

- Department of Microbiology, University of Washington, Seattle, WA, USA
Kaposi’s sarcoma (KS) is a highly vascularized tumor supporting large amounts of neo-angiogenesis. The major cell type in KS tumors is the spindle cell, a cell that expresses markers of lymphatic endothelium. KSHV, the etiologic agent of KS, is found in the spindle cells of all KS tumors. Considering the extreme extent of angiogenesis in KS tumors at all stages it has been proposed that KSHV directly induces angiogenesis in a paracrine fashion. In accordance with this theory, KSHV infection of endothelial cells in culture induces a number of host pathways involved in activation of angiogenesis and a number of KSHV genes themselves can induce pathways involved in angiogenesis. Spindle cells are phenotypically endothelial in nature, and therefore, activation through the induction of angiogenic and/or lymphangiogenic phenotypes by the virus may also be directly involved in spindle cell growth and tumor induction. Accordingly, KSHV infection of endothelial cells induces cell autonomous angiogenic phenotypes to activate host cells. KSHV infection can also reprogram blood endothelial cells to lymphatic endothelium. However, KSHV induces some blood endothelial specific genes upon infection of lymphatic endothelial cells creating a phenotypic intermediate between blood and lymphatic endothelium. Induction of pathways involved in angiogenesis and lymphangiogenesis are likely to be critical for tumor cell growth and spread. Thus, induction of both cell autonomous and non-autonomous changes in angiogenic and lymphangiogenic pathways by KSHV likely plays a key role in the formation of KS tumors.
Kaposi’s Sarcoma and KSHV
Kaposi’s sarcoma (KS) was first described in the 1800s as a rare, fairly indolent tumor of specific populations. This form of KS, now referred to as classic KS, usually presents on the skin of the lower extremities of elderly men of specific European regions and religious origins. In the middle of the twentieth century KS became endemic in parts of Africa and is currently one of the most common tumors in parts of central Africa (Wabinga et al., 1993). In the late twentieth century KS became one of the first AIDS defining illnesses and is the most common tumor of AIDS patients world-wide. AIDS associated KS is generally far more aggressive than classic KS, arising on the skin in many parts of the body as well as in the oral cavity and can occur on internal organs where it is often fatal.
While they differ in aggressiveness, all forms of KS are relatively indistinguishable at the histological level. Grossly, the tumors have a characteristic red to purple hue, indicative of the high vascularization of the tumor. Histologically, the tumor exhibits large vascular slits lined by flattened endothelium; the slits are often, but not always, filled with blood cells. There are discernable levels of extravasated red blood cells and infiltrating lymphocytes in the tumor. While a number of cell types are present, the tumor is predominantly made up of elongated spindle cells. The spindle cells express endothelial cell markers on their surface including CD31 and CD34, but express low levels of factor VIIIRa (Russell Jones et al., 1995). Recent expression data and array studies have found that spindle cells most closely resemble lymphatic endothelium, expressing VEGF receptor 3 (VEGFR3), a specific marker of lymphatic endothelial cells (Jussila et al., 1998; Skobe et al., 1999; Weninger et al., 1999; Wang et al., 2004a). Other lymphatic endothelial cell specific markers, including LYVE-1, podoplanin, and Prox-1, are also expressed by the spindle cells (Carroll et al., 2004; Hong et al., 2004; Wang et al., 2004a).
Based on the epidemiology and the multicentric nature of the tumor, KS was predicted to have an infectious cause (Beral et al., 1990). In 1994, KSHV was discovered associated with all KS tumors and is now considered to be the etiologic agent (Chang et al., 1994). KSHV was the eighth human herpesvirus discovered and is sub-classified as a gamma herpesvirus. Like all herpesviruses KSHV has both a lytic and latent lifecycle. During lytic replication all of the greater than 90 viral genes are expressed, the virus replicates rapidly, produces infectious virions and ultimately causes cell death likely due to a combination of host cell shut off and virus production. During viral latency in endothelial cells, a limited number of genes are expressed from a single locus. This locus includes LANA, vCyc, vFLIP, a family of proteins from a repeat region called the Kaposins, and 12 pre-microRNAs encoding 17 or more mature miRNAs. These latent genes are responsible for the maintenance of the latent viral episome as well as the survival of latently infected cells.
In later-stage KS tumors, all of the spindle cells maintain infection with KSHV (Boshoff et al., 1995; Staskus et al., 1997; Dupin et al., 1999). As expected the virus is predominantly found in the latent state in spindle cells where the limited number of latent genes and miRNAs are expressed (Staskus et al., 1997; Marshall et al., 2007). However, approximately 1–5% of the spindle cells express lytic viral genes and produce infectious virus. In addition to spindle cells, KSHV is also found in other cell types in the KS lesion including monocytes (Blasig et al., 1997). However, only very low levels of these cells are infected in the tumor. KSHV can only sporadically be detected in the endothelium lining the vascular slits in the KS tumor (Dupin et al., 1999). KSHV is also associated with two lymphoproliferative diseases, primary effusion lymphoma (PEL) a pleural cavity solid B-cell lymphoma, and plasmablastic multicentric Castleman’s disease, a lymph node B-cell growth (Cesarman et al., 1995; Soulier et al., 1995).
Because KS is an angioproliferative disease and the KS tumors are highly vascularized even at early stages, it has been proposed that KSHV may directly induce angiogenesis. Angiogenesis is a tightly regulated process. Endothelial cells of the vascular system are normally maintained in a quiescent, non-proliferating state. However, during solid tumor formation, the secretion of pro-angiogenic cytokines by tumor cells can activate nearby endothelial cells to induce new blood vessel formation. Many of the vascular slits identified in histological sections of early stage KS lesions are lined by uninfected endothelium, suggesting they are formed by endothelial cells activated in a paracrine fashion (Dupin et al., 1999). These uninfected cells may later become infected, as KSHV has, in some cases, been detected in the cells surrounding the vascular spaces of later-stage nodular KS (Boshoff et al., 1995; Dupin et al., 1999).
Despite the evidence for paracrine activation of uninfected endothelial cells, KSHV also likely activates infected endothelial cells in an autocrine or cell autonomous fashion. Because KS spindle cells are endothelial in origin, induction of the KS tumor cell is similar to the processes of angiogenesis. Many of the characteristics of activated endothelial cells and angiogenesis are also associated with oncogenesis, including proliferation, migration, and metalloprotease expression. These same phenotypes are induced in KSHV-infected endothelial cells. This review discusses the recent evidence that suggests that (1) KSHV promotes continual neovascularization through paracrine factors and (2) KSHV may drive tumor cell growth through autocrine and cell autonomous activation of angiogenic phenotypes.
Paracrine Induction of Angiogenesis by KSHV
The vascular endothelial growth factor (VEGF) family of cytokines plays a prominent role in regulation of angiogenesis (Breen, 2007). VEGF-A and its receptors are required for embryonic vascular development and are important for vascular permeability, proliferation, and survival of newly formed vasculature. Several studies have explored the role of VEGF-A in KS pathogenesis. VEGF-A expression is detected in spindle cells of KS lesions, and its secretion is known to be increased by inflammatory cytokines that are present in the KS lesions (Samaniego et al., 1998). VEGF-A is also expressed by KSHV-infected PEL cell lines and conditioned media from these cells is sufficient to induce capillary morphogenesis by endothelial cells (Liu et al., 2001; Akula et al., 2005; Subramanian et al., 2010). Infection of endothelial cells with KSHV directly leads to increased expression of VEGF-A (Masood et al., 2002; Sivakumar et al., 2008; Wang and Damania, 2008). Further, KSHV conditioned media has been shown to regulate angiogenic phenotypes in endothelial cells (Sharma-Walia et al., 2010). Therefore, KSHV induction of VEGF-A is likely to be critical for both the induction of angiogenesis as well as the activation of infected spindle cells.
