

- Department of Microbiology, B103 Chemical and Life Science Laboratory, University of Illinois at Urbana-Champaign, Urbana, IL, USA
The discovery of the third domain of life, the Archaea, is one of the most exciting findings of the last century. These remarkable prokaryotes are well known for their adaptations to extreme environments; however, Archaea have also conquered moderate environments. Many of the archaeal biochemical processes, such as methane production, are unique in nature and therefore of great scientific interest. Although formerly restricted to biochemical and physiological studies, sophisticated systems for genetic manipulation have been developed during the last two decades for methanogenic archaea, halophilic archaea and thermophilic, sulfur-metabolizing archaea. The availability of these tools has allowed for more complete studies of archaeal physiology and metabolism and most importantly provides the basis for the investigation of gene expression, regulation and function. In this review we provide an overview of methods for genetic manipulation of Methanosarcina spp., a group of methanogenic archaea that are key players in the global carbon cycle and which can be found in a variety of anaerobic environments.
Introduction
Life on earth, as currently perceived by mankind, is encompassed in three major domains Archaea, Eukarya and Bacteria. The recognition of Archaea as a distinct phylogenetic lineage is a fairly recent discovery (Woese et al., 1990). Initially it was assumed that Archaea were strictly anaerobic and mostly extremophiles, inhabiting environmental niches hostile to most other organisms, such as submarine volcanic vents, solfataric hot springs, or soda lakes. It is known today that Archaea are ubiquitous, represent a significant portion of the global biomass and play important roles in global ecosystems and biochemical cycling (Delong and Pace, 2001; Jarrell et al., 2011).
Archaea share metabolic and physiologic features with Eukarya and Bacteria, but they are also unique in many ways (Jarrell et al., 2011; Jun et al., 2011; White, 2011). In many cases Archaea are uniquely positioned to carry out biochemical reactions that are of significant interest for industrial and biotechnological applications. Prominent examples are: methanogenesis, the reduction of CO2 to methane; or the application of Bacteriorhodopsin, a light driven proton pump, in solar cells and radiation sensors (Thavasi et al., 2009; Ahmadi and Yeow, 2011; De Vrieze et al., 2012).
Biochemical, structural and physiological studies have provided significant insight into the third domain of life over the last three decades (Cavicchioli, 2011). However, our understanding of Archaea still lags behind our knowledge of Eukarya and Bacteria. From a scientific and biotechnological point of view this gap in knowledge needs to be filled urgently. An important step toward achieving this goal was the development of systems for genetic manipulation of members of the methanogenic archaea, the halophilic archaea and the thermophilic, sulfur-metabolizing archaea (Rother and Metcalf, 2005; Buan et al., 2011; Leigh et al., 2011).
The study of methanogenic Archaea has a long-standing history because of their central role in the global carbon cycle and their potential application in biofuel production (Fox et al., 1977; Jarrell et al., 2011; De Vrieze et al., 2012). Methanogenic metabolism converts a limited number of one-carbon (C-1) compounds and acetic acid to methane through a series of coenzyme-bound intermediates in a process that drives the generation of ATP (Thauer et al., 2008). Methanogenic Archaea are represented in five orders, the Methanococcales, the Methanosarcinales, the Methanobacteriales, the Methanomicrobiales and the Methanopyrales (Liu and Whitman, 2008). Genetic systems are available for organisms in the first two orders (Rother and Metcalf, 2005; Leigh et al., 2011). Three classes of methanogens have been proposed based on physiological, biochemical and genomic traits (Anderson et al., 2009). Class I methanogens, Methanococcales, Methanobacteriales and Methanopyrales, and class II methanogens, Methanomicrobiales, are usually hydrogenotrophic; they use H2/CO2 and sometimes formate as substrates for methanogenesis (Anderson et al., 2009). Class III methanogens, Methanosarcinales metabolize a variety of substrates, H2/CO2, C-1 compounds, such as methylamines, methylsulfides or methanol, and acetate in four different methanogenic pathways (Thauer et al., 2008; Anderson et al., 2009). Three organisms, Methanosarcina bakeri Fusaro and Methanosarcina acetivorans C2A and Methanosarcina mazei Gö1 are well established in methanogenesis research and the complete genome sequences of all three organisms are publicly available. The major difference between these organisms lies within their ability to utilize methanogenic substrates, M. bakeri Fusaro and M. mazei Gö1 use all known substrates, whereas M. acetivorans C2A lacks the ability to grow on H2 and CO2 (Thauer et al., 2008; Guss et al., 2009; Kulkarni et al., 2009).
