 Burcu Ünal1
Burcu Ünal1 Verlin Ryan Perry2†
Verlin Ryan Perry2† Mili Sheth2† Vicente Gomez-Alvarez1
Mili Sheth2† Vicente Gomez-Alvarez1 Kuk-Jeong Chin2
Kuk-Jeong Chin2 Klaus Nüsslein1*
Klaus Nüsslein1*- 1 Department of Microbiology, University of Massachusetts, Amherst, MA, USA
- 2 Department of Biology, Georgia State University, Atlanta, GA, USA
Microbial methane from coal beds accounts for a significant and growing percentage of natural gas worldwide. Our knowledge of physical and geochemical factors regulating methanogenesis is still in its infancy. We hypothesized that in these closed systems, trace elements (as micronutrients) are a limiting factor for methanogenic growth and activity. Trace elements are essential components of enzymes or cofactors of metabolic pathways associated with methanogenesis. This study examined the effects of eight trace elements (iron, nickel, cobalt, molybdenum, zinc, manganese, boron, and copper) on methane production, on mcrA transcript levels, and on methanogenic community structure in enrichment cultures obtained from coal bed methane (CBM) well produced water samples from the Powder River Basin, Wyoming. Methane production was shown to be limited both by a lack of additional trace elements as well as by the addition of an overly concentrated trace element mixture. Addition of trace elements at concentrations optimized for standard media enhanced methane production by 37%. After 7 days of incubation, the levels of mcrA transcripts in enrichment cultures with trace element amendment were much higher than in cultures without amendment. Transcript levels of mcrA correlated positively with elevated rates of methane production in supplemented enrichments (R2 = 0.95). Metabolically active methanogens, identified by clone sequences of mcrA mRNA retrieved from enrichment cultures, were closely related to Methanobacterium subterraneum and Methanobacterium formicicum. Enrichment cultures were dominated by M. subterraneum and had slightly higher predicted methanogenic richness, but less diversity than enrichment cultures without amendments. These results suggest that varying concentrations of trace elements in produced water from different subsurface coal wells may cause changing levels of CBM production and alter the composition of the active methanogenic community.
Introduction
Coal bed methane (CBM) is a form of natural gas that is trapped at large quantities in deep unmineable coal bed basins and produced from boreholes (Thielemann et al., 2004). CBM is held at coal internal surfaces by the combination of physical sorption and hydrostatic pressure of subsurface water. Gas extraction is accomplished by pumping water from subsurface coal bed basins to the surface through drilled access wells that end within the coal bed. As water is pumped to the surface, methane is no longer held by the hydrostatic pressure in micro pore structures of coal beds and migrates from the subsurface coal into the water stream flowing to the well. At the surface, methane is separated from produced water and extracted to be used as an energy source (Wheaton and Donato, 2004). Seventy percentage of the world’s CBM production is supplied by the United States while the remainder is produced by Australia, India, Canada, China, United Kingdom, Columbia, Russia, Ukraine, and Austria primarily from shallow to deep subsurface coal beds (Flores et al., 2008).
The production of CBM has increased dramatically, and currently supplies about 10% of the total natural gas produced annually in the U.S. (Fletcher, 2005; Petzet, 2005). The most active methane producing coal seams found in the U.S. are located in Alabama/Mississippi, Southern Colorado/New Mexico, and Montana/Wyoming (Harris et al., 2008). CBM is produced either via microbial degradation of organic molecules (i.e., biogenic gas) or via transformation of organic matter by geochemical processes (i.e., thermogenic gas; Flores et al., 2008). Coal deposits of the Powder River Basin (PRB) in Wyoming have emerged as having some of the most active biogenic gas production, with methane as the main constituent (85–90%; Flores et al., 2008; Strąpoć et al., 2008). Coal beds in the PRB basin are entirely continental, and are deposited in fresh water systems with insignificant concentrations of chloride (Van Voast, 2003). The values and associated fractionation factors indicate microbial CO2 reduction as the overwhelming pathway for methane generation in the PRB (Flores et al., 2008).
Current knowledge of the indigenous microbiota residing in coal bed basins, of the role they play in mediating coal biodegradation to methane, and of the biogeochemical and physical factors that may influence biological methanogenesis is limited. Jones et al. (2010) recently reported variations in the rates of biogenic methane production among CBM wells in the Gulf Coast Basin, Texas. Possible factors causing this interwell variability remain unknown. Only recently have subsurface microbial communities been characterized that contribute to methane production from unmined or abandoned coal beds (Shimizu et al., 2007; Green et al., 2008; Klein et al., 2008; Li et al., 2008; Midgley et al., 2010; Penner et al., 2010; Beckmann et al., 2011; Guo et al., 2012). Other studies determined the potential of these microbes to generate methane (Green et al., 2008; Harris et al., 2008; Jones et al., 2008; Ulrich and Bower, 2008), as well as to characterize the metabolic pathways involved in generating methane in subsurface systems (Strąpoć et al., 2008; Jones et al., 2010).
Analyses of archaeal diversity based on 16S rRNA genes in subsurface coal bed basins have revealed the presence of diverse assemblages of acetoclastic, methylotrophic, and hydrogenotrophic methanogens. For example, the hydrogenotrophic genus Methanoculleus was found together with the methylotrophic genus Methanolobus in groundwater samples from Ishikari coal field, Hokkaido, Japan (Shimizu et al., 2007), and Methanolobus dominated production water in the Ordos Basin, China (Guo et al., 2012). Members of Methanosarcina spp., known to use a wide range of substrates (H2/CO2, acetate, methanol, and methylamines) were detected in a methanogenic enrichment from coal beds in Alberta, Canada (Penner et al., 2010). While hydrogenotrophic Methanobacterium ssp. were found in enrichment cultures from the Jharia coal field, India (Singh et al., 2012), and Methanosarcinales (Green et al., 2008), Methanobacteriales, and Methanococcales (Klein et al., 2008) were discovered in the PRB, Wyoming. Some studies questioned whether members of the methanogenic community could be enhanced for methane production by manipulating temperature, pH, coal particle surface, electron acceptors or donors, or rapid release of organic substrates (Green et al., 2008; Harris et al., 2008; Jones et al., 2010; Penner et al., 2010). However, these attempts involved addition of electron acceptors or donors that might limit methanogenesis by diverting electrons away from methanogens, or, in case of enhanced organic matter release might limit methanogens by the creation of toxic conditions for methane generation within coal beds (Jones et al., 2010).
Microbial communities in subsurface coal beds likely feed on organic and inorganic sources either deposited within the coal during coalification (coal formation process) or those that have migrated from the surface along groundwater flow paths (Krumholz et al., 1997; Ulrich and Bower, 2008). Furthermore, trace elements (e.g., iron, nickel, cobalt, molybdenum, copper, zinc, tungstate, selenium, and boron) are crucial along with macronutrients (C, N, P, etc.), for proper growth and metabolism of microorganisms (Goodwin et al., 1990; Takashima et al., 1990; Lengeler et al., 1999). Various trace element demands of microorganisms are determined by the enzymes and the proteins needed for their particular metabolisms such as CO2 assimilation, catabolism of organic compounds, and respiration (Zhang and Gladyshev, 2009). Iron (Fe), nickel (Ni), cobalt (Co), zinc (Zn), copper (Cu), molybdenum (Mo), and tungstate (W) are known as crucial trace elements for enzymatic activity in the methanogenic system (Takashima et al., 1990; Zhang and Gladyshev, 2010; Glass and Orphan, 2012). Requirement and optimal concentrations of trace elements for diverse microbial metabolisms are commonly studied with pure cultures only, which are orders of magnitude higher than in situ concentrations in most natural environments (Glass and Orphan, 2012). Although, some studies tested the effects of trace elements on methanogenesis in different anaerobic environments such as peatlands, maize silage, food industrial waste, and activated waste water sludge (Burgess et al., 1999; Gonzalez-Gil et al., 1999; Basiliko and Yavitt, 2001; Feng et al., 2010; Pobeheim et al., 2010), no studies have investigated whether methanogenesis in coal bed basins is influenced by trace elements. Two geochemical investigations of trace element chemistry across the PRB in Wyoming did not find consistent trends of trace element concentrations in CBM production waters (McBeth et al., 2003; Jackson and Reddy, 2007).
The aim of this study was to investigate the effect of trace element amendments on methanogenic activity and diversity in an enrichment culture from subsurface produced CBM water. This study was designed in three phases to understand (i) the effect of varying concentrations of trace element mixtures on methane production, (ii) the effect of trace element addition on transcript levels of the gene mcrA, which encodes the α-subunit of methyl coenzyme M reductase, and (iii) the effect of trace element addition on the community structure of metabolically active methanogens.
Materials and Methods
Field Sampling, Analytical Chemistry, and DNA Extraction of CBM Produced Water
Three separate samples of CBM produced water were collected from a CBM well that is located within the Wall coal seam formation at a depth of 376 m below surface in the southern PRB, Wyoming, USA. The water sample was filtered through a series of 0.45 and 0.22 μm filters into gas tight Nalgene sample bottles, and acidified to pH <2 with 1% nitric acid (Trace metal grade, Fisher Scientific, Pittsburgh, PA, USA). Trace element concentrations in filtered water samples were quantified by inductively coupled plasma mass spectrometry (ICP-MS).
For the extraction of DNA 200 ml of water sample were drawn into a sterile 0.22 μm cellulose acetate filter (Millipore, Billerica, MA, USA) inside a presterilized 25-mm Swinnex filter holder, fast frozen on dry ice, and transported to the laboratory for storage at −80°C until further processing. Total DNA was extracted from the filters with the PowerWater DNA isolation kit according to manufacturer’s instructions (MoBio Laboratories, CA, USA).
Microcosms
Microcosms were initiated immediately after sampling from an actively gas-producing well stream by inoculating CBM produced water with pea-sized sub-bituminous coal sampled from the PRB. Ten replicates were set up with 30 ml of produced water and 5 g of coal in 120 ml anoxic serum vials capped with butyl rubber stoppers under a headspace of N2/CO2 [80:20 (v/v)] and incubated at 37°C without agitation. Yeast extract was not added, as coal is the only organic matter to be used for microbial growth. A separate set of microcosms was initiated from the same well stream by inoculating only CBM produced water in basal salt medium. All microcosms were set up anaerobically (Balch and Wolfe, 1976) in 160 ml of anoxic serum vials capped with butyl rubber stoppers. Upon arrival at the laboratory, various electron acceptors were tested and microbial growth and methane production were observed in microcosms incubated at 37°C without agitation. The electron donor H2 (H2/CO2 (80/20 (v/v)) was replaced with 50 mM sodium acetate or 5 mM methanol with N2/CO2 as the headspace gas.
