

- Biological Sciences Division, Pacific Northwest National Laboratory, Richland, WA, USA
Analysis of the growth-limiting factor or environmental stressors affecting microbes in situ is of fundamental importance but analytically difficult. Microbes can reduce in situ limiting nutrient concentrations to sub-micromolar levels, and contaminated ecosystems may contain multiple stressors. The patterns of gene or protein expression by microbes in nature can be used to infer growth limitations, because they are regulated in response to environmental conditions. Experimental studies under controlled conditions in the laboratory provide the physiological underpinnings for developing these physiological indicators. Although regulatory networks may differ among specific microbes, there are some broad principles that can be applied, related to limiting nutrient acquisition, resource allocation, and stress responses. As technologies for transcriptomics and proteomics mature, the capacity to apply these approaches to complex microbial communities will accelerate. Global proteomics has the particular advantage that it reflects expressed catalytic activities. Furthermore, the high mass accuracy of some proteomic approaches allows mapping back to specific microbial strains. For example, at the Rifle IFRC field site in Western Colorado, the physiological status of Fe(III)-reducing populations has been tracked over time. Members of a “subsurface clade” within the Geobacter predominated during carbon amendment to the subsurface environment. At the functional level, proteomic identifications produced inferences regarding (i) temporal changes in anabolism and catabolism of acetate, (ii) the onset of N2 fixation when N became limiting, and (iii) expression of phosphate transporters during periods of intense growth. The application of these approaches in situ can lead to discovery of novel physiological adaptations.
Introduction
A fundamental question in field-based studies of microbial communities is “what nutrient resource or environmental stressor is the limiting factor?” The concept of limiting factors goes back to Liebig’s Law of the Minimum in 1840, and takes into account not only the absolute amount of one resource but the relative ratios of all required nutrients (Sterner and Elser, 2002). With respect to nutrient resources, limitation may refer either to a constraint on the standing crop of biomass or on the specific growth rate of the microbes. Aquatic ecologists refer to cases in which a nutrient resource limits standing crop as under “bottom-up” control (Pernthaler and Amann, 2005). Usually, the concentration of limiting resource is reduced to a level where specific growth rate is less than the maximum. If loss factors such as predation or viral lysis maintain biomass levels below what nutrient resources could yield, the microbial populations are under “top-down” control, and their specific growth rate is probably near their potential maximum under the ambient conditions of temperature and pH. However, other chemical factors may act as environmental stressors to constrain growth rate.
Determining the growth-limiting factor in natural habitats is analytically difficult. Under bottom-up control, the microbial population will reduce the in situ concentration of the limiting nutrient to low micromolar or nanomolar concentrations (Egli, 2010; Lin et al., 2010), which can be too low for analytical chemistry to accurately measure. Experimental additions of nutrients to natural samples collected from nature (bioassays) often produce only modest biomass increases (Elser et al., 1990; Allgeier et al., 2011). As these reviews suggest, this occurs because the addition of the limiting element can quickly drive the system to exhaustion of another required resource, which is present in only a modest excess relative to the limiting one.
An alternative strategy is to query the physiological state of the microbes. Macromolecular composition and patterns of gene expression are regulated by microbes in response to the environmental conditions that they experience. Some of these physiological responses can be identified by cultivating microbes in the laboratory under specific limiting conditions; the responses that are associated with specific physicochemical conditions can be utilized as “physiological indicators” in assaying biomass from natural populations to infer or exclude growth-limiting conditions (Figure 1).
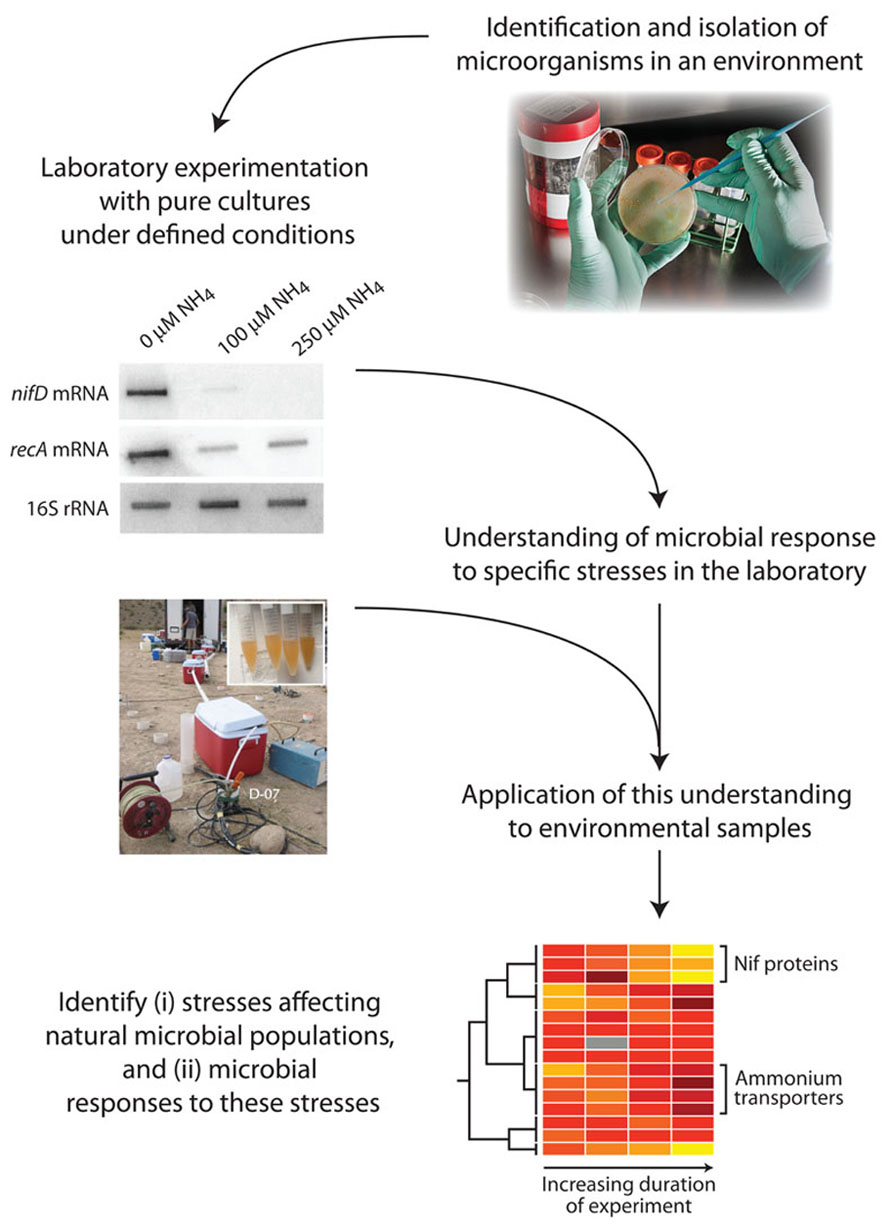
FIGURE 1. Steps involved in the identification of physiological indicators and their application to natural microbial communities. A pure culture isolated from the environment is exposed to a range of physicochemical conditions hypothesized to be relevant to the in situ environment. Physiological responses specific to particular limitations or stressors are elucidated. Natural samples are then tested for the presence of these specific physiological indicators, to infer that the relevant limitations/stressors are acting in situ.