Although the mechanisms by which KSHV induces VEGF-A expression and secretion are still unclear, several potential pathways have been uncovered. Hypoxia induced factor (HIF)-1α is a transcription factor that has been shown to be important for upregulation of VEGF-A (Sodhi et al., 2000; Shin et al., 2008). HIF-1α is readily degraded during normal oxygen conditions. However, during hypoxia, HIF-1α is stabilized and can induce expression of genes through hypoxia responsive elements, including VEGF-A. Interestingly, KSHV infection of endothelial cells induces the expression of HIF-1α during normoxia which leads to increased HIF transcriptional activity (Carroll et al., 2006). Other studies have shown that KSHV encodes proteins that can lead to increased stability of HIF-1. The KSHV latency associated nuclear protein (LANA-1) can stabilize HIF-1α, through both degradation of its suppressors, von Hippel–Lindau protein and p53 (Cai et al., 2006), and through direct interaction between HIF-1α and LANA-1 (Cai et al., 2007). Additionally, the virally encoded interferon response factor (vIRF3) can, like LANA-1, interact with and stabilize HIF-1α, leading to increased VEGF-A expression (Shin et al., 2008). The KSHV viral G-protein coupled receptor (vGPCR) is able to increase the activity of HIF-1α as a transcription factor through activation of the MAPK and p38 signaling pathways and subsequent phosphorylation of HIF-1α (Sodhi et al., 2000). These pathways are depicted in Figure 1. While induction of HIF mRNA expression by KSHV infection has been shown, stabilization of HIF directly by KSHV infection of endothelial cells has yet to be clearly shown.
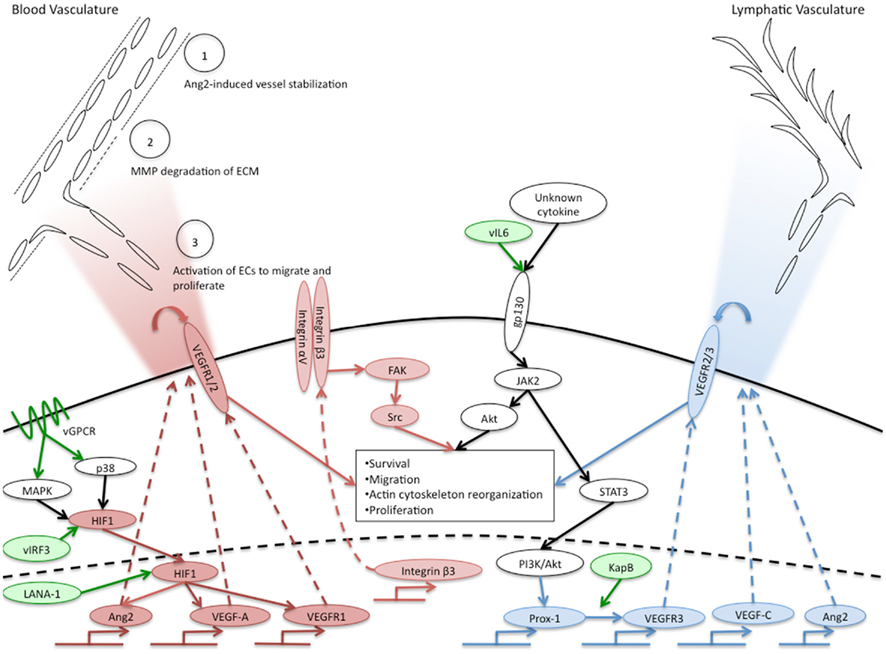
Figure 1. Overview of KSHV induction of angiogenic and lymphangiogenic pathways. Pro-angiogenic factors that are induced by KSHV are indicated in red ovals, pro-lymphangiogenic factors induced by KSHV are indicated in blue ovals. White ovals indicate the activated pathway necessary for KSHV induction of blood to lymphatic endothelial cell differentiation. KSHV genes known to induce angiogenic or lymphangiogenic pathways are indicated in green ovals. The upper left corner indicates phenotypes of angiogenesis that are activated in normal endothelium by KSHV-infected cells and the gradient of cytokines secreted by KSHV-infected cells is indicated by the red gradient. The blue gradient in the upper right hand corner indicates the gradient of lymphangiogenic cytokines induced by KSHV-infected cells that could induce lymphangiogenesis. The central box indicates angiogenic phenotypes induced by KSHV in a cell autonomous fashion.
Other host proteins have been shown to be involved in the induction of VEGF-A during KSHV infection of endothelial cells as well. For example, Emmprin is a membrane-associated glycoprotein that promotes matrix metalloproteinase expression. Its expression in KSHV-infected cells promotes cell invasiveness through activation of the PI3K/Akt and MAPK pathways (Qin et al., 2010; Dai et al., 2011). These pathways are also necessary for emmprin-induced VEGF-A expression. Further work is ongoing in multiple labs to determine the cellular pathways essential for KSHV induction of VEGF-A.
Several KSHV genes expressed during lytic replication have been implicated in regulation of VEGF-A expression (Table 1). In BCBL-1 cells (a pleural effusion lymphoma cell line), glycoprotein B (gB) and K8.1 are required for enhanced VEGF expression (Subramanian et al., 2010). Treatment of these cells with siRNA or neutralizing antibodies to gB or K8.1 significantly reduced VEGF-A production. vGPCR is a constitutively active signaling receptor that has been linked to a variety of cell survival and pro-angiogenic signaling pathways (Arvanitakis et al., 1997; Bais et al., 1998, 2003; Sodhi et al., 2000; Montaner et al., 2001; Shan et al., 2007). When injected into mice, NIH3T3 cells expressing vGPCR form highly vascularized tumors with some similarities to KS and this may be due, at least in part, to increased VEGF-A secretion (Bais et al., 1998; Guo et al., 2003). vGPCR upregulates VEGF-A through activation of MAPK and p38, which, as described above, promotes HIF-1α activity (Sodhi et al., 2000). Transgenic mice expressing vGPCR also form highly vascularized tumors that are reminiscent of KS tumors. However, cell lines derived from these tumors expressed the lymphatic growth factor VEGF-C, rather than VEGF-A (Guo et al., 2003). Increased VEGF-A expression in cells expressing vGPCR is associated with constitutive activation of its receptor, VEGFR2/KDR and downstream activation of PI3K/Akt, contributing to endothelial cell survival (Montaner et al., 2001; Bais et al., 2003). However, gB, K8.1, and vGPCR have only been detected in cells supporting lytic KSHV infection whereas the bulk of the tumor cells are latently infected. The KSHV glycoprotein K1 also induces increased VEGF-A expression in endothelial cells and is capable of immortalizing primary endothelial cells (Wang et al., 2004b, 2006). While there is evidence that K1 is expressed at very low levels during latency, the majority of its expression occurs during lytic infection (Chandriani and Ganem, 2010). In summary, the lytic phase of KSHV infection might play a role in the paracrine induction of angiogenesis through increased secretion of VEGF-A into the tumor milieu.

Table 1. KSHV genes involved in the induction of angiogenesis.
In addition to VEGF-A, KSHV-infected endothelial cells also express other angiogenic cytokines. Angiopoietin-1 and -2 are ligands for the receptor tyrosine kinase Tie2. Although less is known about the functions of angiopoietins and Tie2, their signaling is required for proper vascular development during embryogenesis (Dumont et al., 1994). Angiopoietin-1 is an agonist for the Tie2 receptor, promoting endothelial cell survival and stability. In contrast, Ang2 acts as an antagonist for Tie2, although its role is context-dependent. Ang2 is upregulated during pathologic angiogenesis and this expression is thought to destabilize endothelial cells, allowing them to be activated by other pro-angiogenic stimuli, such as VEGF, see Figure 1, circle 1 (Gale et al., 2002). Interestingly, Ang2 is expressed in KS lesions, and is upregulated in endothelial cells infected with KSHV (Brown et al., 2000; Wang et al., 2004a; Vart et al., 2007; Ye et al., 2007). This induction can be activated by the KSHV genes vGPCR and vIL-6, and can occur through a paracrine mechanism (Vart et al., 2007). Another study suggests that the MAPK pathway activation of transcription factors AP-1 and Ets-1 is involved (Ye et al., 2007). In addition to Ang2, cells transfected with the vGPCR gene expressed increased levels of angiopoietin-like 4, a member of the angiopoietin-like proteins that may play a role in vascular permeability and angiogenesis (Ma et al., 2010).
