
 Coh-ichi Nihei3
Coh-ichi Nihei3- 1 Division of Cancer Stem Cell, National Cancer Center Research Institute, Tokyo, Japan
- 2 Neurovirology Project, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan
- 3 Institute of Microbial Chemistry, Tokyo, Japan
In humans, paralytic poliomyelitis results from the invasion of the central nervous system (CNS) by circulating poliovirus (PV) via the blood–brain barrier (BBB). After the virus enters the CNS, it replicates in neurons, especially in motor neurons, inducing the cell death that causes paralytic poliomyelitis. Along with this route of dissemination, neural pathway has been reported in humans, monkeys, and PV-sensitive human PV receptor (hPVR/CD155)-transgenic (Tg) mice. We demonstrated that a fast retrograde axonal transport process is required for PV dissemination through the sciatic nerve of hPVR-Tg mice and that intramuscularly inoculated PV causes paralysis in a hPVR-dependent manner. We also showed that hPVR-independent axonal transport of PV exists in hPVR-Tg and non-Tg mice, indicating that several different pathways for PV axonal transport exist in these mice. Circulating PV after intravenous inoculation in mice cross the BBB at a high rate in a hPVR-independent manner. We will implicate an involvement of a new possible receptor for PV to permeate the BBB based on our recent findings.
Introduction
Poliovirus (PV), known to be the causative agent of poliomyelitis, is a human enterovirus that belongs to the Picornaviridae. After oral ingestion of PV in humans, the virus may suffer the low pH in stomach to decrease its viability (Ohka et al., 2007; Figure 1). Then, the virus multiplies in the alimentary mucosa (Bodian, 1955; Sabin, 1956), and moves into the bloodstream. The circulating virus invades the central nervous system (CNS) and replicates in motor neurons (MNs). Paralytic poliomyelitis occurs as a result from destruction of MNs by PV replication. In terms of routes for PV dissemination, there are three main routes; transmission from the alimentary mucosa to blood (Figure 1A; Bodian, 1955; Sabin, 1956), permeation through the blood–brain barrier (BBB) into brain (Figure 1B; Yang et al., 1997), and transmission via peripheral MNs from muscle into brain (Figure 1C; Howe and Bodian, 1942; Nathanson and Langmuir, 1963; Ren and Racaniello, 1992b; Ohka et al., 1998). The species specificity of this virus is governed by a specific cell surface molecule that serves as the PV receptor (PVR). Indeed, transgenic (Tg) mice carrying the human PVR (hPVR/CD155) gene show susceptibility to PV, although mice are generally not susceptible to the virus (Ren et al., 1990; Koike et al., 1991). Tg mice are susceptible to PV when the mice are infected via intravenous and intramuscular routes but not via oral routes (Ren et al., 1990; Koike et al., 1991, 1994a, b; Ren and Racaniello,1992a, b; Horie et al.,1994; Zhang and Racaniello, 1997). The Tg mice became susceptible to oral infection when interferon (IFN)-α/β receptor gene is disrupted (Ohka et al., 2007). The two dissemination routes through BBB and MNs are also functional in PV-sensitive Tg mice, whereas the routes through alimentary mucosa is not functional in PV-sensitive Tg mice with normal IFN-α/β response.