There is a rich history of use of physiological indicators over the past 30 years, particularly in the field of phytoplankton ecology (Healey and Hendzel, 1980; Davies et al., 2010). In these cases, assays of enzyme levels, cell composition, or transport capacity were generally made upon bulk samples. As a result, discrete analyses of the physiological state of specific taxa was not possible. The development of nucleic acid and proteomics sequencing technologies and analyses can generate the means to target specific, adaptive responses made by individual populations within a complex community.
Theoretical Bases for Physiological Indicators
There is an extensive literature from laboratory studies on pure cultures of diverse microbes, that demonstrate the regulatory responses in gene expression and protein activity that organisms make to specific nutrient limitations or stress factors (Tempest et al., 1983; Koch, 1997; Chung et al., 2006; Ferenci, 2008; Zhou et al., 2011). There can be significant differences in the molecular details and effects of regulatory mechanisms that have evolved in specific taxa; catabolite repression (Goerke and Stulke, 2008) represents but one example. However, it is also possible to extract some general principles that may be applied in developing targets for physiological indicators.
These principles have emerged both from empirical studies of microbes (particularly in continuous culture (Gottschal, 1990; Bull, 2010) and from theoretical analyses of nutrient-limited microbial growth (Button, 1994; Koch, 1997).
The physiological adaptations to nutrient limitation can include the following:
- Maximize uptake of the rate-limiting element. Molecular mechanisms include derepression of genes encoding the permeases for the limiting nutrient, and induction of new high-affinity permeases.
- Derepress genes encoding permeases or enzymes that transport or make available alternative forms of the resource. For example, nitrate and amino acids represent alternative N sources to ammonia.
- Down-regulate cellular assimilation so that rates do not exceed the inward flux of the limiting nutrient. This can entail lowering fluxes through pathways that generate monomers for macromolecular synthesis and (most importantly) reducing the capacity for protein synthesis (for example, by reducing ribosome content).
- Rearrange metabolism to circumvent bottlenecks caused by the limitation.
Of equal (or greater) importance over a longer time period, microbes may evolve a range of mechanisms that increase their efficiency for dealing with long-term nutrient limitation. Some examples include N-cost minimization and reduction in genome and/or cell size (Grzymski and Dussaq, 2012).
Microbial responses to environmental stressors may be less clear-cut, because environmental stressors may damage and/or denature macromolecules such as DNA, RNA, proteins, and lipids as well as impart changes in the cytoplasmic membrane with consequent effects upon substrate transport and energy generation. As a result, stressors may elicit multiple responses by cells, and those responses may not be specific to (and hence useful to identify) a particular stressor. Some responses that have been documented, particularly in a few “model” organisms include:
- Induction of general stress responses, such as heat shock proteins, that function as molecular chaperones to refold damaged proteins or phage shock proteins (Lund, 2009; Joly et al., 2010).
- Detoxification mechanisms. A strategy broadly deployed across the microbial world is unidirectional efflux pumps – proteins that can transport toxic chemicals out of the cell. Although there are cases where pumps have specificity for particular toxicants, the so-called MDR (multidrug resistance) pumps have broad substrate ranges that extend beyond antibiotic efflux (Segura et al., 1999; Krulwich et al., 2005; Higgins, 2007). In other cases, detoxification occurs in the cytoplasm, via reactions that metabolize the chemical to a less toxic form or synthesize metabolites that counter the toxic effects (Tam et al., 2006).
- Alteration of cell composition (for example, by modifying the cell wall and membrane) in ways that reduce toxicity of the stressor. This involves changes in biosynthetic gene expression and metabolic fluxes (Nicolaou et al., 2010).
Recent work (Borden and Papoutsakis, 2007; Brynildsen and Liao, 2009; Alsaker et al., 2010; Rutherford et al., 2010) that has applied analysis of the transcriptome and proteome in response to stressors has observed responses in genes of unknown function – hence, there are many facets of stress responses in microbes whose biochemical basis are unknown.
Applying Physiological Principles to In Situ Microbial Communities
The principles listed above have arisen from analyzing the physiological ecology of individual microbes. However, in a microbial community, the relative abundance of diagnostic genes, transcripts, or proteins for specific physiological processes can provide insights into the dominant metabolic processes in the community. Lin et al. (2012) documented changes in the relative abundance of anaerobic respirers capable of using sulfate vs. nitrate as terminal electron acceptors across a vertical depth profile in Hanford subsurface sediments. Geochemical analyses demonstrated that the availability of these acceptors varied and quantitative PCR of dsrA and nosZ gene copies was related to these changes. Their analysis (at the DNA level) indicated potential for a specific respiratory process; it would be more insightful to carry out analysis of gene or protein expression that would be related to actual physiological activity.
