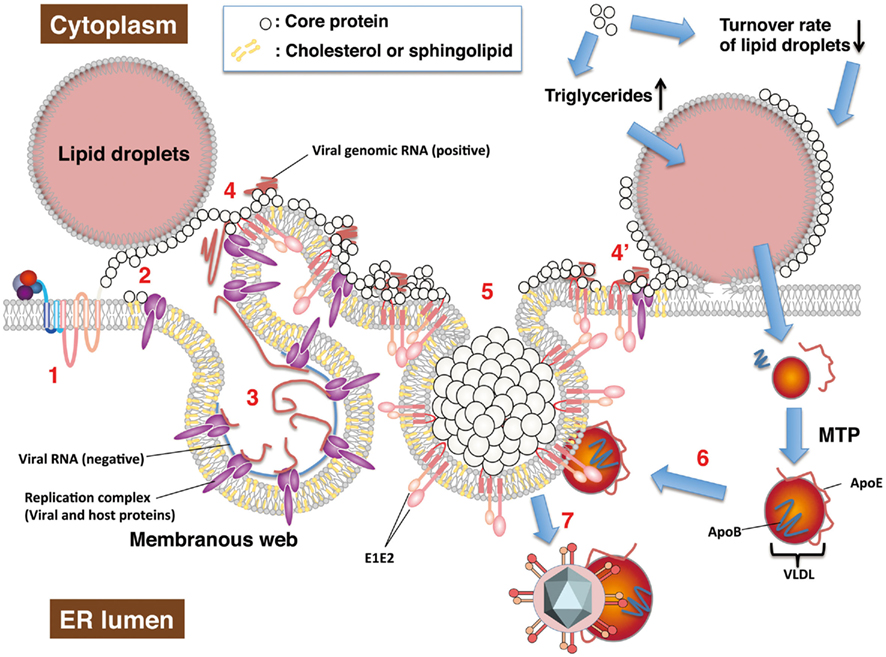
Figure 3. Hepatitis C virus RNA replication and particle production on lipid droplets and the ER membrane. HCV RNA replication and viral particle production are thought to be carried out on the lipid droplets and ER membrane. The viral proteins take advantage of host lipid metabolism and intracellular compartments to produce viral components as described in the text. Each step is described below. (1) A newly synthesized or uncoated vial genomic RNA is translated to a viral polyprotein followed by a cleaving polyprotein. (2) Matured non-structural proteins form replication a complex with host factors. The core protein is translocated to lipid droplets by DGAT1. In cells infected with the highly infectious strain (e.g., Jc1), the core protein may be retained on ER membrane by cooperating with non-structural proteins. (3) NS4B is involved in the convoluted membrane structure dubbed the membranous web. Negative and positive strands of viral RNA are synthesized by the replication complex based on each other. (4) The positive strand of genomic RNA is enclosed with the core proteins, cooperating non-structural proteins. Envelope proteins, NS2, and p7 may determine recruitment of the core protein from lipid droplets to the ER membrane along microtubules. Lipid droplets hemi-fused with ER membrane (Wolins et al., 2006) may be involved in recruitment of core protein on the ER membrane (4′). (5) The core proteins enclose the viral RNA genome, and are then surrounded by envelopes. Non-structural proteins and other factors may support formation of nucleocapsid and budding. (6) VLDL binds to viral particles to form LVP before or after budding. (7) The viral particles egress into the ER lumen with VLDL including ApoE. The core protein upregulates synthesis or storage of triglycerides and cholesteryl ester, cooperating with host factors including DGAT1, and so on.
Although no effective vaccine for HCV has been developed, antiviral drugs targeting to the viral and host factors have been reported recently. The HCV replicon system was reported for a screening system based on cultured cells (Lohmann et al., 1999) and has been improved by modification of cell lines and marker genes and introduction of adaptive mutations in the region of the viral RNA genome for high efficiency of viral replication (Blight et al., 2000; Krieger et al., 2001; Lohmann et al., 2001; Ikeda et al., 2002; Pietschmann et al., 2002). The complete infectious cycle of HCV in cultured cells was established in a highly permissive cell line by using the genotype 2a strain JFH1 or its chimeric recombinant virus (Lindenbach et al., 2005; Wakita et al., 2005; Zhong et al., 2005). A system based on the cultured cell line has been an exclusive drug-screening system for finding antiviral compounds that interfere with the process of viral RNA replication under intracellular conditions. NS3 forms a complex with cofactor NS4A (Failla et al., 1994, 1995; Koch et al., 1996). This interaction stabilizes NS3 and retains it on the ER where it acquires the ability of a protease against viral polyprotein (Wolk et al., 2000) and host protein IPS-1/MAVS (Foy et al., 2005; Loo et al., 2006), which is a molecule downstream of the RIG-I sensor molecule (Sumpter et al., 2005; Loo et al., 2008). NS3 serine protease activity is a target of the direct acting HCV antiviral drugs known as telaprevir and boceprevir, which are available clinically by combination therapy with PEG-INF and ribavirin (Hofmann and Zeuzem, 2011). The RNA helicase activity of NS3 and NS5B RNA-dependent RNA polymerase are also used for drug-screening in particular (Hicham Alaoui-Ismaili et al., 2000; Dhanak et al., 2002; Borowski et al., 2003; De Francesco et al., 2003; Boguszewska-Chachulska et al., 2004; Maga et al., 2005; Najda-Bernatowicz et al., 2010). Combination therapy using several compounds targeting host and viral factors may be able to completely eradicate the virus and suppress the pathogenicity induced by HCV infection.
Liver steatosis, which is characterized by accumulation of lipid droplets in hepatocytes, is significantly associated with the incidence of hepatocellular carcinoma in HCV-infected patients (Ohata et al., 2003). Severe liver steatosis has been frequently found in patients infected with the genotype 3a virus (Rubbia-Brandt et al., 2000; Adinolfi et al., 2001). Successful clearance of HCV reduces steatosis in genotype 3a patients, suggesting an association between genotype 3a and severe steatosis. Furthermore, HCV core protein derived from genotype 1 also induced liver steatosis in mouse and cultured cells (Barba et al., 1997; Moriya et al., 1997; Hope and McLauchlan, 2000). Lipid droplets containing triglycerides and cholesteryl ester are increased in cells expressing core protein and are surrounded by the core protein (Hope and McLauchlan, 2000). Non-structural proteins associate with the lipid droplets surrounded by HCV core proteins to supply the synthesized viral genome for viral assembly (Miyanari et al., 2007). Other lipid components are reported to be involved in formation of viral particles and the viral RNA replication as described below. This review mainly summarizes the viral and host factors that are associated with lipid metabolism with regard to HCV replication and pathogenicity.
The Role of VLDL in HCV Infection
Hepatitis C virus replicates in a convoluted membrane structure as a membranous web (Egger et al., 2002; Gosert et al., 2005; Ferraris et al., 2010) and assembles in the area of the ER membrane-associated with lipid droplets surrounded by the core protein (Miyanari et al., 2007). The LDLR has also been proposed to function as one of entry factors described above for HCV entry, in which interaction between LDLR and HCV particles is facilitated though interaction of the virus with host lipoprotein components (Monazahian et al., 1999; Chang et al., 2007; Huang et al., 2007; Miyanari et al., 2007; Gastaminza et al., 2008). HCV RNA containing particles derived from infected human serum were fractionated in densities with a value of 1.03–1.25 g/ml (Thomssen et al., 1992, 1993). The HCV RNA particles of the fraction with a density of lower than 1.06 g/ml possessed infectivity against chimpanzees, while HCV RNA derived from fractions with a higher density showed poor infectious ability (Bradley et al., 1991; Hijikata et al., 1993). The infectious HCV particles form a LDL–virus complex in the sera of human patients (Andre et al., 2002). An LDL–virus complex was found in the fractions with very low to low buoyant densities (1.03–1.25 g/ml), which varied with the stage of infection (Pumeechockchai et al., 2002; Carabaich et al., 2005). HCV particles prepared from infected human serum forms a complex with lipoproteins designated as lipo-viro-particles (LVP; Figure 3; Andre et al., 2002; Nielsen et al., 2006). LVP includes triglycerides, HCV RNA, core protein, and apolipoproteins B and E (Andre et al., 2002), which are components of VLDLs and LDL (Brodsky et al., 2004).
Very low-density lipoprotein is formed with a hydrophobic particle composed of triglycerides and cholesteryl ester that is surrounded by a surface coat containing phospholipid, free cholesterol, and two dominant lipoproteins, apoB and apoE (review to see Havel, 2000). Both apoB and apoE were found in a low-density fraction of HCV RNA particles (Andre et al., 2002; Chang et al., 2007). HCV virions could also be precipitated with antibodies against apoB or apoE (Andre et al., 2002; Chang et al., 2007). ApoB and microsomal triglyceride transfer protein (MTP) are required for HCV assembly and production, since knockdown of apoB or a specific antibody to MTP could inhibit HCV production (Huang et al., 2007; Gastaminza et al., 2008). However, another report suggests that knockdown of apoB or antibodies to apoB exhibited no significant effect on HCV infectivity and production (Jiang and Luo, 2009). The monoclonal antibodies against apoE neutralized HCV infection in cultured cells (Chang et al., 2007; Jiang and Luo, 2009), while knockdown of apoE markedly reduced HCV infectivity and infectious viral production without affecting viral entry and replication (Chang et al., 2007; Berger et al., 2009; Jiang and Luo, 2009). Hishiki et al. (2010) suggested that the isoforms 3 and 4, but not 2, of apoE are critical for HCV infectivity dependent of affinity to LDLR. Furthermore, NS5A could interact with apoE in infected cells and colocalization of both proteins supports the notion of intracellular interaction in infected cells (Benga et al., 2010). The C-terminal alpha-helix region spanning from residue 205 to 280 was critical for NS5A–apoE interaction and viral production (Cun et al., 2010). ApoE included in LVP may directly bind to LDLR or LDLR-related proteins in hepatocytes (Figure 2), since apoE is a ligand for all members of the LDLR gene family (see review described by Herz et al., 2009). These results suggest that apoE is an essential host factor for HCV entry.