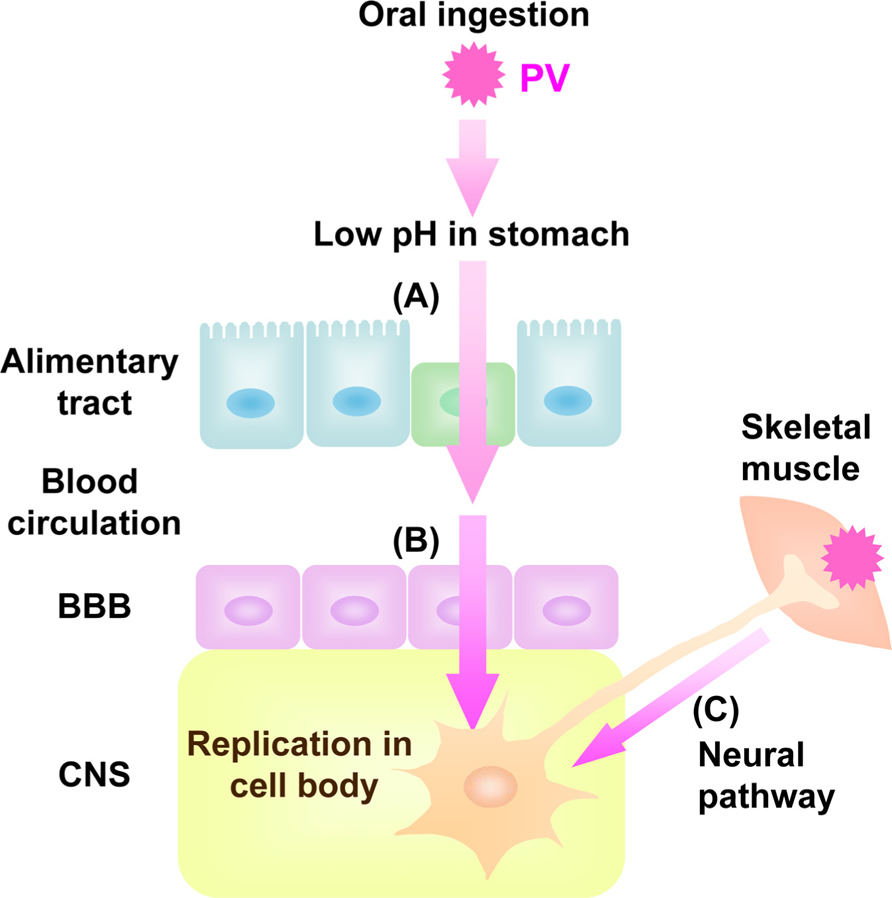
FIGURE 1. Dissemination pathway for PV in human. Oral ingested PV invades into blood through alimentary tract (A) followed by viremia. The virus in the blood permeates BBB into CNS (B). PV also invades into CNS directly by neural pathway through MNs from skeletal muscle to CNS (C).
PVR-Dependent and -Independent Retrograde Axonal Transport From Muscle to CNS
Axonal transport is a cellular process responsible for movement of mitochondria, lipids, synaptic vesicles, proteins, and organelles to and from a neuron’s cell body, through the cytoplasm of its axon. It is well known that some viruses, such as Rabies virus, hijacks the retrograde axonal transport, i.e., from synapse to cell body, to invade the CNS (Warrell and Warrell, 2004). Using a PV-sensitive Tg mouse line, we have shown that intramuscularly inoculated PV is taken up by endocytosis at synapses (Figure 1C; Ohka et al., 2004). PV is able to invade the CNS by traveling on this cargo. The vesicles containing intact PV particles are transported retrogradely along with the axon without initiating uncoating (Ohka et al., 1998). The uncoating event takes place at the cell body of the MN. We demonstrated that a fast retrograde axonal transport process is required for PV dissemination through the sciatic nerve of hPVR-Tg mice and that intramuscularly inoculated PV causes paralytic disease in a hPVR-dependent manner (Ohka et al., 1998). We also showed that hPVR-independent axonal transport of PV exists in hPVR-Tg and non-Tg mice, indicating that several different pathways for PV axonal transport exist in these mice (Ohka et al., 2009). The velocity of hPVR-independent axonal transport of PV (~0.5 μm/s) is slower than hPVR-dependent axonal transport (>0.5 to ~3.7 μm/s) in vivo. Using primary MNs isolated from these mice or rats, we demonstrated that the axonal transport of PV requires several kinetically different motor machineries (Ohka et al., 2009). The fast speed component was ≥ 1.4 μm/s, whereas the slow component was centered between 0.4 and 1.0 μm/s. Using a mutant hPVR, that has a lower affinity to cytoplasmic dynein than intact hPVR, we revealed that the fast transport relies on a system involving cytoplasmic dynein (Ohka et al., 2004).