Identifying and measuring either the presence or shifts in abundance of proteins or gene transcripts that can be linked to specific physiological activities has emerged as a powerful tool to probe environmental stressors not detectable using other means. While metagenomic analysis of a microbial assemblage can offer insights into strain level community composition and functional potential, the coupling of a genomic foundation with either metatranscriptomics or metaproteomics has allowed expressed function to be interrogated. The first such integrated ‘omics approach utilized a combined metagenomics and proteomics technique, and focused on a microbial biofilm population within an acid mine drainage (AMD) system (Tyson et al., 2004; Ram et al., 2005). These studies allowed the effects of nitrogen limitation to be studied at the community level, rather than just for an individual microorganism. Metagenomic analysis indicated that a low abundance strain, Leptospirillum group III was able to fix nitrogen, while the dominant biofilm community member, Leptospirillum group II, was unable to carry out this process. Likewise, community members Ferroplasma types I and II were similarly unable to fix nitrogen, but contained a large number of ammonia permeases and amino acid transporters, indicating an N scavenging role for these strains. Subsequent proteomic analyses confirmed the presence of Nif proteins that could be assigned to Leptospirillum group III, and ammonia uptake systems in Leptospirillum group II, consistent with this strain’s inability to fix atmospheric nitrogen. In addition, this combined proteogenomic approach revealed mechanisms through which biomass was able to survive in metal-rich low pH AMD systems, utilizing a suite of chaperones, thioredoxins and proteins involved in radical defense.
Similar techniques have subsequently been applied to a range of environmental systems. Here, we will focus on two environments where nutrient limitation and other stresses play a key role in determining microbial community structure and expressed functionality: oligotrophic ocean environments, and saturated subsurface aquifers.
Planktonic Marine Environments
Survival and growth in oligotrophic oceanic surface waters poses a range of challenges for microorganisms due to the combined effects of nitrogen or phosphorous limitation plus exposure to intense light. With increasing depth, light irradiance decreases exponentially, and inorganic nutrient concentrations increase. Ammonia/ammonium is an important compound in both surface and deeper waters, while in deeper waters more oxidized forms of N such as nitrite and nitrate are available at higher concentrations. Numerous pure culture studies have investigated the mechanisms that allow the proliferation of biomass within these systems; this understanding has then been applied to the analysis of environmental ‘omics datasets. Research involving ocean-dwelling Prochlorococcus strains in the lab has revealed niche-specific mechanisms for survival (Rocap et al., 2003); despite only varying 3% at the 16S rRNA level, Prochlorococcus strain MED4 is adapted for surface water conditions, while strain MET9313 is adapted for deeper waters by way of differing mechanisms for nitrogen usage, phosphorous uptake, and sensitivity to trace metals (Martiny et al., 2006; Tolonen et al., 2006). The deep-water adapted strain MET9313 is able to utilize nitrite via a series of nitrite transport and reduction genes, whereas MED4 does not possess this metabolic capability (Moore et al., 2005). Using a transcript-based microarray approach, Martiny et al. (2006) demonstrated the expression of phosphorous-acquisition genes (such as components of the pho operon) under P-limitation conditions in strain MED4. At the proteome level, Lv et al. (2008) identified a correlation between the depth of water from which a Prochlorococcus strain had been isolated, and the atomic composition of the proteome of that strain. Strains isolated from deeper marine waters had higher nitrogen and sulfur contents than those from shallower waters, indicating long-term adaptation to environmental conditions. In spite of these long-term adaptations, Gilbert and Fagan (2010) hypothesized that fluctuating nutrient availability frequently encountered in surface waters favors mechanisms involving short-term down-regulation of nutrient-rich proteins. By comparing global mRNA transcripts from MED4 and MET9313 strains grown under a range of N and P conditions, it was shown that MED4 responded rapidly to nitrogen limitation by down-regulating nitrogen-rich ribosomal proteins. Contrastingly, the low-light, high-nutrient adapted MET9313 strain was shown to exhibit slow but sustained down-regulation of the same proteins.
The effects of sample depth in oceanic systems on microbial strategies for survival were recently investigated using a combined metagenomic and metatranscriptomic approach (Shi et al., 2011). Biomass from 25, 75, 125, and 500 m depths in the North Pacific was recovered, and revealed high expression of nitrogen metabolism genes (ammonium transporter, amt; dissimilatory nitrite reductase, nirK; urea transporter, urt; ammonia monooxygenase subunits, amoABC). The ubiquitous expression of Prochlorococcus-related amt and urt genes by Prochlorococcus populations was noted in contrast to the expression of the assimilatory nitrite reductase gene by only a small proportion of the population, consistent with previous observations of niche-differentiation within these microorganisms. Other environmental transcriptomic-based approaches have investigated the role of N2-fixing diazotrophic cyanobacterium species in surface waters. In addition to detecting expression of the nitrogenase cluster, this strain was found to express the iron-stress-induced protein A (isiA) that is involved in protecting photosystem I from high-light-induced damage (Hewson et al., 2009).
The oligotrophic Sargasso Sea has proved a fertile environment for investigating microbial responses to nutrient limitations and environmental stresses; a metaproteomic study (Sowell et al., 2009) concluded that bacteria from the SAR11 clade dedicated a large proportion of their proteome (67% of total spectra) to transport functions, such as ABC transporters involved in substrate uptake. Given the low concentrations of P that can occur in the Sargasso Sea (<5 nm), the detection of the highly abundant periplasmic phosphate-binding protein PstS and proteins involved in regulation of the pst operon (PhoU, PhoR, and PhoB) mirrored laboratory and field observations discussed earlier (Martiny et al., 2006). For SAR11 strains, a role for assimilation of inorganic nitrogen via ammonium, and incorporation into glutamine and glutamate was inferred from the detection of proteins involved in that pathway (AmtB, GlnA, GlnT, and GltB). Proteomic data indicated that Synechococcus were able to utilize a wider range of nitrogen sources including urea, nitrate, nitrite, and ammonium, whilst high-light adapted Prochlorococcus strains preferentially used ammonium and organic sources of nitrogen, correlating with previously discussed transcriptomic data (Shi et al., 2011).
Finally, this study addressed some of the potential mechanisms with which these microorganisms in surface waters survive continual exposure to environmental stresses; the detection of chaperone proteins such as GroEL, GroES, and DnaK together with the membrane protease HflKC at high abundances indicated a role for proteolysis and protein re-folding in maintaining cell viability.
In other marine systems, Morris et al. (2010) carried out metaproteomic analyses of multiple biomass samples recovered from oligotrophic regions of the South Atlantic. Again, transport proteins for urea and ammonia were found to dominate the metaproteome in these regions, illustrating the importance of competing (Button, 1994) for the small concentrations of available N in these environments. In a contrasting nutrient-rich environment off the Oregon coast however (Sowell et al., 2011), no phosphate transporters were detected at high abundance, although the abundance of transporters for amino acids, taurine, and polyamines led the authors to hypothesize that carbon and nitrogen were more limiting in this system.