In the past 15 years, a number of techniques that allow the study of gene function in vivo have been developed for these Methanosarcina species. This review focuses on the genetic manipulation of Methanosarcina spp. and provides an overview about established methods including, random and targeted mutagenesis, complementation, and reporter gene fusions.
Transcription, Translation, and DNA Repair in Archaea
The transcription, translation, and DNA repair systems play an important role during genetic manipulation and therefore need to be considered for method development. The proteins involved in DNA repair facilitate the incorporation of cloned DNA into the genome of the target organisms, while the expression of cloned genes and selective markers is driven by the transcription and translation system.
Like bacteria, known Archaea store their genetic information on a circular chromosome and in many cases on additional plasmids (Keeling et al., 1994; Koonin and Wolf, 2008). In addition, transcriptional units often comprise operons (Brown et al., 1989). Nevertheless, the basal archaeal transcription apparatus including the 11- to 13-subunit DNA-dependent RNA Polymerase (RNAP) is closely related to the eukaryotic Pol II system and some rRNAs and tRNAs were found to be characteristic for Archaea (Woese et al., 1978; Jun et al., 2011).
The archaeal promoter is very similar to that of eukaryotes and includes a TATA box element located approximately 30 bp upstream of the transcriptional start (Hausner et al., 1991; Jun et al., 2011). At least two factors, homologues of the eukaryotic TATA-binding protein (TBP) and Transcription Factor II B (TFIIB), are required for transcription initiation. TFIIB interacts with the B recognition element (BRE), a purine rich sequence located directly upstream of the TATA-box (Jun et al., 2011). The majority of the transcriptional regulators identified to date are homologous to known bacterial regulators and transcriptional regulation generally seems to follow the bacterial model. This is especially evident from the mechanisms of actions described for transcriptional repressors that bind DNA close to the promoter and either occlude the TATA box and the BRE element or inhibit the recruitment of the RNAP (Bell, 2005; Jun et al., 2011; Malys and McCarthy, 2011). However, archaeal histones were shown to also be involved in transcription regulation similar to what is known for Eukaryotes (Reeve, 2003; Jun et al., 2011).
Archaeal mRNAs do not posses a 5′ cap structure and long poly-A tails (Brown and Reeve, 1985, 1986; Hennigan and Reeve, 1994). Translation can occur in a Shine-Dalgarno-dependent or -independent fashion; mRNAs lacking a Shine-Dalgarno sequence are either leaderless or are led by a 5′ untranslated region (Dennis, 1997; Malys and McCarthy, 2011).
DNA repair is achieved through double stranded break repair and homologous recombination. Mechanistically homologous recombination follows the eukaryotic model and the key enzymes involved, Mre11, RadA, and Rad50, are also found in Eukaryotes (White, 2011).
Cultivation of Methanosarcina spp.
Successfully working with any organism requires the ability to satisfy their nutritional and environmental needs. Methanosarcina spp. and methanogens, in general, are very sensitive to oxygen. All experimental manipulations and the cultivation of strains must be performed under strict anaerobic conditions. Media are commonly bicarbonate buffered minimal media with a pH from 6.8 to 7.0 and must have a redox potential of at least –300 mV to keep Methanosarcina metabolically functional (Balch et al., 1979; Sowers et al., 1993; Metcalf et al., 1998).