Localization of the Core Protein on Both ER and Lipid Droplets in Infected Cells
Hepatitis C virus core protein is located at the N-terminus of the HCV polyprotein (Figure 1). The HCV core protein is cleaved from a precursor polyprotein by a SP, releasing it from an envelope E1 protein. Then, the C-terminal transmembrane region of the core protein is further processed by a SPP (McLauchlan et al., 2002). The intramembrane processing of the HCV core protein by SPP is critical for the production of infectious viral particles (Okamoto et al., 2008). The C-terminal end of the mature HCV core protein expressed in insect and human cell lines was determined to be Phe177 (Ogino et al., 2004; Okamoto et al., 2008). Randall et al. (2007) reported that the introduction of an siRNA targeted to SPP (called HM13) reduced the production of infectious HCV particles, suggesting that SPP is required for HCV particle production. Our previous report (Okamoto et al., 2008) showed that the production of HCV in cells persistently infected with the JFH1 strain was impaired by treatment with an SPP inhibitor and that JFH1 viruses bearing SPP-resistant mutations in the core protein failed to propagate in a permissive cell line. These data suggest that intramembrane processing of HCV core protein by SPP is required for viral propagation. Matured core protein was found in a detergent-resistant membrane fraction, which was distinct from the classical lipid rafts (Matto et al., 2004). Our data also suggest that cleavage of HCV core protein by SPP is required for localization of HCV core protein in detergent-resistant membrane fractions including cholesterol and sphingolipid (Figure 3, step 4 and 5). Detergent-resistant membrane fractions may be derived from the membranous web where the viral replication complex synthesizes the viral RNA genome, since the replication complex is fractionated in lipid raft fractions including Vesicle-associated membrane protein-associated protein (VAP)-A, cholesterol, and sphingolipid (Figure 3, step 3; Shi et al., 2003; Aizaki et al., 2004; Gao et al., 2004; Sakamoto et al., 2005). Furthermore, an HCV core protein mutation resistant to SPP results in delayed localization of HCV core protein on lipid droplets and reduction of virus production (Targett-Adams et al., 2008). These reports suggest that cleavage of HCV core protein by SPP is required for its suitable intracellular localization for the viral assembly. Sequence analysis of the core protein suggests that high hydrophobicity is found in the region from amino acid residues 119 to 174, which is called domain 2 (Hope and McLauchlan, 2000). Domain 2 is critical for localization of the core protein on lipid droplets and shares common features with the core protein of GBV-B, but not of other viruses belonging to the Flaviviridae family (Hope et al., 2002). When three hydrophobic amino acids, Leu139, Val140, and Leu144, in the most hydrophobic peak in domain 2 were replaced with Ala to reduce hydrophobicity, the triple mutations in the core protein led to resistance to SPP cleavage, dislocation of the detergent-resistant membrane, and a reduction in virus production (Okamoto et al., 2004, 2008). Furthermore, comparative analysis between JFH1 and Jc1 suggest that binding strength of domain 2 of core for lipid droplets determines efficiency of virus assembly (Shavinskaya et al., 2007). These results suggest that hydrophobicity of domain 2 in the core protein is required for lipid droplet localization, SPP cleavage, and virus production.
Host lipid biogenesis is responsible for replication and assembly. HCV core protein contributes to the accumulation and production of host lipid components and is detected on the surface of lipid droplets (Hope and McLauchlan, 2000). The core protein is translocated into the lipid droplets near the replication complex and encloses newly synthesized viral RNA to form the nucleocapsid (Figure 3, step 2–4 or 4′), egresses into the lumen side of the ER, then is surrounded with host lipid components and viral envelope proteins (Figure 3, step 5; Miyanari et al., 2007). HCV core protein interacts with diacylglycerol acyl transferase 1 (DGAT1), which is required for the trafficking of core protein to lipid droplets (Figure 3, step 2; Herker et al., 2010). However, the translocation of the core protein to lipid droplets may not be required for efficient production of viral particles. The recombinant virus Jc1 exhibits a higher virus titer than the JFH1 strain (Lindenbach et al., 2006; Pietschmann et al., 2006). The core protein of the Jc1 strain is hardly detected on lipid droplets in infected cells and is mainly localized on ER membranes, together with envelope protein E2 (Miyanari et al., 2007; Shavinskaya et al., 2007; Boson et al., 2011). Expression of p7 increases the ER localization of core protein in the absence of envelope proteins (Boson et al., 2011). However, Miyanari et al. (2007) reported that the core protein of the Jc1 strain was mainly localized with envelope proteins on ER in cells transfected with a complete viral genome, but on lipid droplets in cells that were transfected with the viral genome lacking envelope protein genes. Expression of envelope proteins and p7 may determine intracellular localization of the core protein with regard to viral assembly (Figure 3, step 2 and 4 or 4′).
NS2 has been reported to be involved in the assembly process of HCV particles (Jones et al., 2007; Jirasko et al., 2008; Dentzer et al., 2009). NS2, which is composed of three transmembrane regions and a cytoplasmic domain in order after p7 (Lorenz et al., 2006), is known as the autoprotease of which C-terminal cytoplasmic domain is involved in cis cleavage at the NS2–NS3 junction (Santolini et al., 1995; Yamaga and Ou, 2002; Lorenz et al., 2006). Genetic interaction was implied between the N-terminal region of NS2 and the upstream structural proteins, since the first transmembrane of NS2 was identified as a genetic determinant for infectivity by construction of chimeric HCV with various genotypes (Pietschmann et al., 2006). Analyses by co-immunoprecipitation and imaging microscopy for interaction between NS2 and other viral proteins in cultured cells suggest that NS2 interacts with p7 and E2 on the ER-derived dotted structure closed to lipid droplets that are surrounded by HCV core protein (Popescu et al., 2011). NS2 also interacts with NS3/4A to recruit the core protein from lipid droplets to the cytoplasmic motile puncta along microtubules (Counihan et al., 2011). HCV p7 is a short hydrophobic protein composed of 63 amino acids and is encoded between the structural and non-structural proteins (Carrere-Kremer et al., 2002). The cytoplasmic loop of p7 is located between the N-terminal and C-terminal transmembrane regions (Carrere-Kremer et al., 2002). HCV p7 is known as a viroprotein that forms homooligomerize to be a ion channel, which is then involved in assembly and release of virus particle in infected cells by modulating pH equilibration in intracellular vesicles (Carrere-Kremer et al., 2002; Jones et al., 2007; Steinmann et al., 2007; Wozniak et al., 2010). Mutations of conserved amino acids required for ion channel activity impaired the production of infectious virus (Jones et al., 2007). However, recruitment of HCV core protein from lipid droplets to the ER assembly site was independent of the ion channel activity of HCV p7 (Boson et al., 2011). HCV p7 enhanced ER localization of the core protein without other viral proteins regardless of viral genotype, although compatibilities between two transmembrane regions of p7 and the first transmembrane domain of NS2 are responsible for ER localization of core protein and infection (Boson et al., 2011). The second transmembrane region of p7, rather than the first, is critical for compatibilities with NS2 regarding recruitment of core protein to the ER assembly site, although both transmembrane regions of p7 are important to sustain infectivity (Boson et al., 2011). These reports speculate that localization of the core protein on lipid droplets may contribute to suppression of virus production and maintenance of persistent HCV infection, while localization of the core protein on ER may positively support virus production under the fulminant condition.
Regulation of Host Lipid Metabolism by the Core Protein
The mechanisms by which the core protein can induce liver diseases and extrahepatic manifestations are unknown. Liver steatosis, which is one of the characteristics associated with persistent HCV infection, develops by accumulation of triglyceride-rich lipids in hepatocytes. However, the precise functions of HCV proteins in the development of fatty liver remain unknown due to the lack of an adequate system to investigate the pathogenesis of HCV. HCV core protein expression has been shown to induce lipid droplets in cell lines and hepatic steatosis and hepatocellular carcinoma in transgenic mice (Barba et al., 1997; Moriya et al., 1997; Hope and McLauchlan, 2000). The lipid composition of the core-transgenic mouse is similar to that of a hepatitis C patient (Koike et al., 2010; Miyoshi et al., 2011). These reports suggest that HCV core protein plays an important role in the development of various types of liver failure, including steatosis and hepatocellular carcinoma. Biosynthesis of triglycerides is mainly regulated by the sterol regulatory element-binding protein (SREBP)-1c. It has been reported that many genes regulated by SREBPs were induced during the early stage of HCV infection in the livers of chimpanzees (Bigger et al., 2004). Our study has demonstrated that the core protein enhances the binding activity of the LXRα–RXRα complex to the srebp-1c promoter in a PA28γ-dependent manner, resulting in upregulation of SREBP-1c and its regulating genes (Moriishi et al., 2007). The activation may be mediated by the direct interaction between the core protein and RXRα (Tsutsumi et al., 2002). Another mechanism is thought to be suppression of lipid secretion. Reduced serum levels of cholesterol and apolipoprotein B have been reported in patients with severe hepatitis C and core-transgenic mice (Perlemuter et al., 2002). The MTP regulates the assembly and secretion of VLDLs consisting of apolipoprotein E, cholesterol, and triglycerides. In core-transgenic mice, MTP-specific activity is significantly decreased (Perlemuter et al., 2002). In addition, DGAT1, which plays an important role in trafficking core protein from lipid droplets to the ER membrane (Herker et al., 2010), was reported to delay the turnover of lipid droplets that are coated by the core protein (Harris et al., 2011; Figure 3). Furthermore, increases in saturated and monounsaturated fatty acids enhance HCV RNA replication (Kapadia and Chisari, 2005). The core protein can enhance the production of reactive oxygen species (ROS) by induction of induced nitric oxide synthetase (iNOS) or by damage to the mitochondrial electron transport system, contributing to the emergence of hepatocellular carcinoma (Moriya et al., 2001; Okuda et al., 2002; Nunez et al., 2004), suggesting that accumulation of lipids hastens the occurrence of hepatocellular carcinoma by enhancing ROS production. The core protein is reported to be degraded by PA28gamma-dependent, but ubiquitin-independent, proteasome activity, and directly binds to PA28gamma (Moriishi et al., 2003; Suzuki et al., 2009). PA28gamma knockdown diminished liver steatosis, hepatocellular carcinoma, and insulin resistance induced by HCV core protein in the mouse liver (Moriishi et al., 2007). After our reports, several groups found that PA28gamma plays an important role in cell cycling by degradation of SRC-3, p16, p19, and p53 (Li et al., 2006; Chen et al., 2007; Zhang and Zhang, 2008). Furthermore, HCV propagation in a cell culture system is potently suppressed by PA28gamma knockdown, regardless of cell growth (Moriishi et al., 2010). One possibility is that E6AP-dependent ubiquitination of the core protein in cytoplasm is competitively suppressed by peptide fragments deduced from nuclear core protein. However, there is still the possibility of an indirect effect of PA28gamma, since potent reduction of PA28gamma, but not intermediate reduction, can induce nuclear accumulation of HCV core protein in cultured cells and the mouse liver, but both potent and intermediate reductions could suppress viral production (Moriishi et al., 2007, 2010; Cerutti et al., 2011). Further study will be required to clarify the mechanism by which PA28gamma regulates core-induced liver diseases and the HCV life cycle.