Subsurface Sediments
Understanding the stress–responses and physiology of microbial communities in subsurface environments has long proved difficult. In situ measurements are hindered by the localization of biomass in small discrete pore networks within the subsurface. Despite these obstacles, at the Rifle Integrated Field Research Challenge (IFRC) site in Western Colorado, a series of biostimulation experiments via acetate amendment to the subsurface has allowed the study of microbial community responses to excess organic carbon. Given that target acetate concentrations during these experiments can range from 5 to 15 mM (Williams et al., 2011), other nutrient limitations may play a role constraining the specific growth rate of the indigenous microbial population. At the community level, a suite of molecular microbiological tools in numerous studies have indicated that Fe(III)-reducing Geobacter strains are greatly enriched upon carbon addition to the subsurface (Anderson et al., 2003).
Investigations into the physiological state of these bacteria have progressed over the past decade that field experiments have been performed at the site, with data gleaned from single-gene mRNA studies being subsequently utilized in more complex metatranscriptomic and metaproteomic investigations. Both microarray and targeted RNA-based approaches have identified Geobacter-specific transcripts that exhibit abundance shifts in response to (i) nitrogen limitation, (ii) phosphate limitation, (iii) acetate limitation, and (iv) oxidative stress. Holmes et al. (2004) tracked mRNA transcript levels of nifD, the gene that encodes the alpha subunit of the dinitrogenase protein that fixes atmospheric N2; increased expression of this gene was linked to ammonium limitation in both chemostat cultures and sediments from the Rifle IFRC site. Similar trends were observed in experiments using another important subsurface microorganism, Dehalococcoides ethenogenes (Lee et al., 2009). N’Guessan et al. (2010) demonstrated the up-regulation in Geobacter of genes within the phosphate uptake pst (phosphate specific transport)-pho (phosphate) operon under phosphate-limiting chemostat conditions, and there was increased expression of acetate uptake genes in response to acetate limitation (Risso et al., 2008; Elifantz et al., 2010). Mouser et al. (2009a) quantified transcript abundances for two genes [sodA (superoxide dismutase) and cydA (cytochrome d ubiquinol oxidase, subunit I)] involved in Geobacter oxidative stress response during in situ biostimulation at the Rifle IFRC site. Perhaps reflecting the complex responses that cells may utilize when exposed to environmental stresses, high transcript abundances for these two genes did not fully correlate with observed reducing geochemical conditions in the subsurface. Less targeted approaches have utilized microarray-based techniques to identify gene expression patterns under specific environmental conditions. Using a microarray platform constructed from predicted coding sequences of the Geobacter sulfurreducens genome, 106 genes from G. sulfurreducens were shown to exhibit shifts in expression under nitrogen fixing conditions, including increasing abundances of nif genes and components of the Ntr response. However, a significant percentage of the up-regulated genes encoded hypothetical proteins, or proteins of unknown function. Similarly, a comparative metatranscriptomic study using a Geobacter uraniireducens whole-genome microarray detailed the up-regulation of both nitrogen fixation and phosphate uptake genes when this strain was grown on sediments (Holmes et al., 2009). These studies have paved the way for identification of stress or nutrient limitation biomarkers in more broad shotgun ‘omics analyses of microbial communities.
Wilkins et al. (2009) applied global shotgun proteogenomic techniques to the analysis of planktonic Geobacter populations recovered during in situ acetate amendment. Tandem mass spectrometry data was searched against a database of predicted peptides from eight concatenated Geobacter genomes, and revealed the expression of proteins that had been predicted from previously described laboratory experiments such as NifD, NifH, and NifK. Measured ammonium concentrations in bulk groundwater samples were in excess of what has been reported as necessary for Geobacter growth (Mouser et al., 2009b); despite this, the abundances of proteins involved in nitrogen fixation indicated that this energetically expensive process was occurring within the planktonic Geobacter population. Given that laboratory RNA and microarray studies (Holmes et al., 2004; Methe et al., 2005) had reported the rapid inverse response of nifD mRNA transcript levels to ammonium concentrations, this suggested that N limitation existed in pore spaces within the aquifer. Proteins involved in phosphate acquisition were less abundant across time points, suggesting less of role for P limitation in controlling cell proliferation at the site.
Metal-stress responses typically induce the up-regulation of efflux pumps with which to remove toxic concentrations of metal ions that may build up within a cell wall. The Rifle IFRC site is located at a former uranium and vanadium milling operation, and a range of metals remain adsorbed to subsurface sediments. It is believed that some of these metals may be adsorbed onto Fe(III) phases and subsequently mobilized as reductive dissolution of Fe(III) minerals occurs. Within metaproteomic datasets generated from Rifle IFRC groundwater biomass, heavy-metal efflux pumps have been detected, although the non-specific nature of the genome annotation limits the information that can be gleaned from these identifications (Wilkins et al., 2009). The more extreme highly acidic metal contaminated subsurface conditions at the Oak Ridge IFRC site have proved an ideal environment to investigate the community wide genomic potential for dealing with these stresses. Metagenomic analysis of the indigenous groundwater microbial community at the site revealed that the bacteria had evolved a range of mechanisms for existing in that environment (Hemme et al., 2010). Nearly 150 genes were identified as being involved in heavy-metal resistance systems (heavy-metal translocating ATPases, mer operon, Co2+/Zn2+/Cd2+ efflux), while proton and small ion transport between the cytoplasm and periplasm was suggested as a mechanism for tolerating pH stress.
Future Directions
Understanding the environmental factors limiting microbial growth in nature has presented a range of problems that remain more than 30 years after they were articulated (Healey and Hendzel, 1980; Tempest et al., 1983). Although the introduction of molecular tools into microbial ecology over the past 20 years has illuminated the diversity of complex microbial communities, the more recent application of ‘omics technologies requires further refinements over the examples presented here in order to comprehensively reveal the limiting nutrient or environmental stressors impacting natural populations. With an appropriate sampling design and method of data analysis, these tools may be able to go beyond the transfer of laboratory-generated insights to field populations, and uncover novel mechanisms whereby microorganisms survive and/or proliferate under stressful environmental conditions (Figure 2).