DNA Delivery, Exchangeable Promoters, Selectable, and Counterselectable Markers
The successful genetic manipulation of any organism depends on four basic requirements: (1) The ability to grow clonal colonies from single cells, (2) a DNA delivery system, (3) promoters for the expression of cloned genes, and (4) selectable genetic markers.
Methanosarcina often grow in multicellular packets containing from a few, up to tens of thousands of cells. Aggregates of Methanosarcina cells are encapsulated by methanochondroitin, a positively charged exopolysaccharide composed of N-acetyl-D-galactosamine, galacturonic- and glucuronic-acid that interacts with the S-layer, a proteinaceous matrix that is found immediately adjacent to the cell membrane (Kreisl and Kandler, 1986; Ellen et al., 2010). Cell aggregates represent a physical barrier for exogenous DNA and therefore impede genetic manipulation. The production of methanochondroitin seems to be environmentally regulated and it is not produced under conditions of high osmolarity, resulting in unicellular growth (Sowers et al., 1993). Based on this discovery high salt media were developed that allow the growth of clonal populations from single cells and render Methanosarcina more easily accessible for genetic manipulations (Sowers et al., 1993).
Liposome- and polyethylene glycol (PEG)-mediated transformation methods have been developed for Methanosarcina (Metcalf et al., 1997; Oelgeschlager and Rother, 2009). The former achieves high transformation frequencies, up to 2 × 108 transformants per μ g DNA, representing about 20% of the CFU for M. acetivorans. Frequencies in other Methanosarcina spp. are slightly lower but still sufficient for most purposes (Metcalf et al., 1997; Ehlers et al., 2005).
Selectable or testable phenotypes are mandatory to monitor DNA up-take or exchange. The basic requirement, to achieve the establishment of a tractable phenotype through a selectable or counterselectable marker, is a reliable gene expression system. Two exchangeable transcription systems are routinely used for the expression of cloned genes in Methanosarcina spp. Both systems comprise a strong constitutive promoter, pmcr or pmcrB, that drives the transcription of the methyl-reductase operons in Methanococcus voltae and Methanosarcina barkeri Fusaro, respectively, and its corresponding ribosomal binding sites (RBS) and transcriptional terminators (Metcalf et al., 1997; Zhang et al., 2000).
Two antibiotic resistance markers have been developed for Methanosarcina. Resistance to the protein synthesis inhibitor Puromycin is easily achievable, by introducing the pac (puromycin transacetylase) gene from Streptomyces alboniger controlled by pmcr (Gernhardt et al., 1990; Metcalf et al., 1997). A second selectable marker, that codes for resistance to pseudomonic acid was created by mutagenesis of the isoleucyl-tRNA synthetase gene (ileS12) from M. barkeri Fusaro (Boccazzi et al., 2000).
A counterselectable marker system for the construction of M. acetivorans C2A and M. barkeri Fusaro mutants was developed based on the deletion of the hpt gene, encoding a hypoxanthine phosphoribosyltransferase (Pritchett et al., 2004; Guss et al., 2008). The Hpt protein is part of the purine salvage pathway and catalyzes the phosphorylation of various purines to the corresponding monophosphate. Toxic purine analogs like 8-aza-2,6-diaminopurine (8ADP) also serve as substrate for Hpt. Incorporation of these toxic bases into DNA can be lethal (Bowen and Whitman, 1987; Bowen et al., 1996). A M. acetivorans C2A Δhpt strain is approximately 35-fold more resistant to 8ADP than the wild type; 8ADP sensitivity is restored upon reintroduction of the hpt gene. This phenotype has proven useful for the creation of unmarked deletion mutants and therefore a series of Δhpt parental strains were constructed (see below; Pritchett et al., 2004; Guss et al., 2008).
Shuttle Vectors
A series of autonomously replicating Escherichia coli/Methanosarcina shuttle vectors have been developed based on the native M. acetivorans pC2A plasmid that is present in about six copies per cell (Sowers and Gunsalus, 1988; Metcalf et al., 1997). These constructs contain a pac cassette for selection and the pC2A replicon for replication in Methanosarcina. The plasmids can be used for a variety of Methanosarcina spp. The β-lactamase gene enables selection and oriR6Kγ allows replication in E. coli. The variety of plasmids provide different multi cloning sites and some include the lacZ α gene for blue and white screening to allow facile identification of recombinant plasmids in E. coli (Metcalf et al., 1997).