Enrichments
Long term methanogenic enrichment cultures were established from CBM produced water by transferring cultures grown on H2/CO2 [80/20 (v/v)] after 50–80 days of incubation during a period of 2.5 years. Enrichment cultures were established in 160 ml serum vials with H2/CO2 [80/20 (v/v)] and basal salt medium consisting of the following components; 15 ml of KH2PO4 (27.2 g/l), 15 ml Na2HPO4·2H2O (35.6 g/l), 12.5 ml of a mixture of NH4Cl (24 g/l), MgCl2·6H2O (8 g/l), and NaCl (24 g/l), and 40 ml of NaHCO3 (100 g/l); 10 ml vitamin solution (Biotin 2.0 mg, Folic acid 2.0 mg, Pyridoxine-HCl 10.0 mg, Thiamine-HCl·2 H2O 5.0 mg, Riboflavin 5.0 mg, Nicotinic acid 5.0 mg, D-Ca-pantothenate 5.0 mg, p-aminobenzoic acid 5.0 mg, Thioctic acid 5.0 mg/l). To this 1 ml of a well defined trace element solution (SL-10) was added, that contained (per liter) 1.5 g of FeCl2·4H2O, 70 mg of ZnCl2, 100 mg of MnCl2·4H2O, 6 mg of H3BO3, 190 mg of CoCl2·6H2O, 2 mg of CuCl2·2H2O, 24 mg of NiCl2·6H2O, 36 mg of Na2MoO4·2H2O, 15 mg of Na2WO4, 15 mg of Na2SeO3·5H2O, and 10 ml of 25% HCl (Widdel et al., 1983). Resazurin (2.0 g/l) was added as a reducing indicator. The medium pH was adjusted to 7.2 ± 0.2 with dilute HCl. Cysteine at 0.5 mg ml−1 HCl was added into medium as the sole reductant. The commonly used Na2S·9H2O was not preferred as a reductant to avoid a reaction with metals in the medium. It was observed that there is no significant effect on methane production without addition of Na2S·9H2O (data not shown).
Trace Element-Amended Enrichment Cultures
In the first phase of this study, the effects of varying concentrations of trace elements on methane production were evaluated. Initially, 10% (v/v) actively growing log-phase enrichment was incubated in 160 ml of anoxic serum vials capped with butyl rubber stoppers with fresh medium described in the Section “Enrichments.” Yeast extract was not added, since it is a source of trace elements and its element content is unknown. The headspace of serum bottles was aseptically flushed with H2/CO2 [80/20 (v/v)]. No external electron acceptors were supplied and CO2 was the sole electron acceptor. Increasing concentrations of trace element mix solutions (1×-TES, 2.5×-TES, and 5×-TES) were added into enrichment cultures prepared in triplicates. 1×-TES is the concentration of the SL-10 trace element solution for a mixture of eight trace elements (iron, nickel, cobalt, molybdenum, zinc, manganese, boron, and copper), which is given in detail above. Enrichments and abiotic controls were incubated for 6 weeks under strict anaerobic condition in the dark at 37 ± 1°C without agitation. Glassware utilized in the study was pre-cleaned and treated with 10% nitric acid over night and baked at 550°C for 6 h.
For the second and third phases of this study, 10% (v/v) actively growing log-phase enrichment was incubated into fresh medium free of trace elements three times over a period of four and a half months. The last transfer culture was used as seed for 1×-TES amended and unamended enrichment cultures. Medium preparation and incubation conditions were the same as in phase one. Enrichments were set up in 10 replicates and incubated over 5 weeks. The medium pH was adjusted to 7.2 ± 0.2 with dilute HCl, and checked every week. Every week two duplicate incubations were sacrificed for RNA isolation and total cell counting.
Total Cell Enumeration with DAPI Staining
Total cell numbers were estimated with 4′, 6-diamidino-2-phenylindole (DAPI) nucleic acid staining (Becerra et al., 2009) in 1×-TES amended and unamended enrichment cultures and in CBM produced water samples. A sequence of 40 randomly chosen microscopic fields across each slide was examined under 1000× oil-immersion magnification using an epifluorescence microscope (Nikon Eclipse E400) equipped with a Hamamatsu digital camera to determine the average number of cells in the samples.
Detection of Methane Production
Methane accumulation in the headspace was monitored and quantified for all microcosms and enrichments by gas chromatography using a GC-17A (Shimadzu, Co., Kyoto) equipped with an Equity-1 column (30 m × 0.53 mm ID, 3.0 μm, Supelco, St. Louis, MO, USA) and a flame ionization detector with helium as the carrier gas. The injector and column temperatures were adjusted to 100°C and the detector was held at 125°C. Certificated standard CH4 (Fisher Scientific, Pittsburgh, PA, USA) was used for calibration. Methane concentration in the headspace was calculated by comparing peak areas of sample and standard.
Extraction of Total RNA and mRNA
Total RNA was extracted from 1×-TES amended and unamended enrichment cultures at 2, 7, 14, 21, and 28 days using the method described by Chin et al. (2008) with some modifications. The complete procedure was performed at 2°C or on ice. A volume of 2.0 ml of enrichment culture was anaerobically removed from a serum bottle and transferred to 2.0 ml RNase-free screw-cap tubes. Tubes were centrifuged for 5 min and supernatant was decanted. Approximately 0.6 g of 0.1 mm diameter glass beads, 0.6 ml TPM buffer (50 mM Tris–HCl, 1.7% polyvinylpyrrolidon, 20 mM MgCl2·6H2O, in DEPC-treated water) and 2 μl yeast tRNA (mRNA carrier) were added to the cell pellet. The tubes were shaken upside down for 10 min, followed by ballistic cell destruction for 1 min at 4200 rpm using a mini-bead beater (BioSpec Products; Bartlesville, OK, USA). Cell debris and glass beads were concentrated by centrifugation for 3 min. The supernatant was transferred to a tube containing 1 μl RNase inhibitor (Ambion, Austin, TX, USA). The pellet was resuspended in 0.6 ml phenol-saturated lysis buffer [50 mM Tris/HCl (pH 7.0), 50 mM EDTA, 1% (w/v) sodium dodecyl sulfate (Ambion), and 6% water-saturated phenol. After an additional round of bead beating at the same conditions, the tubes were exposed to centrifugation for 5 min and the supernatant was pooled with the supernatant of the first round of bead-beating. A volume of 0.6 ml phenol (pH 4.3) was added to the pooled supernatant, the tubes were vortexed for 40 s and exposed to centrifugation for 5 min. The supernatant was extracted with phenol–chloroform–isoamylalcohol [pH 7.8–8.2; 25:24:1 (v/v)] and chloroform–isoamylalcohol [24:1 (v/v)]. The aqueous phase was transferred to a fresh 2 ml tube containing 1/10th volume of 3 M NaAc (pH 5.2) and 2 μl of linear acrylamide (5 mg/ml; Ambion), and filled with 100% cold ethanol up to a final volume of 2 ml. The tubes were invert mixed and contents concentrated by centrifugation for 30 min. The pellets were washed with 0.5 ml 70% cold ethanol, invert mixed, and again centrifuged for 10 min. The pellet was dried in a desiccator for 5 min and resuspended in water. RNA was treated with RNase free-DNase (Ambion) to remove DNA contamination. DNase treated RNA was further purified by precipitating with 1/10th volume 3 M NaAc (pH 5.2), and 4 volumes of cold ethanol followed by centrifugation for 20 min. The pellet was washed twice with 0.5 ml 70% cold ethanol, dried, and resuspended in water. Enrichment of mRNA was performed with DNase treated and purified RNA using the MICROB Express purification system (Ambion). DNA contamination was checked with agarose gel electrophoresis following reverse transcription-polymerase chain reaction (RT-PCR) by performing control experiments in which no reverse transcriptase was added to the isolated RNA before the PCR step. RNA concentration was determined by absorption at 260 nm with a Biophotometer (Eppendorf, Hamburg, Germany). Purified RNA was stored at -80°C.
Reverse Transcription-Polymerase Chain Reaction
The cDNA of mRNA for the gene mcrA was generated with the primer mcrA-rev, (Steinberg and Regan, 2008) with 0.5 μg mRNA as the template. Multiscribe Reverse Transcriptase™ (250 U; Applied Biosystems) was used according to the manufacturer’s instructions. The conditions used in the cDNA synthesis program were as follows: initial incubation at 25°C for 10 min, incubation at 37°C for 120 min, enzyme inactivation at 85°C for 5 s, followed by rapid cooling to 4°C. cDNA samples were stored at −20°C until use. Methyl-coenzyme M reductase (MCR) is the essential and unique enzyme for methane production. It catalyzes the final step of methanogenesis, which reduces a methyl group linked to coenzyme M to methane (Ellerman et al., 1988). Because this enzyme is found in all known methanogens and is absent from non-methanogenic Archaea and Bacteria (Chistoserdova et al., 1998; Thauer, 1998; Bapteste et al., 2005), the mcrA gene is used as a specific functional marker to detect methanogens (Weil et al., 1988; Hallam et al., 2003). Previous studies have suggested that the phylogeny of mcrA follows the 16S rRNA phylogeny within various environments, and thus has been used as an alternative phylogenetic tool to detect and identify methanogens (Springer et al., 1995; Lueders et al., 2001; Luton et al., 2002; Friedrich, 2005). The MCR operons are found in two forms; MCRI (mcrBDCGA) which encodes mcrA and MCRII (mrtBDGA or mrtBGA) which encodes mrtA. Although, MCRI exists in all methanogens, MCRII is only found in members of the orders Methanobacteriales and Methanococcales (Lehmacher and Klenk, 1994; Bult et al., 1996; Reeve et al., 1997).