NS3/4A and Lipid Droplets
The NS3 also cleaves the host adaptor proteins IPS-1/MAVS and TRIF to modulate TLR and RIG-I signaling, resulting in inhibition of type I interferon production (Ferreon et al., 2005; Li et al., 2005a,b; Cheng et al., 2006; Loo et al., 2006). It is speculated that NS3 suppresses the activation of host innate immunity induced by HCV RNA and then contributes to persistent infection with HCV. NS3/4A may be responsible for not only the replication, but also the virus assembly and production by interaction with viral and host proteins on a region close to lipid droplets/ER assembly site. NS3/4A interacts with NS2 cooperating with p7 and E2 to recruit the core protein from lipid droplets to the cytoplasmic motile puncta along microtubules (Boson et al., 2011; Counihan et al., 2011; Popescu et al., 2011). HCV NS3/4A also interacts with host protein Y-box-binding protein-1 (YB-1) and influences the equilibrium between viral replication and infectious particle production (Chatel-Chaix et al., 2011). Knockdown of YB-1 impaired HCV RNA replication, regardless of the viral genotype, but did not affect NS3/4A autoprocessing and MAVS cleavage (Chatel-Chaix et al., 2011). JFH1 infection allowed YB-1 to translocate to lipid droplets containing core protein and NS3 (Chatel-Chaix et al., 2011), although knockdown of YB-1 enhanced the production of viral infectious particles (Chatel-Chaix et al., 2011). YB-1 may cooperate with NS3/4A to negatively regulate the steps after replication and to positively regulate viral replication.
NS5A and Cyclophilins
The peptide bond cis/trans isomerase converts between cis and trans peptide bonds leading to correct folding of the protein substrate. Peptidyl prolyl cis/trans isomerase (PPIase) includes the families of cyclophilin (Fischer et al., 1989), FK506-binding proteins (FKBP; Siekierka et al., 1989a,b) and parvulins (Rahfeld et al., 1994), and the secondary amide peptide bond cis/trans isomerase (Schiene-Fischer et al., 2002). Cyclophilin and FKBP are categorized as immunophilins, which are targeted by the immunosuppressants cyclosporin and FK506, respectively (Liu et al., 1991). Some cyclophilins and FKBP8 were shown to interact with NS5B and/or NS5A and to regulate HCV replication (Watashi et al., 2005; Okamoto et al., 2006), suggesting that immunophilins could lead to promising therapies for chronic hepatitis C, as discussed below.
Cyclosporin A and its derivatives, which target cyclophilins, were shown to impair HCV RNA replication and to exhibit efficacy in hepatitis C patients (Watashi et al., 2003; Ishii et al., 2006). Inoue et al. (2003) reported cyclosporin A treatment of HCV in a clinical trial. Cyclosporin derivatives lacking the ability to interact with cyclophilin lost their inhibitory effect on HCV replication (Watashi et al., 2005). Cyclophilin B is reported to be a 20-kDa secreted neurotropic factor for spinal cord cells in chick embryos (Spik et al., 1991), and is secreted into human milk and blood (Spik et al., 1991; Allain et al., 1994). Cyclophilin B specifically interacts with NS5B, the HCV RNA-dependent RNA polymerase around the ER of the HCV replicon cells, and promotes NS5B’s association with viral RNA (Watashi et al., 2005). Cyclosporin A (CsA) was shown to disrupt the interaction between NS5B and cyclophilin B (Watashi et al., 2005). Treatment with cyclosporin A and knockdown of cyclophilin B suppressed the replication of HCV. However, several groups reported that interaction between NS5A and cyclophilin A is more important for HCV replication than interaction between NS5B and cyclophilin B. There is a growing consensus that cyclophilin A in particular is a crucial factor during HCV replication. A number of point mutations in both NS5A and NS5B have been reported to be associated with in vitro resistance to cyclophilin A (Yang et al., 2008; Chatterji et al., 2009; Kaul et al., 2009). Direct interaction between cyclophilin A and NS5B or NS5A has been observed (Yang et al., 2008). Several CsA-analogs, i.e., NIM811 (Ma et al., 2006), DEB025, and SCY-635 (Hopkins et al., 2010), are currently in preclinical and clinical development. DEB025 disrupts the interaction between NS5A and cyclophilin A and suppresses cyclophilin A isomerase activity. Although experimental differences in cell lines and replicons may affect employment of cyclophilins in HCV replication, the main molecule targeted by the cyclosporin analogs used clinically so far seems to be cyclophilin A.
The treatment with CsA has been associated with increased susceptibility to atherosclerosis and the development of hyperlipidemia (reviewed by Kockx et al., 2010). Treatment with CsA upregulated activity of cholesteryl ester transfer protein and suppressed lipoprotein lipase activity (Tory et al., 2008). Upregulation of cholesteryl ester by cholesteryl ester transfer protein could lead to accumulation of lipoprotein with cholesteryl ester. The report by Anderson et al. (2011) suggests that cyclophilin A and cyclophilin 40 are important for not only viral replication, but also the release of infectious viral particles. NIM811 treatment suppresses virus production and viral RNA replication (Goto et al., 2006). NIM811 treatment led to enlargement of lipid droplets and apoB crescent formation in replicon cells, but not naïve Huh7 cell line, while decreasing apoB secretion and the number of lipid droplets, rendering NS5A dislocation with apoB (Anderson et al., 2011). Knockdown of cyclophilins A and 40 in replicon cells showed the similar changes in lipid droplets size and apoB localization, comparing with NIM811 treatment (Anderson et al., 2011). Cyclophilins A and 40 may regulate lipid trafficking in the presence of HCV proteins to support secretion of viral particles.
NS5A/B and Membrane-Associated Proteins
Host lipids are well known to be essential components in the viral life cycle, including the assembly, budding, and replication of various viruses (Chen et al., 2005; Giese et al., 2006; Mannova et al., 2006; Oomens et al., 2006). In the case of HCV, several types of lipids are required for the HCV life cycle. Saturated and monounsaturated fatty acids, but not polyunsaturated fatty acids, enhance HCV RNA replication (Kapadia and Chisari, 2005), suggesting that lipid biogenesis is involved in HCV replication. HCV particles bind to lipoprotein receptors for entry (Agnello et al., 1999; Scarselli et al., 2002) and are believed to exist with lipoproteins in the serum of infected patients (Thomssen et al., 1992). There is also evidence that HCV uses the VLDL assembly and secretion pathway for maturation and secretion of viral particles (Huang et al., 2007; Gastaminza et al., 2008). Cholesterol and sphingolipids are employed for virion maturation and infectivity, since depletion of cholesterol or down-regulation of sphingomyelin reduces infectivity (Aizaki et al., 2008). Accumulation of lipid components in the liver leads to liver steatosis, and is associated with progression to liver fibrosis and hepatocellular carcinoma, as described above.
Hepatitis C virus replication is suppressed by an inhibitor of geranylgeranyl transferase I, but not by that of farnesyl transferase (Ye et al., 2003). Geranylgeranylate is known as an intermediate found in the mevalonate pathway and is covalently bound to various cellular proteins that are associated with plasma or the intracellular membrane (Horton et al., 2002). Immunoprecipitation analysis revealed that NS5A interacts with FBL2 (Wang et al., 2005a). The F-box motif is located in the N-terminus of FBL2, followed by 11 leucine-rich repeats (Ilyin et al., 1999) and the CAAX motif, which is thought to be modified by geranylgeranylation (Wang et al., 2005a). The F-box motif is generally essential for the formation of the ubiquitin ligase complex (Ilyin et al., 1999), suggesting that FBL2 regulates the ubiquitination of host or viral proteins through the interaction with NS5A. Another possibility is that FBL2 retains the viral replication complex by interacting with NS5A (Figure 3, step 3).
Screening of a genome-wide siRNA library revealed phosphatidylinositol 4-kinase III alpha (PI4KA) and COPI vesicle coat complex as a human gene associated with HCV replication (Bigger et al., 2004; Borawski et al., 2009; Li et al., 2009; Tai et al., 2009; Trotard et al., 2009; Vaillancourt et al., 2009; Reiss et al., 2011). Phosphatidylinositol 4-phosphate, which is associated with oxysterol binding protein (OSBP) and CERT (Peretti et al., 2008; Banerji et al., 2010) as described below, is increased by HCV infection (Bigger et al., 2004; Hsu et al., 2010; Reiss et al., 2011; Tai and Salloum, 2011). PI4KA is co-localized with NS5A and double stranded RNA in the replication platform composed of detergent-resistant lipid components, known as a membranous web, and is critical for HCV replication at posttranslational stages in the membranous web (Berger et al., 2009). NS5A can interact with PI4KA (Berger et al., 2011; Lim and Hwang, 2011; Reiss et al., 2011) and recruit PI4KA to the membranous web (Berger et al., 2009; Tai et al., 2009; Reiss et al., 2011; Tai and Salloum, 2011). Furthermore, PI4KA, but not phosphatidylinositol 4-kinase III beta, induces the membranous web structure under the non-replicative condition (Berger et al., 2011; Lim and Hwang, 2011; Reiss et al., 2011). Biosynthesis of phosphatidylinositol 4-phosphate by PI4KA that is recruited by NS5A in the membranous web may be required for HCV replication and can be an endogenous biomarker of the membranous web (Figure 3, step 3).
Vesicle-associated membrane protein-associated proteins were originally identified as proteins that bind to vesicle-associated membrane protein (VAMP) in the nematode Aplysia and were designated as VAMP-associated protein 33 kDa (later renamed VAP-A; Skehel et al., 1995). Furthermore, one homolog and its splicing variant were reported as VAP-B and -C, respectively (Nishimura et al., 1999). VAP is classified as a type II membrane protein, and is composed of three functional domains: major sperm protein (MSP), which occupies the N-terminal half region, the coiled-coil domain, and the transmembrane domain. VAP-A shares 60% identity with VAP-B, while VAP-C is the splicing variant of VAP-B that lacks the coiled-coil and transmembrane domains (Nishimura et al., 1999). GST pull-down and immunoprecipitation analyses revealed that NS5A and NS5B interact with human VAP-A/B and that the N-terminal MSP domain and the coiled-coil domain of VAP-A/B are responsible for the binding to NS5B and NS5A, respectively (Tu et al., 1999; Hamamoto et al., 2005). In addition, systematic RNAi screening revealed that 62 target host genes are involved in HCV RNA or proteins including VAP-A/B (Randall et al., 2007). Several reports suggest that HCV replication takes place on the membranous web (Shi et al., 2003; Gao et al., 2004; Sakamoto et al., 2005). NS4B is predominantly associated with a lipid-raft-like detergent-resistant fraction equivalent to the membranous web, and both NS5A and NS5B were co-localized in a similar fraction in the presence of NS4B (Sakamoto et al., 2005). VAP-A was also localized in a detergent-resistant fraction, suggesting that it plays an important role in HCV replication because the dominant negative mutant of VAP-A suppressed the replication of HCV RNA (Gao et al., 2004). VAP-B forms a homodimer and heterodimer with VAP-A, and knockdown of VAP-A or VAP-B led to substantial suppression of HCV replication (Hamamoto et al., 2005). These findings suggest that VAP-A and -B positively regulate HCV replication by binding to NS5A/B.
