Abstract
Ammonia-oxidizing archaea (AOA) have recently been recognized as a significant component of many microbial communities and represent one of the most abundant prokaryotic groups in the biosphere. However, only few AOA have been successfully cultivated so far and information on the physiology and genomic content remains scarce. We have performed a metagenomic analysis to extend the knowledge of the AOA affiliated with group I.1b that is widespread in terrestrial habitats and of which no genome sequences has been described yet. A fosmid library was generated from samples of a radioactive thermal cave (46°C) in the Austrian Central Alps in which AOA had been found as a major part of the microbial community. Out of 16 fosmids that possessed either an amoA or 16S rRNA gene affiliating with AOA, 5 were fully sequenced, 4 of which grouped with the soil/I.1b (Nitrososphaera-) lineage, and 1 with marine/I.1a (Nitrosopumilus-) lineage. Phylogenetic analyses of amoBC and an associated conserved gene were congruent with earlier analyses based on amoA and 16S rRNA genes and supported the separation of the soil and marine group. Several putative genes that did not have homologs in currently available marine Thaumarchaeota genomes indicated that AOA of the soil group contain specific genes that are distinct from their marine relatives. Potential cis-regulatory elements around conserved promoter motifs found upstream of the amo genes in sequenced (meta-) genomes differed in marine and soil group AOA. On one fosmid, a group of genes including amoA and amoB were flanked by identical transposable insertion sequences, indicating that amoAB could potentially be co-mobilized in the form of a composite transposon. This might be one of the mechanisms that caused the greater variation in gene order compared to genomes in the marine counterparts. Our findings highlight the genetic diversity within the two major and widespread lineages of Thaumarchaeota.
Introduction
Molecular surveys of the last two decades have revealed the ubiquitous distribution of an abundant group of archaea that were initially thought to be associated with hyperthermophilic Crenarchaeota (DeLong, 1992; Fuhrman et al., 1992; Schleper et al., 2005). These “mesophilic” Crenarchaeota, or Group I Crenarchaeota (DeLong, 1998) have more recently been assigned to a new archaeal phylum, the Thaumarchaeota (Brochier-Armanet et al., 2008; Spang et al., 2010). Culture-independent metagenomic studies (Venter et al., 2004; Treusch et al., 2005) led to the identification of genes encoding two subunits (A and B) of a putative ammonium monooxygenase associated with these organisms. The physical linkage of an archaeal 16S rRNA gene to the amo genes on a genomic fragment from soil indicated that group I archaea might indeed have the ability to oxidize ammonia to nitrite (Schleper et al., 2005). This physiology was confirmed by the cultivation of a chemolithoautotrophic ammonia-oxidizing archaeon, Nitrosopumilus maritimus, isolated from a marine aquarium in Seattle (Könneke et al., 2005), and more recently of Nitrososphaera viennensis isolated from Viennese garden soil (Tourna et al., 2011). The enrichment cultures of Nitrososphaera gargensis from a hot spring (Hatzenpichler et al., 2008), of Nitrosotalea devanaterra from an acidic environment (Lehtovirta-Morley et al., 2011), and of Nitrosopumilus koreensis from korean agricultural soil (Jung et al., 2011) all confirm growth of these Thaumarchaeota by ammonia oxidation.
Based on a recently updated phylogenetic analyses of available archaeal amoA genes the two earlier identified major groups of Thaumarchaeota (group I.1a, group I.1b) have been extended to a total of five clusters (Pester et al., 2012). The Nitrosopumilus cluster (corresponding to the earlier group I.1a; Pester et al., 2012) is the dominant archaeal group in world’s oceans which accounts for up to 20% of planktonic prokaryotic cells (Fuhrman and Ouverney, 1998; Karner et al., 2001; Herndl et al., 2005). The Nitrososphaera cluster (corresponding to group I.1b) dominate archaeal communities in most soils where they typically account for 1–5% of total 16S rRNA gene abundance (Buckley et al., 1998; Ochsenreiter et al., 2003; Leininger et al., 2006; Bates et al., 2011). Affiliated sequences have been found in other environments including, e.g., hot springs, freshwater, and freshwater sediments (Hershberger et al., 1996; Ochsenreiter et al., 2003; Weidler et al., 2007). Likewise Group I.1a (Nitrosopumilus cluster) has also been found in non-marine environments including freshwater, freshwater sediments as well as soil and terrestrial hot springs (MacGregor et al., 1997; Ochsenreiter et al., 2003; Spear et al., 2007; Weidler et al., 2007, 2008; Ghai et al., 2011).
Although all so far cultivated Thaumarchaeota are capable of generating energy from the oxidation of ammonia and fixing inorganic carbon, several indications for a greater physiological potential have been found. For example, incorporation of organic carbon compounds has been suggested based on studies in the marine realm (Ouverney and Fuhrman, 2000; Herndl et al., 2005; Ingalls et al., 2006; Teira et al., 2006). It remains currently unclear though, if specific marine lineages are dependent on organic carbon or if Nitrosopumilus-type organisms can switch to mixotrophic or heterotrophic growth under certain conditions. Different growth modes have, however, clearly been demonstrated for organisms of the soil cluster (or I.1b group). For example, N. viennensis grows about 10 times faster when low amounts of organic substrates (e.g., pyruvate) are added to the medium, although only a small percentage of carbon from pyruvate is incorporated into biomass (Tourna et al., 2011). Mussmann et al. (2011) demonstrated that growing populations of certain group I.1b organisms in a waste water treatment plant did not incorporate any detectable inorganic carbon, indicating a purely heterotrophic growth mode even without ammonia oxidation. In addition, Jia and Conrad (2009) got indications for growth of group I.1b organisms in an environmental study when no nitrification activity was occurring. Thaumarchaeota that dominate in soil habitats, might thus have greater metabolic diversity compared to organisms of the Nitrosopumilus cluster.
The arrangement of the ammonia monooxygenase gene cluster reveals also differences between the diverse lineages of ammonia-oxidizing Thaumarchaeota, while all ammonia-oxidizing bacteria (AOB) possess an amo operon with conserved CAB subunit arrangement (Norton et al., 2002; Klotz and Stein, 2008). In the marine Thaumarchaeota, e.g., N. maritimus (Walker et al., 2010) and N. limnia (Blainey et al., 2011) amoA, B, and C genes occur within one cluster (AxCB), but the gene arrangement in organisms of the Nitrososphaera and Nitrosocaldus clusters seem more variable, and always lack a closely associated amoC gene (Treusch et al., 2005; de la Torre et al., 2008; Tourna et al., 2011).
While several large scale metagenomic studies (Venter et al., 2004; DeLong et al., 2006; Martin-Cuadrado et al., 2007) and four complete genome sequences have been made available for Thaumarchaeota of the Nitrosopumilus cluster (group I.1a), genomic information on archaea of the Nitrososphaera cluster (I.1b group) are limited to a few metagenomic fragments (Quaiser et al., 2002; Treusch et al., 2004, 2005; Bartossek et al., 2010).
Here we expand genomic insights into group I.1b organisms, by analyzing metagenomic fragments from organisms closely related to Nitrososphaera sp.
Samples from a thermal (46°C) and slightly radioactive spring in Bad Gastein (Austria) were investigated, in which a previous survey had shown a high diversity of bacteria and the presence of 16S rRNA gene sequences of group I.1b Thaumarchaeota, to the exclusion of group I.1a (Weidler et al., 2007) and of bacterial ammonia oxidizers (Weidler et al., 2008). Our study revealed further differences separating the two major lineages of ammonia-oxidizing archaea (AOA) including specific gene sets and the presence of distinct putative cis-regulatory sequences found to be conserved in the promoter regions of amoA, B, C, and associated genes.
Materials and Methods
Sampling, cloning of PCR-products, and sequencing
Spring water, slides (incubated in the cave water for 14 days), and biofilm were sampled in early spring 2006 from Franz Josef Quelle as described previously (Weidler et al., 2007). The cave water was filtered through a 0.22 μm filter before the DNA was isolated using the FastDNA spin kit for soil (MP Biomedicals, Heidelberg) according to manufacturer’s instructions. PCR-products for amoA gene clone libraries of every sample were produced using primers amo19F and amo643R and PCR conditions as described earlier (Leininger et al., 2006). Subsequent cloning into pCR 4-TOPO vector (Invitrogen, Karlsruhe, Germany) and sequencing of transformants was performed as described earlier (Bartossek et al., 2010).
Preparation of high molecular weight DNA from cave material and library construction
For the construction of a fosmid library with high molecular weight DNA, biofilm, and 70 L of water were collected under sterile conditions (Weidler et al., 2007). The water was filtered through a 0.22 μm pore size filter and filter and biofilm were stored at 4°C until further processing. For isolation of purified high molecular weight DNA we used a modified protocol from Zhou et al. (1996) using a CTAB buffer as previously described (Quaiser et al., 2003) with some further optimization (Reigstad et al., 2011). Briefly, the cells were washed off the filter with DNA extraction buffer (Zhou et al., 1996) and pooled with the biofilm to increase the amount of biomass. For lysis, lysozyme (1 mg/mL final concentration) was added and the solution was incubated for 1 h at 37°C. Subsequently a second lysis step with proteinase K (250 μg/mL final concentration) was performed at 40°C for 1 h. SDS was added to a concentration of 1.4% and the mixture was incubated at 65°C for 2 h with inversion every 15 min. The DNA was extracted with phenol-chloroform and precipitated with 2-propanol. Purification of the DNA was performed using a pulsed-field electrophoresis procedure as described earlier (Quaiser et al., 2002) that involved the use of polyvinylpyrrolidone to remove residual polyphenolic compounds such as humic and fulvic acids.
The library was constructed by ligation of the DNA into the fosmid vector pEpiFOS-5 (Epicentre) as described earlier (Quaiser et al., 2002). Around 40,000 clones were obtained for the library Josef01 and 29.874 clones were arrayed in microtiter plates.
Screening for genomic fragments of Thaumarchaeota
The fosmid library was screened for the presence of archaeal amoA and 16S rRNA genes by PCR using the primers amo19F and amo643R (Leininger et al., 2006) and 20F and 958R (DeLong, 1992), respectively. The screening was performed as described earlier (Bartossek et al., 2010). Briefly, DNA pools of every 384-well plate were screened for the presence of either amoA or 16S rRNA genes. 16S rRNA PCR-products of the correct size (950 bp) were obtained from pools 01, 02, 05, 17, and 19. amoA PCR-products of the correct size (647 bp) were obtained from the pools 10, 13, 16, 17, 19, 22, and 24. To identify the positive clone in the plate, the positive 384-well plates were replicated in further microtiter plates and the cells of each column were pooled (24 pools per plate) and lysed by heat. One microliter of the supernatant was used for PCR. To identify the final coordinate 1 μL of the crude cell extract (without lysis) was used in the subsequent PCR.