KSHV induces a number of other cytokines known to be involved in angiogenesis in other systems. These include interleukin 6 (IL-6), Monocyte chemoattractant protein-1 (MCP-1), PAX-1, and prostaglandin E2 (Schwarz and Murphy, 2001; Polson et al., 2002; Xie et al., 2005; Caselli et al., 2007; Fonsato et al., 2008). Cyclooxygenase enzymes catalyze the rate limiting step in the conversion of arachidonic acid into prostaglandins. Prostaglandins signal through g-protein coupled receptors to regulate a variety of functions, including metabolic, neuronal, and immune functions. Cyclooxygenase-2 (COX-2) expression is induced early during KSHV infection of endothelial cells and plays a role in the establishment of latency (Naranatt et al., 2004; Sharma-Walia et al., 2006). This expression is associated with increased secretion of prostaglandin E2, which promotes inflammatory cytokine expression, cell survival, and angiogenesis (Sharma-Walia et al., 2010). An additional cellular factor associated with angiogenesis, angiogenin, is induced in endothelial cells by the latent protein, LANA-1. Angiogenin was recently shown to aid in induction of angiogenesis by both VEGF and basic fibroblast growth factor (Sadagopan et al., 2009). KSHV-induced angiogenin was able to promote endothelial cell migration and capillary morphogenesis (Sadagopan et al., 2009). Since angiogenin is internalized by both infected and uninfected cells, the authors suggested angiogenin may work in both paracrine and autocrine fashions. In fact, all KSHV-induced cytokines that act on endothelial cells have the potential to promote angiogenesis-like phenotypes on the endothelial-derived spindle cells.
Regulation of angiogenesis involves coordinated expression of both pro- and anti-angiogenic factors. KSHV not only upregulates pro-angiogenic cytokines, it may also promote angiogenesis through repression of angiogenic inhibitors. The KSHV latent locus encodes for 17 miRNAs which may play a role in downregulation of angiogenic gene expression (Cai et al., 2005; Pfeffer et al., 2005; Samols et al., 2005). Expression of 10 of these miRNAs in 293 cells altered the expression of 81 genes (Samols et al., 2007). Interestingly, one of these genes is the natural angiogenic inhibitor thrombospondin-1. Thrombospondin-1 plays multiple roles in the repression of angiogenesis, however one of its main functions is activation of the anti-angiogenic growth factor transforming growth factor-β (TGF-β). This study found that thrombospondin-1 contains 34 putative miRNA binding sites, and can be downregulated by multiple KSHV miRNAs (Samols et al., 2007). Downregulation of thrombospondin-1 by KSHV miRNAs corresponds to decreased TGF-β signaling. Therefore, downregulation of anti-angiogenic factors may be an important way by which KSHV promotes continual neovascularization.
The KSHV genome itself encodes for cytokine and chemokine-like factors that activate endothelial cells and stimulate angiogenesis (Table 1). Among these factors are three genes with homology to the cellular chemokine macrophage inflammatory protein, the vMIPs I–III (Boshoff et al., 1997; Stine et al., 2000). In addition to having chemoattractant properties, these proteins promoted neovascularization in the chick chorio-allantoic membrane angiogenesis assay (Boshoff et al., 1997; Stine et al., 2000). KSHV also encodes a viral homolog of interleukin 6 (IL-6), a pro-inflammatory and pro-angiogenic cytokine. This cytokine, when expressed in NIH3T3 cells, promoted secretion of VEGF-A (Aoki et al., 1999). Furthermore, when these cells were injected into mice, they gave rise to tumors more quickly than control cells and the tumors were more highly vascularized (Aoki et al., 1999). Expression of the vMIPs has been predominantly shown to occur during lytic infection. The viral IL-6 (vIL-6) is mostly detected in endothelial cells and spindle cells undergoing lytic replication but like K1 it has been shown to be expressed at very low levels in latently infected PEL cells and to be induced during latency only under specific conditions (Chatterjee et al., 2002; Chandriani et al., 2010).
In summary, conditioned media from KSHV-infected cells can induce angiogenic phenotypes in uninfected endothelial cells as indicated by the red gradient in Figure 1. KSHV infection of endothelial cells induces expression of a number of cytokines that are capable of inducing angiogenesis in a paracrine fashion. Paramount among these is VEGF-A, an angiogenic cytokine that is induced by KSHV infection of endothelial cells. While the predominant viral mechanism of VEGF-A induction is unknown, a number of lytic KSHV genes are sufficient to induce VEGF-A when overexpressed. KSHV-infected cells also produce a number of other angiogenic cytokines of cellular and viral origin that likely play a role in the induction of angiogenesis. Taken together, all of the cytokines and induced pathways likely create a milieu that is beneficial to the induction of new blood vessels and play a significant role in the high vascularization of KS tumors.
KSHV Induction of Angiogenic Phenotypes within the Infected Cell
The predominant tumor cell of KS lesions is the endothelial-derived spindle cell. Oncogenesis in endothelial cells and angiogenesis have many phenotypes in common. Therefore KS tumor formation is likely to include increased angiogenic capacity of the spindle cells. There is growing evidence demonstrating the manipulation of host cell phenotypes by KSHV and the role of these changes in the promotion of angiogenesis related phenotypes. These infected cell phenotypes include increased stability of tubules formed by macrovascular endothelial cells, induction of capillary morphogenesis in low growth factor conditions, and enhanced migration and invasion (Qian et al., 2007; Sadagopan et al., 2007, 2009; Wang and Damania, 2008; Couty et al., 2009; DiMaio et al., 2011). Additionally, KSHV induces the expression of VEGF receptors on the surface of infected endothelial cells as discussed below.
Endothelial cells lining the vasculature form coordinated junctions to maintain barrier function. Breakdown of these junctions is necessary for initiation of angiogenesis, immune cell extravasation, and tumor cell metastasis. Interestingly, several studies have evaluated the adherens junctions of KSHV-infected endothelial cells and found them to be perturbed (Mansouri et al., 2008; Qian et al., 2008; Guilluy et al., 2011). This may result from the degradation of VE-cadherin (Mansouri et al., 2008; Qian et al., 2008) as well as disruption of VE-cadherin/beta-catenin signaling (Guilluy et al., 2011). Therefore, KSHV infection can directly initiate a key angiogenic step, the breakdown of cell–cell adherence. While the direct mechanism of KSHV-induced disruption of adherens junctions during latency is not known, there are a number of candidate KSHV genes that could be involved (Table 1). The KSHV-encoded ubiquitin ligase protein, K5, targets VE-cadherin for degradation (Mansouri et al., 2008). Overexpression of the KSHV vGPCR induces endothelial cell permeability and downregulation of cell surface VE-cadherin as well (Dwyer et al., 2011). K5 also targets other cellular proteins, including platelet/endothelial cell adhesion molecule-1 (PECAM-1, CD31), a transmembrane protein important for endothelial cell–cell communication, which could contribute to barrier dysfunction and increased permeability (Tomescu et al., 2003; Mansouri et al., 2006). K1, a primarily lytic protein that may also be expressed at low levels during latency was also shown to initiate signaling similar to that required for disruption of Cadherin signaling (Guilluy et al., 2011). While the exact viral mechanism of disruption of adherens junctions by KSHV infection is not known, the virus encodes multiple genes capable of altering Cadherin.