The physiological function of VAPs was reported to be trafficking of ceramide and cholesterol between ER and the Golgi apparatus. Several VAP-interacting proteins share the FFAT motif (two phenylalanines in an acidic tract), which has the consensus amino acid sequence EFFDAxE, as determined by a comparison among oxysterol binding proteins, OSBP-related proteins (ORPs; Loewen et al., 2003), and the ceramide transport protein CERT (Hanada et al., 2003; Kawano et al., 2006), contributing to the regulation of lipid metabolism. OSBP binds and transports cholesterol or hydroxycholesterol from ER to the Golgi (Ridgway et al., 1992; Wang et al., 2005b), while CERT binds and transports ceramide from ER to the Golgi, where the ceramide is converted to sphingolipids (Kumagai et al., 2005). Altering the sphingomyelin/ceramide ratio of the plasma membrane can effect HCV entry via the cell surface expression of CD81 (Voisset et al., 2008). OSBP mediates HCV secretion while binding to NS5A and VAP-A (Amako et al., 2009). Inhibition of CERT function effectively suppressed HCV release regardless of RNA replication (Aizaki et al., 2008). Phosphorylation of CERT and OSBP by protein kinase D negatively regulates VAPs binding to CERT and OSBP resulting in an effect on HCV infection (Amako et al., 2011). HCV NS5A may allow VAP-A/B to provide ceramide and cholesterol to replication complexes for upregulation of virus propagation (Figure 3, step 3).
The VAP-B-splicing variant VAP-C interacts with NS5B via the short form of the MSP domain and then suppresses the HCV replication by disrupting binding of other VAPs to NS5B (Kukihara et al., 2009). Expression of VAP-C is observed in various tissues except for the liver, suggesting that tissue distribution of VAP-C determines the tropism of HCV infection (Kukihara et al., 2009). These findings suggest that VAP-C negatively regulates HCV replication by inhibiting the interaction between VAP-A/B and NS5B. Furthermore, expression of VAP-C was negligible in B cells prepared from chronic hepatitis C patients, in whom B cells included HCV particles (Ito et al., 2010), and expression of the full HCV genome in B cells induced B-cell lymphoma in a conditional transgenic mouse (Kasama et al., 2010), suggesting that HCV infection increases the chance of developing B-cell lymphomas via dysregulation of lipid metabolism.
Conclusion
This review summarizes several recently reported viral and host factors that exploit lipid components to support HCV infection. The mechanism by which HCV proteins cooperate with host factors to exploit lipid components and to regulate lipid metabolism in the infection has not been elucidated completely. The aim of identifying host factors is effective and stable therapy; targeting the host factors might be done to prevent the emergence of resistant viruses. Cyclosporin analogs will be used clinically in the near future. Wide screening and proteomics analyses have revealed novel host factors that are required for HCV replications over the past decade. The mechanism by which HCV infection induces formation of membranous web in infected cells has been unknown yet, although NS4B is involved in formation of membranous web (Egger et al., 2002; Gosert et al., 2005; Ferraris et al., 2010). We also found several host proteins to be NS4B-associating host factors by proteomics analysis based on the TargetMine program (Tripathi et al., 2010). Further study will be required to identify the prominent factors essential for lipid metabolism that are associated with each step in the HCV life cycle and to develop effective and stable therapies for hepatitis C.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank R. Tanaka for her secretarial work. Our original work cited here were supported in part by grants-in-aid from the Ministry of Health, Labor, and Welfare (Research on Hepatitis); the Ministry of Education, Culture, Sports, Science, and Technology.
References
Adinolfi, L. E., Gambardella, M., Andreana, A., Tripodi, M. F., Utili, R., and Ruggiero, G. (2001). Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology 33, 1358–1364.
Agnello, V., Abel, G., Elfahal, M., Knight, G. B., and Zhang, Q. X. (1999). Hepatitis C virus and other Flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. U.S.A. 96, 12766–12771.
Aizaki, H., Lee, K. J., Sung, V. M., Ishiko, H., and Lai, M. M. (2004). Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology 324, 450–461.
Aizaki, H., Morikawa, K., Fukasawa, M., Hara, H., Inoue, Y., Tani, H., Saito, K., Nishijima, M., Hanada, K., Matsuura, Y., Lai, M. M., Miyamura, T., Wakita, T., and Suzuki, T. (2008). Critical role of virion-associated cholesterol and sphingolipid in hepatitis C virus infection. J. Virol. 82, 5715–5724.
Albecka, A., Belouzard, S., De Beeck, A. O., Descamps, V., Goueslain, L., Bertrand-Michel, J., Terce, F., Duverlie, G., Rouille, Y., and Dubuisson, J. (2012). Role of LDL receptor in the hepatitis c virus life cycle. Hepatology. doi: 10.1002/hep.25501
Allain, F., Denys, A., and Spik, G. (1994). Characterization of surface binding sites for cyclophilin B on a human tumor T-cell line. J. Biol. Chem. 269, 16537–16540.
Amako, Y., Sarkeshik, A., Hotta, H., Yates, J. III, and Siddiqui, A. (2009). Role of oxysterol binding protein in hepatitis C virus infection. J. Virol. 83, 9237–9246.
Amako, Y., Syed, G. H., and Siddiqui, A. (2011). Protein kinase D negatively regulates hepatitis C virus secretion through phosphorylation of oxysterol-binding protein and ceramide transfer protein. J. Biol. Chem. 286, 11265–11274.
Anderson, L. J., Lin, K., Compton, T., and Wiedmann, B. (2011). Inhibition of cyclophilins alters lipid trafficking and blocks hepatitis C virus secretion. Virol. J. 8, 329.
Andre, P., Komurian-Pradel, F., Deforges, S., Perret, M., Berland, J. L., Sodoyer, M., Pol, S., Brechot, C., Paranhos-Baccala, G., and Lotteau, V. (2002). Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 76, 6919–6928.
Banerji, S., Ngo, M., Lane, C. F., Robinson, C. A., Minogue, S., and Ridgway, N. D. (2010). Oxysterol binding protein-dependent activation of sphingomyelin synthesis in the golgi apparatus requires phosphatidylinositol 4-kinase IIalpha. Mol. Biol. Cell 21, 4141–4150.
Barba, G., Harper, F., Harada, T., Kohara, M., Goulinet, S., Matsuura, Y., Eder, G., Schaff, Z., Chapman, M. J., Miyamura, T., and Brechot, C. (1997). Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc. Natl. Acad. Sci. U.S.A. 94, 1200–1205.
Barth, H., Schafer, C., Adah, M. I., Zhang, F., Linhardt, R. J., Toyoda, H., Kinoshita-Toyoda, A., Toida, T., Van Kuppevelt, T. H., Depla, E., Von Weizsacker, F., Blum, H. E., and Baumert, T. F. (2003). Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 278, 41003–41012.
Bartosch, B., Vitelli, A., Granier, C., Goujon, C., Dubuisson, J., Pascale, S., Scarselli, E., Cortese, R., Nicosia, A., and Cosset, F. L. (2003). Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 278, 41624–41630.
Benga, W. J., Krieger, S. E., Dimitrova, M., Zeisel, M. B., Parnot, M., Lupberger, J., Hildt, E., Luo, G., Mclauchlan, J., Baumert, T. F., and Schuster, C. (2010). Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology 51, 43–53.
Berger, K. L., Cooper, J. D., Heaton, N. S., Yoon, R., Oakland, T. E., Jordan, T. X., Mateu, G., Grakoui, A., and Randall, G. (2009). Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. U.S.A. 106, 7577–7582.
Berger, K. L., Kelly, S. M., Jordan, T. X., Tartell, M. A., and Randall, G. (2011). Hepatitis C virus stimulates the phosphatidylinositol 4-kinase III alpha-dependent phosphatidylinositol 4-phosphate production that is essential for its replication. J. Virol. 85, 8870–8883.
Bigger, C. B., Guerra, B., Brasky, K. M., Hubbard, G., Beard, M. R., Luxon, B. A., Lemon, S. M., and Lanford, R. E. (2004). Intrahepatic gene expression during chronic hepatitis C virus infection in chimpanzees. J. Virol. 78, 13779–13792.
Blanchard, E., Belouzard, S., Goueslain, L., Wakita, T., Dubuisson, J., Wychowski, C., and Rouille, Y. (2006). Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 80, 6964–6972.
Blight, K. J., Kolykhalov, A. A., and Rice, C. M. (2000). Efficient initiation of HCV RNA replication in cell culture. Science 290, 1972–1974.
Boguszewska-Chachulska, A. M., Krawczyk, M., Stankiewicz, A., Gozdek, A., Haenni, A. L., and Strokovskaya, L. (2004). Direct fluorometric measurement of hepatitis C virus helicase activity. FEBS Lett. 567, 253–258.
Borawski, J., Troke, P., Puyang, X., Gibaja, V., Zhao, S., Mickanin, C., Leighton-Davies, J., Wilson, C. J., Myer, V., Cornellataracido, I., Baryza, J., Tallarico, J., Joberty, G., Bantscheff, M., Schirle, M., Bouwmeester, T., Mathy, J. E., Lin, K., Compton, T., Labow, M., Wiedmann, B., and Gaither, L. A. (2009). Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J. Virol. 83, 10058–10074.
Borowski, P., Deinert, J., Schalinski, S., Bretner, M., Ginalski, K., Kulikowski, T., and Shugar, D. (2003). Halogenated benzimidazoles and benzotriazoles as inhibitors of the NTPase/helicase activities of hepatitis C and related viruses. Eur. J. Biochem. 270, 1645–1653.
Boson, B., Granio, O., Bartenschlager, R., and Cosset, F. L. (2011). A concerted action of hepatitis C virus p7 and nonstructural protein 2 regulates core localization at the endoplasmic reticulum and virus assembly. PLoS Pathog. 7, e1002144. doi:10.1371/journal.ppat.1002144
Bradley, D., Mccaustland, K., Krawczynski, K., Spelbring, J., Humphrey, C., and Cook, E. H. (1991). Hepatitis C virus: buoyant density of the factor VIII-derived isolate in sucrose. J. Med. Virol. 34, 206–208.
Brodsky, J. L., Gusarova, V., and Fisher, E. A. (2004). Vesicular trafficking of hepatic apolipoprotein B100 and its maturation to very low-density lipoprotein particles; studies from cells and cell-free systems. Trends Cardiovasc. Med. 14, 127–132.
Carabaich, A., Ruvoletto, M., Bernardinello, E., Tono, N., Cavalletto, L., Chemello, L., Gatta, A., and Pontisso, P. (2005). Profiles of HCV core protein and viremia in chronic hepatitis C: possible protective role of core antigen in liver damage. J. Med. Virol. 76, 55–60.
Carrere-Kremer, S., Montpellier-Pala, C., Cocquerel, L., Wychowski, C., Penin, F., and Dubuisson, J. (2002). Subcellular localization and topology of the p7 polypeptide of hepatitis C virus. J. Virol. 76, 3720–3730.