Sequence analysis
Five fosmids in total were sequenced using either Sanger or 454 sequencing, with fosmid 10c16 sequenced using both approaches. Additionally, Sanger end-sequences of the fosmids and the 16S rRNA gene sequences were needed to be able to assemble the fosmids received in the 454 approach.
Fosmids 10c16, 19c08, and 22i07 were sequenced by preparing individual subclone libraries. Fosmid DNA was fragmented by mechanical shearing and 2–3 kb fragments were cloned into pDrive vector (Qiagen). The ends of the cloned DNA fragments were sequenced with vector primers SP6 and M13R using an ABI PRISM 377 capillary sequencer (ABI). Remaining sequence gaps were closed by primer walking with sequence-derived oligonucleotides and the resulting reads assembled using the PHRED/PHRAP/CONSED package (Ewing et al., 1998; Gordon et al., 1998).
A pyrosequencing shotgun approach was used for sequencing fosmids 02j05, 05d18, and 10c16. DNA from each fosmid was pooled (125 ng each) with DNA derived from 37 other fosmids (derived from a separate study), and sequenced using a GS 20 sequencer (Roche Applied Sciences/454 Life Sciences, Branford, CT, USA). Additionally, end-sequencing of EpiFos fosmids was performed using vector-primer T7, pEF FP, and pEF RP as recommended by the manufacturer (Epicentre). 16S rRNA gene sequencing was performed using primers 20F and 958R (DeLong, 1992). Sequencing reactions and cycling conditions were performed as described previously1. Briefly, 6 μL of fosmid DNA, 2 μL of BigDye Terminator 3.1, 1 μL of ABI 5× sequencing buffer and 10 pmole primer were mixed and amplified using the following thermocycling procedure: 5 min of 95°C, followed by 100 cycles of 95°C for 30 s 50°C for 10 s and 60°C for 4 min. Sequencing was performed using an ABI PRISM 377 capillary sequencer (ABI).
Pyrosequencing data and additional Sanger reads from fosmids 02j05, 05d18, and 10c16 were assembled using Newbler (454 Life Sciences) and the resulting contigs were edited using Consed (Gordon et al., 1998) using sub-assemblies and ensuring that only Sanger reads associated with the correct fosmid were included in each assembly. The resulting contig assemblies were examined further for quality control by manual inspection and verification using the program HawkEye (Schatz et al., 2007).
Sequence annotation
Protein coding sequences were identified using Glimmer v3 (Delcher et al., 2007), blastx searches of public protein databases (GenBank nr and env_nr), the IMG/M metagenome analysis system (Markowitz et al., 2008) and the pedant software system2. Annotations were manually curated as described earlier (Bartossek et al., 2010). IS elements were additionally screened for by IS_finder3.
Phylogenetic analysis
Phylogenetic analysis of 16S rRNA and amo gene sequences was performed using PHyML (Guindon and Gascuel, 2003) for maximum likelihood analysis and MEGA4 (Tamura et al., 2007) for distance analysis. After alignment and using unambiguously aligned positions only, all trees were constructed using ML-estimated gamma-distributed, variable sites, and GTR or Tamura 3-parameter correction for ML and distance analyses, respectively. Bootstrap support was calculated using 100 replicates for ML analyses and 1000 replicates for distance analysis.
Identities of amoABC“X” genes were calculated with the EMBOSS pairwise alignment tool after removing unalignable regions.
Results and Discussion
Sampling and construction of a metagenomic fosmid library
Samples were taken from the “Franz Josef Quelle,” a thermal spring located near Bad Gastein, Salzburg, Austria. Spring water (46°C, pH 8) and surrounding biofilm material were sampled, together with material grown in situ on glass slides which have been incubated several weeks to enable microbial colonization from the spring water. An initial analysis of the three different sample types was performed to determine whether there were differences in the archaeal community structures. DNA was extracted and after PCR of amoA genes, small clone libraries were constructed and 28 clones sequenced. The phylogenetic tree (Figure 1) shows that the amoA sequences from PCR-products of all sample types formed three clusters, two belonged to group I.1b amoA, and one to group I.1a. Since no differences in the population structure were observed among the samples, biomass from all samples was pooled to obtain as much initial chromosomal DNA as possible for the construction of a fosmid library. High molecular weight (hmw) DNA was extracted using the method based on Zhou et al. (1996) after some optimizations (Reigstad et al., 2011). A fosmid library with 29,874 clones was produced and archived. The average insert size was approximately 40 kb as determined by restriction analysis and pulse field gel electrophoresis.
Figure 1
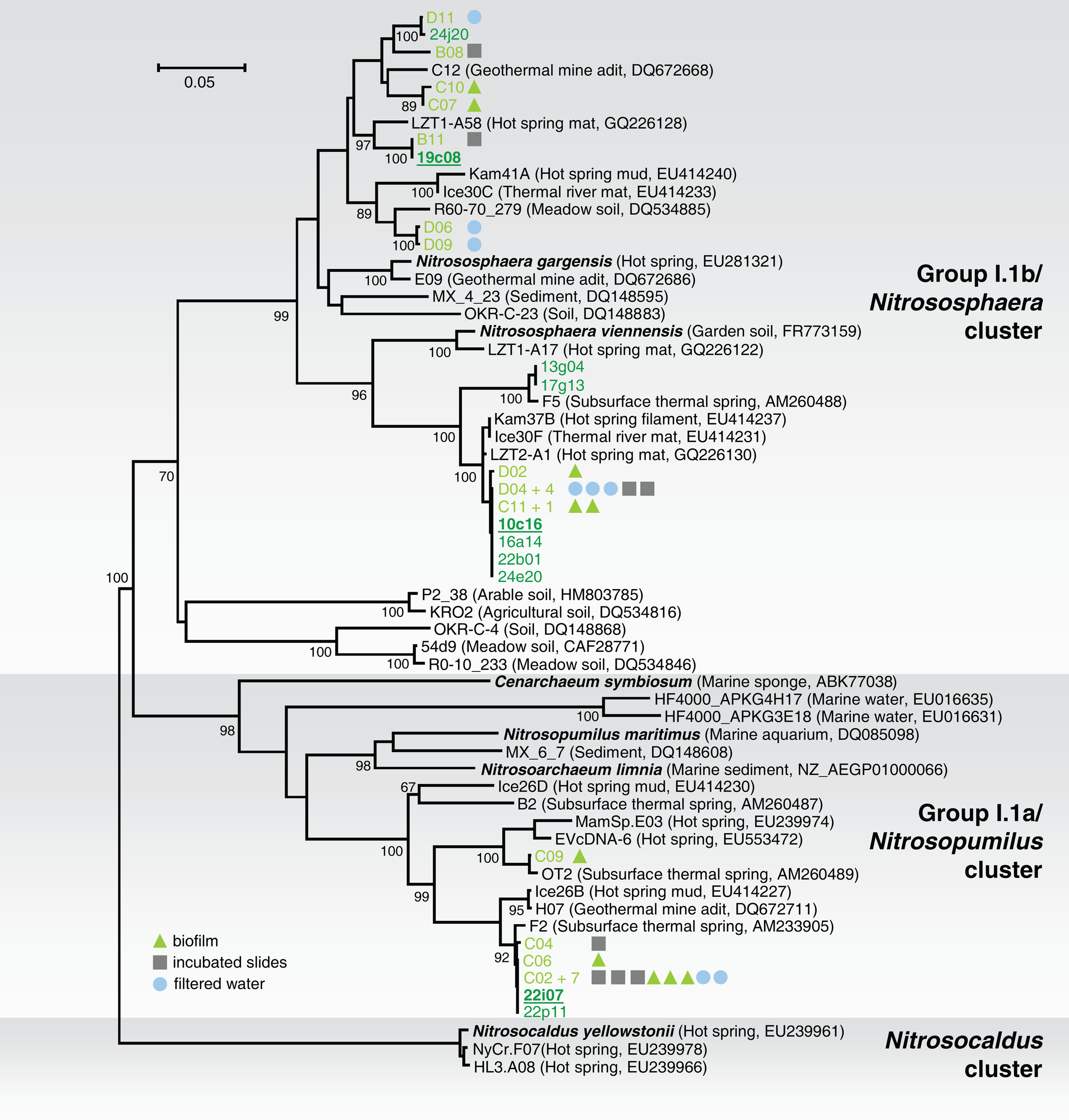
Phylogenetic analysis of archaeal amoA nucleic acid sequences from the thermal spring Franz Josef Quelle (FJQ) and selected reference sequences from “soil” (Nitrososphaera cluster), “marine” (Nitrosopumilus cluster), and ThAOA (Nitrosocaldus cluster) lineages. Sequences from this study are derived from partial (green) and fully sequenced fosmids (green underlined) and cloned PCR-products (light green) from incubated slides (gray square), biofilm (green triangle), and filtered water (blue circle). The tree was constructed using estimated variable sites only (68% of 478 positions). The tree represents a distance analysis with values at nodes with the most conservative value from either ML or distance analysis (100 and 1000 replicates, respectively). The scale bar represents 0.05 changes per nucleotide position.
Isolation of archaeal amoA and 16S rRNA genes from the metagenomic library
Twenty-five microtiter plates (9575 clones) were screened by PCR for the presence of archaeal 16S rRNA and ammonia monooxygenase subunit A (amoA) genes. Six fosmids were positive for 16S rRNA genes (01b24, 02j05, 02g06, 05d18, 17e16, and 19k20) and 10 fosmids were positive for amoA genes (10c16, 13g04, 16a14, 17g13, 19c08, 22b01, 22i07, 22p11, 24e20, 24j20), with no individual fosmid containing both a 16S rRNA and amoA gene. Assuming that an average prokaryotic genome is around 4 Mb in size (Liles et al., 2003) and that all AOA genomes contain only one 16S rRNA and amoA gene, this would indicate that between 6 and 10% of the genomes in the 25 screened plates were of archaeal origin.
Phylogenetic analysis of 16S rRNA and amoA gene sequences from thaumarchaeal fosmid clones
Phylogenetic analysis of six 16S rRNA gene sequences associated with fosmids 01b24, 02j05, 02g06, 05d18, 17e16, and 19k20 was performed with reference sequences including 16S rRNA sequences from environmental thaumarchaeal clones and from six representatives in pure or enrichment culture: N. maritimus, N. limnia, C. symbiosum, N. yellowstonii, N. viennensis, and N. gargensis (Figure 2). Five fosmid sequences were affiliated with group I.1b (“soil group”) and were placed within a lineage dominated by sequences from subsurface or thermophilic environments. Together, they formed a monophyletic group (with all sequences sharing ≥97% identity) with 16S rRNA gene sequences from the same habitat (Franz Josef spring – FJQ sequences in Figure 2; Weidler et al., 2007), the moderate thermophile N. gargensis (Hatzenpichler et al., 2008), soil archaeon N. viennensis (Tourna et al., 2011), sequence SAGMA-2 from a South African Gold mine (Takai et al., 2001), and Gitt-GR sequences recovered from a uranium mining waste in Germany (Selenska-Pobell et al., unpublished). The close relationship to the latter sequences is interesting as, like the Franz Josef Spring, the source of “Gitt” samples contain low amounts of radioactivity (radium, radon, and uranium; Heinen and Lauwers, 1988). Fosmid 19k20 was placed within the “marine” group I.1a. Organisms from this lineage have not been identified in previous studies of the archaeal communities in the Franz Josef spring (Weidler et al., 2007). This sequence showed high similarity to SAGMA-8 (99%) and HAuD-MA13 (97%), both of which were also recovered from subsurface environments in South Africa and Japan, respectively (Takai et al., 2001; Nunoura et al., 2005).