Axonal Transport of PV In Vivo and in Cultured MNs
Unexpectedly, the hPVR-independent incorporation of PV followed by the axonal transport was not observed in cultured MNs. Thus, PV transport machineries in cultured MNs and in vivo differ in their hPVR requirements (Ohka et al., 2009). Figure 2 summarizes the PV transports depending on the cell types. In hPVR-Tg mice, most of the hPVR-containing vesicles are retrogradely transported with fast kinetics in an hPVR-dependent manner. PV is also transported in a hPVR-independent manner with slow kinetics by an unknown mechanism. Indeed, PV is transported with slow kinetics in non-Tg mice. In isolated MNs, only hPVR-dependent PV endocytosis is detected. No endocytosis of PV is detected in control cultured MNs lacking hPVR. These results suggest that the axonal trafficking of PV is carried out by several distinct pathways, and that MNs in culture and in the sciatic nerve in vivo are fundamentally different in the uptake and axonal transport of PV.
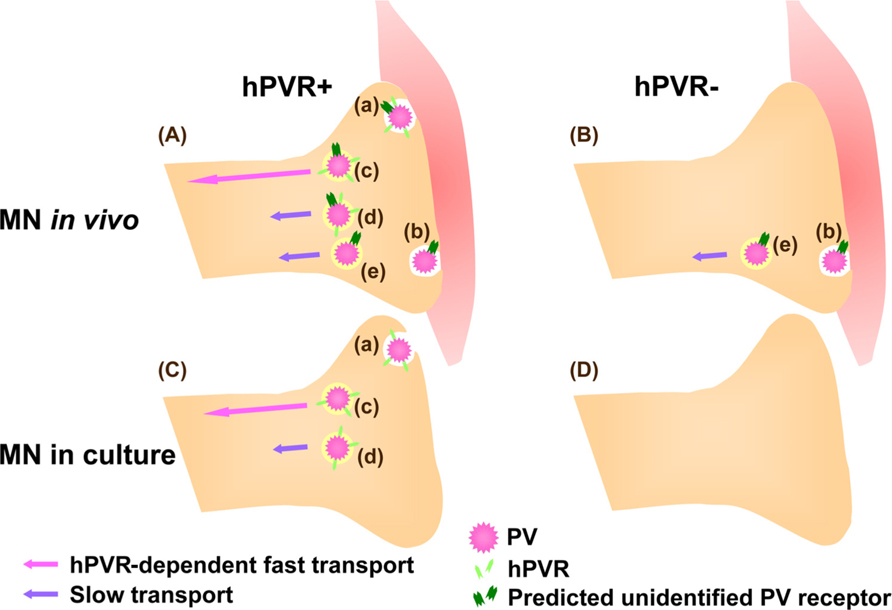
FIGURE 2. Mechanisms for hPVR-dependent and -independent transport of PV in MNs. (A) In hPVR-Tg mice, PV is endocytosed after interacting to hPVR (Aa). Most of the hPVR-containing vesicles are retrogradely transported in an hPVR-dependent manner with fast kinetics (Ac). hPVR-independent endocytosis (Ab) and transport (Ae) of PV also occur. The hPVR-independent endocytosis is possibly mediated by an unidentified PV receptor expressed at neuromuscular junctions. Alternatively, hPVR-independent endocytosis may be promoted by synaptic activity in vivo. PV-containing vesicles with or without hPVR can be retrogradely transported in an hPVR-independent manner with slow kinetics (Ad and Ae). (B) In non-Tg mice, PV is endocytosed and transported with slow kinetics in an hPVR-independent manner (Be). The hPVR-independent endocytosis is possibly mediated by the unidentified PV receptor expressed at neuromuscular junctions (Bb). (C) In cultured MNs, only hPVR-dependent PV endocytosis is detected (Ca).The hPVR-dependent transport shows fast kinetics (Cc). It might be possible that the slow component in cultured MNs (Cd) requires hPVR for endocytosis, whereas its transport may be hPVR-independent. (D) No endocytosis of PV is detected in cultured MNs lacking hPVR, possibly due to the absence of the unidentified PV receptor.