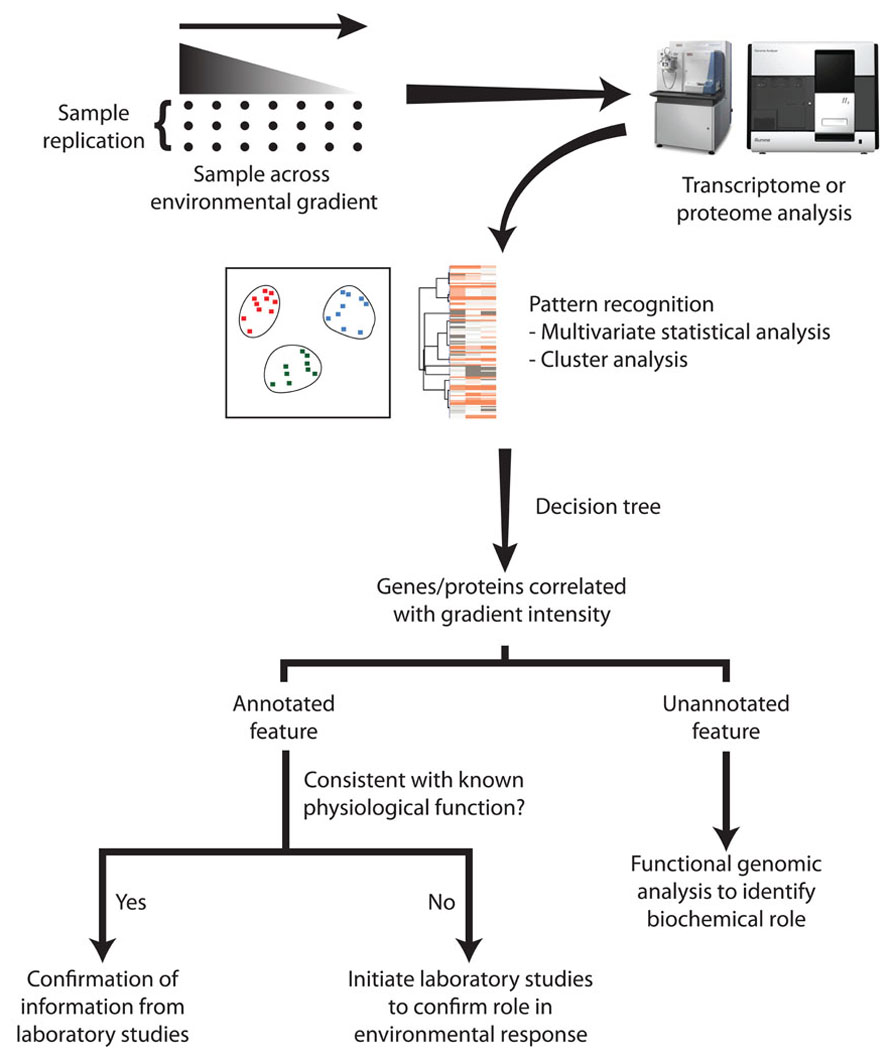
FIGURE 2. The discovery of novel physiological responses via data mining of transcriptome or proteome data. Replicate environmental samples are collected along an environmental gradient where a measured physicochemical factor is known to vary. Multivariate statistical analyses of the molecular sequences and the associated environmental metadata identify genes or proteins whose abundance is related to environmental characteristics. Some of these may represent annotated genes that previously have been identified as physiological indicators. If this data mining suggests that there is an annotated gene that has not previously been recognized as part of the adaptive response, laboratory experiments should be undertaken to confirm its role under well-controlled conditions. However, the most powerful outcome of this approach may be the identification of unannotated genes that contribute to specific physiological adaptations. This generates a motivation for detailed functional genomic analysis, and subsequent biochemical experimentation to elucidate its novel function.
The advances we foresee in this field by the further application of ‘omics techniques will be consequences of innovations in technology, computational analysis, and the application of sophisticated experimental design coupled to novel data mining approaches.
Technological advances in mass spectrometry should allow improved sensitivity and quantitation with regards to proteomic measurements, resulting in “deeper” analysis of samples, while newer peptide-specific techniques such as Multiple Reaction Monitoring (MRM) may be utilized for tracking the absolute abundances of specific biomarker peptides indicative of a particular stress in an environment (Werner et al., 2009).
Methodologies for nucleic acid sequencing have undergone dramatic changes that have reduced their cost by a factor of 50,000 over the past 10 years. However, the recent advances have come with an associated “cost,” i.e., a reduction in read length to a small fraction of a gene’s worth of information. The fundamental unit for analysis relevant to microbial ecology is not the base pair but rather the gene, and greater ecological insights regarding genes of unknown function can be made by genome context analysis, as genes of similar function are often closely linked in Bacteria (Morales and Holben, 2011). Advances in computational methods may yield the extraction and reconstruction of complete or almost-complete bacterial genomes from complex environmental datasets (Iverson et al., 2012), and we anticipate that the future generations of nucleic acid sequencing machines will yield increasingly longer read lengths that may span multiple genes and thereby ease genome context analysis.
The iteration between laboratory experiments under specific environmental conditions to identify physiological responses and their application to natural samples to interpret limiting conditions will continue to be important. However, future improvements in ‘omics technology and analysis can provide the opportunity to extend beyond this current formula, to the discovery of novel stress resistance mechanisms by comparative analysis of natural communities. Environmental genomicists recognize that ecosystems contain “dark matter.” Although this term has been applied to sequencing the genomes of microbes that have not yet been cultivated, we believe that its deeper (and more challenging) meaning is the presence of significant proportions of functionally ambiguous or misannotated genes, as well as genes/proteins of unknown function, whether in uncultivated or cultivated microbes. Many metagenomic analyses have made use of much less than 50% of the sequence reads (Mackelprang et al., 2011); we suggest that this occurs because a high proportion of the remaining reads come from genes that encode proteins which play important roles in situ, but not under controlled conditions of pure culture. These traits may include physiological responses to nutrient limitation, stress, or intercellular communication.
Experimental strategies to discover novel or unrecognized functions from transcriptomic or proteomic data would be heavily dependent upon selection of natural systems and application of appropriate sampling design (Figure 2). For example, comparative spatial analysis (Anderson-Glenna et al., 2008; Kageyama et al., 2008; Lowell et al., 2009; Meyer-Dombard et al., 2011) across environmental gradients of stress factors such as salinity, temperature, heavy metals, or xenobiotics would generate correlations between environmental factors and the up- or down-regulation of genes or proteins. Statistical methodologies that identify genes comprising metabolic pathways within metagenomic datasets, and correlating these with environmental data have recently been described (Gianoulis et al., 2009). These would then become targets for deeper analysis of biochemical function. Techniques such as common garden or reciprocal transplant experiments that juxtapose environmental conditions (Reed and Martiny, 2007) can generate environmental forcing factors by which transcriptomics and proteomics may illuminate organism-specific responses.