Figure 2
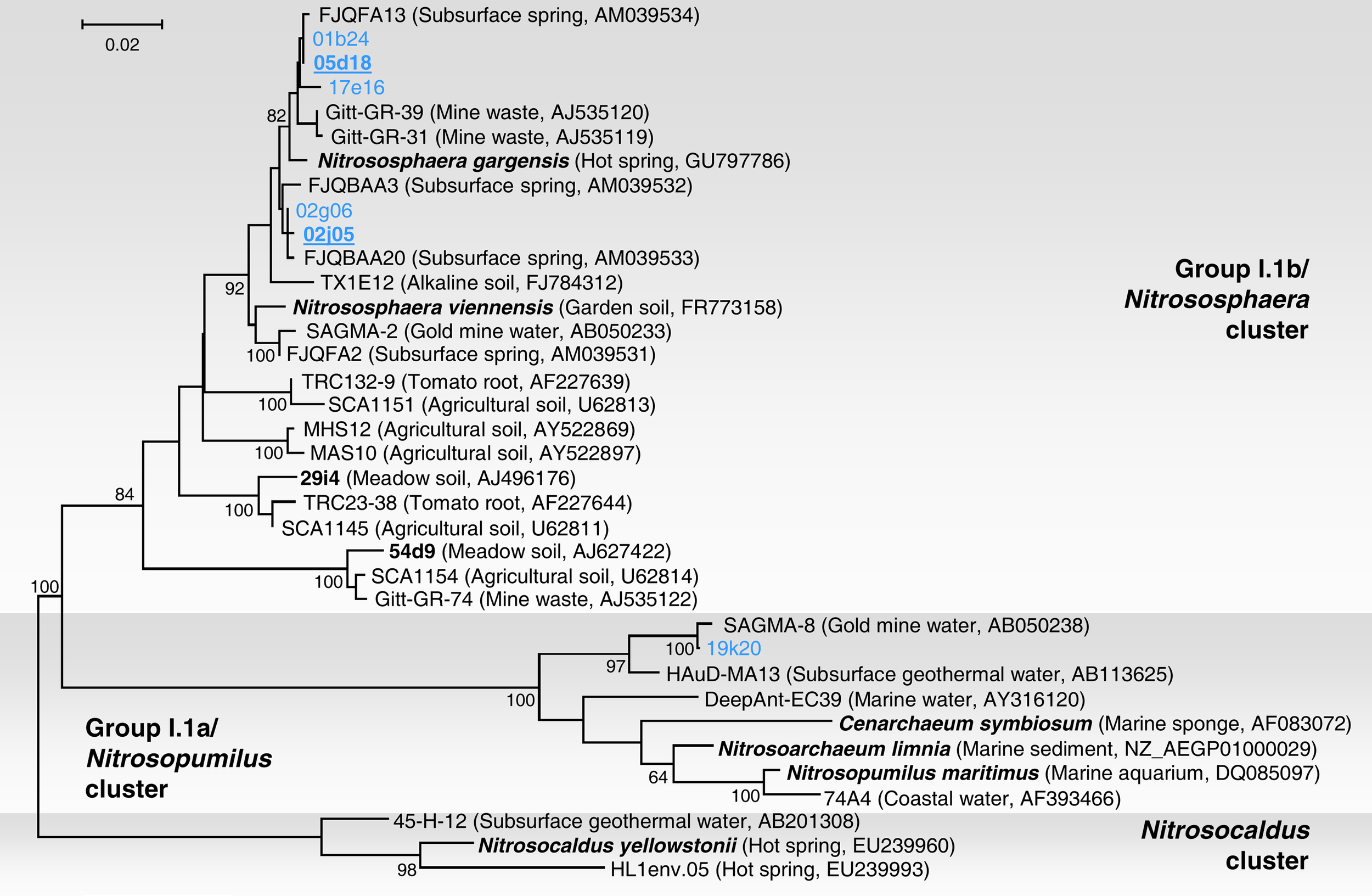
Phylogenetic analysis of archaeal 16S rRNA nucleic acid sequences form the thermal spring FJQ and selected reference sequences from Group I.1b/“soil” (Nitrososphaera cluster), Group I.1a/“marine” (Nitrosopumilus cluster), and “ThAOA” (Nitrosocaldus cluster) lineages. Sequences from this study are derived from partial (blue) and fully sequenced fosmids (bold blue underlined). The tree was constructed using estimated variable sites only (57% of 733 positions), it represents a distance analysis with the most conservative bootstrap values at nodes from either ML or distance analysis (100 and 1000 replicates, respectively). The scale bar represents 0.02 changes per nucleotide position.
Phylogenetic analysis of the amoA gene sequences associated with 10 fosmids (10c16, 13g04, 16a14, 17g13, 19c08, 22b01, 22i07, 22p11, 24e20, 24j20) and cloned amoA PCR-products showed a similar phylogenetic distribution like 16S rRNA genes (Figure 1). Again, the majority of the fosmids affiliated with the soil (“Nitrososphaera”-) group showed high similarity to sequences of N. viennensis and N. gargensis (8 out of 10). Two of the 10 fosmids possessed amoA gene sequences which clustered with the marine (“Nitrosopumilus”-) group, including C. symbiosum and N. maritimus.
The soil lineage amoA gene sequences could be placed within one of two distinct clusters, and despite their relatively disparate phylogenetic distribution (sequence similarity ranging between 83 and 100% on aa level), all sequences showed highest similarity to sequences from other thermo-, terrestrial, or subsurface habitats including a hot spring microbial mat in China (Jiang et al., 2010), a geothermal mine adit in Colorado (Spear et al., 2007), or hot springs from Iceland and Kamchatka (Reigstad et al., 2008). The same was found for the marine group. The majority of sequences clustered together, with one sequence separated from the cluster, and again all sequences showed highest similarity to those previously described from the same location (Weidler et al., 2007) or to sequences from before mentioned thermal habitats. This indicates that although these organisms are within widely distributed groups associated with soil and marine habitats, they are specifically affiliated with organisms from thermal environments.
Identification and characterization of genomic fragments from Thaumarchaeota
The amoA-containing fosmids 10c16, 19c08, and 22i07 were selected for full sequencing in order to best cover the assumed diversity based on the phylogenetic studies. Similarly, from six possible 16S rRNA gene containing fosmids, one from each cluster in the I.1b group (02j05 and 05d18) was selected. General information on the fosmid sequences is given in Table 1 and Figure 3.
Table 1
| Fosmid | 02j05 | 05d18 | 10c16 | 19c08 | 22i07 |
|---|---|---|---|---|---|
| Length (bp) | 45,998 | 40,266 | 43,181 | 31,692 | 42,801 |
| Gene fosmid was identified with | 16S rRNA | 16S rRNA | amoA | amoA | amoA |
| G + C content (%) | 50.8 | 49.9 | 54.5 | 51.2 | 42.8 |
| Coding density (%) | 76.7 | 91.8 | 91.6 | 82.3 | 89.9 |
| Number of ORFs | 59 | 48 | 47 | 42 | 50 |
| Hypothetical proteins | 26 | 17 | 18 | 11 | 5 |
| Uncharacterized conserved proteins | 13 | 13 | 13 | 6 | 18 |
| Proteins with assigned function | 20 | 19 | 16 | 25 | 27 |
| Proteins with most similar ortholog in Thaumarchaeota* | 24 | 23 | 21 | 15 | 45 |
| Proteins with ortholog only in Thaumarchaeota | 5 | 7 | 11 | 2 | 16 |
General characteristics of the five archaeal genome fragments.
*Including the proteins with orthologs only in Thaumarchaeota.
Figure 3
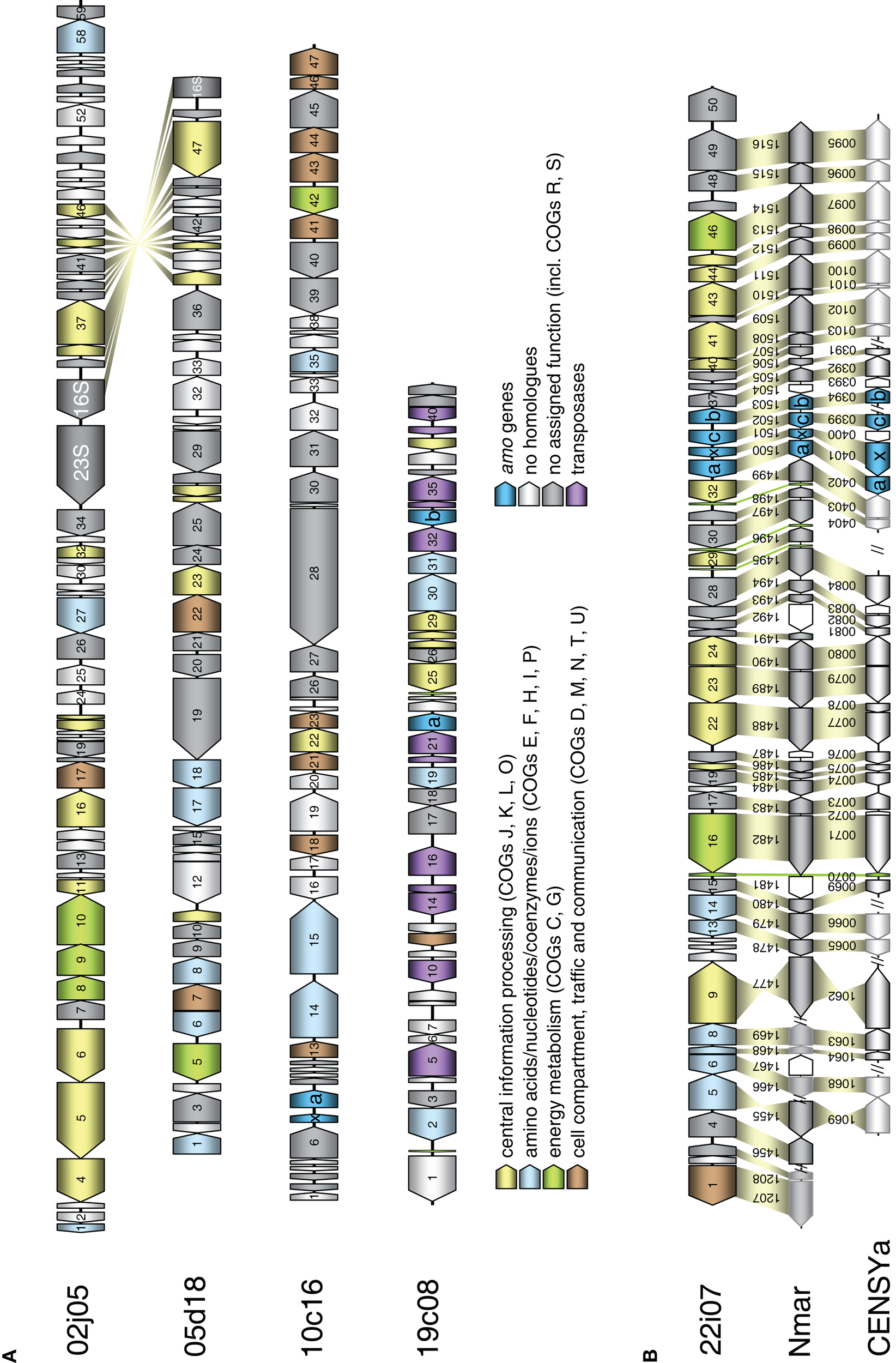
Schematic representation of fosmids from the FJQ spring containing archaeal 16S rRNA or amoA genes. Open reading frames are represented by arrowed boxes. The color code refers to grouped functional COG classification. Yellow: Central information processing (J: Translation, ribosomal structure, and biogenesis; K: Transcription; L: DNA replication, recombination, and repair; O: Posttranslational modification, protein turnover, chaperones). Blue: Amino acids/nucleotides/coenzymes/ions (E: Amino acid transport and metabolism; F: Nucleotide transport and metabolism; H: Coenzyme metabolism; I: Lipid metabolism; P: Inorganic ion transport and metabolism). Green: Energy metabolism (C: Energy production and conversion; G: Carbohydrate transport and metabolism). Brown: Cell compartment, traffic, and communication (D: cell cycle control, cell-division, chromosome partitioning; M: Cell envelope biogenesis, outer membrane; N: Cell motility and secretion; T: Signal transduction mechanisms; U: Intracellular trafficking, secretion, and vesicular transport). Turquoise: amo genes. Violet: transposases. Gray: no homologs. Dark gray: no assigned function (including COGs R: general function prediction only. S: function unknown). Homologous genes between fosmids are connected by yellow shading. (A) Sequenced fosmids 02j05, 05d18, 10c16, and 19c08 from I.1b/“soil” (Nitrososphaera) group. (B) Schematic representation of marine group fosmid 22i07 containing an archaeal amoA gene. Homologous genes in the genomes of Cenarchaeum symbiosum (CENSYa) and Nitrosopumilus maritimus (Nmar) are indicated below with a connecting yellow shading, tRNA with a green shading, non-homologous genes are white. Color code as in (A).