Cloning, Sequencing, and Phylogenetic Analysis
Extracted genomic DNA was used as template for PCR amplification of 16S rRNA genes using archaea-specific primers ARC21F (DeLong, 1992) and 1492R (Lane, 1991). PCR cycles were as follows: 1 cycle at 94°C for 5 min, 30 cycles at 94°C for 1 min, 58°C for 1.5 min and 72°C for 2 min, and a final extension step at 72°C for 10 min. In addition, three mRNAs clone libraries were generated from mcrA-specific RT-PCR amplicons (491 bp length amplified with primers mlas and mcrA-rev; Steinberg and Regan, 2008) obtained from trace element amended (TES-T1) and unamended enrichment cultures (NO-TES) after 7 days of incubation, and from startup enrichment cultures designated as time zero (T0). The purified RT-PCR amplicons from enrichment cultures and Archaeal 16S rRNA genes from produced water were ligated into the pGEM®-T Easy Vector (Promega) and transformed into Escherichia coli JM109. Positive clones were verified by PCR amplification with vector specific primers M13F and M13R according to the protocol supplied by manufacturer. Clones were randomly selected, and plasmid inserts were sequenced with the M13F primer by Sanger sequencing. Sequences were checked manually and edited for ambiguous bases using the software Chromas sequence viewer (Technelysium, Tewantin, Australia), and multiple sequence alignments were created using ClustalX v.1.83 (Thompson et al., 1997). Archaeal 16S rRNA gene sequences were assigned to an Operational Taxonomic Unit (OTU) using the software package DOTUR based on ≥97% similarity (Schloss and Handelsman, 2005). Phylogenetic and taxonomic analyses were conducted using a representative sequence from each OTU. Phylogenetic trees based on the mcrA amino acid sequence were constructed using the Neighbor-Joining method (Saitou and Nei, 1987) with the “percentage of acceptance mutations distance correction model (PAM)” provided by the software package MEGA4.1 (Kumar et al., 2008).
To estimate the confidence of tree topology, bootstrap resampling analysis for 1000 replicates was performed with the Neighbor-Joining method. After sequences were assigned to OTUs, the frequency data at species level were used to construct rarefaction curves and to calculate diversity and richness indices using DOTUR. Estimates of phylotype richness were calculated according to the Chao1 estimator that takes into account the number of different OTUs in a sample, and the abundance-based coverage estimate (ACE). Species richness and evenness were calculated using the Shannon diversity index (HSHANNON) and the Simpson’s index (HSIMPSON), which takes into account the level of phylotype dominance.
Real-Time PCR Quantification
Dilution series of purified mcrA RT-PCR amplicons were used as calibration standards for the real-time PCR quantification. The purified RT-PCR products were quantified and prepared for serial dilution as described previously (Chin et al., 2004b). The detection limits were determined from two independent measurements with dilution series of purified RT-PCR products (standards) in real-time PCR (108–101 target molecules per reaction). The copy numbers of unknown samples were calculated after real-time amplification from the linear regression of the standard curve. The cDNA generated with mcrA gene-specific primers was quantified with real-time quantitative PCR using SYBR Green. The levels of mcrA transcripts (per μg mRNA) were determined in enrichment cultures for which methane production was measured in parallel. All reactions were carried out in 20 μl reaction volume containing 0.02 units of iProof High Fidelity polymerase (BioRad), buffer, 2.5 pmol of each dNTP, bovine serum albumin, and 20 pmol of each primer of the pair mlas-mcrArev (Steinberg and Regan, 2008). The temperature profile was composed of an initial incubation at 98°C for 1 min, followed by 35 cycles of 98°C for 10 s, 58°C annealing for 10 s, 65°C extension for 32 s, and a final extension at 65°C for 6 min. Dissociation curve analysis to detect the presence of primer dimers and non-specific amplification peaks was performed after the final extension. The size of the PCR products was checked with agarose gel electrophoresis, and the specificity of PCR products was verified by sequence analysis of the clone library. Quantitative analysis of the cDNA was carried out with the 7500 Real-Time PCR System (Applied Biosystem, Foster City, CA, USA) using 7500 Real-Time PCR System Sequence Detection software (v 1.3.1). The precision as well as the reproducibility of quantification were carefully optimized, and PCR products were checked for their correct lengths as described previously (Chin et al., 2004b).
Nucleotide Sequence Accession Numbers
The sequences for mcrA mRNA clones and for archaeal 16S rRNA gene clones generated in this study were deposited in the GenBank database under the following accession numbers: JQ917180 to JQ917194 for all mcrA sequences, and JQ917195 to JQ917203 for archaeal 16S rRNA sequences.
Statistical Analyses
Statistical comparisons were performed using one-way ANOVA followed by the all pairwise multiple comparison procedures (Holm-Sidak method) on the probability level P < 0.001. Data analyses were performed using the software package SYSTAT 11 (Systat Software, 2004).
Results
CBM Produced Water Characterization
In situ conditions at well depth indicated a pH value of 6.7 and a temperature of 29.6°C. Physicochemical analysis of the CBM produced water included: dissolved inorganic carbon (DIC) 13.161 mm/kg; F, 0.68 mM; Cl, 0.31 mM; SO4, 0.07 mM; Na, 6.24 mM; Mg, 0.12 mM; K, 0.13 mM; Ca, 0.26 mM; Ba, 0.365 mM; total cations at 7.11 mM, and total anions at 6.65 mM. Trace element concentrations (μg/l) were B, 32.05; Al, 0.100; V, 0.49; Mn, 6.84; Fe, 14.92; Co, 0.03; Ni, 0.76; Cu, 0.48; Zn, 21.61; As, 0.07; Mo, 0.62; Ag, 0.07; Ba, 329.100; Tl, 0.01; Pb, 0.02; and U, 0.01. The concentrations of trace elements in 1×-TES, which was the concentration of trace elements in the SL-10 solution, was far greater than in situ concentrations except for boron, which was at a lower concentration in trace element enrichment cultures (21.3 μg/l). The calculated concentrations of elements in 1×-TES enrichment cultures were 785 times more in Fe, 132 times more in Ni, four orders magnitude more in Co, 476 times more Mo, 73 times more Mn, 28 times more Zn and 31 times more Cu than in situ concentrations.
Methanogenic Potential and Phylogenetic Analysis of Methanogens in CBM Produced Water
Total cell number in the CBM produced water sample was determined at 3.2 × 105 by DAPI staining, which is within midrange of cell numbers observed in other deep aquifers (Pedersen, 1993). Significant amounts of methane were produced in microcosms inoculated with pie-size coals (9.2 ± 0.6 μmol/ml) after 6 months of incubation. This indicates that the methanogenic community had the ability to degrade coal into methane.
Since coal is potentially the source of trace elements (Jackson and Reddy, 2007), we did not use coal as a carbon source to test the effects of trace elements on methanogenesis. For controlled and reproducible conditions, the CBM produced water sample was supplemented with a methanogen specific medium and amended with electron acceptors such as acetate, methanol, or CO2. After 5 months of incubation, methane production was detected only in microcosms with H2/CO2 (80/20 v/v) at the cumulative amount of 7.7 μmol/ml, and subsequent enrichments were continued based on this set of cultures.
An archaeal 16S RNA gene clone library was constructed to identify the methanogens in the CBM produced water sample. Nine OTUs were identified within the 67 clonal sequences based on a “cut-off” value of ≥97% nucleotide sequence identity. The archaeal community was dominated by the genus Methanobacterium sp. (60%) with 40% identified as uncultured archaea (Table 1). A total of 25 sequences were affiliated with Methanobacterium sp. GH (96–99% similarity) which was previously isolated from sediments of the Gahai Lake in China (Zhu et al., 2011). Fifteen sequences were affiliated with Methanobacterium sp. SA-12 (97%) found in a subsurface gold mine (unpublished data, GenBank accession number AY695843.1) and to an uncultured clone (97%) from a minerotrophic fen (Cadillo-Quiroz et al., 2008). Five clones were affiliated with an uncultured archaeon clone (93%) from a gas hydrate production research well, Canada (unpublished data); four clones were affiliated (92–97%) with clones obtained from the deep aquifer of gold mines, South Africa (Takai et al., 2001); 10 clones were affiliated (96%) with clones from deep subsurface sediments in the Pacific (Biddle et al., 2006; Zhang et al., 2010); and eight clones were affiliated with a clone (91%) from hydrothermal sediments in Japan (Nunoura et al., 2010).
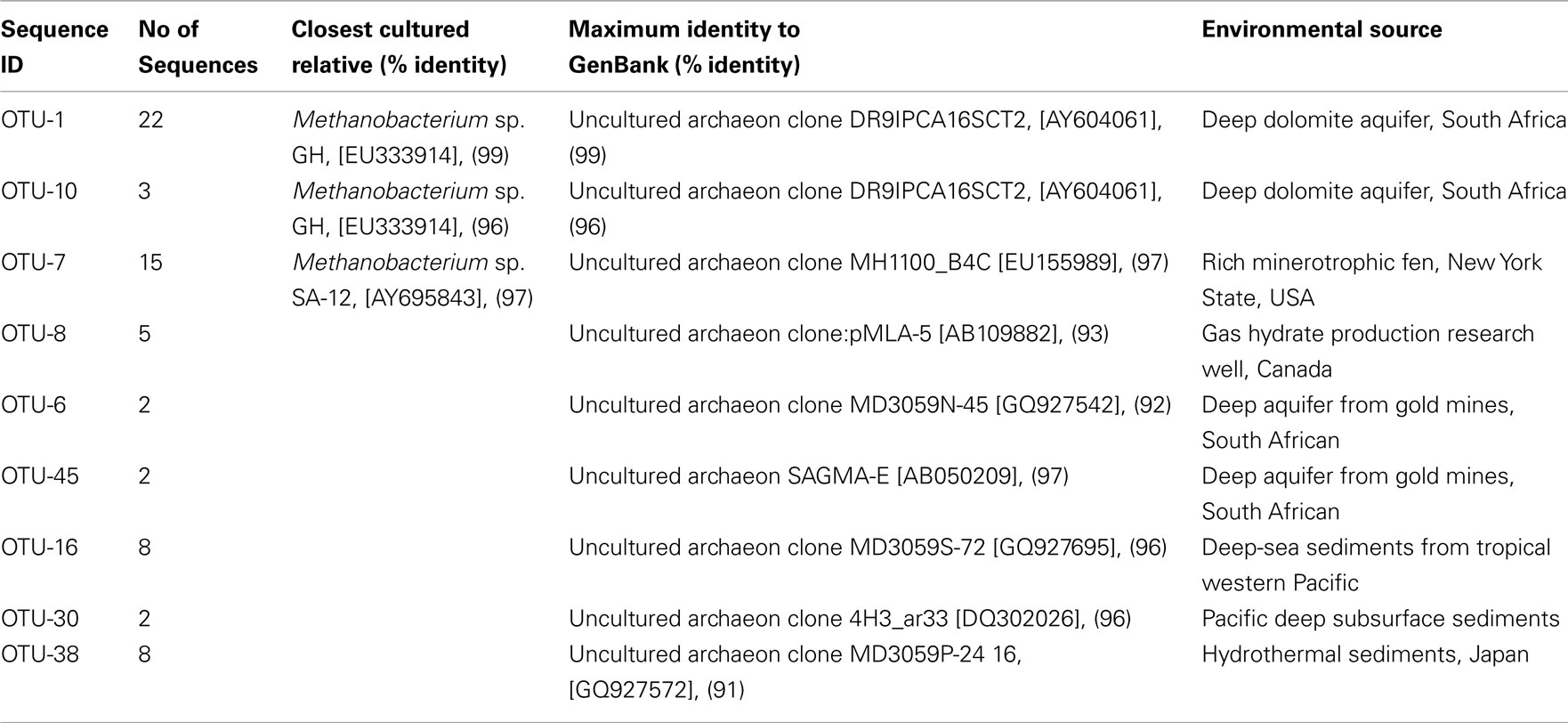
Table 1. Phylogenetic affiliations of archaeal phylotypes based on 16S rRNA gene sequences in the produced water from a CBM well in the PRB.