Cerutti, A., Maillard, P., Minisini, R., Vidalain, P. O., Roohvand, F., Pecheur, E. I., Pirisi, M., and Budkowska, A. (2011). Identification of a functional, CRM-1-dependent nuclear export signal in hepatitis C virus core protein. PLoS ONE 6, e25854. doi:10.1371/journal.pone.0025854
Chang, K. S., Jiang, J., Cai, Z., and Luo, G. (2007). Human apolipoprotein E is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 81, 13783–13793.
Chatel-Chaix, L., Melancon, P., Racine, M. E., Baril, M., and Lamarre, D. (2011). Y-box-binding protein 1 interacts with hepatitis C virus NS3/4A and influences the equilibrium between viral RNA replication and infectious particle production. J. Virol. 85, 11022–11037.
Chatterji, U., Bobardt, M., Selvarajah, S., Yang, F., Tang, H., Sakamoto, N., Vuagniaux, G., Parkinson, T., and Gallay, P. (2009). The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J. Biol. Chem. 284, 16998–17005.
Chen, B. J., Takeda, M., and Lamb, R. A. (2005). Influenza virus hemagglutinin (H3 subtype) requires palmitoylation of its cytoplasmic tail for assembly: M1 proteins of two subtypes differ in their ability to support assembly. J. Virol. 79, 13673–13684.
Chen, X., Barton, L. F., Chi, Y., Clurman, B. E., and Roberts, J. M. (2007). Ubiquitin-independent degradation of cell-cycle inhibitors by the REGgamma proteasome. Mol. Cell 26, 843–852.
Cheng, G., Zhong, J., and Chisari, F. V. (2006). Inhibition of dsRNA-induced signaling in hepatitis C virus-infected cells by NS3 protease-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. U.S.A. 103, 8499–8504.
Codran, A., Royer, C., Jaeck, D., Bastien-Valle, M., Baumert, T. F., Kieny, M. P., Pereira, C. A., and Martin, J. P. (2006). Entry of hepatitis C virus pseudotypes into primary human hepatocytes by clathrin-dependent endocytosis. J. Gen. Virol. 87, 2583–2593.
Counihan, N. A., Rawlinson, S. M., and Lindenbach, B. D. (2011). Trafficking of hepatitis C virus core protein during virus particle assembly. PLoS Pathog. 7, e1002302. doi:10.1371/journal.pone.0025854
Cun, W., Jiang, J., and Luo, G. (2010). The C-terminal alpha-helix domain of apolipoprotein E is required for interaction with nonstructural protein 5A and assembly of hepatitis C virus. J. Virol. 84, 11532–11541.
De Francesco, R., Tomei, L., Altamura, S., Summa, V., and Migliaccio, G. (2003). Approaching a new era for hepatitis C virus therapy: inhibitors of the NS3-4A serine protease and the NS5B RNA-dependent RNA polymerase. Antiviral Res. 58, 1–16.
Dentzer, T. G., Lorenz, I. C., Evans, M. J., and Rice, C. M. (2009). Determinants of the hepatitis C virus nonstructural protein 2 protease domain required for production of infectious virus. J. Virol. 83, 12702–12713.
Dhanak, D., Duffy, K. J., Johnston, V. K., Lin-Goerke, J., Darcy, M., Shaw, A. N., Gu, B., Silverman, C., Gates, A. T., Nonnemacher, M. R., Earnshaw, D. L., Casper, D. J., Kaura, A., Baker, A., Greenwood, C., Gutshall, L. L., Maley, D., Delvecchio, A., Macarron, R., Hofmann, G. A., Alnoah, Z., Cheng, H. Y., Chan, G., Khandekar, S., Keenan, R. M., and Sarisky, R. T. (2002). Identification and biological characterization of heterocyclic inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Biol. Chem. 277, 38322–38327.
Egger, D., Wolk, B., Gosert, R., Bianchi, L., Blum, H. E., Moradpour, D., and Bienz, K. (2002). Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 76, 5974–5984.
Evans, M. J., Von Hahn, T., Tscherne, D. M., Syder, A. J., Panis, M., Wolk, B., Hatziioannou, T., Mckeating, J. A., Bieniasz, P. D., and Rice, C. M. (2007). Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446, 801–805.
Failla, C., Tomei, L., and De Francesco, R. (1994). Both NS3 and NS4A are required for proteolytic processing of hepatitis C virus nonstructural proteins. J. Virol. 68, 3753–3760.
Failla, C., Tomei, L., and De Francesco, R. (1995). An amino-terminal domain of the hepatitis C virus NS3 protease is essential for interaction with NS4A. J. Virol. 69, 1769–1777.
Ferraris, P., Blanchard, E., and Roingeard, P. (2010). Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J. Gen. Virol. 91, 2230–2237.
Ferreon, J. C., Ferreon, A. C., Li, K., and Lemon, S. M. (2005). Molecular determinants of TRIF proteolysis mediated by the hepatitis C virus NS3/4A protease. J. Biol. Chem. 280, 20483–20492.
Fischer, G., Wittmann-Liebold, B., Lang, K., Kiefhaber, T., and Schmid, F. X. (1989). Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 337, 476–478.
Foy, E., Li, K., Sumpter, R. Jr., Loo, Y. M., Johnson, C. L., Wang, C., Fish, P. M., Yoneyama, M., Fujita, T., Lemon, S. M., and Gale, M. Jr. (2005). Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl. Acad. Sci. U.S.A. 102, 2986–2991.
Gao, L., Aizaki, H., He, J. W., and Lai, M. M. (2004). Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J. Virol. 78, 3480–3488.
Gastaminza, P., Cheng, G., Wieland, S., Zhong, J., Liao, W., and Chisari, F. V. (2008). Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 82, 2120–2129.
Giese, S. I., Woerz, I., Homann, S., Tibroni, N., Geyer, M., and Fackler, O. T. (2006). Specific and distinct determinants mediate membrane binding and lipid raft incorporation of HIV-1(SF2) Nef. Virology 355, 175–191.
Gosert, R., Jendrsczok, W., Berke, J. M., Brass, V., Blum, H. E., and Moradpour, D. (2005). Characterization of nonstructural protein membrane anchor deletion mutants expressed in the context of the hepatitis C virus polyprotein. J. Virol. 79, 7911–7917.
Goto, K., Watashi, K., Murata, T., Hishiki, T., Hijikata, M., and Shimotohno, K. (2006). Evaluation of the anti-hepatitis C virus effects of cyclophilin inhibitors, cyclosporin A, and NIM811. Biochem. Biophys. Res. Commun. 343, 879–884.
Grakoui, A., Mccourt, D. W., Wychowski, C., Feinstone, S. M., and Rice, C. M. (1993a). Characterization of the hepatitis C virus-encoded serine proteinase: determination of proteinase-dependent polyprotein cleavage sites. J. Virol. 67, 2832–2843.
Grakoui, A., Mccourt, D. W., Wychowski, C., Feinstone, S. M., and Rice, C. M. (1993b). A second hepatitis C virus-encoded proteinase. Proc. Natl. Acad. Sci. U.S.A. 90, 10583–10587.
Grakoui, A., Wychowski, C., Lin, C., Feinstone, S. M., and Rice, C. M. (1993c). Expression and identification of hepatitis C virus polyprotein cleavage products. J. Virol. 67, 1385–1395.
Hamamoto, I., Nishimura, Y., Okamoto, T., Aizaki, H., Liu, M., Mori, Y., Abe, T., Suzuki, T., Lai, M. M., Miyamura, T., Moriishi, K., and Matsuura, Y. (2005). Human VAP-B is involved in hepatitis C virus replication through interaction with NS5A and NS5B. J. Virol. 79, 13473–13482.
Hanada, K., Kumagai, K., Yasuda, S., Miura, Y., Kawano, M., Fukasawa, M., and Nishijima, M. (2003). Molecular machinery for non-vesicular trafficking of ceramide. Nature 426, 803–809.
Harada, S., Watanabe, Y., Takeuchi, K., Suzuki, T., Katayama, T., Takebe, Y., Saito, I., and Miyamura, T. (1991). Expression of processed core protein of hepatitis C virus in mammalian cells. J. Virol. 65, 3015–3021.
Harris, C., Herker, E., Farese, R. V. Jr., and Ott, M. (2011). Hepatitis C Virus Core Protein Decreases Lipid Droplet Turnover: a mechanism for core-induced steatosis. J. Biol. Chem. 286, 42615–42625.
Herker, E., Harris, C., Hernandez, C., Carpentier, A., Kaehlcke, K., Rosenberg, A. R., Farese, R. V. Jr., and Ott, M. (2010). Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 16, 1295–1298.
Herz, J., Chen, Y., Masiulis, I., and Zhou, L. (2009). Expanding functions of lipoprotein receptors. J. Lipid Res. 50(Suppl.), S287–S292.
Hicham Alaoui-Ismaili, M., Gervais, C., Brunette, S., Gouin, G., Hamel, M., Rando, R. F., and Bedard, J. (2000). A novel high throughput screening assay for HCV NS3 helicase activity. Antiviral Res. 46, 181–193.
Hijikata, M., Kato, N., Ootsuyama, Y., Nakagawa, M., and Shimotohno, K. (1991). Gene mapping of the putative structural region of the hepatitis C virus genome by in vitro processing analysis. Proc. Natl. Acad. Sci. U.S.A. 88, 5547–5551.
Hijikata, M., Shimizu, Y. K., Kato, H., Iwamoto, A., Shih, J. W., Alter, H. J., Purcell, R. H., and Yoshikura, H. (1993). Equilibrium centrifugation studies of hepatitis C virus: evidence for circulating immune complexes. J. Virol. 67, 1953–1958.
Hishiki, T., Shimizu, Y., Tobita, R., Sugiyama, K., Ogawa, K., Funami, K., Ohsaki, Y., Fujimoto, T., Takaku, H., Wakita, T., Baumert, T. F., Miyanari, Y., and Shimotohno, K. (2010). Infectivity of hepatitis C virus is influenced by association with apolipoprotein E isoforms. J. Virol. 84, 12048–12057.
Hofmann, W. P., and Zeuzem, S. (2011). A new standard of care for the treatment of chronic HCV infection. Nat. Rev. Gastroenterol. Hepatol. 8, 257–264.
Hope, R. G., and McLauchlan, J. (2000). Sequence motifs required for lipid droplet association and protein stability are unique to the hepatitis C virus core protein. J. Gen. Virol. 81, 1913–1925.
Hope, R. G., Murphy, D. J., and Mclauchlan, J. (2002). The domains required to direct core proteins of hepatitis C virus and GB virus-B to lipid droplets share common features with plant oleosin proteins. J. Biol. Chem. 277, 4261–4270.
Hopkins, S., Scorneaux, B., Huang, Z., Murray, M. G., Wring, S., Smitley, C., Harris, R., Erdmann, F., Fischer, G., and Ribeill, Y. (2010). SCY-635, a novel nonimmunosuppressive analog of cyclosporine that exhibits potent inhibition of hepatitis C virus RNA replication in vitro. Antimicrob. Agents Chemother. 54, 660–672.