The four fosmids associated with the Nitrososphaera lineage or the soil group had a G + C content greater than 49.9% (Table 1), while fosmid 22i07 of the marine group had a lower G + C content of 42.8%. The latter fosmid had remarkably high synteny with the genome of N. maritimus sharing gene order in 36 out of 50 ORFs (Figure 3B). It also contained the highest number of ORFs with homologs in known Thaumarchaeota and metagenomic thaumarchaeal sequences (45 of 50), many of which were exclusively found in this archaeal lineage (16 out of 50). In comparison, fosmids 02j05, 05d18, 10c16, and 19c08 showed a higher number of proteins with most similar orthologs in bacteria, euryarchaeotes, or crenarchaeotes and a higher amount of hypothetical proteins (Table 1).
In line with this finding, only 12–37% of the ORFs encoded on the four group I.1b fosmids had a best blastP hit with marine Thaumarchaeota like N. maritimus, N. limnia, C. symbiosum, N. koreensis, and marine metagenomic sequences, while 90% of the ORFs on fosmid 22i07 had a best blastP hit with marine Thaumarchaeota (Table S1 in Supplementary Material). A syntenic region spanning 12 genes was present between fosmid 02j05 and 05d18 in the region containing the 16S rRNA gene sequence (Figure 3A). No synteny could be detected between the fosmids containing the amo gene cluster in this survey.
A total of 90 protein encoding genes were exclusively found in Thaumarchaeota but absent from other archaeal genomes (e-value ≤10−5, analysis included the draft genomes of N. viennensis and N. gargensis, data not shown), most of which had no assigned function. Eight of these ORFs belonged to the amo gene cluster. Several genes with homology in other archaea or bacteria may be directly or indirectly involved in carbon metabolism. For example aconitate hydratase encoded by ORF 16 on fosmid 22i07 and a putative fumarase encoded by ORF 1 on fosmid 5d18 are enzymes involved in the tricarboxylic acid (TCA) cycle. Furthermore, a homolog of a bifunctional glucose-6-phosphase/mannose-6-phosphase isomerase (Hansen et al., 2004) is present on fosmid 10c16 (ORF 42) which is involved in the conversion of fructose-6-phosphate to glucose-6-phosphate in the last step of gluconeogenesis.
As in other Thaumarchaeota genomes, genes for proteins involved in two different potential systems for cell-division were identified on the fosmids: Firstly, FtsZ a GTPase and major player in cell-division in most bacteria and euryarchaeotes and secondly a homolog of ESCRT-III like proteins, that have been shown to be involved in cell-division in Crenarchaeota (Samson and Bell, 2011) and N. maritimus (Pelve et al., 2011; ORFs 17 and 13 on fosmids 02j05 and 10c16, respectively). Further genes identified encoded several ribosomal proteins, tRNA synthetases, genes involved in regulation of transcription, uvrABC homologs, genes encoding proteins potentially involved in sugar conversion, cofactor biosynthesis and ion transport (Table S1 in Supplementary Material).
Fosmid 10c16 contained a cluster of six genes (ORF 41, 42, 43, 44, 46, 47) with some similarity to genes involved in monosaccharide metabolism. Some of these are related to the rmlADBC or rfbADCB operons (according to Salmonella or Streptococcus annotation, respectively) responsible for the conversion of glucose-1-phosphate to dTDP-l-rhamnose which is an important precursor for exopolysaccharide (EPS) biosynthesis in bacteria (Samuel and Reeves, 2003). Homologous of these genes are also found in other archaea. ORF 41 shows remote similarity to galE that encodes a potential uridine diphosphate (UDP)-galactose-4-epimerase involved in the reversible conversion of UDP-galactose to UDP-glucose. Interestingly, this enzymatic step is important for the biosynthesis of EPS in bacteria (Niou et al., 2009).
The presence of co-encoding rmlA-D and galE genes might suggest the ability to synthesize exopolysaccharides, which are important in biofilm formation. Rhamnose has been reported as a cell wall polymer in only a few archaea, such as the methanogenic Methanobacterium formicicum and M. ruminantium (Kandler and Konig, 1978) and one halophilic archaeon, Haloferax volcanii (Kuntz et al., 1997). Homologs of rmlA-D are present in M. ruminantium (Leahy et al., 2010) and H. volcanii. However, a connection between those genes and the rhamnose occurrence in the cell has not yet been demonstrated.
fix ABCX gene cluster on fosmid 02j05
A cluster of three genes (ORF 8–10) with high similarity to the fixABCX operon found on thaumarchaeal soil fosmid 29i4 (Quaiser et al., 2002) and in N. viennensis and N. gargensis (A. Spang and C. Schleper unpublished data) has been identified on fosmid 02j05. Homologs occur as well in halobacteria and Sulfolobales, and in various bacteria. FixAB and fixCX genes code for electron transferring flavoproteins (Etf) and Etf-quinone reductase, respectively, that might be involved in the transfer of electrons to nitrogenase in some diazotrophic bacteria (Arigoni et al., 1991; Tsai and Saier, 1995; Weidenhaupt et al., 1996). No homologs could be found in marine Thaumarchaeota or on metagenomic fragments. Since homologs of fixABCX are found in archaea such as Sulfolobus solfataricus and Thermoplasma acidophilum for which nitrogen fixation has not been demonstrated, functional studies are required to determine the physiological role of these putative electron transfer proteins.
uvr ABC gene cluster on fosmid 02j05
Fosmid 02j05 contains uvrABC genes (ORFs 04 through 06) coding for a potential UvrABC excinuclease, an enzyme complex mediating DNA excision repair (Selby and Sancar, 1990). Homologs were found in all published thaumarchaeal genomes (with the exception of N. limnia which possesses only a partial cluster (uvrAB) at the end of an incomplete genomic contig). Additionally, UvrABC homologs can be found in halophilic and methanogenic Euryarchaeota and bacteria, but not in hyperthermophilic Euryarchaeota or Crenarchaeota (Goosen and Moolenaar, 2008). Interestingly, only in Thaumarchaeota the uvrABC genes occur in the conserved order uvrBAC, indicating that these organisms might have maintained an ancestral operon structure.
Association of amo genes and transposases on Fosmid 19c08
Fosmid 19c08 encodes ten putative transposases that indicate the presence of insertion sequence (IS) elements (Tables S1 and S2 in Supplementary Material). This fosmid could either be derived from a genome region of high IS element activity, or could be an indicator of a high amount of transposases in the original archaeal genome. There is a precedent for archaeal genomes containing a high density of mobile elements (IS and transposable elements) with S. solfataricus containing 344 mobile elements, which constitute over 10% of the entire genome (Brugger et al., 2004) and are comprised of 650 annotated transposases (including 89 partial transposases in the GenBank database for S. solfataricus P2).
Seven of the transposases showed highest amino acid similarity to bacterial transposases, that are derived from a number of IS element families, i.e., ISH3, IS1595, IS110, and IS630 (Table S2 in Supplementary Material). However, homologs of nearly all IS elements can also be found in archaea. Only two of the transposases, ORF 21 and ORF 35 (Figure 4C), which are almost identical to each other (97.9% identical on aa level) belong to an uncharacterized IS family. These putative transposases appear specific to archaea and have highest similarity to the archaeal fosmid gzfos1d1 (Hallam et al., 2004). The upstream ORFs 20 and 34 are almost identical to each other (98.8% on aa level) and are homologs to genes on the before mentioned archaeal fosmid gzfos1d1. In contrast to ORF 20 and 21 that are separated by 65 nt, the transposases encoded by ORF 34 and 35 are separated by a single −1 frameshift, which is a common feature of diverse IS element families and represents a means to regulate translation of transposase genes (Figure 4C).