Effect of Increasing Concentrations of Trace Elements on Methane Production
The effect of increasing concentrations of trace elements (1×-TES, 2.5×-TES, and 5×-TES) on methane production in CBM produced water enrichments was tested weekly for a total duration of 42 days. The differences in the average values of cumulative methane production among the enrichment cultures amended with 1× and 2.5× trace element concentrations were statistically significant (P < 0.001), when compared with unamended enrichment cultures. At the end of the incubation period, the level of methane production was significantly lower (8.52 μmol/ml) in enrichment cultures without addition of trace elements than in enrichment cultures with addition of trace elements (11.7 μmol/ml; Figure 1A). This suggests that methanogenic activity became diminished under trace element limitation. It was also observed that there was a significant negative effect on methane production in enrichment cultures with higher concentration of trace elements (5×-TES) during 42 days of incubation (P = 0.822). Here we determined that the undiluted, 1×-TES concentration of trace element solution was the optimum concentration for methane production. Consequently, we used this concentration for further experiments to investigate the effect of trace element addition on mcrA transcript level and on the composition of methanogenic communities in the enrichment cultures.
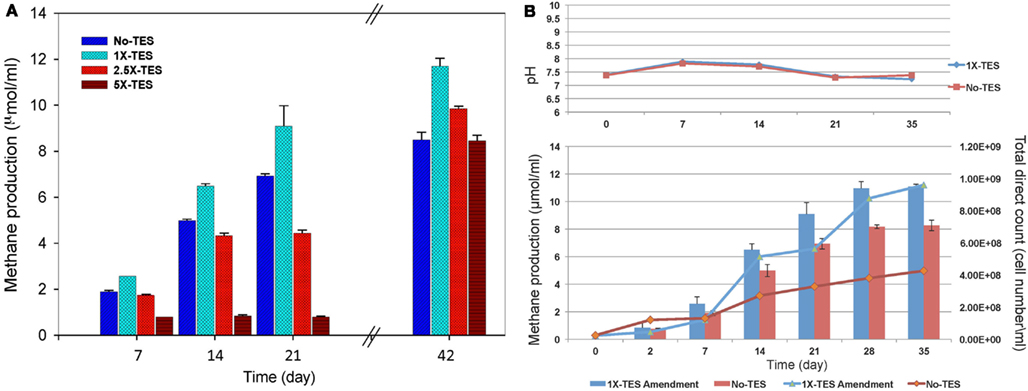
Figure 1. (A) Effect of increasing concentration of trace elements on cumulative methane production over time. No-TES is the enrichment without addition of any trace elements, 1×-TES, 2.5×-TES, and 5×-TES are the enrichments amended with increasing concentrations of trace element mix solutions. 1×-TES is detailed in the Section “Materials and Methods.” Error bars represent standard deviations (n = 3). Probability values were calculated for cumulative methane production at the end of 6 weeks. P < 0.001 for 1×-TES and 2.5×-TES; and P = 0.822 for 5×-TES. (B) Changes in pH value and total cell number (lines; DAPI total direct counts), and cumulative methane production (bars) over time in 1× -TES-amended and unamended cultures (No-TES) (n = 3).
Methane production rate during the first week in 1×-TES enrichment cultures, was the highest (2.57 μmol ml−1 week−1). While, methane production rate in unamended enrichment cultures reached its highest level at the end of the second week of incubation (2.64 μmol ml−1 week−1; Table 2). The cumulative methane production was significantly lower (P < 0.001) than 1×-TES enrichments over 6 weeks of incubation (Figure 1A). The methane production rate in 5×-TES enrichment cultures was low and remained stable during the first 3 weeks. However, after 6 weeks it reached the highest level (10.70 μmol ml−1 week−1; Table 2), which might be the result of adaptation over time of the methanogenic community to higher amounts of trace element concentrations.
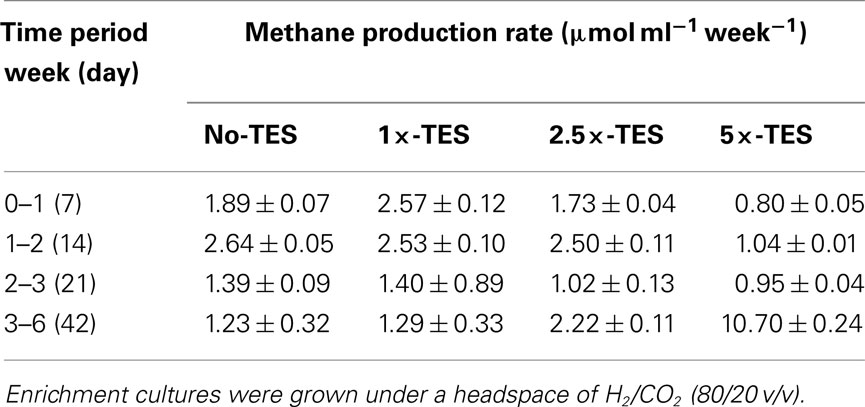
Table 2. Comparison of methane production rates between enrichments amended with increasing concentrations of trace elements (1×-TES, 2.5×-TES, 5×-TES) and enrichments without addition of trace elements over 6 weeks.
Direct cell counts were obtained over 35 days to document cell growth in 1×-TES amended and unamended cultures (Figure 1B). Amended and unamended studies were started with the addition of 2.3 × 107 and 2.5 × 107 cells/ml, respectively. At the end of the incubation time, cell densities had increased to 9.61 × 108 and to 4.26 × 108, respectively. Total cell numbers were ∼125% higher in trace element amended than in unamended enrichment cultures after 5 weeks of incubation. Since these are mixed cultures, growth trends were not well resolved for lag, log, and stationary phases (Meyer-Dombard et al., 2012). Calculated generation times were ∼70 h during the first 168 h (day 7) for amended and unamended cultures. After 7–14 days, the generation times for amended cultures were ∼80 h and two times longer (∼159 h) for unamended cultures. Total cell numbers increased steadily during incubation time for unamended cultures with increasing doubling times ∼622, ∼791, and ∼1019 h for 14–21, 21–28, and 28–35 days, respectively. However, in amended cultures cell numbers fluctuated. Generation times were increased to ∼1281 h between 14 and 21 days, dropped back to ∼264 h between 21 and 28 days and were calculated as ∼1273 h between 28 and 35 days. Changes in pH values were observed in all incubations, but there was no significant difference between enrichment cultures during the entire incubation time (Figure 1B).
Quantifying mcrA Transcripts
The levels of mcrA transcripts in 1×-TES amended and unamended enrichments were quantified in weekly time intervals using quantitative RT-PCR over 35 days of incubation. After 1 week, a quicker and stronger response was observed for mcrA transcript levels in amended enrichments compared to unamended enrichments. At this time point, the addition of trace elements enhanced mcrA transcript levels to its maximum (2.18 × 107 copy numbers/μg mRNA), while the level of mcrA transcripts remained much lower in enrichment cultures without addition of trace elements (7.03 × 106 copy numbers/μg mRNA). Unamended cultures reached their maximum levels at 14 days of incubation (1.3 × 107), but it was still lower than amended enrichment cultures after 7 days of incubation (Figure 2). After 35 days of incubation, mcrA transcript numbers were relatively low, with 3.05 × 104 and 1.05 × 104 for amended and unamended enrichment cultures, respectively.
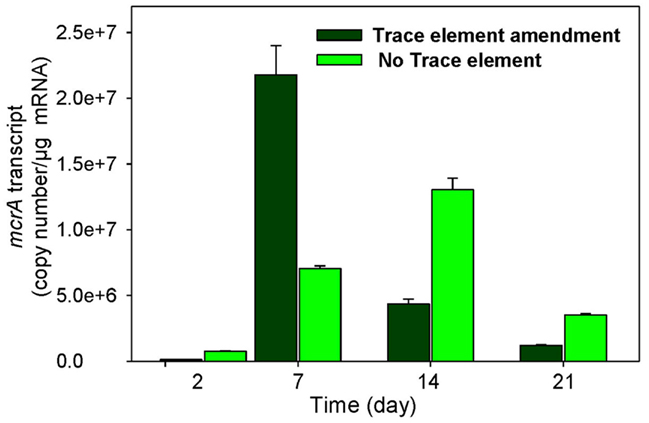
Figure 2. Levels of mcrA transcripts in enrichment cultures with (trace element amendment) and without (no trace element) addition of trace elements over time.
The increased levels of mcrA transcripts in response to trace element addition correlated positively with elevated rates of methane production (R2 = 0.95), while a very weak correlation was determined in unamended cultures (R2 = 0.69; Figure 3). Additionally, the rate of methane production in unamended enrichments was lower than in amended enrichments, which correlates with mcrA transcript levels over the first 7 days of incubation (Table 2; Figure 2).
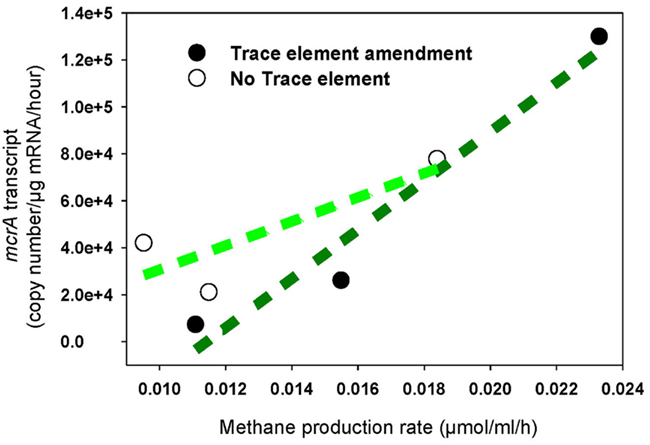
Figure 3. Methane production rate correlated with mcrA transcription in enrichment cultures with addition of trace elements (regression coefficient R2 = 0.95) and without addition of trace elements (regression coefficient R2 = 0.69). Black circle: Enrichment cultures amended with trace element mix solution, white circle: enrichment cultures without addition of any trace elements.