For elucidating the precise mechanisms of infection in MNs, it is important to establish a culture system closely mimicking in vivo. MNs in culture and in the sciatic nerve in vivo are different in the uptake and axonal transport of PV (Ohka et al., 2009). The differences might be due to the lack of neuromuscular junctions in MNs in culture. Furthermore, MNs are infected both at synapse side and cell body side in conventional culture system, whereas the natural infection occurs mainly at the synapse side in vivo. To reflect the conditions in vivo, we applied microfluidic culture platform made of polydimethylsiloxane (PDMS) that polarizes the growth of neural axons fluidically isolated environment (Taylor et al., 2005; Park et al., 2006), which enables infection with PV only from synapse side or cell body side. Using the system, we indicated that the infection from the synapse side intrinsically differ from the infection from the cell body side (author’s unpublished data). This system may enable co-culture of the synapse side of MNs with muscular cells to form neuromuscular junctions, which more closely mimic in vivo, and will shed light on the precise analysis of early infection with infectious agents in MNs in culture.
Transferrin Receptor-Dependent BBB Permeation
Vascular endothelial cells make up the walls that are sealed together at their edges by tight junctions. BBB does not allow the free transportation of materials including pathogens between the bloodstream and parenchyma of the CNS and blocks viral infection in the CNS. Evidences that support PV invasion of BBB by infecting endothelial cells in vivo have not been reported. PV is believed to invade the CNS through the BBB. PV permeation through BBB is hPVR-independent and fast as cationized rat serum albumin which is known to permeate the BBB at a high rate (Yang et al., 1997). Similarly, using transwell in vitro BBB model involving mouse brain capillary endothelial cells (MBEC4), we demonstrated that BBB permeation of PV is hPVR-independent and fast as that of transferrin (Figure 3, author’s unpublished data). These evidences strongly suggest the presence of a new PV receptor for BBB permeation. To elucidate the new PV receptor which interacts with PV, we analyzed the PV-interacting protein by mass spectrometry. As a result, we identified mouse transferrin receptor 1 (TfR1), a membrane bound receptor for transferrin, as a binding protein to PV. Furthermore, peptide sequences of mouse TfR1 and PV involved in the binding were identified (author’s unpublished data; the data will be published on another paper). The identified peptide of PV had a permeation activity in in vitro BBB model using MBEC4 cells cultured in transwell, and the permeation was fast as transferrin (Figure 3). Therefore, mouse TfR1 is a possible receptor of PV for the BBB permeation. As for the homology of TfR1 between mouse and human, both mouse TfR1 and human TfR1 conserve function of iron delivery and the binding site of TfR1 has higher sequence homology (89%) between human and mouse TfR1 than the full length TfR1 molecule (77%). The homology indicates that human TfR1 may function as a PV receptor for the BBB permeation in natural infection in human.
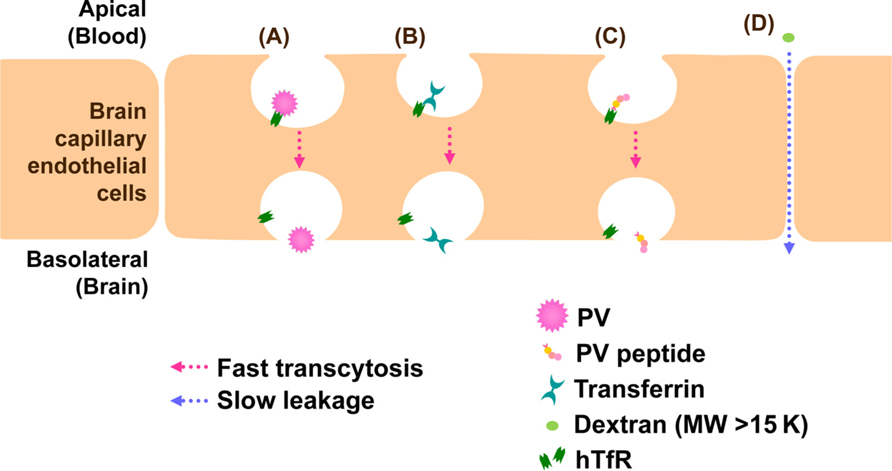
FIGURE 3. Models for BBB permeation of PV. TfR1 attaches to PV (A), transferrin (B), or PV peptide (C) and transported through brain capillary endothelial cells into brain by a fast process. On the other hand, dextran (MW > 15 K) is not transcytosed by TfR1 and leaks between cell junctions by a slow process (D).