Figure 4
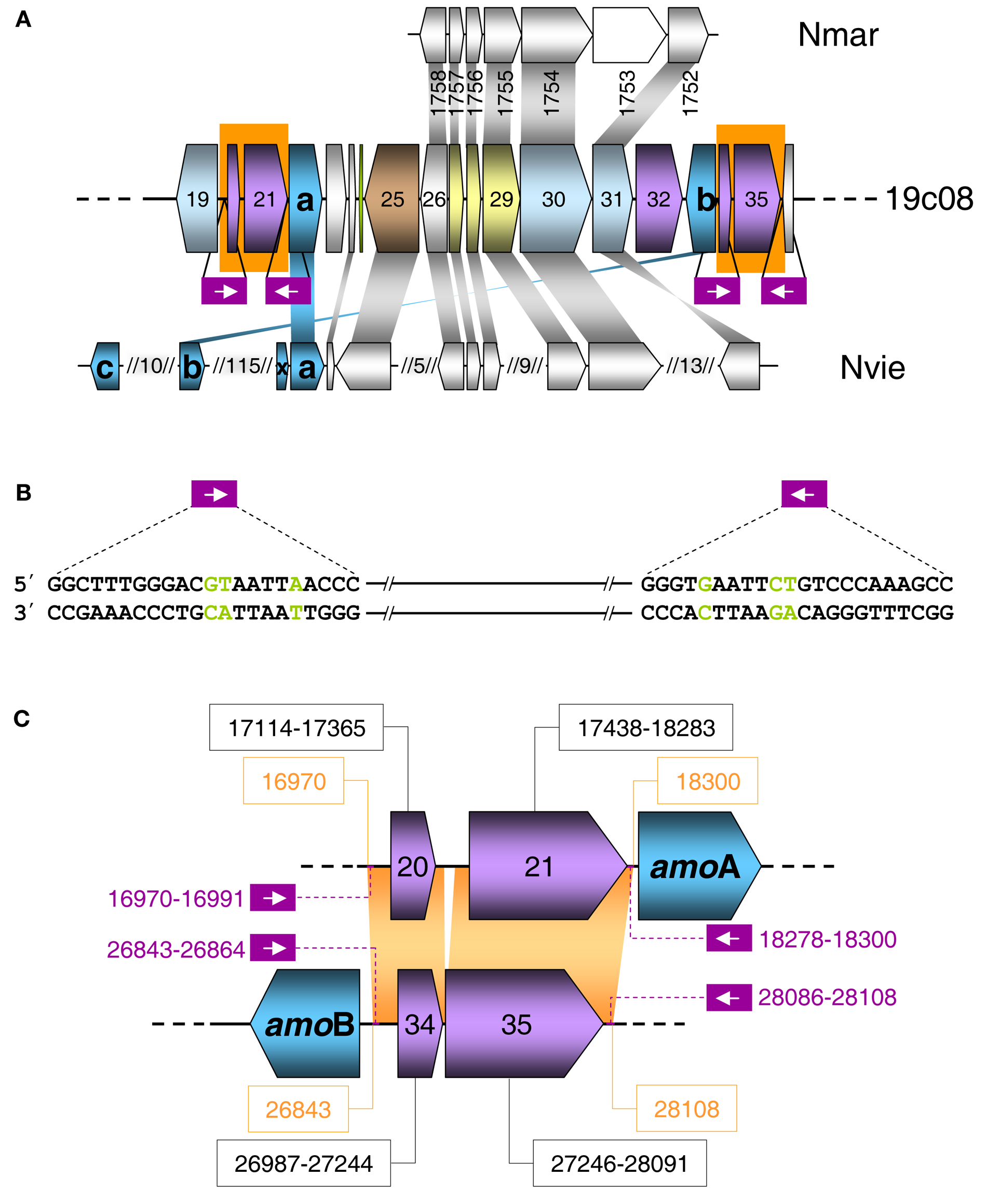
(A) Schematic representation of a section of fosmid 19c08. The color code refers to grouped functional COG classification according to Figure 3. Homologous genes in the genomes of Nitrosopumilus maritimus (Nmar) and Nitrososphaera viennensis (Nvie) are indicated above and below with a connecting gray shading. Non-homologous genes are white. AmoAB genes are turquoise. Purple boxes indicate the position of the repeats and inverted repeats. (B) Sequence of the repeat and the inverted repeat of the transposases shown in a) on fosmid 19c08. Green letters are the mismatches in the repeats. (C) Schematic representation of ORFs 20–22 and 33–35 of fosmid 19c08 with identical sequences shaded in orange. Violet ORFs 20, 21, and 34, 35 code for potential transposases. The sequence position of the ORFs are in black boxes. The purple numbering shows the position of the repeats and inverted repeats.
The fact that both IS Elements (ORFs 20+21 and 34+35) including their flanking sequences and inverted repeats are identical and even share inverted repeats (with three mismatches) (Figure 4B) indicates that they might be able to form a composite transposon, which could potentially mobilize all ORFs in the region from ORF 20 to ORF 35, including amoA and amoB.
Transposition could also provide an explanation for the finding that amoA on this fosmid is disconnected from the gene encoding a conserved hypothetical protein which is usually found next to amoA (Figure 5) such as ORF 38 in fosmid 54d9 (Treusch et al., 2005). Since this hypothetical gene has always been detected in association with other amo genes in all other currently described AOA genomes or genome fragments, it seems likely that it is associated with the archaeal ammonia monooxygenase and we thus refer to this conserved hypothetical gene as “amoX.” Fosmid 19c08 represents the only known example for the separation of amoA from “amoX.” Transposition (or recombination) may also explain the substantial variation in gene order observed within amo gene clusters and the opposite orientation of amoA and amoB in 19c08 compared to other amo gene clusters (Figure 5). However, whether amoAB genes might really be mobilized can only be investigated experimentally or by further genomic sequencing.
Figure 5
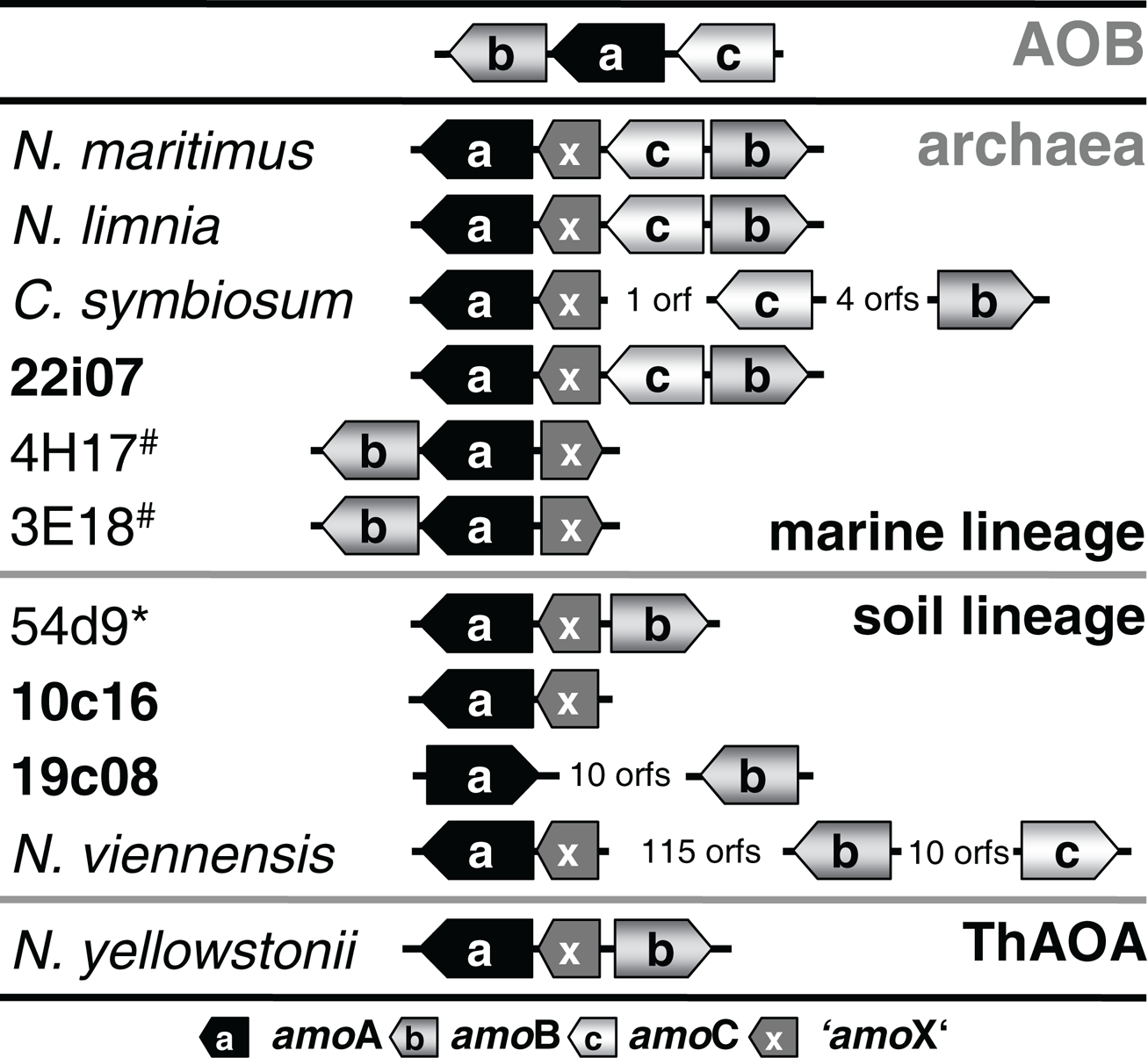
Schematic representation of the gene arrangement of ammonia monooxygenase subunits in ammonia-oxidizing bacteria (AOB) and amoA carrying Thaumarchaeota from marine, soil, and ThAOA lineages. “amoX” is a conserved hypothetical protein associated with the amo gene cluster. Fosmids from this study are highlighted in bold. 54d9* is a fosmid isolated from a calcareous grassland (AJ627422) and 3E18# and 4H17# are fosmids isolated from (4000 m depth) sea water (EU016631, EU016635).
Interestingly, the region flanked by the transposases is syntenic to other thaumarchaeotal genomes including N. maritimus, N. liminia and N. viennensis (Figure 4A).
Comparison of amo gene sequences and conserved upstream promoter regions
The amoA, amoB, amoC, and “amoX” gene sequences including upstream regions from the three cave fosmids 10c16, 19c08, and 22i07 were compared with those from thaumarchaeal (meta)genomes (N. gargensis, N. viennensis, N. yellowstonii, N. maritimus, N. limnia, C. symbiosum) and fosmid sequences (54d9, HF4000_APKG3E18 and HF4000_APKG4H17) where several subunits were known. Phylogenetic trees of each gene, i.e., amoB (22i07, 19c08), amoC (22i07), and “amoX” (22i07, 10c16) showed the same overall topology as amoA and 16S RNA gene phylogenies (Figure 6). AmoB, amoC, and “amoX” genes of fosmid 22i07 cluster with the marine/Group I.1a lineage whereas those of fosmid 19c08 and 10c16 branch within the soil/Group I.1b lineage, respectively (Figure 6).