During angiogenesis, endothelial cells migrate from the pre-existing vasculature toward the site of angiogenic stimulus. Endothelial cells exhibit enhanced migration and invasion following latent KSHV infection (DiMaio et al., 2011; Wu et al., 2011). This has been demonstrated by more rapid motility through transwell dishes. KSHV-infected cells also express increased levels of the matrix metalloproteinases MMP-1, -2, and -9 (Qian et al., 2007). MMP proteins break down the extracellular matrix supporting stable vasculature to allow for invasion and migration of endothelial cells during angiogenesis (Figure 1, circle 2). Expression of MMP proteins induced by KSHV allows for increased invasion of both infected and uninfected endothelial cells (Wang et al., 2004b; Qian et al., 2007; Shan et al., 2007). While these processes constitute one component of angiogenesis, they are also known to play roles in oncogenesis (Gialeli et al., 2011) indicating that KSHV activation of angiogenic phenotypes in endothelial cells may lead to enhanced oncogenesis as well.
Endothelial cells grown in three-dimensional culture will migrate and organize into capillary-like structures. This activity is dependent, at least in part, on growth factors and cytokines present in the matrix or growth media. KSHV-infected cells are able to undergo capillary morphogenesis in low growth factor conditions to a greater extent than uninfected cells (Wang and Damania, 2008). This could be partially due to increased cytokine secretion from KSHV-infected cells. In fact, when endothelial cells are cultured in the presence of conditioned media from KSHV-infected BCBL-1 cells, their ability to organize into capillary-like structures is increased (Wang and Damania, 2008). However, the effect of BCBL-1 conditioned media was greater on KSHV-infected endothelial cells than on mock-infected cells, suggesting that infected cells are more receptive to angiogenic growth factors. In addition, this same study found that capillary-like structures formed by KSHV-infected endothelial cells are more persistent than mock-infected cells, indicative of the promotion of cell survival and continual angiogenesis by KSHV (Wang and Damania, 2008 and our unpublished results).
KSHV latent infection of endothelial cells also induces VEGF receptor expression, which may allow infected cells to respond more robustly to VEGFs. There are three main receptors for VEGFs. VEGF receptors 1 and 2 play roles in angiogenesis while 2 and 3 play a role in lymphangiogenesis (described below). While KSHV infection has not been reported to alter the expression levels of VEGFR2 (KDR), VEGFR1 expression is significantly increased following KSHV endothelial cell infection (Carroll et al., 2004). Drugs that inhibited HIF-1 activation and signaling also inhibited VEGFR1 upregulation (Carroll et al., 2006). VEGFR1 has been described as both a positive and negative regulator of angiogenesis depending on the context. VEGFR1 mouse knockouts have higher levels of angiogenesis (Fong et al., 1995). However, in cell culture models, VEGFR1 has been shown to potentiate angiogenesis (Cao, 2009). More studies will be needed to determine the importance of increased VEGFR1 expression in KSHV infection and KS tumor formation. Interestingly, expression of VEGFR3, the main receptor for VEGF-C and D is also significantly increased by KSHV infection (Carroll et al., 2004; Hong et al., 2004). VEGFR3, a receptor specific to lymphatic endothelium and critical for lymphangiogenesis will be discussed below. Importantly, endothelial tip cells at the leading edge of new vascular protrusions are the only main adult cell type known to express both the blood endothelial cell receptor, VEGFR1, and the lymphatic endothelial cell receptor, VEGFR3 (Tammela et al., 2008): KSHV infection of endothelial cells directly induces expression of both of these receptors.
The mechanisms by which KSHV induces angiogenic phenotypes in latently infected cells are largely unknown. A number of angiogenic phenotypes are likely to be a direct result of the cytokine milieu of the infected cells. As described above, KSHV-infected cells secrete both viral and host cytokines that are sufficient to induce angiogenic phenotypes. These paracrine factors surely play a role in the induction of tumor cells. However, it is also apparent that some of the angiogenic effects seen in latently infected cells are cell autonomous, independent of either paracrine or autocrine factors. As described above, conditioned media from PEL cells had stronger effects on tubule formation of KSHV-infected endothelial cells (Akula et al., 2005). We have also recently found that KSHV infection induces the pro-angiogenic integrin, integrin β3, during latent infection (DiMaio et al., 2011). Induction of integrin β3 leads to increased cell surface expression of the αVβ3 integrin heterodimer. We have further shown that latently infected endothelial cells become more adherent to the integrin ligands fibronectin and vitronectin, and are more migratory than mock-infected cells. These induced phenotypes require RGD-binding integrins, specifically integrin β3. Although both uninfected and infected cells organize in three-dimensional cultures in complete media, infected cells are more sensitive to inhibitors of integrin β3 and its downstream signaling molecules, such as Src kinase (DiMaio et al., 2011). This suggests that during latent KSHV infection there is a shift in endothelial cell signaling that results in a more angiogenic phenotype dependent on αVβ3 expression on the surface of the cell (Figure 1, center). Therefore, KSHV alteration of endothelial cell signaling pathways can dramatically affect how the cell responds to intra- and extra-cellular signals. These changes that lead to alterations in angiogenic properties are likely to play a role in the growth and cell–cell interactions of infected cells, thereby playing a role in KS tumor formation.
Angiogenesis vs. Lymphangiogenesis
During development of the vascular system, a subset of endothelial cells in the cardinal vein begin to express markers of lymphatic differentiation, including the master regulatory gene, prox-1. These cells then bud from the cardinal vein, differentiate into lymphatic endothelial cells, and form the lymphatic vascular system (Wigle and Oliver, 1999). The mechanisms regulating lymphangiogenesis are in general less well understood when compared to angiogenesis. Immunohistochemistry of KS tumors showed that spindle cells express markers of lymphatic endothelium, suggesting these cells may arise from primary infection of lymphatic endothelial cells, rather than blood endothelial cells (Jussila et al., 1998; Skobe et al., 1999; Weninger et al., 1999; Pyakurel et al., 2006). An alternative hypothesis is that KSHV infection of blood endothelial cells drives differentiation toward a more lymphatic phenotype. This idea is supported by evidence that KSHV infection of blood endothelial cells promotes expression of lymphatic-specific genes, including prox-1, VEGFR3, podoplanin, and LYVE-1, effectively driving the reprogramming of blood endothelial cells to become lymphatic endothelium (Carroll et al., 2004; Hong et al., 2004; Wang et al., 2004a). Microarray analysis comparing KSHV-infected blood endothelial cells to blood and lymphatic endothelial cells indicate that KSHV-infected blood endothelial cells have gene expression profiles that align more closely to lymphatic endothelial cells than that of blood endothelial cells (Carroll et al., 2004; Hong et al., 2004; Wang et al., 2004a).
The mechanism by which KSHV induces lymphatic differentiation is not completely clear. The KSHV latent gene Kaposin B can directly promote the stability of Prox-1 mRNA (Yoo et al., 2010) leading to increased expression of Prox-1. However, this effect was not sufficient to induce Prox-1 expression in blood endothelial cells. We recently found that induction of blood to lymphatic endothelial cell reprogramming requires signaling through the cellular receptor gp130. Endothelial cells that are latently infected with KSHV have increased expression and signaling of gp130 (Morris et al., 2008). This leads to activation of the JAK/STAT3 pathway and the PI3K/AKT pathway leading to expression of lymphatic-specific genes starting with Prox-1. Inhibition of this pathway by siRNAs that target gp130 or AKT or pharmacological inhibitors that block PI3 kinase or Jak2/STAT3 signaling is sufficient to block lymphatic differentiation (see Figure 1, center). The cytokine responsible for activating gp130 is currently not known. KSHV vIL-6 is sufficient to induce gp130 activation and we recently found that vIL-6 is sufficient to induce lymphatic differentiation (Morris et al., 2012). However, KSHV lacking vIL-6 is still able to cause blood to lymphatic endothelial cell differentiation, indicating that KSHV has evolved multiple strategies to activate gp130 and induce blood to lymphatic endothelial cell differentiation (Morris et al., 2008).