Surprisingly, the majority of the PV related materials after the BBB permeation were intact infectious particles in the brain of hPVR-Tg mice and non-Tg mice (Yang et al., 1997). Moreover, the permeated materials contained infectious particles in in vitro BBB model (author’s unpublished data). These evidences indicate that PV retains infectivity during the BBB permeation, the transcytosis through capillary endothelial cell layers. At present, it cannot be excluded that TfR1 could actually be the long sought after molecule involved in the hPVR-independent axonal transport of PV in vivo. TfR1 is expressed in MNs in mouse spinal cord (Jeong et al., 2011), supporting the hypothesis that TfR1 exists in MNs of sciatic nerve and serves to facilitate hPVR-independent axonal transport. MNs extending neurite in culture express only a small amount of TfR1 (Nakamura et al., 2011), which is consistent with the lack of PV incorporation into MNs in culture (Ohka et al., 2009).
Dissemination Through Alimentary Mucosa
The mechanism of the dissemination through alimentary mucosa is unclear (Figure 1A). The instability of the virus in the gastric environment decreases the efficiency of the oral infection in mice (Ohka et al., 2001). Furthermore, we have shown that IFN-α/β plays an essential role in preventing PV from replicating in the intestines using hPVR-Tg mice with or without IFN-α/β receptor expression (Ohka et al., 2001). Recently, Kuss et al. (2011) reported that intestinal bacteria promote PV replication and systemic pathogenesis using hPVR-Tg mice without IFN-α/β receptor expression, indicating that intestinal microbes may enhance PV intestinal infection even in human. In terms of the receptor localization in polarized epithelial cells, hPVRα, one of the two identified membrane bound forms of hPVR, shows basolateral localization due to its basolateral sorting signal, whereas hPVRδ, another membrane bound form of hPVR, non-polarized localization (Ohka et al., 2001). hPVRs mainly localizes to the basolateral in human colon carcinoma (Caco-2) epithelia cells (Ohka et al., 2001) and human Peyer’s patches including human microfold cells (M cells), that are distinctive mucosal epithelial cells mediating immune surveillance (Iwasaki et al., 2002). Nevertheless, PV efficiently permeates alimentary mucosa from the apical side into bloodstream, suggesting the possibility that a small amount of apical hPVR or another PV receptor besides hPVR contributes to the transport. It should be noted that PV does not replicate in the alimentary tracts of hPVR-Tg mice that express high levels of hPVR in the intestinal epithelial cells after oral administration (Zhang and Racaniello, 1997) in contrast to its behavior in humans. Much work should be done to elucidate mechanisms of PV infection in and dissemination from the alimentary tract.