Figure 6
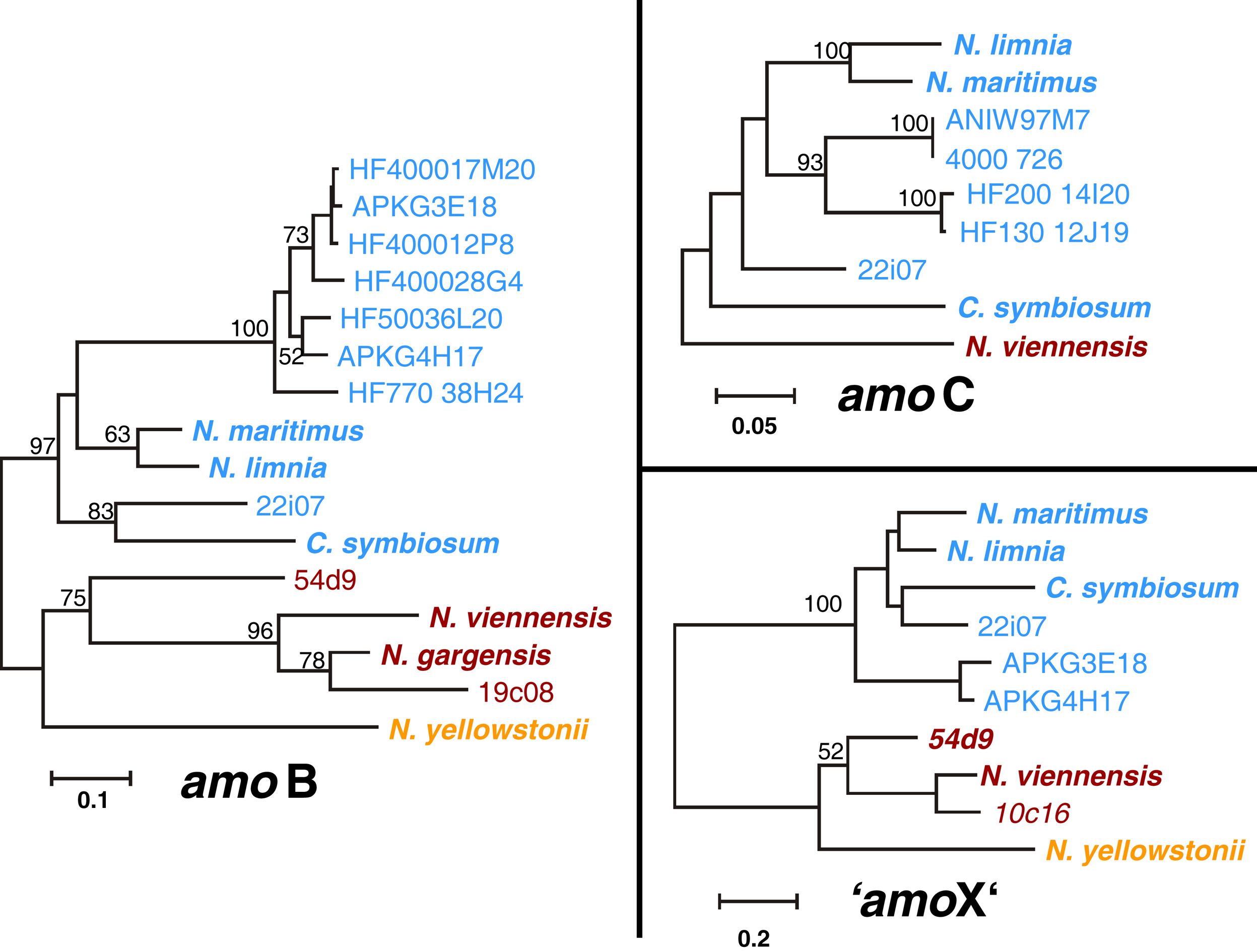
Phylogenetic analysis of amoB, amoC, and “amoX” nucleic acid sequences from the thermal spring FJQ and selected reference sequences from Group I.1b/“soil” (red), Group I.1a/“marine” (blue), and “ThAOA” (orange) lineages. The tree represents a distance analysis with values at nodes with the most conservative value from either ML or distance analysis (100 and 1000 replicates, respectively).
amoA
The 12 compared amoA gene sequences were identical in length except for that of N. yellowstonii, which has an additional codon (de la Torre et al., 2008). The range of identity at the nucleic acid level was between 68% (10c16 and HF4000_APKG3E18) and 91% (N. viennensis and 10c16). Assuming an ATG translational start codon at the same relative position (and differing from those published for 54d9 and the HF4000-fosmids), conserved motifs that are typically found in basal archaeal (and eukaryotic) promoters (Bell and Jackson, 2001), i.e., a TATA box and parts of a BR element (transcription factor B recognition element) can be identified 23 nt or 24 nt upstream of all sequences (Figure 7A, boxed sequences). This finding indicates that the amoA gene in AOA is not co-transcribed with upstream located genes of the cluster, but rather separately from its own promoter. As demonstrated in other archaea (Torarinsson et al., 2005), the transcriptional and translational start sites might be located on the same nucleotide and codon, respectively. Furthermore, both upstream and downstream of the basal promoter elements additional conserved sequence motifs specific for either representatives of group I.1a or group I.1b lineages, respectively, could be identified (underlined and bold sequences in Figure 7). These are candidates for cis-regulatory sequences that could serve for binding of specific transcriptional regulators.
Figure 7

Alignment of start and upstream region of the four amo gene sequences [(A)amoA, (B)amoB, (C)amoC and (D)amo’X’] from fosmids and cultivated thaumarchaeal representatives from the soil and marine group and one thermophilic Thaumarchaeota. The ATG start codon and TATA box/BR elements are highlighted (Bell et al., 1999). Identical positions conserved in 85% of the sequences are shaded in gray. Sequences in blue are upstream ORFs. Sequences in violet are upstream transposase ORFs. Sequences in bold and underlined are common motifs potentially involved in regulation. Amo gene sequences are from fosmids 54d9, 19c08, 22i07, HF4000_APKG3E18 (3E18), HF4000_APKG4H17 (4E17), HF4000_Aniw97M7 (97M7), and from Nitrososphaera viennensis (Nvie), Nitrosopumilus maritimus (Nmar), Nitrosoarchaeum limnia (Nlim), Cenarchaeum symbiosum (CensyA), Nitrosocaldus yellowstonii (Nyel) where gene information was available.
amoB
Comparison of 10 amoB sequences showed that they were less conserved than amoA, i.e., between 54% (19c08 and HF4000_APKG3E18) and 88% (N. maritimus and N. limnia) identity. In contrast to amoA genes, it was impossible to define a common translational start. Nevertheless, a common TATA and BR element could be identified for all sequences, though the distance to the potential translational start codon was different (Figure 7B). In fosmids HF4000_APKG3E18 and HF4000_APKG4H17 (which are highly syntenic to each other; Konstantinidis and DeLong, 2008) only four or five nucleotides separate the stop codon of amoA from the start codon of amoB and no typical TATA or BR element was observed. These two fosmids are the only published examples where the amoA ORF is directly upstream of amoB, and “amoX” is in reverse orientation to amoA (Figure 5). It is possible that these organisms co-transcribe amoA and amoB genes as a single polycistronic RNA.
Additional short conserved sequences upstream of amoB were observed among group I.1a and I.1b lineages, respectively, representing putative regulatory sequences. In addition, the sequence motif GGGAT was found in almost all published amoB sequences directly upstream of the potential translational start codon (Figure 7B).
amoC
amoC has a range of identity at the nucleic acid level of 76% (N. viennensis and HF4000_ANIW97M7) to 87% (N. maritimus and 22i7). AmoC is absent from the amoA“X”B gene cluster on cave fosmids 10c16 and 19c08, as well as on the marine fosmids HF4000_APKG4H17 and HF4000_APKG3E18, soil fosmid 54d9 and N. yellowstonii (Figure 5). Although, automatic genome annotations have led to the selection of different start positions for diverse amoC genes, the high conservation and the presence of a start codon 24 nt downstream from a potential TATA box in all known amoC sequences indicates a common ATG start differing from that predicted previously for C. symbiosum, N. maritimus, and HF4000_ANIW97M7 (Figure 7C). Conserved sequence motifs, that represent potential regulatory elements could not be identified for amoC genes.
‘amoX’
Comparison of 10 sequences reveals that “amoX” is the least conserved of the four amo genes (A, B, C, and “X”). Identity at the nucleic acid level ranges between 40% (N. yellowstonii and C. symbiosum) and 85% (N. viennensis and 10c16). A common start codon (ATG) and a common TATA box and BR element can be identified for all “amoX” genes (Figure 7D). Additionally, short conserved potential cis-regulatory sequences could be identified between the TATA boxes and start codons. The deduced proteins of the marine organisms were longer, encoding an additional 25 AA spanning transmembrane domain beside the two transmembrane domains predicted for all other “AmoX” proteins.
Conclusion
The cultivation of Thaumarchaeota is challenging as seen by the limited number of representatives in laboratory culture. The production of large-insert libraries therefore still remains a relevant tool for exploring the genomic content of specific lineages. The fosmids derived from a thermal cave expand the knowledge on AOA of group I.1b (Nitrososphaera cluster) of Thaumarchaeota and highlight some differences in the genomic content between organisms of different lineages. Analysis of the upstream sequences of amoA, amoB, amoC, and “amoX” genes revealed potential cis-regulatory elements that might be suitable targets for the study of gene regulation in these ammonia oxidizers. Furthermore, it remains to be investigated to what extend mobile genetic elements such as IS elements as identified in this study might have favored the spread of amo genes among organisms or might influence the architecture of thaumarchaeal genomes and their evolution.
Accession Numbers
All sequences have been deposited in the GenBank database with Accession No. JQ768055 to JQ768096.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/Terrestrial_Microbiology/10.3389/fmicb.2012.00208/abstract
Supplementary Table S1Predicted RNA and protein encoding genes in thaumarchaeal fosmids 02j05, 05d18, 10c16, 19c08, and 22i07.
Supplementary Table S2List of blast results of IS elements/transposable elements of fosmid 19c08 with IS-Finder and GenBank.