Induction of lymphatic differentiation by KSHV is only part of the story, however. Despite the expression of lymphatic-specific genes, blood endothelial cells infected with KSHV retain expression of some blood specific markers (Wang et al., 2004a). Additionally, infection of lymphatic endothelial cells with KSHV induces expression of blood specific markers (Wang et al., 2004a). KSHV miRNAs were found to target the transcription factor MAF (Hansen et al., 2010). Downregulation of MAF in lymphatic endothelial cells by siRNA restored expression of blood endothelial markers, such as VEGFR1 and CXCR4. Thus, infection of blood or lymphatic endothelial cells by KSHV alters host gene expression to an intermediate state between the two cell types. As described above, this intermediate phenotype with both VEGFR1 and R3 expression is only present in the leading tip of endothelial cells involved in active neo-angiogenesis. In the KS lesions only LANA+ cells expressed Prox-1, indicating that this effect requires KSHV gene expression (Hong et al., 2004). This suggests that differentiation toward lymphatic endothelial cells may specifically allow the spindle cells to respond to lymphangiogenic growth factors. In fact, KSHV infection of endothelial cells induces both VEGF-A and VEGF-C (Sivakumar et al., 2008). VEGF-C is a key regulator of lymphangiogenesis. Therefore, induction of both VEGFR1 and R3 allow KSHV-infected cells to respond to key angiogenic and lymphangiogenic factors in the tumor environment. The direct role of KSHV reprogramming of blood endothelial cells to lymphatic in induction of angiogenic and lymphangiogenic phenotypes is still under investigation.
Summary
The highly vascular nature of KS tumors and the large amounts of neo-angiogenesis in the tumor led to the proposal that the etiologic agent of the tumor might directly induce angiogenesis. In accordance with this hypothesis KSHV infection of endothelial cells, the main tumor cell type, induces host cell cytokines involved in angiogenesis. In particular, KSHV induces the expression of VEGF-A and -C and other cytokines as well as encoding angiogenic cytokines from its own genome (Boshoff et al., 1997; Aoki et al., 1999; Brown et al., 2000; Stine et al., 2000; Schwarz and Murphy, 2001; Masood et al., 2002; Polson et al., 2002; Wang et al., 2004b; Xie et al., 2005; Caselli et al., 2007; Vart et al., 2007; Ye et al., 2007; Fonsato et al., 2008; Sivakumar et al., 2008; Wang and Damania, 2008; Sadagopan et al., 2009; Ma et al., 2010; Sharma-Walia et al., 2010). Therefore, KSHV may induce seeding of new blood vessels to the tumor milieu. Additionally, because the tumor cell is endothelial in nature, induction of angiogenic cytokines may also activate the tumor cells and aid in the growth of KS tumors. KSHV also induces angiogenic phenotypes directly in latently infected cells in a cell autonomous fashion, indicating that angiogenic activation of the infected endothelial cell may directly play a role in tumor formation.
While KSHV activates many growth-signaling properties and in general the induction of angiogenic phenotypes supports endothelial cell proliferation, in most cultures KSHV does not induce increases in endothelial cell proliferation. It is possible that the cell culture milieu simply does not match the tumor milieu. KS spindle cells are not fully transformed ex vivo and, except in very rare cases, have a limited life span indicating that factors in the tumor environment that come from other cells types could be necessary to maintain KS spindle cell growth. The increase in growth could also be masked by the fact that endothelial cells in culture are rapidly dividing and therefore do not need additional growth signals. Along those lines, mature endothelium in vivo is relatively quiescent. That being said, the endothelial cell transforming potential of KSHV in culture can be unmasked given specific conditions. Dermal microvascular endothelial cells that were immortalized with the E6 and E7 genes from papillomavirus are readily transformed by KSHV, including increased proliferation (Moses et al., 1999). Therefore, KSHV activation of endothelial cells can induce a proliferative advantage in the correct genetic environment. However, it is unknown if viral induction of angiogenic phenotypes is necessary for the growth in the E6/E7 immortalized endothelial cells.
In general, viruses do not evolve to cause cancer, as it is likely a dead end for transmission. KSHV likely evolved to activate the cell where it is maintained to ensure survival and spread of the virus. A major side effect of this activation may be providing an ideal environment for angiogenesis leading to increased vascularization of small tumor growths and expansion of KS tumors. While the study of viral induction of angiogenesis can lead to a better understanding of how KSHV causes endothelial cell tumors, information gleaned from the study of viral mechanisms of induction of angiogenesis and lymphangiogenesis will also lead to a better understanding of endothelial cell activation and tumor angiogenesis in general. Thus, the study of KSHV infection of endothelial cells provides a controlled system for analyzing the regulation and induction of angiogenic phenotypes that will likely shed light on the field of tumor angiogenesis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Terri A. DiMaio is supported by a post-doctoral fellowship from the American Cancer Society (119029-PF-10-166-01-MPC). Michael Lagunoff is supported by grants from the NCI and the NIDCR (RO1CA097934 and PO1DE021954).
References
Akula, S. M., Ford, P. W., Whitman, A. G., Hamden, K. E., Bryan, B. A., Cook, P. P., and McCubrey, J. A. (2005). B-Raf-dependent expression of vascular endothelial growth factor-A in Kaposi sarcoma-associated herpesvirus-infected human B cells. Blood 105, 4516–4522.
Aoki, Y., Jaffe, E. S., Chang, Y., Jones, K., Teruya-Feldstein, J., Moore, P. S., and Tosato, G. (1999). Angiogenesis and hematopoiesis induced by Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6. Blood 93, 4034–4043.
Arvanitakis, L., Geras-Raaka, E., Varma, A., Gershengorn, M. C., and Cesarman, E. (1997). Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature 385, 347–350.
Bais, C., Santomasso, B., Coso, O., Arvanitakis, L., Raaka, E. G., Gutkind, J. S., Asch, A. S., Cesarman, E., Gershengorn, M. C., Mesri, E. A., and Gerhengorn, M. C. (1998). G-protein-coupled receptor of Kaposi’s sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature 391, 86–89.
Bais, C., Van Geelen, A., Eroles, P., Mutlu, A., Chiozzini, C., Dias, S., Silverstein, R. L., Rafii, S., and Mesri, E. A. (2003). Kaposi’s sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/ KDR. Cancer Cell 3, 131–143.
Beral, V., Peterman, T. A., Berkelman, R. L., and Jaffe, H. W. (1990). Kaposi’s sarcoma among persons with AIDS: a sexually transmitted infection? Lancet 335, 123–128.
Blasig, C., Zietz, C., Haar, B., Neipel, F., Esser, S., Brockmeyer, N. H., Tschachler, E., Colombini, S., Ensoli, B., and Stürzl, M. (1997). Monocytes in Kaposi’s sarcoma lesions are productively infected by human herpesvirus 8. J. Virol. 71, 7963–7968.
Boshoff, C., Endo, Y., Collins, P. D., Takeuchi, Y., Reeves, J. D., Schweickart, V. L., Siani, M. A., Sasaki, T., Williams, T. J., Gray, P. W., Moore, P. S., Chang, Y., and Weiss, R. A. (1997). Angiogenic and HIV-inhibitory functions of KSHV-encoded chemokines. Science 278, 290–294.
Boshoff, C., Schulz, T. F., Kennedy, M. M., Graham, A. K., Fisher, C., Thomas, A., McGee, J. O., Weiss, R. A., and O’Leary, J. J. (1995). Kaposi’s sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat. Med. 1, 1274–1278.