The shuttle plasmid vectors gave rise to a series of molecular tools that make forward and reverse genetics possible in Methanosarcina. All plasmids for the genetic manipulation of Methanosarcina are designed to be maintained in appropriate E. coli hosts. Ampicillin- (bla), chloramphenicol- (cat) or kanamycin-resistance (aph) genes serve as selective markers. Replication and copy number control are dependent on either the high copy number control pMB1ori, the medium copy pir-dependent oriR6Kγ or the single copy number maintenance oriS in combination with the inducible high copy number maintenance oriV-TrfA system (Metcalf et al., 1997; Zhang et al., 2000; Pritchett et al., 2004; Guss et al., 2008).
Forward Genetics
A transposon system derived from the mariner transposable element Himar1, which transposes to random sites in the genome at high frequency, is available for mutagenesis in M. acetivorans C2A (Zhang et al., 2000). The transposition of mariner elements is solely dependent on their cognate transposases and does not require host factors, hence they can be used for in vivo mutagenesis of a wide variety of eukaryotic and prokaryotic organisms (Lampe et al., 1996; Plasterk, 1996; Tosi and Beverley, 2000). The modified mini-Himar1 transposon carries elements for selection in Methanosarcina (pac cassette) and in E. coli (aph) as well as the oriR6Kγ, for identification of the mutated gene through cloning of the insertion in E. coli. A suicide vector serves as the delivery plasmid. The mariner transposase (tnp) gene, controlled by the pmcrB, is not part of the transposable element, but resides on the plasmid and therefore perishes with the vector. This construct assures the stability of the insert, since the Tnp mediated transposition is fully reversible (Zhang et al., 2000).
Reverse Genetics
Integration of a cloned DNA fragment into the chromosome of Methanosarcina through homologous recombination was first achieved by Conway De Macario et al. (1996). Gene replacement or disruption can be facilitated by simply introducing the linearized cloning vector pBluescript (Stratagene) containing the appropriate homologous Methanosarcina DNA fragment and selective marker (Rother et al., 2005). As described above, only two selective markers, the pac and ileS12 genes, are available for Methanosarcina. Thus, markerless deletions are preferred, because this allows repetitive use of both markers.
The first unmarked mutant constructed was a M. acetivorans C2A Δhpt strain that served as the parent for subsequent deletion strains, since the loss of a functional hpt gene provides a convenient counter selective system (Pritchett et al., 2004). This principle was extended in a series of M. acetivorans C2A and M. bakeri Fusaro strains engineered to harbor a highly efficient site-specific recombination system at the Δhpt locus that allows integration of genes into the chromosome (Guss et al., 2008).
This system utilizes the host factor independent ΦC31 integrase of the Streptomyces bacteriophage ΦC31, encoded by the int gene, and the ΦC31 phage integration sites (Thorpe and Smith, 1998; Guss et al., 2008). The Int protein catalyzes site-specific recombination between the ΦC31 site at the Δhpt locus (attB or attP) and the corresponding ΦC31 (attP or attB) of a plasmid (Figure 1). The reaction is unidirectional and results in the stabile integration of the entire plasmid. This allows the constitutive expression of the ΦC31 integrase from pmcrB without destabilizing the insert. Some constructs contain an artificial tetR-int operon, expressed from pmcrB (Guss et al., 2008). In general, the tetR gene codes for the TetR transcriptional repressor that binds the tetO operator of the target promoter and prevents transcription. TetR releases the promoter upon binding tetracycline and gene expression is initiated. This tetracycline regulated promoter system allows the tight regulation of cloned genes and is used for different applications in Methanosarcina (see below; Beck et al., 1982; Guss et al., 2008).