Statements
Acknowledgments
The authors would like to thank Thomas Rattei and Thomas Weinmayer for support with the bioinformatic analysis, Graeme W. Nicol for support with the phylogenetic analysis, and Daniela Teichmann for helpful discussions. Anja Spang was supported by a docfForte fellowship of the Austrian Academy of Sciences. This work is supported by grant P23000 to Christa Schleper of the Austrian Science Fund (FWF).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Arigoni F. Kaminski P. A. Hennecke H. Elmerich C. (1991). Nucleotide sequence of the fixABC region of Azorhizobium caulinodans ORS571: similarity of the fixB product with eukaryotic flavoproteins, characterization of fixX, and identification of nifW. Mol. Gen. Genet.225, 514–520.10.1007/BF00261695
2
Bartossek R. Nicol G. W. Lanzen A. Klenk H. P. Schleper C. (2010). Homologues of nitrite reductases in ammonia-oxidizing archaea: diversity and genomic context. Environ. Microbiol.12, 1075–1088.10.1111/j.1462-2920.2010.02153.x
3
Bates S. T. Berg-Lyons D. Caporaso J. G. Walters W. A. Knight R. Fierer N. (2011). Examining the global distribution of dominant archaeal populations in soil. ISME J.5, 908–917.10.1038/ismej.2010.171
4
Bell S. D. Jackson S. P. (2001). Mechanism and regulation of transcription in archaea. Curr. Opin. Microbiol.4, 208–213.10.1016/S1369-5274(00)00190-9
5
Bell S. D. Kosa P. L. Sigler P. B. Jackson S. P. (1999). Orientation of the transcription preinitiation complex in archaea. Proc. Natl. Acad. Sci. U.S.A.96, 13662–13667.10.1073/pnas.96.24.13662
6
Blainey P. C. Mosier A. C. Potanina A. Francis C. A. Quake S. R. (2011). Genome of a low-salinity ammonia-oxidizing archaeon determined by single-cell and metagenomic analysis. PLoS ONE6, e16626.10.1371/journal.pone.0016626
7
Brochier-Armanet C. Boussau B. Gribaldo S. Forterre P. (2008). Mesophilic Crenarchaeota: proposal for a third archaeal phylum, the Thaumarchaeota. Nat. Rev. Microbiol.6, 245–252.10.1038/nrmicro1852
8
Brugger K. Torarinsson E. Redder P. Chen L. Garrett R. A. (2004). Shuffling of Sulfolobus genomes by autonomous and non-autonomous mobile elements. Biochem. Soc. Trans.32, 179–183.10.1042/BST0320179
9
Buckley D. H. Graber J. R. Schmidt T. M. (1998). Phylogenetic analysis of nonthermophilic members of the kingdom Crenarchaeota and their diversity and abundance in soils. Appl. Environ. Microbiol.64, 4333–4339.
10
de la Torre J. R. Walker C. B. Ingalls A. E. Konneke M. Stahl D. A. (2008). Cultivation of a thermophilic ammonia oxidizing archaeon synthesizing crenarchaeol. Environ. Microbiol.10, 810–818.10.1111/j.1462-2920.2007.01506.x
11
Delcher A. L. Bratke K. A. Powers E. C. Salzberg S. L. (2007). Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics23, 673–679.10.1093/bioinformatics/btm009
12
DeLong E. F. (1992). Archaea in coastal marine environments. Proc. Natl. Acad. Sci. U.S.A.89, 5685–5689.10.1073/pnas.89.12.5685
13
DeLong E. F. (1998). Everything in moderation: archaea as “non-extremophiles.”Curr. Opin. Genet. Dev.8, 649–654.10.1016/S0959-437X(98)80032-4
14
DeLong E. F. Preston C. M. Mincer T. Rich V. Hallam S. J. Frigaard N. U. Martinez A. Sullivan M. B. Edwards R. Brito B. R. Chisholm S. W. Karl D. M. (2006). Community genomics among stratified microbial assemblages in the ocean’s interior. Science311, 496–503.10.1126/science.1120250
15
Ewing B. Hillier L. Wendl M. C. Green P. (1998). Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res.8, 175–185.
16
Fuhrman J. A. Mccallum K. Davis A. A. (1992). Novel major archaebacterial group from marine plankton. Nature356, 148–149.10.1038/356148a0
17
Fuhrman J. A. Ouverney C. C. (1998). Marine microbial diversity studied via 16S rRNA sequences: cloning results from coastal waters and counting of native archaea with fluorescent single cell probes. Aquatic Ecol.32, 3–15.10.1023/A:1009974817127
18
Ghai R. Rodriguez-Valera F. Mcmahon K. D. Toyama D. Rinke R. Cristina Souza de Oliveira T. Wagner Garcia J. Pellon De Miranda F. Henrique-Silva F. (2011). Metagenomics of the water column in the pristine upper course of the Amazon river. PLoS ONE6, e23785.10.1371/journal.pone.0023785
19
Goosen N. Moolenaar G. F. (2008). Repair of UV damage in bacteria. DNA Repair (Amst.)7, 353–379.10.1016/j.dnarep.2007.09.002
20
Gordon D. Abajian C. Green P. (1998). Consed: a graphical tool for sequence finishing. Genome Res.8, 195–202.
21
Guindon S. Gascuel O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol.52, 696–704.10.1080/10635150390235520
22
Hallam S. J. Putnam N. Preston C. M. Detter J. C. Rokhsar D. Richardson P. M. Delong E. F. (2004). Reverse methanogenesis: testing the hypothesis with environmental genomics. Science305, 1457–1462.10.1126/science.1100025
23
Hansen T. Wendorff D. Schönheit P. (2004). Bifunctional phosphoglucose/phosphomannose isomerases from the archaea Aeropyrum pernix, and Thermoplasma acidophilum constitute a novel enzyme family within the phosphoglucose isomerase superfamily. J. Biol. Chem.279, 2262–2272.10.1074/jbc.M402452200
24
Hatzenpichler R. Lebedeva E. V. Spieck E. Stoecker K. Richter A. Daims H. Wagner M. (2008). A moderately thermophilic ammonia-oxidizing crenarchaeote from a hot spring. Proc. Natl. Acad. Sci. U.S.A.105, 2134–2139.10.1073/pnas.0708857105
25
Heinen W. Lauwers A. M. (1988). Leaching of silica and uranium and other quantitative aspects of the lithobiontic colonization in a radioactive thermal spring. Microb. Ecol.15, 135–149.10.1007/BF02011708
26
Herndl G. J. Reinthaler T. Teira E. Van Aken H. Veth C. Pernthaler A. Pernthaler J. (2005). Contribution of archaea to total prokaryotic production in the deep Atlantic Ocean. Appl. Environ. Microbiol.71, 2303–2309.10.1128/AEM.71.5.2303-2309.2005
27
Hershberger K. L. Barns S. M. Reysenbach A. L. Dawson S. C. Pace N. R. (1996). Wide diversity of Crenarchaeota. Nature384, 420–420.10.1038/384420a0
28
Ingalls A. E. Shah S. R. Hansman R. L. Aluwihare L. I. Santos G. M. Druffel E. R. Pearson A. (2006). Quantifying archaeal community autotrophy in the mesopelagic ocean using natural radiocarbon. Proc. Natl. Acad. Sci. U.S.A.103, 6442–6447.10.1073/pnas.0510157103
29
Jia Z. Conrad R. (2009). Bacteria rather than archaea dominate microbial ammonia oxidation in an agricultural soil. Environ. Microbiol.11, 1658–1671.10.1111/j.1462-2920.2009.01891.x
30
Jiang H. Huang Q. Dong H. Wang P. Wang F. Li W. Zhang C. (2010). RNA-based investigation of ammonia-oxidizing archaea in hot springs of Yunnan Province, China. Appl. Environ. Microbiol.76, 4538–4541.10.1128/AEM.00143-10
31
Jung M. Y. Park S. J. Min D. Kim J. S. Rijpstra W. I. Sinninghe Damste J. S. Kim G. J. Madsen E. L. Rhee S. K. (2011). Enrichment and characterization of an autotrophic ammonia-oxidizing archaeon of mesophilic crenarchaeal group I.1a from an agricultural soil. Appl. Environ. Microbiol.77, 8635–8647.10.1128/AEM.05787-11
32
Kandler O. Konig H. (1978). Chemical composition of the peptidoglycan-free cell walls of methanogenic bacteria. Arch. Microbiol.118, 141–152.10.1007/BF00415722
33
Karner M. B. Delong E. F. Karl D. M. (2001). Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature409, 507–510.10.1038/35054051
34
Klotz M. G. Stein L. Y. (2008). Nitrifier genomics and evolution of the nitrogen cycle. FEMS Microbiol. Lett.278, 146–156.10.1111/j.1574-6968.2007.00970.x
35
Könneke M. Bernhard A. E. de la Torre J. R. Walker C. B. Waterbury J. B. Stahl D. A. (2005). Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature437, 543–546.10.1038/nature03911
36
Konstantinidis K. T. DeLong E. F. (2008). Genomic patterns of recombination, clonal divergence and environment in marine microbial populations. ISME J.2, 1052–1065.10.1038/ismej.2008.62
37
Kuntz C. Sonnenbichler J. Sonnenbichler I. Sumper M. Zeitler R. (1997). Isolation and characterization of dolichol-linked oligosaccharides from Haloferax volcanii. Glycobiology7, 897–904.10.1093/glycob/7.7.897
38
Leahy S. C. Kelly W. J. Altermann E. Ronimus R. S. Yeoman C. J. Pacheco D. M. Li D. Kong Z. Mctavish S. Sang C. Lambie S. C. Janssen P. H. Dey D. Attwood G. T. (2010). The genome sequence of the rumen methanogen Methanobrevibacter ruminantium reveals new possibilities for controlling ruminant methane emissions. PLoS ONE5, e8926.10.1371/journal.pone.0008926
39
Lehtovirta-Morley L. E. Stoecker K. Vilcinskas A. Prosser J. I. Nicol G. W. (2011). Cultivation of an obligate acidophilic ammonia oxidizer from a nitrifying acid soil. Proc. Natl. Acad. Sci. U.S.A.108, 15892–15897.10.1073/pnas.1107196108
40
Leininger S. Urich T. Schloter M. Schwark L. Qi J. Nicol G. W. Prosser J. I. Schuster S. C. Schleper C. (2006). Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature442, 806–809.10.1038/nature04983
41
Liles M. R. Manske B. F. Bintrim S. B. Handelsman J. Goodman R. M. (2003). A census of rRNA genes and linked genomic sequences within a soil metagenomic library. Appl. Environ. Microbiol.69, 2684–2691.10.1128/AEM.69.5.2684-2691.2003
42
MacGregor B. J. Moser D. P. Alm E. W. Nealson K. H. Stahl D. A. (1997). Crenarchaeota in Lake Michigan sediment. Appl. Environ. Microbiol.63, 1178–1181.