Brown, L. F., Dezube, B. J., Tognazzi, K., Dvorak, H. F., and Yancopoulos, G. D. (2000). Expression of Tie1, Tie2, and angiopoietins 1, 2, and 4 in Kaposi’s sarcoma and cutaneous angiosarcoma. Am. J. Pathol. 156, 2179–2183.
Cai, Q., Murakami, M., Si, H., and Robertson, E. S. (2007). A potential alpha-helix motif in the amino terminus of LANA encoded by Kaposi’s sarcoma-associated herpesvirus is critical for nuclear accumulation of HIF-1alpha in normoxia. J. Virol. 81, 10413–10423.
Cai, Q. L., Knight, J. S., Verma, S. C., Zald, P., and Robertson, E. S. (2006). EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog. 2, e116. doi:10.1371/journal.ppat.0020116
Cai, X., Lu, S., Zhang, Z., Gonzalez, C. M., Damania, B., and Cullen, B. R. (2005). Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. U.S.A. 102, 5570–5575.
Cao, Y. (2009). Positive and negative modulation of angiogenesis by VEGFR1 ligands. Sci. Signal. 2, re1.
Carroll, P. A., Brazeau, E., and Lagunoff, M. (2004). Kaposi’s sarcoma-associated herpesvirus infection of blood endothelial cells induces lymphatic differentiation. Virology 328, 7–18.
Carroll, P. A., Kenerson, H. L., Yeung, R. S., and Lagunoff, M. (2006). Latent Kaposi’s sarcoma-associated herpesvirus infection of endothelial cells activates hypoxia-induced factors. J. Virol. 80, 10802–10812.
Caselli, E., Fiorentini, S., Amici, C., Di Luca, D., Caruso, A., and Santoro, M. G. (2007). Human herpesvirus 8 acute infection of endothelial cells induces monocyte chemoattractant protein 1-dependent capillary-like structure formation: role of the IKK/NF-kappaB pathway. Blood 109, 2718–2726.
Cesarman, E., Chang, Y., Moore, P. S., Said, J. W., and Knowles, D. M. (1995). Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332, 1186–1191.
Chandriani, S., and Ganem, D. (2010). Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 84, 5565–5573.
Chandriani, S., Xu, Y., and Ganem, D. (2010). The lytic transcriptome of Kaposi’s sarcoma-associated herpesvirus reveals extensive transcription of noncoding regions, including regions antisense to important genes. J. Virol. 84, 7934–7942.
Chang, Y., Cesarman, E., Pessin, M. S., Lee, F., Culpepper, J., Knowles, D. M., and Moore, P. S. (1994). Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266, 1865–1869.
Chatterjee, M., Osborne, J., Bestetti, G., Chang, Y., and Moore, P. S. (2002). Viral IL-6-induced cell proliferation and immune evasion of interferon activity. Science 298, 1432–1435.
Couty, J. P., Lupu-Meiri, M., Oron, Y., and Gershengorn, M. C. (2009). Kaposi’s sarcoma-associated herpesvirus-G protein-coupled receptor-expressing endothelial cells exhibit reduced migration and stimulated chemotaxis by chemokine inverse agonists. J. Pharmacol. Exp. Ther. 329, 1142–1147.
Dai, L., Bratoeva, M., Toole, B. P., Qin, Z., and Parsons, C. (2011). KSHV activation of VEGF secretion and invasion for endothelial cells is mediated through viral upregulation of emmprin-induced signal transduction. Int. J. Cancer. doi:10.1002/ijc.26428
DiMaio, T. A., Gutierrez, K. D., and Lagunoff, M. (2011). Latent KSHV infection of endothelial cells induces integrin beta3 to activate angiogenic phenotypes. PLoS Pathog. 7, e1002424. doi:10.1371/journal.ppat.1002424
Dumont, D. J., Gradwohl, G., Fong, G. H., Puri, M. C., Gertsenstein, M., Auerbach, A., and Breitman, M. L. (1994). Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes Dev. 8, 1897–1909.
Dupin, N., Fisher, C., Kellam, P., Ariad, S., Tulliez, M., Franck, N., van Marck, E., Salmon, D., Gorin, I., Escande, J. P., Weiss, R. A., Alitalo, K., and Boshoff, C. (1999). Distribution of human herpesvirus-8 latently infected cells in Kaposi’s sarcoma, multicentric Castleman’s disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. U.S.A. 96, 4546–4551.
Dwyer, J., Le Guelte, A., Galan Moya, E. M., Sumbal, M., Carlotti, A., Douguet, L., Gutkind, J. S., Grange, P. A., Dupin, N., and Gavard, J. (2011). Remodeling of VE-cadherin junctions by the human herpes virus 8 G-protein coupled receptor. Oncogene 30, 190–200.
Fong, G. H., Rossant, J., Gertsenstein, M., and Breitman, M. L. (1995). Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 376, 66–70.
Fonsato, V., Buttiglieri, S., Deregibus, M. C., Bussolati, B., Caselli, E., Di Luca, D., and Camussi, G. (2008). PAX2 expression by HHV-8-infected endothelial cells induced a proangiogenic and proinvasive phenotype. Blood 111, 2806–2815.
Gale, N. W., Thurston, G., Hackett, S. F., Renard, R., Wang, Q., McClain, J., Martin, C., Witte, C., Witte, M. H., Jackson, D., Suri, C., Campochiaro, P. A., Wiegand, S. J., and Yancopoulos, G. D. (2002). Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by angiopoietin-1. Dev. Cell 3, 411–423.
Gialeli, C., Theocharis, A. D., and Karamanos, N. K. (2011). Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 278, 16–27.
Guilluy, C., Zhang, Z., Bhende, P. M., Sharek, L., Wang, L., Burridge, K., and Damania, B. (2011). Latent KSHV infection increases the vascular permeability of human endothelial cells. Blood 118, 5344–5354.
Guo, H. G., Sadowska, M., Reid, W., Tschachler, E., Hayward, G., and Reitz, M. (2003). Kaposi’s sarcoma-like tumors in a human herpesvirus 8 ORF74 transgenic mouse. J. Virol. 77, 2631–2639.
Hansen, A., Henderson, S., Lagos, D., Nikitenko, L., Coulter, E., Roberts, S., Gratrix, F., Plaisance, K., Renne, R., Bower, M., Kellam, P., and Boshoff, C. (2010). KSHV-encoded miRNAs target MAF to induce endothelial cell reprogramming. Genes Dev. 24, 195–205.
Hong, Y. K., Foreman, K., Shin, J. W., Hirakawa, S., Curry, C. L., Sage, D. R., Libermann, T., Dezube, B. J., Fingeroth, J. D., and Detmar, M. (2004). Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat. Genet. 36, 683–685.
Jussila, L., Valtola, R., Partanen, T. A., Salven, P., Heikkilä, P., Matikainen, M. T., Renkonen, R., Kaipainen, A., Detmar, M., Tschachler, E., Alitalo, R., and Alitalo, K. (1998). Lymphatic endothelium and Kaposi’s sarcoma spindle cells detected by antibodies against the vascular endothelial growth factor receptor-3. Cancer Res. 58, 1599–1604.
Liu, C., Okruzhnov, Y., Li, H., and Nicholas, J. (2001). Human herpesvirus 8 (HHV-8)-encoded cytokines induce expression of and autocrine signaling by vascular endothelial growth factor (VEGF) in HHV-8-infected primary-effusion lymphoma cell lines and mediate VEGF-independent antiapoptotic effects. J. Virol. 75, 10933–10940.
Ma, T., Jham, B. C., Hu, J., Friedman, E. R., Basile, J. R., Molinolo, A., Sodhi, A., and Montaner, S. (2010). Viral G protein-coupled receptor up-regulates angiopoietin-like 4 promoting angiogenesis and vascular permeability in Kaposi’s sarcoma. Proc. Natl. Acad. Sci. U.S.A. 107, 14363–14368.