The analysis of DNA sequences from environmental samples has generated tremendous insights into the membership and functional potential of microbial communities. Further maturation of transcriptome and proteome analyses will enhance our mechanistic understanding of the selective forces that are structuring those communities and the physiological responses made by specific organisms within them. With appropriate sampling designs and data mining techniques, we anticipate that these ‘omics techniques can discover unrecognized genes and proteins that perform critical roles in the ecology of microbes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the U.S. Department of Energy (DOE), Office of Biological and Environmental Research (BER), as part of Subsurface Biogeochemistry Research Program’s Scientific Focus Area (SFA), and also by the Microbial Communities Initiative LDRD Program at the Pacific Northwest National Laboratory (PNNL). PNNL is operated for DOE by Battelle under contract DE-AC06-76RLO 1830.
References
Allgeier, J. E., Rosemond, A. D., and Layman, C. A. (2011). The frequency and magnitude of non-additive responses to multiple nutrient enrichment. J. Appl. Ecol. 48, 96–101.
Alsaker, K. V., Paredes, C., and Papoutsakis, E. T. (2010). Metabolite stress and tolerance in the production of biofuels and chemicals: gene-expression-based systems analysis of butanol, butyrate, and acetate stresses in the anaerobe Clostridium acetobutylicum. Biotechnol. Bioeng. 105, 1131–1147.
Anderson, R. T., Vrionis, H. A., Ortiz-Bernad, I., Resch, C. T., Long, P. E., Davyault, R., Karp, K., Marutzky, S., Metzler, D. R., Peacock, A., White, D. C., Lowe, M., and Lovley, D. R. (2003). Stimulating the in situ activity of Geobacter species to remove uranium from the groundwater of a uranium-contaminated aquifer. Appl. Environ. Microbiol. 69, 5884–5891.
Anderson-Glenna, M. J., Bakkestuen, V., and Clipson, N. J. W. (2008). Spatial and temporal variability in epilithic biofilm bacterial communities along an upland river gradient. FEMS Microbiol. Ecol. 64, 407–418.
Borden, J. R., and Papoutsakis, E. T. (2007). Dynamics of genomic-library enrichment and identification of solvent tolerance genes for Clostridium acetobutylicum. Appl. Environ. Microbiol. 73, 3061–3068.
Brynildsen, M. P., and Liao, J. C. (2009). An integrated network approach identifies the isobutanol response network of Escherichia coli. Mol. Syst. Biol. 5, 277.
Bull, A. T. (2010). The renaissance of continuous culture in the post-genomics age. J. Ind. Microbiol. Biotechnol. 37, 993–1021.
Chung, H. J., Bang, W., and Drake, M. A. (2006). Stress response of Escherichia coli. Compr. Rev. Food Sci. Food Saf. 5, 52–64.
Davies, J. M., Nowlin, W. H., Matthews, B., and Mazumder, A. (2010). Temporal discontinuity of nutrient limitation in plankton communities. Aquat. Sci. 72, 393–402.
Elifantz, H., N’Guessan, L. A., Mouser, P. J., Williams, K. H., Wilkins, M. J., Risso, C., Holmes, D. E., Long, P. E., and Lovley, D. R. (2010). Expression of acetate permease-like (apl) genes in subsurface communities of Geobacter species under fluctuating acetate concentrations. FEMS Microbiol. Ecol. 73, 441–449.
Elser, J. J., Marzolf, E. R., and Goldman, C. R. (1990). Phosphorus and nitrogen limitation of phytoplankton growth in the freshwaters of North America – a review and critique of experimental enrichments. Can. J. Fish. Aquat. Sci. 47, 1468–1477.
Ferenci, T. (2008). Bacterial physiology, regulation and mutational adaptation in a chemostat environment. Adv. Microb. Physiol. 53, 169–229.
Gianoulis, T. A., Raes, J., Patel, P. V., Bjornson, R., Korbel, J. O., Letunic, I., Yamada, T., Paccanaro, A., Jensen, L. J., Snyder, M., Bork, P., and Gerstein, M. B. (2009). Quantifying environmental adaptation of metabolic pathways in metagenomics. Proc. Natl. Acad. Sci. U.S.A. 106, 1374–1379.
Gilbert, J. D. J., and Fagan, W. F. (2010). Contrasting mechanisms of proteomic nitrogen thrift in Prochlorococcus. Mol. Ecol. 20, 92–104.
Goerke, B., and Stulke, J. (2008). Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat. Rev. Microbiol. 6, 613–624.
Gottschal, J. C. (1990). Phenotypic response to environmental-changes. FEMS Microbiol. Ecol. 74, 93–102.
Grzymski, J. J., and Dussaq, A. M. (2012). The significance of nitrogen cost minimization in proteomes of marine microorganisms. ISME J. 6, 71–80.
Healey, F. P., and Hendzel, L. L. (1980). Physiological indicators of nutrient deficiency in lake phytoplankton. Can. J. Fish. Aquat. Sci. 37, 442–453.
Hemme, C. L., Deng, Y., Gentry, T. J., Fields, M. W., Wu, L. Y., Barua, S., Barry, K., Tringe, S. G., Watson, D. B., He, Z. L., Hazen, T. C., Tiedje, J. M., Rubin, E. M., and Zhou, J. Z. (2010). Metagenomic insights into evolution of a heavy metal-contaminated groundwater microbial community. ISME J.4, 660–672.
Hewson, I., Poretsky, R. S., Beinart, R. A., White, A. E., Shi, T., Bench, S. R., Moisander, P. H., Paerl, R. W., Tripp, H. J., Montoya, J. P., Moran, M. A., and Zehr, J. P. (2009). In situ transcriptomic analysis of the globally important keystone N2-fixing taxon Crocosphaera watsonii. ISME J. 3, 618–631.
Higgins, C. F. (2007). Multiple molecular mechanisms for multidrug resistance transporters. Nature 446, 749–757.