Conclusion
We demonstrated that axonal transport of PV includes hPVR-dependent and -independent pathways, implying the involvement of a new receptor for PV besides hPVR. We also demonstrated that PV permeates BBB at a high rate in an hPVR-independent manner, and we implicated a new possible receptor for PV to permeate the BBB. Thus, PV receptors including hPVR and new possible receptors play important roles for PV dissemination. It is important to elucidate new possible receptors in the future to clarify the precise mechanisms for PV dissemination pathways. Additionally, we revealed that the discrepancy exists between in vivo and in cultured MNs. It would be useful to establish adequate systems in culture for elucidating the transport mechanisms of PV. The discrepancy itself might be a clue for understanding the event in vivo.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to M. Sakai for analyzing BBB transport of PV in vitro, Y. Takahashi for help in mass spectrography, and Professor T. Fujii, Dr. S. Kaneda, and Dr. K. Mogi for preparing the PDMS device. We appreciate Professor G. Schiavo, S. Bohnert, and K. Deinhardt teaching the techniques for MN culture and helpful discussions. We thank A. Ohmura for breeding the mice and E. Suzuki for help in the preparation of the manuscript. This work was supported in part by Grants-in-Aid for Advanced Medical Science Research by Ministry of Education, Culture, Sports, Science and Technology (MEXT), Grant-in-Aid for Scientific Research on Priority Areas, Grant-in-Aid for Scientific Research (S), Grant-in-Aid for Young Scientists (B), Human Frontier Science Program, Special coordination funds for promoting Science and Technology, Contracted research allowance “Research and Development in a New Converting Field Based on Nanotechnology and Materials Science”by MEXT,Industrial TechnologyResearch Grant Program in ’02 from New Energy and Industrial Technology Development Organization (NEDO) of Japan, The Naito Foundation, a grant from the Ministry of Health, Labour and Welfare of Japan, and Cancer Research UK.
References
Horie, H., Koike, S., Kurata, T., Sato-Yoshida, Y., Ise, I., Ota, Y., Abe, S., Hioki, K., Kato, H., Taya, C., Nomura, T., Hashizume, S., Yonekawa, H., and Nomoto, A. (1994). Transgenic mice carrying the human poliovirus receptor: new animal models for study of poliovirus neurovirulence. J. Virol. 68, 681–688.
Iwasaki, A., Welker, R., Mueller, S., Linehan, M., Nomoto, A., and Wimmer, E. (2002). Immunofluorescence analysis of poliovirus receptor expression in Peyer’s patches of humans, primates, and CD155 transgenic mice: implications for poliovirus infection. J. Infect. Dis. 186, 585–592.
Jeong, S. Y., Crooks, D. R., Wilson-Ollivierre, H., Ghosh, M. C., Sougrat, R., Lee, J., Cooperman, S., Mitchell, J. B., Beaumont, C., and Rouault, T. A. (2011). Iron insufficiency compromises motor neurons and their mitochondrial function in irp2-null mice. PLoS ONE 6, e25404. doi: 10.1371/journal.pone.0025404
Koike, S., Aoki, J., and Nomoto, A. (1994a). “Transgenic mice for the study of poliovirus pathogenicity,” in Cellular Receptors for Animal Viruses, eds E. Wimmer and R. Weiss (New York: Cold Spring Harbor Laboratory Press), 463–480.
Koike, S., Taya, C., Aoki, J., Matsuda, Y., Ise, I., Takeda, H., Matsuzaki, T., Amanuma, H., Yonekawa, H., and Nomoto, A. (1994b). Characterization of three different transgenic mouse lines that carry human poliovirus receptor gene – influence of the transgene expression on pathogenesis. Arch. Virol. 139, 351–363.
Koike, S., Taya, C., Kurata, T., Abe, S., Ise, I., Yonekawa, H., and Nomoto, A. (1991). Transgenic mice susceptible to poliovirus. Proc. Natl. Acad. Sci. U.S.A. 88, 951–955.
Kuss, S. K., Best, G. T., Etheredge, C. A., Pruijssers, A. J., Frierson, J. M., Hooper, L. V., Dermody, T. S., and Pfeiffer, J. K. (2011). Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 334, 249–252.
Nakamura, Y., Nakamichi, N., Takarada, T., Ogita, K., and Yoneda, Y. (2011). Transferrin receptor-1 suppresses neurite outgrowth in neuroblastoma Neuro2A cells. Neurochem. Int. 60, 448–457.
Nathanson, N., and Langmuir, A. D. (1963). The Cutter incident: poliomyelitis following formaldehyde-inactivated poliovirus vaccination in the United States during the spring of 1955. III. Comparison of the clinical character of vaccinated and contact cases occurring after use of high rate lots of Cutter vaccine. Am. J. Hyg. 78, 61–81.