Mansouri, M., Douglas, J., Rose, P. P., Gouveia, K., Thomas, G., Means, R. E., Moses, A. V., and Früh, K. (2006). Kaposi sarcoma herpesvirus K5 removes CD31/PECAM from endothelial cells. Blood 108, 1932–1940.
Mansouri, M., Rose, P. P., Moses, A. V., and Früh, K. (2008). Remodeling of endothelial adherens junctions by Kaposi’s sarcoma-associated herpesvirus. J. Virol. 82, 9615–9628.
Marshall, V., Parks, T., Bagni, R., Wang, C. D., Samols, M. A., Hu, J., Wyvil, K. M., Aleman, K., Little, R. F., Yarchoan, R., Renne, R., and Whitby, D. (2007). Conservation of virally encoded microRNAs in Kaposi sarcoma – associated herpesvirus in primary effusion lymphoma cell lines and in patients with Kaposi sarcoma or multicentric Castleman disease. J. Infect. Dis. 195, 645–659.
Masood, R., Cesarman, E., Smith, D. L., Gill, P. S., and Flore, O. (2002). Human herpesvirus-8-transformed endothelial cells have functionally activated vascular endothelial growth factor/vascular endothelial growth factor receptor. Am. J. Pathol. 160, 23–29.
Montaner, S., Sodhi, A., Pece, S., Mesri, E. A., and Gutkind, J. S. (2001). The Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor promotes endothelial cell survival through the activation of Akt/protein kinase B. Cancer Res. 61, 2641–2648.
Morris, V. A., Punjabi, A. S., and Lagunoff, M. (2008). Activation of Akt through gp130 receptor signaling is required for Kaposi’s sarcoma-associated herpesvirus-induced lymphatic reprogramming of endothelial cells. J. Virol. 82, 8771–8779.
Morris, V. A., Punjabi, A. S., Wells, R. C., Wittkopp, C. J., Vart, R., and Lagunoff, M. (2012). The KSHV Viral IL-6 homolog is sufficient to induce blood to lymphatic endothelial cell differentiation. Virology. (in press).
Moses, A. V., Fish, K. N., Ruhl, R., Smith, P. P., Strussenberg, J. G., Zhu, L., Chandran, B., and Nelson, J. A. (1999). Long-term infection and transformation of dermal microvascular endothelial cells by human herpesvirus 8. J. Virol. 73, 6892–6902.
Naranatt, P. P., Krishnan, H. H., Svojanovsky, S. R., Bloomer, C., Mathur, S., and Chandran, B. (2004). Host gene induction and transcriptional reprogramming in Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: insights into modulation events early during infection. Cancer Res. 64, 72–84.
Pfeffer, S., Sewer, A., Lagos-Quintana, M., Sheridan, R., Sander, C., Grässer, F. A., van Dyk, L. F., Ho, C. K., Shuman, S., Chien, M., Russo, J. J., Ju, J., Randall, G., Lindenbach, B. D., Rice, C. M., Simon, V., Ho, D. D., Zavolan, M., and Tuschl, T. (2005). Identification of microRNAs of the herpesvirus family. Nat. Methods 2, 269–276.
Polson, A. G., Wang, D., DeRisi, J., and Ganem, D. (2002). Modulation of host gene expression by the constitutively active G protein-coupled receptor of Kaposi’s sarcoma-associated herpesvirus. Cancer Res. 62, 4525–4530.
Pyakurel, P., Pak, F., Mwakigonja, A. R., Kaaya, E., Heiden, T., and Biberfeld, P. (2006). Lymphatic and vascular origin of Kaposi’s sarcoma spindle cells during tumor development. Int. J. Cancer 119, 1262–1267.
Qian, L. W., Greene, W., Ye, F., and Gao, S. J. (2008). Kaposi’s sarcoma-associated herpesvirus disrupts adherens junctions and increases endothelial permeability by inducing degradation of VE-cadherin. J. Virol. 82, 11902–11912.
Qian, L. W., Xie, J., Ye, F., and Gao, S. J. (2007). Kaposi’s sarcoma-associated herpesvirus infection promotes invasion of primary human umbilical vein endothelial cells by inducing matrix metalloproteinases. J. Virol. 81, 7001–7010.
Qin, Z., Dai, L., Slomiany, M. G., Toole, B. P., and Parsons, C. (2010). Direct activation of emmprin and associated pathogenesis by an oncogenic herpesvirus. Cancer Res. 70, 3884–3889.
Russell Jones, R., Orchard, G., Zelger, B., and Wilson Jones, E. (1995). Immunostaining for CD31 and CD34 in Kaposi sarcoma. J. Clin. Pathol. 48, 1011–1016.
Sadagopan, S., Sharma-Walia, N., Veettil, M. V., Bottero, V., Levine, R., Vart, R. J., and Chandran, B. (2009). Kaposi’s sarcoma-associated herpesvirus upregulates angiogenin during infection of human dermal microvascular endothelial cells, which induces 45S rRNA synthesis, antiapoptosis, cell proliferation, migration, and angiogenesis. J. Virol. 83, 3342–3364.
Sadagopan, S., Sharma-Walia, N., Veettil, M. V., Raghu, H., Sivakumar, R., Bottero, V., and Chandran, B. (2007). Kaposi’s sarcoma-associated herpesvirus induces sustained NF-kappaB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J. Virol. 81, 3949–3968.
Samaniego, F., Markham, P. D., Gendelman, R., Watanabe, Y., Kao, V., Kowalski, K., Sonnabend, J. A., Pintus, A., Gallo, R. C., and Ensoli, B. (1998). Vascular endothelial growth factor and basic fibroblast growth factor present in Kaposi’s sarcoma (KS) are induced by inflammatory cytokines and synergize to promote vascular permeability and KS lesion development. Am. J. Pathol. 152, 1433–1443.
Samols, M. A., Hu, J., Skalsky, R. L., and Renne, R. (2005). Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 79, 9301–9305.
Samols, M. A., Skalsky, R. L., Maldonado, A. M., Riva, A., Lopez, M. C., Baker, H. V., and Renne, R. (2007). Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog. 3, e65. doi:10.1371/journal.ppat.0030065
Schwarz, M., and Murphy, P. M. (2001). Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor constitutively activates NF-kappa B and induces proinflammatory cytokine and chemokine production via a C-terminal signaling determinant. J. Immunol. 167, 505–513.
Shan, B., Morris, C. A., Zhuo, Y., Shelby, B. D., Levy, D. R., and Lasky, J. A. (2007). Activation of proMMP-2 and Src by HHV8 vGPCR in human pulmonary arterial endothelial cells. J. Mol. Cell. Cardiol. 42, 517–525.
Sharma-Walia, N., Paul, A. G., Bottero, V., Sadagopan, S., Veettil, M. V., Kerur, N., and Chandran, B. (2010). Kaposi’s sarcoma associated herpes virus (KSHV) induced COX-2: a key factor in latency, inflammation, angiogenesis, cell survival and invasion. PLoS Pathog. 6, e1000777. doi:10.1371/journal.ppat.1000777
Sharma-Walia, N., Raghu, H., Sadagopan, S., Sivakumar, R., Veettil, M. V., Naranatt, P. P., Smith, M. M., and Chandran, B. (2006). Cyclooxygenase 2 induced by Kaposi’s sarcoma-associated herpesvirus early during in vitro infection of target cells plays a role in the maintenance of latent viral gene expression. J. Virol. 80, 6534–6552.
Shin, Y. C., Joo, C. H., Gack, M. U., Lee, H. R., and Jung, J. U. (2008). Kaposi’s sarcoma-associated herpesvirus viral IFN regulatory factor 3 stabilizes hypoxia-inducible factor-1 alpha to induce vascular endothelial growth factor expression. Cancer Res. 68, 1751–1759.