Holmes, D. E., Nevin, K. P., and Lovley, D. R. (2004). In situ expression of nifD in Geobacteraceae in subsurface sediments. Appl. Environ. Microbiol. 70, 7251–7259.
Holmes, D. E., O’Neil, R. A., Chavan, M. A., N’Guessan, L. A., Vrionis, H. A., Perpetua, L. A., Larrahondo, M. J., Didonato, R., Liu, A., and Lovley, D. R. (2009). Transcriptome of Geobacter uraniireducens growing in uranium-contaminated subsurface sediments. ISME J. 3, 216–230.
Iverson, V., Morris, R. M., Frazar, C. D., Berthiaume, C. T., Morales, R. L., and Armbrust, E. V. (2012). Untangling genomes from metagenomes: revealing an uncultured class of marine Euryarchaeota. Science 335, 587–590.
Joly, N., Engl, C., Jovanovic, G., Huvet, M., Toni, T., Sheng, X., Stumpf, M. P. H., and Buck, M. (2010). Managing membrane stress: the phage shock protein (Psp) response, from molecular mechanisms to physiology. FEMS Microbiol. Rev. 34, 797–827.
Kageyama, S. A., Posavatz, N. R., Waterstripe, K. E., Jones, S. J., Bottomley, P. J., Cromack, K., and Myrold, D. D. (2008). Fungal and bacterial communities across meadow-forest ecotones in the western Cascades of Oregon. Can. J. For. Res. 38, 1053–1060.
Koch, A. L. (1997). Microbial physiology and ecology of slow growth. Microbiol. Mol. Biol. Rev. 61, 305–318.
Krulwich, T. A., Lewinson, O., Padan, E., and Bibi, E. (2005). Do physiological roles foster persistence of drug/multidrug-efflux transporters? A case study. Nat. Rev. Microbiol. 3, 566–572.
Lee, P. K. H., He, J., Zinder, S. H., and Alvarez-Cohen, L. (2009). Evidence for nitrogen fixation by “Dehalococcoides ethenogenes” strain 195. Appl. Environ. Microbiol. 75, 7551–7555.
Lin, X. J., Kennedy, D., Peacock, A., Mckinley, J., Resch, C. T., Fredrickson, J., and Konopka, A. (2012). Distribution of microbial biomass and potential for anaerobic respiration in Hanford Site 300 Area subsurface sediment. Appl. Environ. Microbiol. 78, 759–767.
Lin, Y. S., Biddle, J. F., Lipp, J. S., Orcutt, B. N., Holler, T., Teske, A., and Hinrichs, K. U. (2010). Effect of storage conditions on archaeal and bacterial communities in subsurface marine sediments. Geomicrobiol. J. 27, 261–272.
Lowell, J. L., Gordon, N., Engstrom, D., Stanford, J. A., Holben, W. E., and Gannon, J. E. (2009). Habitat heterogeneity and associated microbial community structure in a small-scale floodplain hyporheic flow path. Microb. Ecol. 58, 611–620.
Lund, P. A. (2009). Multiple chaperonins in bacteria – why so many? FEMS Microbiol. Rev. 33, 785–800.
Lv, J., Li, N., and Niu, D.-K. (2008). Association between the availability of environmental resources and the atomic composition of organismal proteomes: evidence from Prochlorococcus strains living at different depths. Biochem. Biophys. Res. Commun. 375, 241–246.
Mackelprang, R., Waldrop, M. P., Deangelis, K. M., David, M. M., Chavarria, K. L., Blazewicz, S. J., Rubin, E. M., and Jansson, J. K. (2011). Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 480, 368–371.
Martiny, A. C., Coleman, M. L., and Chisholm, S. W. (2006). Phosphate acquisition genes in Prochlorococcus ecotypes: evidence for genome-wide adaptation. Proc. Natl. Acad. Sci. U.S.A. 103, 12552–12557.
Methe, B. A., Webster, J., Nevin, K., Butler, J., and Lovley, D. R. (2005). DNA microarray analysis of nitrogen fixation and Fe(III) reduction in Geobacter sulfurreducens. Appl. Environ. Microbiol. 71, 2530–2538.
Meyer-Dombard, D. R., Swingley, W., Raymond, J., Havig, J., Shock, E. L., and Summons, R. E. (2011). Hydrothermal ecotones and streamer biofilm communities in the Lower Geyser Basin, Yellowstone National Park. Environ. Microbiol. 13, 2216–2231.
Moore, L. R., Ostrowski, M., Scanlan, D. J., Feren, K., and Sweetsir, T. (2005). Ecotypic variation in phosphorus acquisition mechanisms within marine picocyanobacteria. Aquat. Microb. Ecol. 39, 257–269.
Morales, S. E., and Holben, W. E. (2011). Linking bacterial identities and ecosystem processes: can ‘omic’ analyses be more than the sum of their parts? FEMS Microbiol. Ecol. 75, 2–16.
Morris, R. M., Nunn, B. L., Frazar, C., Goodlett, D. R., Ting, Y. S., and Rocap, G. (2010). Comparative metaproteomics reveals ocean-scale shifts in microbial nutrient utilization and energy transduction. ISME J. 4, 673–685.
Mouser, P. J., Holmes, D. E., Perpetua, L. A., Didonato, R., Postier, B., Liu, A., and Lovley, D. R. (2009a). Quantifying expression of Geobacter spp. oxidative stress genes in pure culture and during in situ uranium bioremediation. ISME J. 3, 454–465.
Mouser, P. J., N’Guessan, A. L., Elifantz, H., Holmes, D. E., Williams, K. H., Wilkins, M. J., Long, P. E., and Lovley, D. R. (2009b). Influence of heterogeneous ammonium availability on bacterial community structure and the expression of nitrogen fixation and ammonium transporter genes during in situ bioremediation of uranium-contaminated groundwater. Environ. Sci. Technol. 43, 4386–4392.
N’Guessan, A. L., Elifantz, H., Nevin, K. P., Mouser, P. J., Methe, B., Lwoodard, T., Manley, K., Williams, K. H., Wilkins, M. J., Larsen, J. T., Long, P. E., and Lovley, D. R. (2010). Molecular analysis of phosphate limitation in Geobacteraceae during the bioremediation of a uranium-contaminated aquifer. ISME J. 4, 253–266.