Ohka, S., Igarashi, H., Nagata, N., Sakai, M., Koike, S., Nochi, T., Kiyono, H., and Nomoto, A. (2007). Establishment of a poliovirus oral infection system in human poliovirus receptor-expressing transgenic mice that are deficient in alpha/beta interferon receptor. J. Virol. 81, 7902– 7912.
Ohka, S., Matsuda, N., Tohyama, K., Oda, T., Morikawa, M., Kuge, S., and Nomoto, A. (2004). Receptor (CD155)-dependent endocytosis of poliovirus and retrograde axonal transport of the endosome. J. Virol. 78, 7186–7198.
Ohka, S., Ohno, H., Tohyama, K., and Nomoto, A. (2001). Basolateral sorting of human poliovirus receptor alpha involves an interaction with the mu1B subunit of the clathrin adaptor complex in polarized epithelial cells. Biochem. Biophys. Res. Commun. 287, 941–948.
Ohka, S., Sakai, M., Bohnert, S., Igarashi, H., Deinhardt, K., Schiavo, G., and Nomoto, A. (2009). Receptor-dependent and -independent axonal retrograde transport of poliovirus in motor neurons. J. Virol. 83, 4995–5004.
Ohka, S., Yang, W. X., Terada, E., Iwasaki, K., and Nomoto, A. (1998). Retrograde transport of intact poliovirus through the axon via the fast transport system. Virology 250, 67–75.
Park, J. W., Vahidi, B., Taylor, A. M., Rhee, S. W., and Jeon, N. L. (2006). Microfluidic culture platform for neuroscience research. Nat. Protoc. 1, 2128–2136.
Ren, R., and Racaniello, V. R. (1992a). Human poliovirus receptor gene expression and poliovirus tissue tropism in transgenic mice. J. Virol. 66, 296–304.
Ren, R., and Racaniello, V. R. (1992b). Poliovirus spreads from muscle to the central nervous system by neural pathways. J. Infect. Dis. 166, 747–752.
Ren, R. B., Costantini, F., Gorgacz, E. J., Lee, J. J., and Racaniello, V. R. (1990). Transgenic mice expressing a human poliovirus receptor: a new model for poliomyelitis. Cell 63, 353–362.
Sabin, A. B. (1956). Pathogenesis of poliomyelitis: reappraisal in the light of new data. Science 123, 1151–1157.
Taylor, A. M., Blurton-Jones, M., Rhee, S. W., Cribbs, D. H., Cotman, C. W., and Jeon, N. L. (2005). A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat. Methods 2, 599–605.
Warrell, M. J., and Warrell, D. A. (2004). Rabies and other lyssavirus diseases. Lancet 363, 959–969.
Yang, W. X., Terasaki, T., Shiroki, K., Ohka, S., Aoki, J., Tanabe, S., Nomura, T., Terada, E., Sugiyama, Y., and Nomoto, A. (1997). Efficient delivery of circulating poliovirus to the central nervous system independently of poliovirus receptor. Virology 229, 421–428.
Keywords: axonal transport, poliovirus, poliovirus receptor, blood–brain barrier
Citation: Ohka S, Nihei C, Yamazaki M and Nomoto A (2012) Poliovirus trafficking toward central nervous system via human poliovirus receptor-dependent and -independent pathway. Front. Microbio. 3:147. doi: 10.3389/fmicb.2012.00147
Received: 15 December 2011; Accepted: 27 March 2012;
Published online: 18 April 2012.
Edited by:
Akio Adachi, The University of Tokushima Graduate School, JapanReviewed by:
Chiaho Shih, Academia Sinica, TaiwanSatoshi Koike, Tokyo Metropolitan Institute of Medical Science, Japan
Minetaro Arita, National Institute of Infectious Diseases, Japan
Copyright: © 2012 Ohka, Nihei, Yamazaki and Nomoto. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits noncommercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Seii Ohka, Neurovirology Project, Tokyo Metropolitan Institute of Medical Science, 2-1-6 Kamikitazawa, Setagaya-ku, Tokyo 156-8506, Japan. e-mail:b2hrYS1zaUBpZ2FrdWtlbi5vci5qcA==