Trace Element Effect on Methanogenic Community Structure and Diversity
Enrichments incubated for 1 week were chosen to construct clone libraries based on their highest methane production rates (2.57 μmol ml−1 week−1) and paralleled by highest mcrA transcript levels (2.18 × 107 copy numbers/μg mRNA) as a proxy of highest methanogenic activity over 6 weeks of incubation (Table 2; Figure 2). Three clone libraries of RT-PCR amplicons of mcrA mRNA were constructed to compare the metabolically active methanogenic taxa in 1×-TES amended (designated T1:TES) and unamended (T1:No TES) enrichment cultures after 1 week of incubation, and in startup enrichment cultures (designated T0). Eighty-one unique sequences were clustered into individual phylotypes (OTUs) to calculate rarefaction and non-parametric estimators (Table 3). Methanobacterium formicicum- and Methanobacterium subterraneum-like sequences were detected in startup, amended, and unamended enrichment cultures.
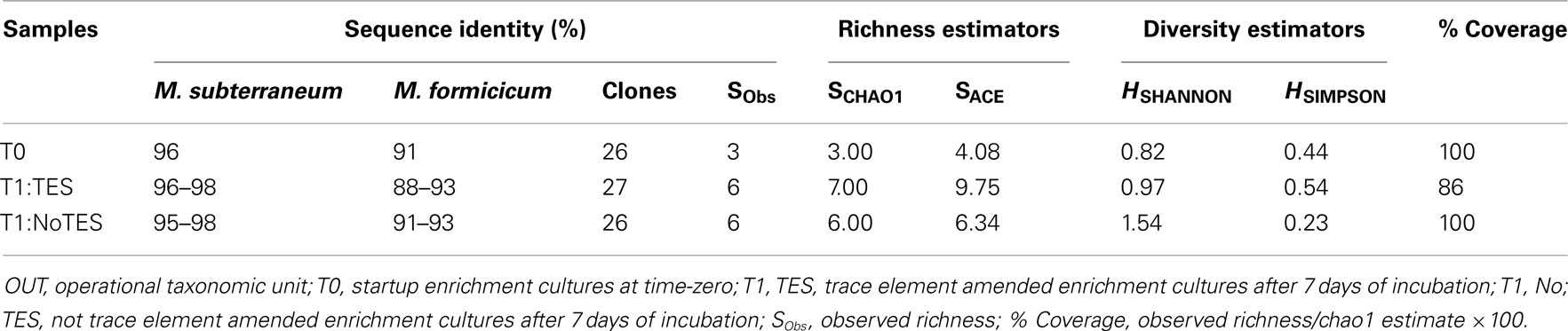
Table 3. Sequence identities of dominant members, clone library sizes, and richness and diversity indices of mcrA sequences based on 97% OTU clusters.
Both richness and diversity of the methanogenic community changed during 7 days of incubation. Enrichment cultures with addition of trace elements had a slightly higher predicted methanogen richness and diversity than enrichment cultures without the addition of trace elements (Table 3).
The effects of trace elements on the relative abundance and distribution of methanogens were monitored. Addition of trace elements contributed specifically to the enhancement of M. subterraneum-like sequences over M. formicicum-like sequences (Figure 4). Amended and unamended enrichments were dominated by clones that grouped closely with the mcrA genes of M. subterraneum-like sequences (81.5% in amended enrichments and 46.2% in unamended enrichments) and M. formicicum-like sequences (18.5% in amended enrichments and 53.8% in unamended enrichments). The methanogenic community composition in the starting enrichment culture (T0) appeared to be unchanged during 1 week of incubation, and mcrA genes from M. subterraneum-like sequences comprised 46.2% and M. formicicum-like sequences 53.8% of the clone library, representing similar ratios between both taxa in enrichment cultures without addition of trace elements (Figure 4).
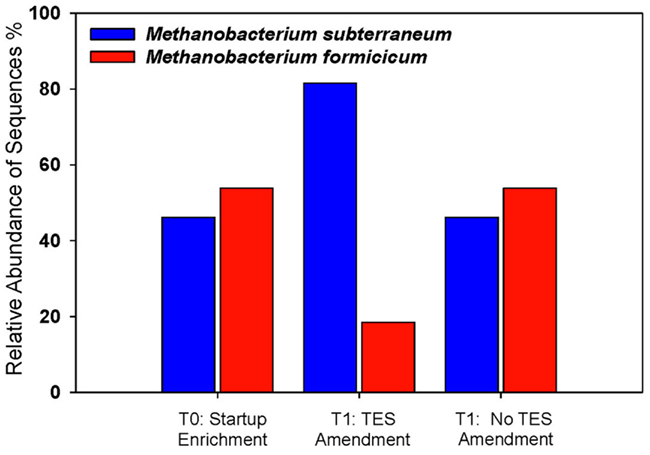
Figure 4. Methanogenic community structure in trace element-amended and unamended enrichments. Startup enrichment culture (T0), trace element amended (T1:TES), and unamended (T1:No TES) enrichments. Blue bars: Methanobacterium subterraneum-like sequences; red bars: Methanobacterium formicicum-like sequences.
Rarefaction analysis based on 97% sequence similarity showed adequate sampling for the startup enrichment, whereas enrichments with and without addition of trace elements did not reach asymptotes (Figure 5). Non-parametric statistical analyses (Table 3) showed that the observed richness covered between 100, 86, and 100% of the estimated total methanogenic richness in startup enrichment, trace element amended and unamended enrichment cultures, respectively.
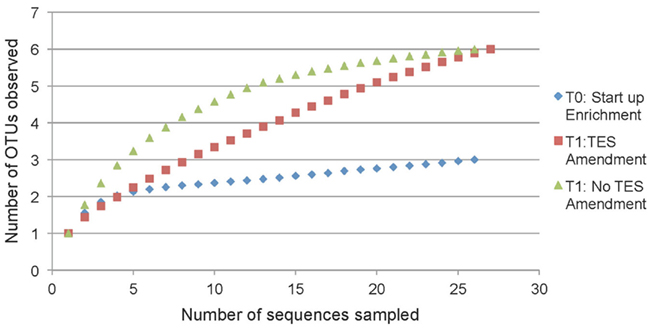
Figure 5. Rarefaction curves indicating mcrA richness within clone libraries derived from the startup enrichment culture (T0), trace element amended (T1:TES), and trace element unamended (T1:No TES) enrichment cultures. Sequences were grouped into phylotypes based on 97% sequence similarity.
Phylogenetic Analyses of Methanogens
A phylogenetic tree based on amino acid sequences of the genes mcrA was constructed using a selection of sequences from cultured methanogens and environmental clones from the public database GenBank. Several clones from T1:TES, T1:No TES, and T0 clone libraries were found to be closely related (95–99%) with two sequences identified in subsurface shale environments (GenBank accession numbers EF117654, EF117630). Clone TES-3 was the most dominant clone in amended enrichments, representing 20 out of 27 clones. It exhibited 97% sequence identity with M. subterraneum which is the nearest isolated relative. Clone No-15 represented 11 of 26 clones and clustered most closely with M. subterraneum with 98% similarity. Only one clone from T1:TES and three clones from T1:No TES were closely related to unpublished clone rlm_673a (97%) which was identified in an anaerobic waste water digester (GenBank accession number JF460370.1). As many as 14 and 12 out of 26 clones in T0 clone libraries have 91 and 95–97% identities to the next closest pure culture, M. formicicum and M. subterraneum, respectively (Figure 6).
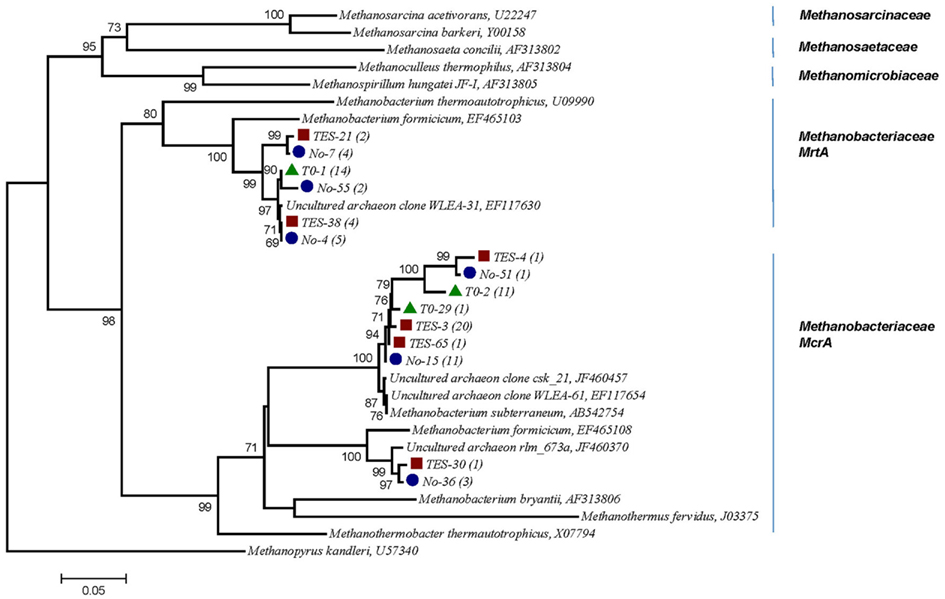
Figure 6. Phylogenetic tree of 15 selected mcrA mRNA sequences retrieved from methanogenic enrichment cultures in this study and their relationship to reference sequences of cultivated methanogens and to other environmental clone sequences. Numbers in parentheses reflect the number of sequences in each OTU. Red square, trace element amended enrichment culture (TES); blue circle, non-amended enrichment culture (No); green triangle, startup enrichment culture (T0). Evolutionary history was inferred using the Neighbor-Joining method. Bootstrap values are shown for nodes in an analysis of 1000 replicates. Evolutionary distances were computed based on amino acid sequences, the scale bar represents 0.05 substitutions per amino acid position. Methanopyrus kandleri, the deepest branching methanogen, was chosen as out-group (Burggraf et al., 1991).
Discussion
This study has enhanced our understanding of trace element effects on methane production, and on the activity and diversity of methanogens in enrichments of water produced from a CBM well. In the present study, we showed that addition of an optimized concentration of trace elements elevated methane production over 37% and increased total cell number by 125%. By comparing enrichment cultures with and without trace element amendments (Fe, Ni, Co, Mo, Co, Zn, B, Mn), we have found not only a correlation between increasing mcrA levels and elevated methane production, but have also demonstrated a shift in the metabolically active methanogenic community from a M. formicicum-like group to a M. subterraneum-like group.