Horton, J. D., Goldstein, J. L., and Brown, M. S. (2002). SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131.
Hsu, M., Zhang, J., Flint, M., Logvinoff, C., Cheng-Mayer, C., Rice, C. M., and Mckeating, J. A. (2003). Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U.S.A. 100, 7271–7276.
Hsu, N. Y., Ilnytska, O., Belov, G., Santiana, M., Chen, Y. H., Takvorian, P. M., Pau, C., Van Der Schaar, H., Kaushik-Basu, N., Balla, T., Cameron, C. E., Ehrenfeld, E., Van Kuppeveld, F. J., and Altan-Bonnet, N. (2010). Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141, 799–811.
Huang, H., Sun, F., Owen, D. M., Li, W., Chen, Y., Gale, M. Jr., and Ye, J. (2007). Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. U.S.A. 104, 5848–5853.
Hussy, P., Langen, H., Mous, J., and Jacobsen, H. (1996). Hepatitis C virus core protein: carboxy-terminal boundaries of two processed species suggest cleavage by a signal peptide peptidase. Virology 224, 93–104.
Ikeda, M., Yi, M., Li, K., and Lemon, S. M. (2002). Selectable subgenomic and genome-length dicistronic RNAs derived from an infectious molecular clone of the HCV-N strain of hepatitis C virus replicate efficiently in cultured Huh7 cells. J. Virol. 76, 2997–3006.
Ilyin, G. P., Rialland, M., Glaise, D., and Guguen-Guillouzo, C. (1999). Identification of a novel Skp2-like mammalian protein containing F-box and leucine-rich repeats. FEBS Lett. 459, 75–79.
Inoue, K., Sekiyama, K., Yamada, M., Watanabe, T., Yasuda, H., and Yoshiba, M. (2003). Combined interferon alpha2b and cyclosporin A in the treatment of chronic hepatitis C: controlled trial. J. Gastroenterol. 38, 567–572.
Ishii, N., Watashi, K., Hishiki, T., Goto, K., Inoue, D., Hijikata, M., Wakita, T., Kato, N., and Shimotohno, K. (2006). Diverse effects of cyclosporine on hepatitis C virus strain replication. J. Virol. 80, 4510–4520.
Ito, M., Masumi, A., Mochida, K., Kukihara, H., Moriishi, K., Matsuura, Y., Yamaguchi, K., and Mizuochi, T. (2010). Peripheral B cells may serve as a reservoir for persistent hepatitis C virus infection. J. Innate Immun. 2, 607–617.
Jacobson, I. M., Cacoub, P., Dal Maso, L., Harrison, S. A., and Younossi, Z. M. (2010). Manifestations of chronic hepatitis C virus infection beyond the liver. Clin. Gastroenterol. Hepatol. 8, 1017–1029.
Jiang, J., and Luo, G. (2009). Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J. Virol. 83, 12680–12691.
Jirasko, V., Montserret, R., Appel, N., Janvier, A., Eustachi, L., Brohm, C., Steinmann, E., Pietschmann, T., Penin, F., and Bartenschlager, R. (2008). Structural and functional characterization of nonstructural protein 2 for its role in hepatitis C virus assembly. J. Biol. Chem. 283, 28546–28562.
Jones, C. T., Murray, C. L., Eastman, D. K., Tassello, J., and Rice, C. M. (2007). Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 81, 8374–8383.
Kapadia, S. B., and Chisari, F. V. (2005). Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. U.S.A. 102, 2561–2566.
Kasama, Y., Sekiguchi, S., Saito, M., Tanaka, K., Satoh, M., Kuwahara, K., Sakaguchi, N., Takeya, M., Hiasa, Y., Kohara, M., and Tsukiyama-Kohara, K. (2010). Persistent expression of the full genome of hepatitis C virus in B cells induces spontaneous development of B-cell lymphomas in vivo. Blood 116, 4926–4933.
Kaul, A., Stauffer, S., Berger, C., Pertel, T., Schmitt, J., Kallis, S., Zayas, M., Lohmann, V., Luban, J., and Bartenschlager, R. (2009). Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 5, e1000546. doi:10.1371/journal.ppat.1000546
Kawano, M., Kumagai, K., Nishijima, M., and Hanada, K. (2006). Efficient trafficking of ceramide from the endoplasmic reticulum to the Golgi apparatus requires a VAMP-associated protein-interacting FFAT motif of CERT. J. Biol. Chem. 281, 30279–30288.
Koch, J. O., Lohmann, V., Herian, U., and Bartenschlager, R. (1996). In vitro studies on the activation of the hepatitis C virus NS3 proteinase by the NS4A cofactor. Virology 221, 54–66.
Kockx, M., Jessup, W., and Kritharides, L. (2010). Cyclosporin A and atherosclerosis – cellular pathways in atherogenesis. Pharmacol. Ther. 128, 106–118.
Koike, K., Tsutsumi, T., Yotsuyanagi, H., and Moriya, K. (2010). Lipid metabolism and liver disease in hepatitis C viral infection. Oncology 78(Suppl. 1), 24–30.
Krieger, N., Lohmann, V., and Bartenschlager, R. (2001). Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J. Virol. 75, 4614–4624.
Kukihara, H., Moriishi, K., Taguwa, S., Tani, H., Abe, T., Mori, Y., Suzuki, T., Fukuhara, T., Taketomi, A., Maehara, Y., and Matsuura, Y. (2009). Human VAP-C negatively regulates hepatitis C virus propagation. J. Virol. 83, 7959–7969.
Kumagai, K., Yasuda, S., Okemoto, K., Nishijima, M., Kobayashi, S., and Hanada, K. (2005). CERT mediates intermembrane transfer of various molecular species of ceramides. J. Biol. Chem. 280, 6488–6495.
Li, K., Foy, E., Ferreon, J. C., Nakamura, M., Ferreon, A. C., Ikeda, M., Ray, S. C., Gale, M. Jr., and Lemon, S. M. (2005a). Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. U.S.A. 102, 2992–2997.
Li, X. D., Sun, L., Seth, R. B., Pineda, G., and Chen, Z. J. (2005b). Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. U.S.A. 102, 17717–17722.
Li, Q., Brass, A. L., Ng, A., Hu, Z., Xavier, R. J., Liang, T. J., and Elledge, S. J. (2009). A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl. Acad. Sci. U.S.A. 106, 16410–16415.
Li, X., Lonard, D. M., Jung, S. Y., Malovannaya, A., Feng, Q., Qin, J., Tsai, S. Y., Tsai, M. J., and O’malley, B. W. (2006). The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REGgamma proteasome. Cell 124, 381–392.
Lim, Y. S., and Hwang, S. B. (2011). Hepatitis C virus NS5A protein interacts with phosphatidylinositol 4-kinase type IIIalpha and regulates viral propagation. J. Biol. Chem. 286, 11290–11298.
Lindenbach, B. D., Evans, M. J., Syder, A. J., Wolk, B., Tellinghuisen, T. L., Liu, C. C., Maruyama, T., Hynes, R. O., Burton, D. R., Mckeating, J. A., and Rice, C. M. (2005). Complete replication of hepatitis C virus in cell culture. Science 309, 623–626.
Lindenbach, B. D., Meuleman, P., Ploss, A., Vanwolleghem, T., Syder, A. J., Mckeating, J. A., Lanford, R. E., Feinstone, S. M., Major, M. E., Leroux-Roels, G., and Rice, C. M. (2006). Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc. Natl. Acad. Sci. U.S.A. 103, 3805–3809.
Liu, J., Farmer, J. D. Jr., Lane, W. S., Friedman, J., Weissman, I., and Schreiber, S. L. (1991). Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66, 807–815.
Loewen, C. J., Roy, A., and Levine, T. P. (2003). A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. EMBO J. 22, 2025–2035.
Lohmann, V., Korner, F., Dobierzewska, A., and Bartenschlager, R. (2001). Mutations in hepatitis C virus RNAs conferring cell culture adaptation. J. Virol. 75, 1437–1449.
Lohmann, V., Korner, F., Koch, J., Herian, U., Theilmann, L., and Bartenschlager, R. (1999). Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285, 110–113.
Loo, Y. M., Fornek, J., Crochet, N., Bajwa, G., Perwitasari, O., Martinez-Sobrido, L., Akira, S., Gill, M. A., Garcia-Sastre, A., Katze, M. G., and Gale, M. Jr. (2008). Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 82, 335–345.
Loo, Y. M., Owen, D. M., Li, K., Erickson, A. K., Johnson, C. L., Fish, P. M., Carney, D. S., Wang, T., Ishida, H., Yoneyama, M., Fujita, T., Saito, T., Lee, W. M., Hagedorn, C. H., Lau, D. T., Weinman, S. A., Lemon, S. M., and Gale, M. Jr. (2006). Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc. Natl. Acad. Sci. U.S.A. 103, 6001–6006.
Lorenz, I. C., Marcotrigiano, J., Dentzer, T. G., and Rice, C. M. (2006). Structure of the catalytic domain of the hepatitis C virus NS2-3 protease. Nature 442, 831–835.
Ma, S., Boerner, J. E., Tiongyip, C., Weidmann, B., Ryder, N. S., Cooreman, M. P., and Lin, K. (2006). NIM811, a cyclophilin inhibitor, exhibits potent in vitro activity against hepatitis C virus alone or in combination with alpha interferon. Antimicrob. Agents Chemother. 50, 2976–2982.
Maga, G., Gemma, S., Fattorusso, C., Locatelli, G. A., Butini, S., Persico, M., Kukreja, G., Romano, M. P., Chiasserini, L., Savini, L., Novellino, E., Nacci, V., Spadari, S., and Campiani, G. (2005). Specific targeting of hepatitis C virus NS3 RNA helicase. Discovery of the potent and selective competitive nucleotide-mimicking inhibitor QU663. Biochemistry 44, 9637–9644.
Mannova, P., Fang, R., Wang, H., Deng, B., Mcintosh, M. W., Hanash, S. M., and Beretta, L. (2006). Modification of host lipid raft proteome upon hepatitis C virus replication. Mol. Cell Proteomics 5, 2319–2325.
Masaki, T., Suzuki, R., Murakami, K., Aizaki, H., Ishii, K., Murayama, A., Date, T., Matsuura, Y., Miyamura, T., Wakita, T., and Suzuki, T. (2008). Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 82, 7964–7976.
Matto, M., Rice, C. M., Aroeti, B., and Glenn, J. S. (2004). Hepatitis C virus core protein associates with detergent-resistant membranes distinct from classical plasma membrane rafts. J. Virol. 78, 12047–12053.
McLauchlan, J., Lemberg, M. K., Hope, G., and Martoglio, B. (2002). Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 21, 3980–3988.
Meertens, L., Bertaux, C., and Dragic, T. (2006). Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J. Virol. 80, 11571–11578.