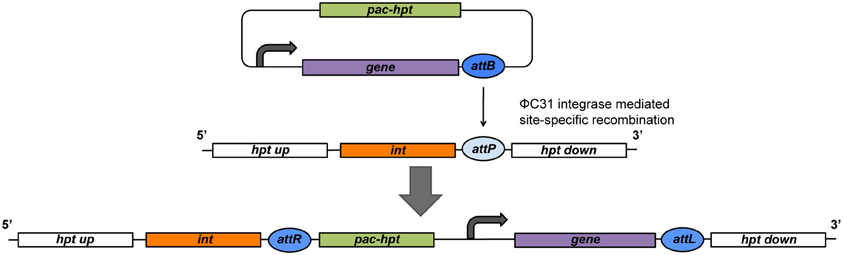
Figure 1. Schematic of a site-specific recombination event at the engineered Δhpt locus of the Methanosarcina chromosome. The ΦC31 integrase (int) is constitutively expressed at the Δhpt locus that also harbors a ΦC31 attachment site (attB in this figure). The integration plasmid contains the gene of interest controlled by a pmcrB derivative (curved arrow) and a ΦC31 attachment site (attP in this figure). pac, gene for puromycin N-acetyltransferase; hpt, gene for hypoxanthine phosphoribosyltransferase.
Markerless Exchange
Two methods, employing different mechanisms, have been established for routine use to generate unmarked mutants in Methanosarcina in the Δhpt background (Pritchett et al., 2004; Welander and Metcalf, 2008). Both methods rely on homologous recombination, using deletion constructs consisting of either the 5′- and 3′- sequences of the gene, resulting in gene disruption, or the flanking up- and downstream sequences, resulting in gene deletion (Pritchett et al., 2004; Welander and Metcalf, 2008).
The first method makes use of the pac selectable marker and hpt counter selectable marker and relies on an unstable merodiploid intermediate state (Figure 2; Pritchett et al., 2004). Plasmid pMP44 does not replicate in Methanosarcina and carries the hpt gene in addition to the pac cassette. Derivatives of pMP44 containing a deletion construct are used to transform the Methanosarcina Δhpt parental strain to puromycin resistance and 8ADP sensitivity during the first recombination event. The plasmid integrates into the chromosome at either the up- or downstream homologous regions, resulting in an unstable merodiploid, which is resolved by a successive second recombination event in the absence of selective pressure. This event removes the vector backbone including the pac and hpt genes and renders the progeny puromycin sensitive and 8ADP resistant (Figure 2). The second step gives rise to two types of 8ADP-resistant progeny, half the offspring will retain the desired mutation and the other half will retain the wild type locus, provided that the target gene is not essential and there is no difference in growth rate between the mutant and the wild type strain. Mutants are then identified via PCR screening or phenotypic testing.
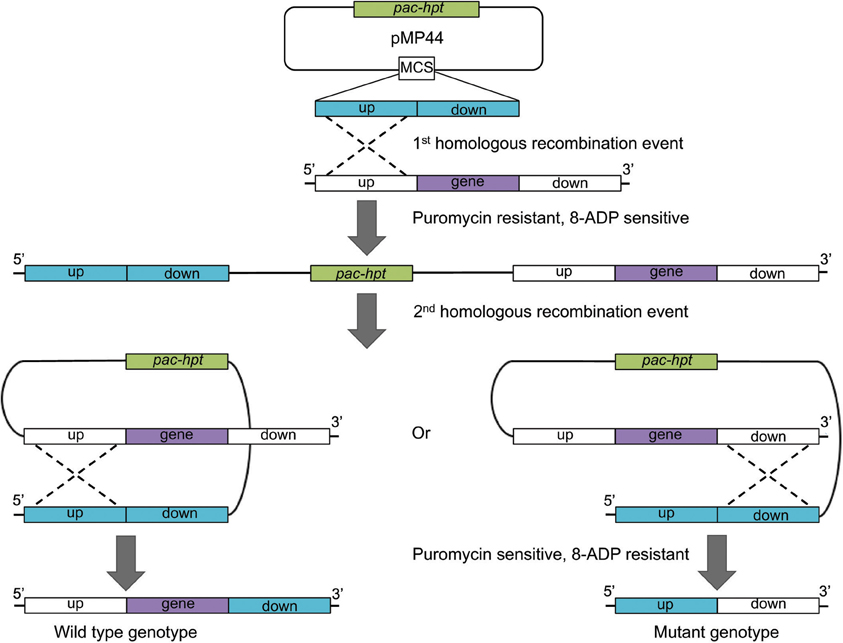
Figure 2. Schematic of the construction of an unmarked deletion mutant strain using pMP44. Plasmid pMP44 carries a deletion construct comprising the homologous up- and downstream regions of the target gene. The circular plasmid is transformed into a Methanosarcina Δ hpt strain. A homologous recombination event (dotted line) creates a puromycin resistant and 8-aza-2,6-diaminopurine (8-ADP) sensitive merodiploid intermediate. The resolution of the instable intermediate either reconstitutes the wild type genotype or results in the deletion of the target gene, both events restore puromycin sensitivity and 8-ADP resistance. pac, gene for puromycin N-acetyltransferase; hpt, gene for hypoxanthine phosphoribosyltransferase.