43
Markowitz V. M. Ivanova N. N. Szeto E. Palaniappan K. Chu K. Dalevi D. Chen I. M. Grechkin Y. Dubchak I. Anderson I. Lykidis A. Mavromatis K. Hugenholtz P. Kyrpides N. C. (2008). IMG/M: a data management and analysis system for metagenomes. Nucleic Acids Res.36, D534–D538.10.1093/nar/gkm869
44
Martin-Cuadrado A. B. Lopez-Garcia P. Alba J. C. Moreira D. Monticelli L. Strittmatter A. Gottschalk G. Rodriguez-Valera F. (2007). Metagenomics of the deep Mediterranean, a warm bathypelagic habitat. PLoS ONE2, e914.10.1371/journal.pone.0000914
45
Mussmann M. Brito I. Pitcher A. Sinninghe Damste J. S. Hatzenpichler R. Richter A. Nielsen J. L. Nielsen P. H. Muller A. Daims H. Wagner M. Head I. M. (2011). Thaumarchaeotes abundant in refinery nitrifying sludges express amoA but are not obligate autotrophic ammonia oxidizers. Proc. Natl. Acad. Sci. U.S.A.108, 16771–16776.10.1073/pnas.1106427108
46
Niou Y. K. Wu W. L. Lin L. C. Yu M. S. Shu H. Y. Yang H. H. Lin G. H. (2009). Role of galE on biofilm formation by Thermus spp. Biochem. Biophys. Res. Commun.390, 313–318.10.1016/j.bbrc.2009.09.120
47
Norton J. M. Alzerreca J. J. Suwa Y. Klotz M. G. (2002). Diversity of ammonia monooxygenase operon in autotrophic ammonia-oxidizing bacteria. Arch. Microbiol.177, 139–149.10.1007/s00203-001-0369-z
48
Nunoura T. Hirayama H. Takami H. Oida H. Nishi S. Shimamura S. Suzuki Y. Inagaki F. Takai K. Nealson K. H. Horikoshi K. (2005). Genetic and functional properties of uncultivated thermophilic crenarchaeotes from a subsurface gold mine as revealed by analysis of genome fragments. Environ. Microbiol.7, 1967–1984.10.1111/j.1462-2920.2005.00881.x
49
Ochsenreiter T. Selezi D. Quaiser A. Bonch-Osmolovskaya L. Schleper C. (2003). Diversity and abundance of Crenarchaeota in terrestrial habitats studied by 16S RNA surveys and real time PCR. Environ. Microbiol.5, 787–797.10.1046/j.1462-2920.2003.00476.x
50
Ouverney C. C. Fuhrman J. A. (2000). Marine planktonic archaea take up amino acids. Appl. Environ. Microbiol.66, 4829–4833.10.1128/AEM.66.11.4829-4833.2000
51
Pelve E. A. Lindas A. C. Martens-Habbena W. de la Torre J. R. Stahl D. A. Bernander R. (2011). Cdv-based cell division and cell cycle organization in the thaumarchaeon Nitrosopumilus maritimus. Mol. Microbiol.82, 555–566.10.1111/j.1365-2958.2011.07834.x
52
Pester M. Rattei T. Flechl S. Grongroft A. Richter A. Overmann J. Reinhold-Hurek B. Loy A. Wagner M. (2012). amoA-based consensus phylogeny of ammonia-oxidizing archaea and deep sequencing of amoA genes from soils of four different geographic regions. Environ. Microbiol.14, 525–539.10.1111/j.1462-2920.2011.02666.x
53
Quaiser A. Ochsenreiter T. Klenk H. P. Kletzin A. Treusch A. H. Meurer G. Eck J. Sensen C. W. Schleper C. (2002). First insight into the genome of an uncultivated crenarchaeote from soil. Environ. Microbiol.4, 603–611.10.1046/j.1462-2920.2002.00345.x
54
Quaiser A. Ochsenreiter T. Lanz C. Schuster S. C. Treusch A. H. Eck J. Schleper C. (2003). Acidobacteria form a coherent but highly diverse group within the bacterial domain: evidence from environmental genomics. Mol. Microbiol.50, 563–575.10.1046/j.1365-2958.2003.03707.x
55
Reigstad L. J. Bartossek R. Schleper C. (2011). Preparation of high-molecular weight DNA and metagenomic libraries from soils and hot springs. Meth. Enzymol.496, 319–344.10.1016/B978-0-12-386489-5.00013-0
56
Reigstad L. J. Richter A. Daims H. Urich T. Schwark L. Schleper C. (2008). Nitrification in terrestrial hot springs of Iceland and Kamchatka. FEMS Microbiol. Ecol.64, 167–174.10.1111/j.1574-6941.2008.00466.x
57
Samson R. Y. Bell S. D. (2011). Cell cycles and cell division in the archaea. Curr. Opin. Microbiol.14, 350–356.10.1016/j.mib.2011.04.005
58
Samuel G. Reeves P. (2003). Biosynthesis of O-antigens: genes and pathways involved in nucleotide sugar precursor synthesis and O-antigen assembly. Carbohydr. Res.338, 2503–2519.10.1016/j.carres.2003.07.009
59
Schatz M. C. Phillippy A. M. Shneiderman B. Salzberg S. L. (2007). Hawkeye: an interactive visual analytics tool for genome assemblies. Genome Biol.8, R34.10.1186/gb-2007-8-3-r34
60
Schleper C. Jurgens G. Jonuscheit M. (2005). Genomic studies of uncultivated archaea. Nat. Rev. Microbiol.3, 479–488.10.1038/nrmicro1159
61
Selby C. P. Sancar A. (1990). Structure and function of the (A)BC excinuclease of Escherichia coli. Mutat. Res.236, 203–211.
62
Spang A. Hatzenpichler R. Brochier-Armanet C. Rattei T. Tischler P. Spieck E. Streit W. Stahl D. A. Wagner M. Schleper C. (2010). Distinct gene set in two different lineages of ammonia-oxidizing archaea supports the phylum Thaumarchaeota. Trends Microbiol.18, 331–340.10.1016/j.tim.2010.06.003
63
Spear J. R. Barton H. A. Robertson C. E. Francis C. A. Pace N. R. (2007). Microbial community biofabrics in a geothermal mine adit. Appl. Environ. Microbiol.73, 6172–6180.10.1128/AEM.00393-07
64
Takai K. Moser D. P. Deflaun M. Onstott T. C. Fredrickson J. K. (2001). Archaeal diversity in waters from deep South African gold mines. Appl. Environ. Microbiol.67, 5750–5760.10.1128/AEM.67.21.5750-5760.2001
65
Tamura K. Dudley J. Nei M. Kumar S. (2007). MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol.24, 1596–1599.10.1093/molbev/msm092
66
Teira E. Van Aken H. Veth C. Herndl G. J. (2006). Archaeal uptake of enantiomeric amino acids in the meso- and bathypelagic waters of the North Atlantic. Limnol. Oceanogr.51, 60–69.10.4319/lo.2006.51.5.2131
67
Torarinsson E. Klenk H. P. Garrett R. A. (2005). Divergent transcriptional and translational signals in archaea. Environ. Microbiol.7, 47–54.10.1111/j.1462-2920.2004.00674.x
68
Tourna M. Stieglmeier M. Spang A. Konneke M. Schintlmeister A. Urich T. Engel M. Schloter M. Wagner M. Richter A. Schleper C. (2011). Nitrososphaera viennensis, an ammonia oxidizing archaeon from soil. Proc. Natl. Acad. Sci. U.S.A.108, 8420–8425.10.1073/pnas.1013488108
69
Treusch A. H. Kletzin A. Raddatz G. Ochsenreiter T. Quaiser A. Meurer G. Schuster S. C. Schleper C. (2004). Characterization of large-insert DNA libraries from soil for environmental genomic studies of archaea. Environ. Microbiol.6, 970–980.10.1111/j.1462-2920.2004.00663.x
70
Treusch A. H. Leininger S. Kletzin A. Schuster S. C. Klenk H. P. Schleper C. (2005). Novel genes for nitrite reductase and Amo-related proteins indicate a role of uncultivated mesophilic Crenarchaeota in nitrogen cycling. Environ. Microbiol.7, 1985–1995.10.1111/j.1462-2920.2005.00906.x
71
Tsai M. H. Saier M. H. Jr. (1995). Phylogenetic characterization of the ubiquitous electron transfer flavoprotein families ETF-alpha and ETF-beta. Res. Microbiol.146, 397–404.10.1016/0923-2508(96)80285-3
72
Venter J. C. Remington K. Heidelberg J. F. Halpern A. L. Rusch D. Eisen J. A. Wu D. Paulsen I. Nelson K. E. Nelson W. Fouts D. E. Levy S. Knap A. H. Lomas M. W. Nealson K. White O. Peterson J. Hoffman J. Parsons R. Baden-Tillson H. Pfannkoch C. Rogers Y. H. Smith H. O. (2004). Environmental genome shotgun sequencing of the Sargasso Sea. Science304, 66–74.10.1126/science.1093857
73
Walker C. B. De La Torre J. R. Klotz M. G. Urakawa H. Pinel N. Arp D. J. Brochier-Armanet C. Chain P. S. Chan P. P. Gollabgir A. Hemp J. Hugler M. Karr E. A. Konneke M. Shin M. Lawton T. J. Lowe T. Martens-Habbena W. Sayavedra-Soto L. A. Lang D. Sievert S. M. Rosenzweig A. C. Manning G. Stahl D. A. (2010). Nitrosopumilus maritimus genome reveals unique mechanisms for nitrification and autotrophy in globally distributed marine crenarchaea. Proc. Natl. Acad. Sci. U.S.A.107, 8818–8823.10.1073/pnas.0913533107
74
Weidenhaupt M. Rossi P. Beck C. Fischer H. M. Hennecke H. (1996). Bradyrhizobium japonicum possesses two discrete sets of electron transfer flavoprotein genes: fixA, fixB and etfS, etfL. Arch. Microbiol.165, 169–178.10.1007/s002030050312
75
Weidler G. W. Dornmayr-Pfaffenhuemer M. Gerbl F. W. Heinen W. Stan-Lotter H. (2007). Communities of archaea and bacteria in a subsurface radioactive thermal spring in the Austrian Central Alps, and evidence of ammonia-oxidizing Crenarchaeota. Appl. Environ. Microbiol.73, 259–270.10.1128/AEM.01570-06
76
Weidler G. W. Gerbl F. W. Stan-Lotter H. (2008). Crenarchaeota and their role in the nitrogen cycle in a subsurface radioactive thermal spring in the Austrian Central Alps. Appl. Environ. Microbiol.74, 5934–5942.10.1128/AEM.02602-07
77
Zhou J. Z. Bruns M. A. Tiedje J. M. (1996). DNA recovery from soils of diverse composition. Appl. Environ. Microbiol.62, 316–322.
Summary
Keywords
metagenomics, fosmids, Thaumarchaeota, ammonia-oxidizing archaea, amoA, promoter motif, transposon
Citation
Bartossek R, Spang A, Weidler G, Lanzen A and Schleper C (2012) Metagenomic Analysis of Ammonia-Oxidizing Archaea Affiliated with the Soil Group. Front. Microbio. 3:208. doi: 10.3389/fmicb.2012.00208
Received
19 February 2012
Accepted
21 May 2012
Published
20 June 2012
Volume
3 - 2012
Edited by
Lisa Y. Stein, University of Alberta, Canada
Reviewed by
Martin G. Klotz, University of North Carolina at Charlotte, USA; Francisco Rodriguez-Valera, Universidad Miguel Hernandez, Spain
Copyright
© 2012 Bartossek, Spang, Weidler, Lanzen and Schleper.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Christa Schleper, Department of Genetics in Ecology, University of Vienna, Althanstraße 14, A-1090 Vienna, Austria. e-mail: christa.schleper@univie.ac.at
This article was submitted to Frontiers in Terrestrial Microbiology, a specialty of Frontiers in Microbiology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.