Trace Element as a Limiting Factor in Subsurface Coal Bed Methane Wells
Microbial methane production in deep coal bed basins indicates the ability to sustain microbial life in these limiting environmental conditions (dark, anoxic, reduced, limited substrate replenishment, etc.). The associated physical, chemical, and biological restrictions may influence activity, function, and diversity of the microbial species in the anaerobic biodegradation of organic matter in coal. It is suggested that distribution and activity of the microbial communities are supported by water–coal interaction in subsurface environments (Green et al., 2008). The organic fraction (e.g., carbon, hydrogen, nitrogen, oxygen, and sulfur) and the inorganic constituents of coal (e.g., trace elements) support growth in this subsurface biosphere, and the geochemical potential determines the energy profile for microbial growth, maintenance, and survival under dormancy. Coal–water interactions are expected to provide trace elements for microorganisms (Finkelman, 1995). Yet, our water chemical analysis from the coal bed well in the PRB demonstrated that the amount of trace elements concentrations at this habitat was far lower than the concentrations required for optimum methanogenesis (Figure 1A).
Biodegradation of coal organic deposits into methane is limited to anoxic and reduced conditions, and by the interaction of at least four metabolically diverse groups of microorganisms, hydrolytic, fermentative, acetogenic, and methanogens (Wawrik et al., 2012). Although the rate-limiting step for coal biodegradation is the initial solubilization of a macromolecular, polycyclic, lignin-derived aromatic network into utilizable substrates by microorganisms (Fakoussa and Hofrichter, 1999), we argued that methanogenesis would be another limiting step in coal biodegradation to methane, if the amount of trace elements is limited. Methanogens are the terminal oxidizers in anaerobic biodegradation of organic matter and they are vital for the resultant production of methane (Strąpoć et al., 2008). This study provides the novel consideration that the availability of optimized trace element concentrations might have an influence on methanogenic community structures and on methanogenesis.
Trace elements in coal bed basins may be concentrated in three different ways: (1) concentration during the life of plants prior to coalification; (2) concentration during the decay of organic substances; and (3) concentration by aqueous solution after burial of plant materials (Goldschmidt, 1935). Trace element concentration should be unique in each coal bed deposit, since the composition of plant material and the water source for each coal bed basin can vary with the geological formation, surrounding geochemical conditions, and adjacent overlying mudstones, siltstones, and sandstones. This geological material might be the source of trace elements, which leach with the groundwater into coal bed basins. Furthermore, the source of CBM produced waters might be water accumulated during coal formations or derived from meteoric waters (Flores et al., 2008). Since coal has trace elements embedded within its structure at varying concentrations, we avoided to add coal to incubations to prevent uncontrolled trace element effects.
Methane Production in Different Concentrations of Added Trace Elements
Optimal concentrations of trace elements exist for cellular activity, and concentrations beyond this optimum can have a toxic effect on cells (Swaine and Goodarzi, 1995). The results of this study demonstrated that an optimized concentration of trace elements added to the enrichment cultures from coal bed produced water is beneficial for methanogenic growth and activity. Some recent studies suggested that the addition of high amounts of trace elements leads to a decrease in methanogenic activity in environmental applications (Chakraborty et al., 2010; Pobeheim et al., 2010). Our results indicate that the lack of added trace elements (No-TES) to enrichment cultures limited methane production in a way similar to adding high concentrations of trace elements (2.5×-TES, and 5×-TES; Figure 1A).
Effect of Methanogenic Community Shift on Methanogenic Activity
In addition, the results from this study demonstrated that addition of trace elements to production water enrichments influenced methanogenic community structure (Table 3). This suggests that trace elements in coal bed basins may be an important factor influencing the methanogenic community structure in coal bed basins in situ. It also indicates that M. subterraneum-like group has a growth advantage over M. formicicum-like group in the applied concentration of trace elements after 7 days of incubation (Figure 4).
Trace elements, utilized by microorganisms, provide proteins with unique coordination, catalytic, and electron transfer properties. Shi et al. (2005) reported that almost one fourth of structurally characterized proteins required a coordinated metal atom. Cobalt (Co), nickel (Ni), iron (Fe), zinc (Zn), molybdenum (Mo), and/or tungsten (W) are known to be crucial for the activity of enzymes in methanogens (Takashima et al., 1990). The concentration and type of trace elements required for metabolism and growth may vary among different species of microorganisms (Basiliko and Yavitt, 2001). Burgess et al. (1999) reported that trace metals influence microbial waste degradation and species diversity within sewage sludge. Gough and Stahl (2010) have shown that individual microbial populations are selected by exposition to varying levels of metals in different regions of an anoxic freshwater lake. Chin et al. (2004) reported that structural changes in the methanogenic community lead to functional changes in methane production in rice fields.
The active microbial community can be represented by RNA rather than DNA analysis (Lloyd et al., 2010), and previous studies suggested a correlation between metabolic activity and the transcript level of key genes (Fleming et al., 1993; Nazaret et al., 1994; Chin et al., 2008). In fact, transcripts for mcrA can be employed to demonstrate that methanogens are metabolically active in the environment (Juottonen et al., 2008; Yuan et al., 2011). The results from this study suggest that addition of trace elements caused the methanogenic community shift in composition, and a range of mcrA transcript levels could be directly associated with a rate of methane production and thus with the activity of methanogens (Figure 3).
In this study, mcrA genes of M. formicicum and M. subterraneum, and the gene mrtA of M. formicicum were identified from both trace elements amended and unamended enrichment cultures (Figure 6). Since there is a high degree of DNA sequence conservation between mcrA and mrtA sequences (Luton et al., 2002), primers designed for detection of mcrA sequences also detect mrtA sequences. Since the gene mrtA has arisen by lateral transfer from the Methanococcales rather than gene duplication (Reeve et al., 1997), they form a distinct cluster on the phylogenetic tree closer to Methanococcales than Methanobacteriales mcrA sequences (Luton et al., 2002).
Methanobacterium ssp. in Subsurface Coal Beds in the PRB
Molecular analyses and cultivation studies demonstrated that methanogens are one of the most abundant groups in subsurface environments such as petroleum reservoirs, faults, and marine sediments (Orphan et al., 2000; Shlimon et al., 2004; Moser et al., 2005). So far, very few methanogenic species were isolated from these environments (Kotelnikova et al., 1998; Lai and Wu, 2011). In this study, both results, the phylogenetic analysis of archaea and the methane production in microcosms with H2/CO2, supported that methane production dominantly occurs via the hydrogenotrophic pathway in the sampled coal bed well. Our result was supported Klein et al. (2008) who found Methanobacterium sp. in CBM produced water and also associated with coal samples from PRB, Wyoming. Furthermore, many other studies also reported findings of Methanobacteriales in a variety of deep subsurface coal basins such as the Jhari Gippsland basin, Australia (Midgley et al., 2010), Alberta coal beds of Canada (Penner et al., 2010), and coal fields located at Parbatpur in India (Singh et al., 2012). After ∼2.5 years of routine transfers of CBM produced water into methanogenic media, the enrichments were highly dominated by two species of Methanobacterium sp., which are M. subterraneum and M. formicicum. It is important to note that enrichment cultures successfully represented the majority of methanogens in the CBM produced water sample (Table 1). M. subterraneum was isolated from deep subterranean granitic aquifers, and its phenotypic and phylogenetic characteristics were described previously (Kotelnikova et al., 1998). M. formicicum was isolated from a domestic sewage sludge digest (Bryant and Boone, 1987) and in a municipal land field sample as an endosymbiont of a ciliate (Finlay and Fenchel, 1991). H2/CO2 and formate are the growth substrates for these two species but they cannot grow on methylamines, acetate, pyruvate, dimethyl sulfide, methanol, or other alcohols plus carbon dioxide. Growth of M. subterraneum strain A08 is inhibited by yeast extract (Kotelnikova et al., 1998). In the present study, H2/CO2, acetate, and methanol were each tested as growth substrate for microcosms, and H2/CO2 was found to be main carbon and energy source. Additionally, yeast extract, which was used in the initial enrichment culture, was not added to the trace element amended enrichments due to the unknown quantities of trace elements in it. Optimum temperature conditions for M. subterraneum strain A08 and M. formicicum are described as 20–40°C and 37–45°C, respectively. Optimum pH values for M. subterraneum strain A08 and M. formicicum are between pH 6.6–6.8 and pH 7.8–8.8, respectively (Kotelnikova et al., 1998). In this study, the pH value in enrichments was adjusted to 7.2 ± 0.2 and no differences were observed during 7 days of incubation at 37°C between trace element amended and non-amended enrichments. Among all growth factors in the enrichments such as temperature, pH, or added substrates, trace element addition was the only variable that could have an effect on methanogenesis.
There are still many unknowns regarding the effect of trace elements, due to their bioavailability and optimal concentration on facilitated methanogenesis, and their toxic concentrations for microbial growth and activity in coal bed basins in situ. Bioavailability and toxicity of trace elements in the environment is under the control of geochemical processes such as mineral dissolution, precipitation, and ion adsorption/desorption. In this study, highly enriched methanogenic cultures were used to investigate the effect of trace elements on methanogenesis. However, in the natural environment, abundance and concentration of trace elements might enhance the rate of carbon source degradation by methanogens either directly or in a more indirect way. For example, it might increase the specific metabolic activity of bacterial groups such as primary or secondary fermenters whose products are the substrates for methanogens. Further study should focus on effects of trace elements on bacterial activity in coal biodegradation pathways and their associated community compositions. Effects of individual trace elements on growth and activity of methanogens and on fermenters might provide further insights to enhance biogenic methane production in coal bed wells. Therefore, better understanding of trace elements as a limiting factor for methanogenic activity in a coal bed basin will have a broader impact on our knowledge about ecology and physiology of methanogens.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. K. Baek for intensive discussions and to Drs. M. Kirk and A. Martini for ICP-MS measurements of trace elements. We thank the Turkish Ministry of Education for a scholarship to Burcu Ünal. This project was supported in part by a Agriculture and Food Research Initiative Competitive Grant no. 2009-35319-05186 and Hatch funds no. MAS00945 from the USDA National Institute of Food and Agriculture to Klaus Nüsslein.
References
Balch, W. E., and Wolfe, R. S. (1976). New approach to the cultivation of methanogenic bacteria: 2-mercaptoethanesulfonic acid (HS-CoM)-dependent growth of Methanobacterium ruminatium in a pressurized atmosphere. Appl. Environ. Microbiol. 32, 781–791.
Bapteste, E., Brochier, C., and Boucher, Y. (2005). Higher-level classification of the Archaea: evolution of methanogenesis and methanogens. Archaea 1, 353–363.