The second established method is based on creating a marked mutant that allows subsequent removal of the selective and counter selective marker (Figure 3A; Rother and Metcalf, 2005; Welander and Metcalf, 2008). Plasmid pJK301 carries an artificial pac-hpt operon, flanked by two Flp recombinase recognition sites (FRT). Two multi cloning sites for the cloning of the desired homologous regions are located directly up- and downstream of the FRT-pac-hpt-FRT cassette. The plasmid, carrying the deletion construct needs to be linearized before transformation to ensure that the wild type gene is replaced or disrupted with the FRT-pac-hpt-FRT cassette through a double-recombination event. The resulting marked mutant is puromycin resistant and 8ADP sensitive. Introducing plasmid pMR55 that expresses the Saccharomyces Flp recombinase creates an unmarked, puromycin sensitive and 8ADP resistant mutant. The Flp recombinase recognizes the FRT sites and excises the pac-hpt cassette, leaving behind a “FRT scar.” The Flp recombinase system is highly effective, but fully reversible (Huang et al., 1997; Schweizer, 2003). Thus, the flp gene is transiently expressed and pMR55 does not replicate in Methanosarcina, in order to stabilize the construct.
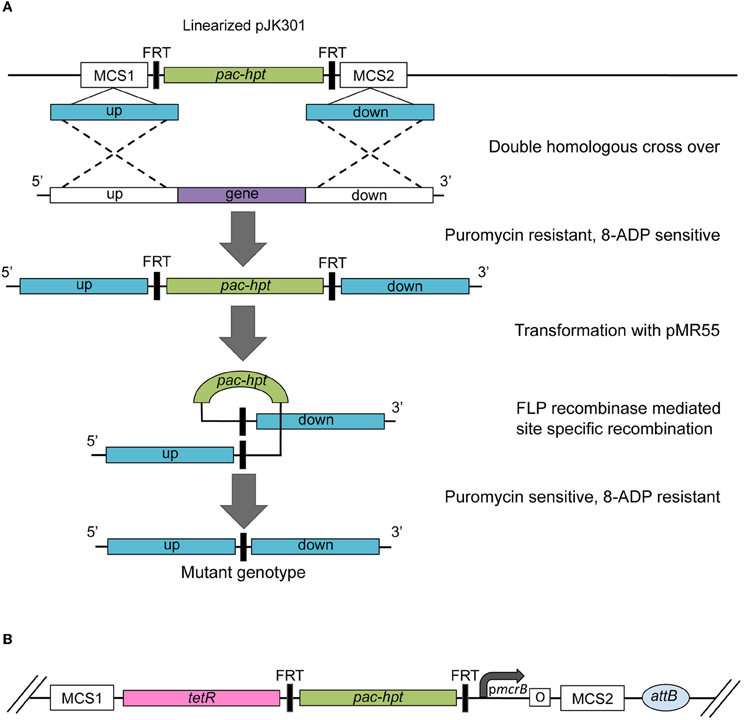
Figure 3. (A) Schematic of the construction of an unmarked deletion mutant strain using pMPJK301 and pMR55. The pac-hpt cassette of plasmid pMJK301 is flanked by the homologous up- and downstream regions of the target gene. The linearized plasmid is transformed into a Methanosarcina Δhpt strain. A double homologous recombination event (dotted line) creates a puromycin resistant and 8-aza-2,6-diaminopurine (8-ADP) marked deletion mutant. Successive transformation with plasmid pMR55 results in the removal of the pac-hpt cassette via site-specific recombination between the FRT sites mediated by the Flp recombinase expressed from pMR55. The resulting mutant strain is puromycin sensitive and 8-ADP resistant. (B) Schematic of a plasmid used for promoter swaps. The pmcrB is represented by a bend arrow and the Tet operator by the letter “o” enclosed in a box. pac, gene for puromycin N-acetyltransferase; hpt, gene for hypoxanthine phosphoribosyltransferase; tetR, gene for the TetR transcriptional repressor.