Merz, A., Long, G., Hiet, M. S., Brugger, B., Chlanda, P., Andre, P., Wieland, F., Krijnse-Locker, J., and Bartenschlager, R. (2011). Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 286, 3018–30132.
Miyanari, Y., Atsuzawa, K., Usuda, N., Watashi, K., Hishiki, T., Zayas, M., Bartenschlager, R., Wakita, T., Hijikata, M., and Shimotohno, K. (2007). The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9, 1089–1097.
Miyoshi, H., Moriya, K., Tsutsumi, T., Shinzawa, S., Fujie, H., Shintani, Y., Fujinaga, H., Goto, K., Todoroki, T., Suzuki, T., Miyamura, T., Matsuura, Y., Yotsuyanagi, H., and Koike, K. (2011). Pathogenesis of lipid metabolism disorder in hepatitis C: polyunsaturated fatty acids counteract lipid alterations induced by the core protein. J. Hepatol. 54, 432–438.
Monazahian, M., Bohme, I., Bonk, S., Koch, A., Scholz, C., Grethe, S., and Thomssen, R. (1999). Low density lipoprotein receptor as a candidate receptor for hepatitis C virus. J. Med. Virol. 57, 223–229.
Moriishi, K., Mochizuki, R., Moriya, K., Miyamoto, H., Mori, Y., Abe, T., Murata, S., Tanaka, K., Miyamura, T., Suzuki, T., Koike, K., and Matsuura, Y. (2007). Critical role of PA28gamma in hepatitis C virus-associated steatogenesis and hepatocarcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 104, 1661–1666.
Moriishi, K., Okabayashi, T., Nakai, K., Moriya, K., Koike, K., Murata, S., Chiba, T., Tanaka, K., Suzuki, R., Suzuki, T., Miyamura, T., and Matsuura, Y. (2003). Proteasome activator PA28gamma-dependent nuclear retention and degradation of hepatitis C virus core protein. J. Virol. 77, 10237–10249.
Moriishi, K., Shoji, I., Mori, Y., Suzuki, R., Suzuki, T., Kataoka, C., and Matsuura, Y. (2010). Involvement of PA28gamma in the propagation of hepatitis C virus. Hepatology 52, 411–420.
Moriya, K., Nakagawa, K., Santa, T., Shintani, Y., Fujie, H., Miyoshi, H., Tsutsumi, T., Miyazawa, T., Ishibashi, K., Horie, T., Imai, K., Todoroki, T., Kimura, S., and Koike, K. (2001). Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 61, 4365–4670.
Moriya, K., Yotsuyanagi, H., Shintani, Y., Fujie, H., Ishibashi, K., Matsuura, Y., Miyamura, T., and Koike, K. (1997). Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J. Gen. Virol. 78 (Pt 7), 1527–1531.
Najda-Bernatowicz, A., Krawczyk, M., Stankiewicz-Drogon, A., Bretner, M., and Boguszewska-Chachulska, A. M. (2010). Studies on the anti-hepatitis C virus activity of newly synthesized tropolone derivatives: identification of NS3 helicase inhibitors that specifically inhibit subgenomic HCV replication. Bioorg. Med. Chem. 18, 5129–5136.
Nielsen, S. U., Bassendine, M. F., Burt, A. D., Martin, C., Pumeechockchai, W., and Toms, G. L. (2006). Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J. Virol. 80, 2418–2428.
Nishimura, Y., Hayashi, M., Inada, H., and Tanaka, T. (1999). Molecular cloning and characterization of mammalian homologues of vesicle-associated membrane protein-associated (VAMP-associated) proteins. Biochem. Biophys. Res. Commun. 254, 21–26.
Nunez, O., Fernandez-Martinez, A., Majano, P. L., Apolinario, A., Gomez-Gonzalo, M., Benedicto, I., Lopez-Cabrera, M., Bosca, L., Clemente, G., Garcia-Monzon, C., and Martin-Sanz, P. (2004). Increased intrahepatic cyclooxygenase 2, matrix metalloproteinase 2, and matrix metalloproteinase 9 expression is associated with progressive liver disease in chronic hepatitis C virus infection: role of viral core and NS5A proteins. Gut 53, 1665–1672.
Ogino, T., Fukuda, H., Imajoh-Ohmi, S., Kohara, M., and Nomoto, A. (2004). Membrane binding properties and terminal residues of the mature hepatitis C virus capsid protein in insect cells. J. Virol. 78, 11766–11777.
Ohata, K., Hamasaki, K., Toriyama, K., Matsumoto, K., Saeki, A., Yanagi, K., Abiru, S., Nakagawa, Y., Shigeno, M., Miyazoe, S., Ichikawa, T., Ishikawa, H., Nakao, K., and Eguchi, K. (2003). Hepatic steatosis is a risk factor for hepatocellular carcinoma in patients with chronic hepatitis C virus infection. Cancer 97, 3036–3043.
Okamoto, K., Mori, Y., Komoda, Y., Okamoto, T., Okochi, M., Takeda, M., Suzuki, T., Moriishi, K., and Matsuura, Y. (2008). Intramembrane processing by signal peptide peptidase regulates the membrane localization of hepatitis C virus core protein and viral propagation. J. Virol. 82, 8349–8361.
Okamoto, K., Moriishi, K., Miyamura, T., and Matsuura, Y. (2004). Intramembrane proteolysis and endoplasmic reticulum retention of hepatitis C virus core protein. J. Virol. 78, 6370–6380.
Okamoto, T., Nishimura, Y., Ichimura, T., Suzuki, K., Miyamura, T., Suzuki, T., Moriishi, K., and Matsuura, Y. (2006). Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 25, 5015–5025.
Okuda, M., Li, K., Beard, M. R., Showalter, L. A., Scholle, F., Lemon, S. M., and Weinman, S. A. (2002). Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology 122, 366–375.
Oomens, A. G., Bevis, K. P., and Wertz, G. W. (2006). The cytoplasmic tail of the human respiratory syncytial virus F protein plays critical roles in cellular localization of the F protein and infectious progeny production. J. Virol. 80, 10465–10477.
Peretti, D., Dahan, N., Shimoni, E., Hirschberg, K., and Lev, S. (2008). Coordinated lipid transfer between the endoplasmic reticulum and the Golgi complex requires the VAP proteins and is essential for Golgi-mediated transport. Mol. Biol. Cell 19, 3871–3884.
Perlemuter, G., Sabile, A., Letteron, P., Vona, G., Topilco, A., Chretien, Y., Koike, K., Pessayre, D., Chapman, J., Barba, G., and Brechot, C. (2002). Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. FASEB J. 16, 185–194.
Pietschmann, T., Kaul, A., Koutsoudakis, G., Shavinskaya, A., Kallis, S., Steinmann, E., Abid, K., Negro, F., Dreux, M., Cosset, F. L., and Bartenschlager, R. (2006). Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U.S.A. 103, 7408–7413.
Pietschmann, T., Lohmann, V., Kaul, A., Krieger, N., Rinck, G., Rutter, G., Strand, D., and Bartenschlager, R. (2002). Persistent and transient replication of full-length hepatitis C virus genomes in cell culture. J. Virol. 76, 4008–4021.
Pileri, P., Uematsu, Y., Campagnoli, S., Galli, G., Falugi, F., Petracca, R., Weiner, A. J., Houghton, M., Rosa, D., Grandi, G., and Abrignani, S. (1998). Binding of hepatitis C virus to CD81. Science 282, 938–941.
Ploss, A., Evans, M. J., Gaysinskaya, V. A., Panis, M., You, H., De Jong, Y. P., and Rice, C. M. (2009). Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457, 882–886.
Popescu, C. I., Callens, N., Trinel, D., Roingeard, P., Moradpour, D., Descamps, V., Duverlie, G., Penin, F., Heliot, L., Rouille, Y., and Dubuisson, J. (2011). NS2 protein of hepatitis C virus interacts with structural and non-structural proteins towards virus assembly. PLoS Pathog. 7, e1001278. doi:10.1371/journal.ppat.1001278
Pumeechockchai, W., Bevitt, D., Agarwal, K., Petropoulou, T., Langer, B. C., Belohradsky, B., Bassendine, M. F., and Toms, G. L. (2002). Hepatitis C virus particles of different density in the blood of chronically infected immunocompetent and immunodeficient patients: implications for virus clearance by antibody. J. Med. Virol. 68, 335–342.
Rahfeld, J. U., Schierhorn, A., Mann, K., and Fischer, G. (1994). A novel peptidyl-prolyl cis/trans isomerase from Escherichia coli. FEBS Lett. 343, 65–69.
Randall, G., Panis, M., Cooper, J. D., Tellinghuisen, T. L., Sukhodolets, K. E., Pfeffer, S., Landthaler, M., Landgraf, P., Kan, S., Lindenbach, B. D., Chien, M., Weir, D. B., Russo, J. J., Ju, J., Brownstein, M. J., Sheridan, R., Sander, C., Zavolan, M., Tuschl, T., and Rice, C. M. (2007). Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. U.S.A. 104, 12884–12889.
Reiss, S., Rebhan, I., Backes, P., Romero-Brey, I., Erfle, H., Matula, P., Kaderali, L., Poenisch, M., Blankenburg, H., Hiet, M. S., Longerich, T., Diehl, S., Ramirez, F., Balla, T., Rohr, K., Kaul, A., Buhler, S., Pepperkok, R., Lengauer, T., Albrecht, M., Eils, R., Schirmacher, P., Lohmann, V., and Bartenschlager, R. (2011). Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9, 32–45.
Ridgway, N. D., Dawson, P. A., Ho, Y. K., Brown, M. S., and Goldstein, J. L. (1992). Translocation of oxysterol binding protein to Golgi apparatus triggered by ligand binding. J. Cell Biol. 116, 307–319.
Rubbia-Brandt, L., Quadri, R., Abid, K., Giostra, E., Male, P. J., Mentha, G., Spahr, L., Zarski, J. P., Borisch, B., Hadengue, A., and Negro, F. (2000). Hepatocyte steatosis is a cytopathic effect of hepatitis C virus genotype 3. J. Hepatol. 33, 106–115.
Sainz, B. Jr., Barretto, N., Martin, D. N., Hiraga, N., Imamura, M., Hussain, S., Marsh, K. A., Yu, X., Chayama, K., Alrefai, W. A., and Uprichard, S. L. (2012). Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. doi: 10.1038/nm.2581
Sakamoto, H., Okamoto, K., Aoki, M., Kato, H., Katsume, A., Ohta, A., Tsukuda, T., Shimma, N., Aoki, Y., Arisawa, M., Kohara, M., and Sudoh, M. (2005). Host sphingolipid biosynthesis as a target for hepatitis C virus therapy. Nat. Chem. Biol. 1, 333–337.
Santolini, E., Migliaccio, G., and La Monica, N. (1994). Biosynthesis and biochemical properties of the hepatitis C virus core protein. J. Virol. 68, 3631–3641.