Sivakumar, R., Sharma-Walia, N., Raghu, H., Veettil, M. V., Sadagopan, S., Bottero, V., Varga, L., Levine, R., and Chandran, B. (2008). Kaposi’s sarcoma-associated herpesvirus induces sustained levels of vascular endothelial growth factors A and C early during in vitro infection of human microvascular dermal endothelial cells: biological implications. J. Virol. 82, 1759–1776.
Skobe, M., Brown, L. F., Tognazzi, K., Ganju, R. K., Dezube, B. J., Alitalo, K., and Detmar, M. (1999). Vascular endothelial growth factor-C (VEGF-C) and its receptors KDR and flt-4 are expressed in AIDS-associated Kaposi’s sarcoma. J. Invest. Dermatol. 113, 1047–1053.
Sodhi, A., Montaner, S., Patel, V., Zohar, M., Bais, C., Mesri, E. A., and Gutkind, J. S. (2000). The Kaposi’s sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1alpha. Cancer Res. 60, 4873–4880.
Soulier, J., Grollet, L., Oksenhendler, E., Cacoub, P., Cazals-Hatem, D., Babinet, P., d’Agay, M. F., Clauvel, J. P., Raphael, M., and Degos, L. (1995). Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 86, 1276–1280.
Staskus, K. A., Zhong, W., Gebhard, K., Herndier, B., Wang, H., Renne, R., Beneke, J., Pudney, J., Anderson, D. J., Ganem, D., and Haase, A. T. (1997). Kaposi’s sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 71, 715–719.
Stine, J. T., Wood, C., Hill, M., Epp, A., Raport, C. J., Schweickart, V. L., Endo, Y., Sasaki, T., Simmons, G., Boshoff, C., Clapham, P., Chang, Y., Moore, P., Gray, P. W., and Chantry, D. (2000). KSHV-encoded CC chemokine vMIP-III is a CCR4 agonist, stimulates angiogenesis, and selectively chemoattracts TH2 cells. Blood 95, 1151–1157.
Subramanian, R., Sehgal, I., D’Auvergne, O., and Kousoulas, K. G. (2010). Kaposi’s sarcoma-associated herpesvirus glycoproteins B and K8.1 regulate virion egress and synthesis of vascular endothelial growth factor and viral interleukin-6 in BCBL-1 cells. J. Virol. 84, 1704–1714.
Tammela, T., Zarkada, G., Wallgard, E., Murtomäki, A., Suchting, S., Wirzenius, M., Waltari, M., Hellström, M., Schomber, T., Peltonen, R., Freitas, C., Duarte, A., Isoniemi, H., Laakkonen, P., Christofori, G., Ylä-Herttuala, S., Shibuya, M., Pytowski, B., Eichmann, A., Betsholtz, C., and Alitalo, K. (2008). Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 454, 656–660.
Tomescu, C., Law, W. K., and Kedes, D. H. (2003). Surface downregulation of major histocompatibility complex class I, PE-CAM, and ICAM-1 following de novo infection of endothelial cells with Kaposi’s sarcoma-associated herpesvirus. J. Virol. 77, 9669–9684.
Vart, R. J., Nikitenko, L. L., Lagos, D., Trotter, M. W., Cannon, M., Bourboulia, D., Gratrix, F., Takeuchi, Y., and Boshoff, C. (2007). Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6 and G-protein-coupled receptor regulate angiopoietin-2 expression in lymphatic endothelial cells. Cancer Res. 67, 4042–4051.
Wabinga, H. R., Parkin, D. M., Wabwire-Mangen, F., and Mugerwa, J. W. (1993). Cancer in Kampala, Uganda, in 1989-91: changes in incidence in the era of AIDS. Int. J. Cancer 54, 26–36.
Wang, H. W., Trotter, M. W., Lagos, D., Bourboulia, D., Henderson, S., Mäkinen, T., Elliman, S., Flanagan, A. M., Alitalo, K., and Boshoff, C. (2004a). Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat. Genet. 36, 687–693.
Wang, L., Wakisaka, N., Tomlinson, C. C., DeWire, S. M., Krall, S., Pagano, J. S., and Damania, B. (2004b). The Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) K1 protein induces expression of angiogenic and invasion factors. Cancer Res. 64, 2774–2781.
Wang, L., and Damania, B. (2008). Kaposi’s sarcoma-associated herpesvirus confers a survival advantage to endothelial cells. Cancer Res. 68, 4640–4648.
Wang, L., Dittmer, D. P., Tomlinson, C. C., Fakhari, F. D., and Damania, B. (2006). Immortalization of primary endothelial cells by the K1 protein of Kaposi’s sarcoma-associated herpesvirus. Cancer Res. 66, 3658–3666.
Weninger, W., Partanen, T. A., Breiteneder-Geleff, S., Mayer, C., Kowalski, H., Mildner, M., Pammer, J., Stürzl, M., Kerjaschki, D., Alitalo, K., and Tschachler, E. (1999). Expression of vascular endothelial growth factor receptor-3 and podoplanin suggests a lymphatic endothelial cell origin of Kaposi’s sarcoma tumor cells. Lab. Invest. 79, 243–251.
Wigle, J. T., and Oliver, G. (1999). Prox1 function is required for the development of the murine lymphatic system. Cell 98, 769–778.
Wu, Y. H., Hu, T. F., Chen, Y. C., Tsai, Y. N., Tsai, Y. H., Cheng, C. C., and Wang, H. W. (2011). The manipulation of miRNA-gene regulatory networks by KSHV induces endothelial cell motility. Blood 118, 2896–2905.
Xie, J., Pan, H., Yoo, S., and Gao, S. J. (2005). Kaposi’s sarcoma-associated herpesvirus induction of AP-1 and interleukin 6 during primary infection mediated by multiple mitogen-activated protein kinase pathways. J. Virol. 79, 15027–15037.
Ye, F. C., Blackbourn, D. J., Mengel, M., Xie, J. P., Qian, L. W., Greene, W., Yeh, I. T., Graham, D., and Gao, S. J. (2007). Kaposi’s sarcoma-associated herpesvirus promotes angiogenesis by inducing angiopoietin-2 expression via AP-1 and Ets1. J. Virol. 81, 3980–3991.
Yoo, J., Kang, J., Lee, H. N., Aguilar, B., Kafka, D., Lee, S., Choi, I., Lee, J., Ramu, S., Haas, J., Koh, C. J., and Hong, Y. K. (2010). Kaposin-B enhances the PROX1 mRNA stability during lymphatic repro-gramming of vascular endothelial cells by Kaposi’s sarcoma herpes virus. PLoS Pathog. 6, e1001046. doi:10.1371/journal.ppat.1001046
Keywords: Kaposi’s Sarcoma, HHV-8, angiogenesis, lymphangiogenesis, viral oncogenesis, Kaposi’s Sarcoma-associated herpesvirus
Citation: DiMaio TA and Lagunoff M (2012) KSHV induction of angiogenic and lymphangiogenic phenotypes. Front. Microbio. 3:102. doi: 10.3389/fmicb.2012.00102
Received: 15 December 2011; Paper pending published: 12 January 2012;
Accepted: 01 March 2012; Published online: 30 March 2012.
Edited by:
Keiji Ueda, Osaka University Graduate School of Medicine, JapanReviewed by:
Keiji Ueda, Osaka University Graduate School of Medicine, JapanBlossom Damania, University of North Carolina at Chapel Hill, USA
Enrique A. Mesri, University of Miami Miller School of Medicine, USA
Copyright: © 2012 DiMaio and Lagunoff. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Michael Lagunoff, Department of Microbiology, University of Washington, Box 357735, Seattle, WA 98195, USA. e-mail:bGFndW5vZmZAdS53YXNoaW5ndG9uLmVkdQ==