Genotypic Complementation and Reporter Gene Fusions
The respective wild type gene(s) can be reintroduced into Methanosarcina mutants through either plasmid based complementation or single integration into the chromosome.
Genotypic Complementation via Multicopy Plasmids and Single Copy Integration
Multicopy expression can be achieved by using the E. coli/Methanosarcina shuttle vector pWM321 (Metcalf et al., 1997; Zhang et al., 2002). The plasmid offers a large multi cloning site but no promoter to drive the expression of the gene of interest and no RBS. A promoter-RBS-gene fusion needs to be cloned into pWM321. This provides the opportunity to tailor the construct to whatever is required by either choosing the native promoter of the gene, a promoter of known strength, or a tetracycline regulated promoter. The desired gene can either be cloned with its native RBS or placed under the control of the RBS of the methyl-reductase operon from M. barkeri Fusaro (pmcrB RBS).
Genotypic Complementation Through Single Copy Integration
A number of plasmids were designed to express genes of interest from either a constitutive or tetracycline regulated promoter (Guss et al., 2008). The pmcrB promoter achieves the highest level of constitutive expression. Lower constitutive expression levels can be obtained from the tetracycline regulated promoters, pmcrB(tetO1), pmcrB(tetO3), and pmcrB(tetO4), which decrease in promoter strength, respectively; if the plasmids are introduced into a host that does not express the tetR gene from the Δhpt locus. All plasmids provide the pmcrB RBS and the pmcrB terminator and carry the FRT-pac-hpt-FRT cassette as well as a ΦC31 phage integration site. They do not replicate in Methanosarcina but integrate at the Δhpt locus through site-specific recombination. Mutants that have successfully inserted the desired plasmid into the chromosome can be identified based on puromycin resistance and 8ADP sensitivity.
Another useful feature of the single copy expression vectors is one of the λ attachment sites (λ attA or λ attB) that can be used for retrofitting with other plasmids via a commercially available λ integrase system (Guss et al., 2008). This system can be used to turn the single copy expression vectors into autonomous, multicopy Methanosarcina plasmids through site-specific recombination with plasmid pAMG40 that carries the pC2A replicon. A Methanosarcina Δhpt strain lacking the int gene and the ΦC31 integration site needs to be used as host for the expression plasmid:pAMG40 constructs to avoid integration into the chromosome.
Gene Expression Studies, Testing for Gene Essentiality and Promoter Swap
Reporter Gene Fusions
The widely used reporter gene uidA, encoding the β-glucu-ronidase from E. coli, is functional in Methanosarcina. Plasmid pAB79 was designed to allow the construction of transcriptional or translational fusions to uidA (Figure 4). Stable single copy fusions are advantageous for expression studies; pAB79 derivatives therefore integrate into the chromosome at the Δhpt locus via ΦC31 mediated recombination (Guss et al., 2008).