Santolini, E., Pacini, L., Fipaldini, C., Migliaccio, G., and Monica, N. (1995). The NS2 protein of hepatitis C virus is a transmembrane polypeptide. J. Virol. 69, 7461–7471.
Scarselli, E., Ansuini, H., Cerino, R., Roccasecca, R. M., Acali, S., Filocamo, G., Traboni, C., Nicosia, A., Cortese, R., and Vitelli, A. (2002). The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21, 5017–5025.
Schiene-Fischer, C., Habazettl, J., Schmid, F. X., and Fischer, G. (2002). The hsp70 chaperone DnaK is a secondary amide peptide bond cis-trans isomerase. Nat. Struct. Biol. 9, 419–424.
Shavinskaya, A., Boulant, S., Penin, F., Mclauchlan, J., and Bartenschlager, R. (2007). The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J. Biol. Chem. 282, 37158–37169.
Shi, S. T., Lee, K. J., Aizaki, H., Hwang, S. B., and Lai, M. M. (2003). Hepatitis C virus RNA replication occurs on a detergent-resistant membrane that cofractionates with caveolin-2. J. Virol. 77, 4160–4168.
Siekierka, J. J., Hung, S. H., Poe, M., Lin, C. S., and Sigal, N. H. (1989a). A cytosolic binding protein for the immunosuppressant FK506 has peptidyl-prolyl isomerase activity but is distinct from cyclophilin. Nature 341, 755–757.
Siekierka, J. J., Staruch, M. J., Hung, S. H., and Sigal, N. H. (1989b). FK-506, a potent novel immunosuppressive agent, binds to a cytosolic protein which is distinct from the cyclosporin A-binding protein, cyclophilin. J. Immunol. 143, 1580–1583.
Skehel, P. A., Martin, K. C., Kandel, E. R., and Bartsch, D. (1995). A VAMP-binding protein from Aplysia required for neurotransmitter release. Science 269, 1580–1583.
Soriano, V., Peters, M. G., and Zeuzem, S. (2009). New therapies for hepatitis C virus infection. Clin. Infect. Dis. 48, 313–320.
Spik, G., Haendler, B., Delmas, O., Mariller, C., Chamoux, M., Maes, P., Tartar, A., Montreuil, J., Stedman, K., Kocher, H. P., Keller, R., Hiestad, P. C., and Movva, N. R. (1991). A novel secreted cyclophilin-like protein (SCYLP). J. Biol. Chem. 266, 10735–10738.
Steinmann, E., Penin, F., Kallis, S., Patel, A. H., Bartenschlager, R., and Pietschmann, T. (2007). Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 3, e103. doi:10.1371/journal.ppat.0030103
Sumpter, R. Jr., Loo, Y. M., Foy, E., Li, K., Yoneyama, M., Fujita, T., Lemon, S. M., and Gale, M. Jr. (2005). Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79, 2689–2699.
Suzuki, R., Moriishi, K., Fukuda, K., Shirakura, M., Ishii, K., Shoji, I., Wakita, T., Miyamura, T., Matsuura, Y., and Suzuki, T. (2009). Proteasomal turnover of hepatitis C virus core protein is regulated by two distinct mechanisms: a ubiquitin-dependent mechanism and a ubiquitin-independent but PA28gamma-dependent mechanism. J. Virol. 83, 2389–2392.
Tai, A. W., Benita, Y., Peng, L. F., Kim, S. S., Sakamoto, N., Xavier, R. J., and Chung, R. T. (2009). A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 5, 298–307.
Tai, A. W., and Salloum, S. (2011). The role of the phosphatidylinositol 4-kinase PI4KA in hepatitis C virus-induced host membrane rearrangement. PLoS ONE 6, e26300. doi:10.1371/journal.pone.0026300
Takikawa, S., Ishii, K., Aizaki, H., Suzuki, T., Asakura, H., Matsuura, Y., and Miyamura, T. (2000). Cell fusion activity of hepatitis C virus envelope proteins. J. Virol. 74, 5066–5074.
Targett-Adams, P., Hope, G., Boulant, S., and Mclauchlan, J. (2008). Maturation of hepatitis C virus core protein by signal peptide peptidase is required for virus production. J. Biol. Chem. 283, 16850–16859.
Thomssen, R., Bonk, S., Propfe, C., Heermann, K. H., Kochel, H. G., and Uy, A. (1992). Association of hepatitis C virus in human sera with beta-lipoprotein. Med. Microbiol. Immunol. 181, 293–300.
Thomssen, R., Bonk, S., and Thiele, A. (1993). Density heterogeneities of hepatitis C virus in human sera due to the binding of beta-lipoproteins and immunoglobulins. Med. Microbiol. Immunol. 182, 329–334.
Tory, R., Sachs-Barrable, K., Hill, J. S., and Wasan, K. M. (2008). Cyclosporine A and rapamycin induce in vitro cholesteryl ester transfer protein activity, and suppress lipoprotein lipase activity in human plasma. Int. J. Pharm. 358, 219–223.
Tripathi, L. P., Kataoka, C., Taguwa, S., Moriishi, K., Mori, Y., Matsuura, Y., and Mizuguchi, K. (2010). Network based analysis of hepatitis C virus core and NS4B protein interactions. Mol. Biosyst. 6, 2539–2553.
Trotard, M., Lepere-Douard, C., Regeard, M., Piquet-Pellorce, C., Lavillette, D., Cosset, F. L., Gripon, P., and Le Seyec, J. (2009). Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. FASEB J. 23, 3780–3789.
Tscherne, D. M., Jones, C. T., Evans, M. J., Lindenbach, B. D., Mckeating, J. A., and Rice, C. M. (2006). Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 80, 1734–1741.
Tsukiyama-Kohara, K., Iizuka, N., Kohara, M., and Nomoto, A. (1992). Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 66, 1476–1483.
Tsutsumi, T., Suzuki, T., Shimoike, T., Suzuki, R., Moriya, K., Shintani, Y., Fujie, H., Matsuura, Y., Koike, K., and Miyamura, T. (2002). Interaction of hepatitis C virus core protein with retinoid X receptor alpha modulates its transcriptional activity. Hepatology 35, 937–946.
Tu, H., Gao, L., Shi, S. T., Taylor, D. R., Yang, T., Mircheff, A. K., Wen, Y., Gorbalenya, A. E., Hwang, S. B., and Lai, M. M. (1999). Hepatitis C virus RNA polymerase and NS5A complex with a SNARE-like protein. Virology 263, 30–41.
Vaillancourt, F. H., Pilote, L., Cartier, M., Lippens, J., Liuzzi, M., Bethell, R. C., Cordingley, M. G., and Kukolj, G. (2009). Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology 387, 5–10.
Vieyres, G., Thomas, X., Descamps, V., Duverlie, G., Patel, A. H., and Dubuisson, J. (2010). Characterization of the envelope glycoproteins associated with infectious hepatitis C virus. J. Virol. 84, 10159–10168.
Voisset, C., Lavie, M., Helle, F., Op De Beeck, A., Bilheu, A., Bertrand-Michel, J., Terce, F., Cocquerel, L., Wychowski, C., Vu-Dac, N., and Dubuisson, J. (2008). Ceramide enrichment of the plasma membrane induces CD81 internalization and inhibits hepatitis C virus entry. Cell. Microbiol. 10, 606–617.
Wakita, T., Pietschmann, T., Kato, T., Date, T., Miyamoto, M., Zhao, Z., Murthy, K., Habermann, A., Krausslich, H. G., Mizokami, M., Bartenschlager, R., and Liang, T. J. (2005). Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11, 791–796.
Wang, C., Gale, M. Jr., Keller, B. C., Huang, H., Brown, M. S., Goldstein, J. L., and Ye, J. (2005a). Identification of FBL2 as a geranylgeranylated cellular protein required for hepatitis C virus RNA replication. Mol. Cell 18, 425–434.
Wang, P. Y., Weng, J., and Anderson, R. G. (2005b). OSBP is a cholesterol-regulated scaffolding protein in control of ERK 1/2 activation. Science 307, 1472–1476.
Watashi, K., Hijikata, M., Hosaka, M., Yamaji, M., and Shimotohno, K. (2003). Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology 38, 1282–1288.
Watashi, K., Ishii, N., Hijikata, M., Inoue, D., Murata, T., Miyanari, Y., and Shimotohno, K. (2005). Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol. Cell 19, 111–122.
Wolins, N. E., Brasaemle, D. L., and Bickel, P. E. (2006). A proposed model of fat packaging by exchangeable lipid droplet proteins. FEBS Lett. 580, 5484–5491.
Wolk, B., Sansonno, D., Krausslich, H. G., Dammacco, F., Rice, C. M., Blum, H. E., and Moradpour, D. (2000). Subcellular localization, stability, and trans-cleavage competence of the hepatitis C virus NS3-NS4A complex expressed in tetracycline-regulated cell lines. J. Virol. 74, 2293–2304.
Wozniak, A. L., Griffin, S., Rowlands, D., Harris, M., Yi, M., Lemon, S. M., and Weinman, S. A. (2010). Intracellular proton conductance of the hepatitis C virus p7 protein and its contribution to infectious virus production. PLoS Pathog. 6, e1001087. doi:10.1371/journal.ppat.1001087
Yamaga, A. K., and Ou, J. H. (2002). Membrane topology of the hepatitis C virus NS2 protein. J. Biol. Chem. 277, 33228–33234.
Yang, F., Robotham, J. M., Nelson, H. B., Irsigler, A., Kenworthy, R., and Tang, H. (2008). Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J. Virol. 82, 5269–5278.
Ye, J., Wang, C., Sumpter, R. Jr., Brown, M. S., Goldstein, J. L., and Gale, M. Jr. (2003). Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc. Natl. Acad. Sci. U.S.A. 100, 15865–15870.
Zhang, Z., and Zhang, R. (2008). Proteasome activator PA28 gamma regulates p53 by enhancing its MDM2-mediated degradation. EMBO J. 27, 852–864.
Keywords: HCV, hepatitis, lipid droplets, host factor
Citation: Moriishi K and Matsuura Y (2012) Exploitation of lipid components by viral and host proteins for hepatitis C virus infection. Front. Microbio. 3:54. doi: 10.3389/fmicb.2012.00054
Received: 01 December 2011;
Accepted: 31 January 2012;
Published online: 14 February 2012.
Edited by:
Yasuko Yokota, National Institute of Infectious Diseases, JapanReviewed by:
Glenn Randall, The University of Chicago, USATetsuro Suzuki, Hamamatsu University School of Medicine, Japan
Copyright: © 2012 Moriishi and Matsuura. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Kohji Moriishi, Division of Medicine, Department of Microbiology, Interdisciplinary Graduate School of Medicine and Engineering, University of Yamanashi, 1110 Shimokato, Chuo-shi, Yamanashi 409-3898, Japan. e-mail:a21vcmlpc2hpQHlhbWFuYXNoaS5hYy5qcA==