Nicolaou, S. A., Gaida, S. M., and Papoutsakis, E. T. (2010). A comparative view of metabolite and substrate stress and tolerance in microbial bioprocessing: from biofuels and chemicals, to biocatalysis and bioremediation. Metab. Eng. 12, 307–331.
Pernthaler, J., and Amann, R. (2005). Fate of heterotrophic microbes in pelagic habitats: focus on populations. Microbiol. Mol. Biol. Rev. 69, 440–461.
Ram, R. J., Verberkmoes, N. C., Thelen, M. P., Tyson, G. W., Baker, B. J., Blake, R. C., Shah, M., Hettich, R. L., and Banfield, J. F. (2005). Community proteomics of a natural microbial biofilm. Science 308, 1915–1920.
Reed, H. E., and Martiny, J. B. H. (2007). Testing the functional significance of microbial composition in natural communities. FEMS Microbiol. Ecol. 62, 161–170.
Risso, C., Methe, B., Elifantz, H., Holmes, D., and Lovley, D. (2008). Highly conserved genes in Geobacter species with expression patterns indicative of acetate limitation. Microbiology 154, 2589–2599.
Rocap, G., Larimer, F. W., Lamerdin, J., Malfatti, S., Chain, P., Ahlgren, N. A., Arellano, A., Coleman, M., Hauser, L., Hess, W. R., Johnson, Z. I., Land, M., Lindell, D., Post, A. F., Regala, W., Shah, M., Shaw, S. L., Steglich, C., Sullivan, M. B., Ting, C. S., Tolonen, A., Webb, E. A., Zinser, E. R., and Chisholm, S. W. (2003). Genome divergence in two Prochlorococcus ecotypes reflects oceanic niche differentiation. Nature 424, 1042–1047.
Rutherford, B. J., Dahl, R. H., Price, R. E., Szmidt, H. L., Benke, P. I., Mukhopadhyay, A., and Keasling, J. D. (2010). Functional genomic study of exogenous n-butanol stress in Escherichia coli. Appl. Environ. Microbiol. 76, 1935–1945.
Segura, A., Duque, E., Mosqueda, G., Ramos, J. L., and Junker, F. (1999). Multiple responses of Gram-negative bacteria to organic solvents. Environ. Microbiol. 1, 191–198.
Shi, Y., Tyson, G. W., Eppley, J. M., and Delong, E. F. (2011). Integrated metatranscriptomic and metagenomic analyses of stratified microbial assemblages in the open ocean. ISME J. 5, 999–1013.
Sowell, S. M., Abraham, P. E., Shah, M., Verberkmoes, N. C., Smith, D. P., Barofsky, D. F., and Giovannoni, S. J. (2011). Environmental proteomics of microbial plankton in a highly productive coastal upwelling system. ISME J. 5, 856–865.
Sowell, S. M., Wilhelm, L. J., Norbeck, A. D., Lipton, M. S., Nicora, C. D., Barofsky, D. F., Carlson, C. A., Smith, R. D., and Giovanonni, S. J. (2009). Transport functions dominate the SAR11 metaproteome at low-nutrient extremes in the Sargasso Sea. ISME J. 3, 93–105.
Sterner, R. W., and Elser, J. J. (2002). Ecological Stoichiometry. Princeton: Princeton University Press.
Tam, L. T., Eymann, C., Albrecht, D., Sietmann, R., Schauer, F., Hecker, M., and Antelmann, H. (2006). Differential gene expression in response to phenol and catechol reveals different metabolic activities for the degradation of aromatic compounds in Bacillus subtilis. Environ. Microbiol. 8, 1408–1427.
Tempest, D., Neijssel, O., and Zevenboom, W. (1983). Properties and performance of microorganisms in laboratory culture; their relevance to growth in natural ecosystems. Symp. Soc. Gen. Microbiol. 34, 119–152.
Tolonen, A. C., Aach, J., Lindell, D., Johnson, Z. I., Rector, T., Steen, R., Church, G. M., and Chisholm, S. W. (2006). Global gene expression of Prochlorococcus ecotypes in response to changes in nitrogen availability. Mol. Syst. Biol. 2, 53.
Tyson, G. W., Chapman, J., Hugenholtz, P., Allen, E. E., Ram, R. J., Richardson, P. M., Solovyev, V. V., Rubin, E. M., Rokhsar, D. S., and Banfield, J. F. (2004). Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428, 37–43.
Werner, J. J., Ptak, A. C., Rahm, B. G., Zhang, S., and Richardson, R. E. (2009). Absolute quantification of Dehalococcoides proteins: enzyme bioindicators of chlorinated ethene dehalorespiration. Environ. Microbiol. 11, 2687–2697.
Wilkins, M. J., Verberkmoes, N. C., Williams, K. H., Callister, S. J., Mouser, P. J., Elifantz, H., N’Guessan, A. L., Thomas, B. C., Nicora, C. D., Shah, M. B., Abraham, P., Lipton, M. S., Lovley, D. R., Hettich, R. L., Long, P. E., and Banfield, J. F. (2009). Proteogenomic monitoring of Geobacter physiology during stimulated uranium bioremediation. Appl. Environ. Microbiol. 75, 6591–6599.
Williams, K. H., Long, P. E., Davis, J. A., Wilkins, M. J., N’Guessan, L. A., Steefel, C. I., Yang, L., Newcomer, D. R., Spane, F. A., Kerkhof, L. J., McGuinness, L. M., Dayvault, R., and Lovley, D. R. (2011). Acetate availability and its influence on sustainable bioremediation of uranium-contaminated groundwater. Geomicrobiol. J. 28, 519–539.
Keywords: physiological indicators, marine habitats, subsurface sediments
Citation: Konopka A and Wilkins MJ (2012) Application of meta-transcriptomics and -proteomics to analysis of in situ physiological state. Front. Microbio. 3:184. doi: 10.3389/fmicb.2012.00184
Received: 26 March 2012; Accepted: 03 May 2012;
Published online: 18 May 2012.
Edited by:
Matthew Fields, Montana State University, USAReviewed by:
Carl James Yeoman, University of Illinois at Champaign-Urbana, USAGiridhar Upadhyaya, University of Michigan, USA
Copyright: © 2012 Konopka and Wilkins. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Allan Konopka, Biological Sciences Division, Pacific Northwest National Laboratory, P.O. Box 999, MSIN J4-18, Richland, WA 99352, USA. e-mail:YWxsYW4ua29ub3BrYUBwbm5sLmdvdg==