Basiliko, N., and Yavitt, J. B. (2001). Influence of Ni, Co, Fe, and Na additions on methane production in Sphagnum-dominated Northern American peatlands. Biogeochemistry 52, 133–153.
Becerra, C. A., Lopez-Luna, E., Ergas, S. J., and Nüsslein, K. (2009). Microcosm-based study of the attenuation of an acid mine drainage-impacted site through biological sulfate and iron reduction, Geomicrobiol. J. 26, 9–20.
Beckmann, S., Lueders, T., Krüger, M., von Netzer, F., Engelen, B., and Cypionka, H. (2011). Appl. Environ. Microbiol. 77, 3749–3756.
Biddle, J. F., Lipp, J. S., Lever, M. A., Lloyd, K. G., Sorensen, K. B., Anderson, R., Fredricks, H. F., Elvert, M., Kelly, T. J., Schrag, D. P., Sogin, M. L., Brenchley, J. E., Teske, A., House, C. H., and Hinrichs, K. U. (2006). Heterotrophic Archaea dominate sedimentary subsurface ecosystems off Peru. Proc Natl. Acad. Sci. U.S.A. 103, 3846–3851.
Bryant, M. P., and Boone, D. R. (1987). Isolation and characterization of Methanobacterium forrnicicum MF. Int. Union Microbiol. Soc. 37, 171.
Bult, C. J., White, O., Olsen, G. J., Zhou, L., Fleischmann, R. D., Sutton, G. G., Blake, J. A., FitzGerald, L. M., Clayton, R. A., Gocayne, J. D., Kerlavage, A. R., Dougherty, B. A., Tomb, J. F., Adams, M. D., Reich, C. I., Overbeek, R., Kirkness, E. F., Weinstock, K. G., Merrick, J. M., Glodek, A., Scott, J. L., Geoghagen, N. S., and Venter, J. C. (1996). Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science 23, 1058–1073.
Burgess, J. E., Quarmby, J., and Stephenson, T. (1999). Role of micronutrients in activated sludge-based biotreatment of industrial effluents. Biotechnol. Adv. 17, 50–70.
Burggraf, S., Stetter, K. O., Rouvière, P., and Woese, C. R. (1991). Methanopyrus kandleri: an archaeal methanogen unrelated to all other known methanogens. Syst. Appl. Microbiol. 14, 346–351.
Cadillo-Quiroz, H., Yashiro, E., Yavitt, J. B., and Zinder, S. H. (2008). Characterization of the archaeal community in a minerotrophic fen and terminal restriction fragment length polymorphism-directed isolation of a novel hydrogenotrophic methanogen. Appl. Environ. Microbiol. 74, 2059–2068.
Chakraborty, N., Chatterjee, M., Sarkar, G. M., and Lahiri, S. C. (2010). Inhibitory effects of the divalent metal ions on biomethanation by isolated mesophilic methanogen in AC21 medium in presence or absence of juices from water hyacinth. Bioenergy Res 3, 314–320.
Chin, K.-J., Lueders, T., Friedrich, M. W., Klose, M., and Conrad, R. (2004a). Archaeal community structure and pathway of methane formation on rice roots. Microb. Ecol. 47, 59–67.
Chin, K.-J., Esteve-Nunez, A., Leang, C., and Lovley, D. R. (2004b). Direct correlation between rates of anaerobic respiration and levels of mRNA for key respiratory genes in Geobacter sulfurreducens. Appl. Environ. Microbiol. 70, 5183–5189.
Chin, K.-J., Sharma, M. L., Russell, L. A., O’Neill, K. R., and Lovley, D. R. (2008). Quantifying expression of a dissimilatory (bi)sulfite reductase gene in petroleum-contaminated marine harbor sediments. Microb. Ecol. 55, 489–499.
Chistoserdova, L., Vorholt, J. A., Thauer, R. K., and Lidstrom, M. E. (1998). C1 transfer enzymes and coenzymes linking methylotrophic bacteria and methanogenic Archaea. Science 281, 99–102.
DeLong, E. F. (1992). Archaea in coastal marine environments. Proc. Natl. Acad. Sci. U.S.A. 89, 5685–5689.
Ellerman, J., Hedderich, R., Bocher, R., and Thauer, R. K. (1988). The final step in methane formation. Investigations with highly purified methyl-CoM reductase (component C) from Methanobacterium thermoautotrophicum (strain Marburg). Eur. J. Biochem. 172, 669–677.
Fakoussa, R. M., and Hofrichter, M. (1999). Biotechnology and microbiology of coal degradation. Appl. Environ. Microbiol. 52, 25–40.
Feng, X. M., Karlsson, A., Svensson, B. H., and Bertilsson, S. (2010). Impact of trace element addition on biogas production from food industrial waste-linking process to microbial communities. FEMS Microbiol. Ecol. 74, 226–240.
Finkelman, R. B. (1995). “Modes of occurrence of environmentally-sensitive trace elements in coal,” in Environmental Aspects of Trace Elements in Coal, eds D. J. Swaine and F. Goodarzi (Dordrecht: Kluwer Academic Publishing), 24–50.
Finlay, B. J., and Fenchel, T. (1991). An anaerobic protozoon, with symbiotic methanogens, living in municipal landfill material. FEMS Microbiol. Lett. 85, 169–179.
Fleming, J. T., Sanseverino, J., and Sayler, G. S. (1993). Quantitative relationship between naphthalene catabolic gene frequency and expression in predicting PAH degradation in soils at town gas manufacturing sites. Environ. Sci. Technol. 27, 1068–1074.
Flores, R. M., Rice, C. A., Stricker, G. D., Warden, A., and Ellis, M. S. (2008). Methanogenic pathways of coal-bed gas in the Powder River Basin, United States: the geologic factor. Int. J. Coal Geol. 76, 52–75.
Friedrich, M. W. (2005). Methyl-coenzyme M reductase genes: unique functional markers for methanogenic and anaerobic methane-oxidizing Archaea. Meth. Enzymol. 397, 428–442.
Glass, J. B., and Orphan, V. J. (2012). Trace metal requirements for microbial enzymes involved in the production and consumption of methane and nitrous oxide. Front. Microbiol. 61, 1–20. doi:10.3389/fmicb.2012.00061
Gonzalez-Gil, G., Kleerebezem, R., and Lettinga, G. (1999). Effects of nickel and cobalt on kinetics of methanol conversion by methanogenic sludge as assessed by on-line ch4 monitoring. Appl. Environ. Microbiol. 65, 1789–1793.
Goodwin, J., Wase, D., and Forster, C. (1990). Effects of nutrient limitation on the anaerobic upflow sludge blanket reactor. Enzyme Microb. Technol. 12, 877–884.
Gough, H. L., and Stahl, D. A. (2010). Microbial community structures in anoxic freshwater lake sediment along a metal contamination gradient. ISME J. 5, 543–558.
Green, M. S., Flanegan, K. C., and Gilcrease, P. C. (2008). Characterization of a methanogenic consortium enriched from a coal bed methane well in the Powder River Basin, U.S.A. Int. J. Coal Geol. 76, 34–45.
Guo, H., Liu, R., Yu, Z., Zhang, H., Yun, J., Li, Y., Liu, X., and Pan, J. (2012). Pyrosequencing reveals the dominance of methylotrophic methanogenesis in a coal bed methane reservoir associated with Eastern Ordos Basin in China. Int. J. Coal Geol. 93, 56–61.
Hallam, S. J., Girguis, P. R., Preston, C. M., Richardson, P. M., and DeLong, E. F. (2003). Identification of methyl coenzyme M reductase A (mcrA) genes associated with methane-oxidizing Archaea. Appl. Environ. Microbiol. 69, 5483–5491.
Harris, S. H., Smith, R. L., and Barker, C. E. (2008). Microbial and chemical factors influencing methane production in laboratory incubations of low-rank subsurface coals. Int. J. Coal Geol. 76, 46–51.
Jackson, R. E., and Reddy, K. J. (2007). Trace element chemistry of coalbed natural gas produced water in the Powder River Basin, Wyoming. Environ. Sci. Technol. 1, 5953–5959.
Jones, E. J. P., Voytek, M. A., Corum, M. D., and Orem, W. H. (2010). Stimulation of methane generation from nonproductive coal by addition of nutrients or a microbial consortium. Appl. Environ. Microbiol. 76, 7013–7022.
Jones, E. J. P., Voytek, M. A., Warwick, P. D., Corum, M. D., Cohn, A., Bunnell, J. E., Clark, A. J., and Orem, W. H. (2008). Bioassay for estimating the biogenic methane-generating potential of coal samples. Int. J. Coal Geol. 76, 138–150.
Juottonen, H., Tuittila, E., Juutinen, S., Fritze, H., and Yrjälä, K. (2008). Seasonality of rDNA- and rRNAderived archaeal communities and methanogenic potential in a boreal mire. ISME J. 2, 1157–1168.
Klein, D. A., Flores, R. M., Venot, C., Gabbert, K., Schmidt, R., Stricker, G. D., Pruden, A., and Mandernack, K. (2008). Molecular sequences derived from Paleocene Fort Union Formation coals vs. associated produced waters: implications for CBM regeneration. Int. J. Coal Geol. 76, 3–13.
Kotelnikova, S., Macario, A. J. L., and Pedersen, A. (1998). Methanobacterium subterraneurn sp. nov., a new alkaliphilic, eurythermic and halotolerant methanogen isolated from deep granitic groundwater. Int. J. Syst. Bacteriol. 48, 357–367.
Krumholz, L. R., McKinley, J. P., Ulrich, G. A., and Suflita, J. M. (1997). Confined subsurface microbial communities in Cretaceous rock. Nature 386, 64–66.
Kumar, S., Nei, M., Dudley, J., and Tamura, K. (2008). MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 9, 299–306.
Lai, M. C., and Wu, S. Y. (2011). Methanogenic Archaea isolated from Taiwan’s Chelungpu fault. Appl. Environ. Microbiol. 77, 830–838.
Lane, D. J. (1991). Nucleic Acid Techniques in Bacterial Systematics. eds E. Stackebrandt and M. Goodfellow (New York: Wiley), 115–175.
Lehmacher, A., and Klenk, H.-P. (1994). Characterization and phylogeny of mcrII, a gene cluster encoding an isoenzyme of methyl coenzyme M reductase from hyperthermophilic Methanothermus fervidus. Mol. Gen. Genet. 243, 198–206.
Lengeler, L., Drews, G., and Schlegel, H. (1999). Biology of the Prokaryotes. Stuttgart: Blackwell Science.
Li, D., Hendry, P., and Faiz, M. (2008). A survey of the microbial populations in some Australian coalbed methane reservoirs. Int. J. Coal Geol. 76, 1–2.
Lloyd, K. G., Albert, D. B., Biddle, J. F., Chanton, J. P., Pizarro, O., and Teske, A. (2010). Spatial structure and activity of sedimentary microbial communities underlying a Beggiatoa spp. Mat in a Gulf of Mexico hydrocarbon seep. PLoS ONE 5, e8738. doi:10.1371/journal.pone.0008738
Lueders, T., Chin, K. J., Condrad, R., and Frederich, M. (2001). Molecular analyses of methyl-coenzyme M reductase alpha-subunit (mcrA) genes in rice field soil and enrichment cultures reveal the methanogenic phenotype of a novel archaeal lineage. Environ. Microbiol. 3, 194–204.
Luton, P. E., Wayne, J. M., Sharp, R. J., and Riley, P. W. (2002). The mcrA gene as an alternative to 16S rRNA in the phylogenetic analysis of methanogen populations in landfill. Microbiology 148, 3521–3530.
McBeth, I., Reddy, K. J., and Skinner, Q. D. (2003). Chemistry of trace elements in coalbed methane product water. Water Res. 37, 884–890.
Meyer-Dombard, D. R., Shock, E. L., and Amend, J. P. (2012). Effects of trace element concentrations on culturing thermophiles. Extremophiles 16, 317–331.
Midgley, D. J., Hendry, P., Pinetown, K. L., Fuentes, D., Gong, S., Mitchell, D. L., and Faiz, M. (2010). Characterization of a microbial community associated with a deep, coal seam methane reservoir in the Gippsland Basin, Australia. Int. J. Coal Geol. 8, 232–239.
Moser, D. P., Gihring, T. M., Brockman, F. J., Fredrickson, J. K., Balkwill, D. L., Dollhopf, M. E., Lollar, B. S., Pratt, L. M., Boice, E., Southam, G., Wanger, G., Baker, B. J., Pfiffner, S. M., Lin, L.-H., and Onstott, T. C. (2005). Desulfotomaculum and Methanobacterium spp. Dominate a 4- to 5-kilometer-deep fault. Appl. Environ. Microbiol. 71, 8773–8783.
Nazaret, S., Jeffrey, H. W., Saouter, E., Haven, R. V., and Barkay, T. (1994). merA gene expression in aquatic environments measured by mRNA production and Hg(II) volatilization. Appl. Environ. Microbiol. 60, 4059–4065.
Nunoura, T., Oida, H., Nakaseama, M., Kosaka, A., Ohkubo, S. B., Kikuchi, T., Kazama, H., Hosoi Tanabe, S., Nakamura, K., Kinoshita, M., Hirayama, H., Inagaki, F., Tsunogai, U., Ishibashi, J., and Takai, K. (2010). Archaeal diversity and distribution along thermal and geochemical gradients in hydrothermal sediments at the Yonaguni Knoll IV hydrothermal field in the Southern Okinawa trough. Appl. Environ. Microbiol. 76, 1198–1211.
Orphan, V. J., Taylor, L. T., Hafenbradl, D., and Delong, L. T. (2000). Culture-dependent and culture-independent characterization of microbial assemblages associated with high-temperature petroleum reservoirs. Appl. Environ. Microbiol. 66, 700–711.
Penner, T. J., Foght, J. M., and Budwill, K. (2010). Microbial diversity of western Canadian subsurface coal beds and methanogenic coal enrichment cultures. Int. J. Coal Geol. 82, 81–93.
Petzet, A. (2005). Resource plays, CBM to fuel drilling upturns in, U.S. Canada. Oil Gas J. 103, 32–34.
Pobeheim, H., Munk, B., Johansson, J., and Guebitz, G. M. (2010). Influence of trace elements on methane formation from a synthetic model substrate for maize silage. Bioresour. Technol. 101, 836–839.
Reeve, J. N., Nolling, J., Morgan, R. M., and Smith, D. R. (1997). Methanogenesis: genes, genomes, and who’s on first? J. Bacteriol. 179, 5975–5986.
Saitou, N., and Nei, M. (1987). The neighbor-joining method-a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Schloss, P. D., and Handelsman, J. (2005). Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 71, 1501–1506.
Shi, W., Zhan, C., Ignatov, A., Manjasetty, B., Marinkovic, N., Sullivan, M., Huang, R., and Chance, M. R. (2005). Metalloproteomics: high-throughput structural and functional annotation of proteins in structural genomics. Structure 13, 1473–1486.
Shimizu, S., Akiyama, M., Naganuma, T., Fujioka, M., Nako, M., and Ishijima, Y. (2007). Molecular characterization of microbial communities in deep coal seam groundwater of northern Japan. Geobiology 5, 423–433.
Shlimon, A. G., Friedrich, M. W., Niemann, H., Ramsing, N. B., and Finster, K. (2004). Methanobacterium aarhusense sp. nov., a novel methanogen isolated from a marine sediment (Aarhus Bay, Denmark). Int. J. Syst. Evol. Microbiol. 54, 759–763.
Singh, D. N., Kumar, A., Sarbhai, M. P., and Tripathi, A. K. (2012). Cultivation-independent analysis of archaeal and bacterial communities of the formation water in an Indian coal bed to enhance biotransformation of coal into methane. Appl. Microbiol. Biotechnol. 93, 1337–1350.
Springer, E., Sachs, M. S., Woese, C. R., and Boone, D. R. (1995). Partial gene sequences for the A subunit of methyl-coenzyme M reductase (mcrI) as a phylogenetic tool for the family Methanosarcinaceae. Int. J. Syst. Bacteriol. 45, 554–559.
Steinberg, L. M., and Regan, J. M. (2008). Phylogenetic comparison of the methanogenic communities from an acidic, oligotrophic fen and an anaerobic digester treating municipal wastewater sludge. Appl. Environ. Microbiol. 73, 6663–6671.
Strąpoć, D., Picardal, F. W., Turich, C., Schaperdoth, I., Macalady, J. L., Lipp, J. S., Lin, Y., Ertefa, T. F., Schubotz, F., Hinrichs, K., Mastalerz, M., and Schimmelmann, A. (2008). Methane-producing microbial community in a coal bed of the Illinois Basin. Environ. Microbiol. 74, 2424–2432.
Swaine, D. J., and Goodarzi, F. (1995). Environmental Aspects of Trace Elements in Coal. Dordrecht, Boston: Kluwer Academic Publishers.
Takai, K., Moser, D. P., DeFlaun, M., Onstott, T. C., and Fredrickson, J. K. (2001). Archaeal diversity in waters from deep South African gold mines. Appl. Environ. Microbiol. 67, 5750–5760.
Takashima, M., Speece, R. E., and Parkin, G. F. (1990). Mineral requirements for methane fermentation. Crit. Rev. Environ. Control 19, 465–479.
Thauer, R. K. (1998). Biochemistry of methanogenesis: a tribute to Marjory Stephenson. Microbiology 144, 2377–2406.
Thielemann, T., Cramer, B., and Schippers, A. (2004). Coalbed methane in the Ruhr basin, Germany: a renewable energy resource? Adv. Org. Geochem. 35, 1537–1549.
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997). The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 15, 4876–4882.
Ulrich, G., and Bower, S. (2008). Active methanogenesis and acetate utilization in power river basin coals, United States. Int. J. Coal Geol. 76, 25–33.
Van Voast, W. A. (2003). Geochemical signature of formation waters associated with coalbed methane. Am. Assoc. Pet. Geol. Bull. 87, 667–676.
Wawrik, B., Mendivelso, M., Parisi, V. A., Suflita, J. M., Davidova, I. A., Marks, C. R., Van Nostrand, J. D., Liang, Y., Zhou, J., Huizinga, B. J., Strąpoć, D., and Callaghan, A. V. (2012). Field and laboratory studies on the bioconversion of coal to methane in the San Juan Basin. FEMS Microbiol. Ecol. 80. doi: 10.1111/j.1574-6941.2011.01272.x
Weil, C. F., Cram, D. S., Sherf, B. A., and Reeve, J. N. (1988). Structure and comparative analysis of the genes encoding component C of methyl coenzyme M reductase in the extremely thermophilic archaebacterium Methanothermus fervidus. J. Bacteriol. 170, 4718–4726.
Wheaton, J., and Donato, T. (2004). Coal bed methane basics: Powder River Basin, Montana. Montana Bureau of Mines and Geology, Information Pam. 5, 1–25.
Widdel, F., Kohring, G. W., and Mayer, F. (1983). Studies on dissimilatory sulfate-reducing bacteria that decompose fatty acids. 111. Characterization of the filamentous gliding Desulfonema limicola. Microbiology 134, 286–294.
Yuan, Y., Conrad, R., and Lu, Y. (2011). Transcriptional response of methanogen mcrA genes to oxygen exposure of rice field soil. Environ Microbiol. Rep. 3, 320–328.
Zhang, W., Saren, G., Li, T., Yu, X., and Zhang, L. (2010). Diversity and community structure of archaea in deep subsurface sediments from the tropical Western pacific. Curr. Microbiol. 60, 439–445.
Zhang, Y., and Gladyshev, V. D. (2009). Comparative genomics of trace elements: emerging dynamic view of trace element utilization and function. Chem. Rev. 4828, 4828–4861.
Zhang, Y., and Gladyshev, V. D. (2010). General trends in trace element utilization revealed by comparative genomic analyses of Co, Cu, Mo, Ni, and Se. J. Biol. Chem. 285, 3393–3405.
Keywords: methanogens, trace elements, coal bed methane, mcrA transcript, enrichments
Citation: Ünal B, Perry VR, Sheth M, Gomez-Alvarez V, Chin K-J and Nüsslein K (2012) Trace elements affect methanogenic activity and diversity in enrichments from subsurface coal bed produced water. Front. Microbio. 3:175. doi: 10.3389/fmicb.2012.00175
Received: 16 November 2011; Accepted: 20 April 2012;
Published online: 10 May 2012.
Edited by:
Andreas Teske, University of North Carolina at Chapel Hill, USAReviewed by:
Martin Krüger, Federal Institute for Geosciences and Natural Resources, GermanyCraig Lee Moyer, Western Washington University, USA
Copyright: © 2012 Ünal, Perry, Sheth, Gomez-Alvarez, Chin and Nüsslein. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Klaus Nüsslein, Department of Microbiology, University of Massachusetts, 203 Morrill Science Center IVN, 639 North Pleasant Street, Amherst, MA 01003, USA. e-mail:bnVzc2xlaW5AbWljcm9iaW8udW1hc3MuZWR1
†Verlin Ryan Perry and Mili Sheth have contributed equally to this work.