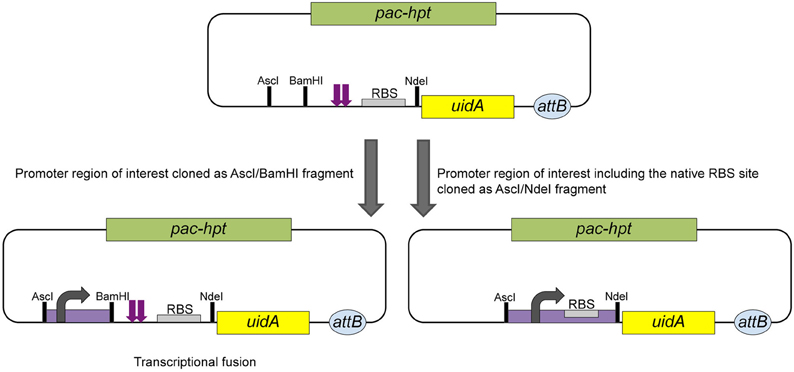
Figure 4. Schematic of the construction of transcriptional and translational reporter gene fusions using pAB79. Transcriptional fusions can be achieved by cloning the promoter region of interest as an AscI/BamHI fragment into pAB79. In this construct transcription is initiated from the cloned promoter (bend arrow). During translation no read through can occur from the cloned fragment into the uidA gene (β-glucuronidase) due the to tandem stopcodons (purple arrows). Translation of the β-glucuronidase is initiated from the pmcrB RBS (gray box). To create a translational fusion the promoter of interest, including its translational elements, is cloned as an AscI/NdeI fragment. pac, gene for puromycin N-acetyltransferase; hpt, gene for hypoxanthine phosphoribosyltransferase; attB, ΦC31 attachment site.
For a transcriptional fusion the (putative) promoter of interest is cloned as an AscI/BamH1 fragment immediately upstream of tandem translational stop codons. The stop codons are followed by the further downstream located pmcrB RBS and uidA gene. In this construct transcription initiation is dependent on the cloned fragment, but translation initiation of the β-glucuronidase is based on the strong and effective pmcrB RBS provided by pAB79. The tandem stop codons guarantee no translational read through from the cloned fragment into the uidA gene (Figure 4).
To create a translational fusion the desired promoter is cloned as an AscI/NdeI fragment and replaces the tandem stop codons and the pmcrB RBS completely. The promoter's native RBS and other translational elements are maintained by converting the start codon of the respective gene into the NdeI site used for cloning. Transcriptional and translational initiation are dependent on the elements provided by the cloned fragment in this construct (Figure 4; Guss et al., 2008).
Testing for Gene Essentiality and Promoter Swap
The methods for gene deletion discussed in the preceding sections cannot be employed to study the function of essential genes. Instead, a promoter swap can be used to create a conditional mutant as a way to investigate gene essentiality. In this method, the native promoter of the target gene is replaced with one of the tetracycline regulated promoters. A series of plasmids is available that are similar in design to the deletion vector pJK301 that carry the FRT-pac-hpt-FRT cassette for selection (Figure 3B; Guss et al., 2008). A tetR cassette is located upstream and a tetracycline dependent promoter downstream of the marker cassette. Two multi cloning sites flank this construct allowing the subsequent cloning of the upstream homologous region of the targeted promoter and the 5′ homologous region of the gene of interest. The promoter swap relies on the same mechanisms as the construction of a deletion mutant using the pJK301/pMR55 system (Figure 3A). If the targeted gene is essential the resulting mutant strain will only be viable in the presence of tetracycline.
Conclusions and Future Directions
The availability of the genetic tools described above has greatly improved our knowledge of the methanogenic process in Methanosarcina species. Recent genetic studies have provided a better understanding of the C1 oxidation/reduction pathway and the energy-conserving electron transport chain of M. bakeri Fusaro (Welander and Metcalf, 2008; Kulkarni et al., 2009). It was also possible to characterize the biological roles of isozymes involved in different methanogenic pathways of M. acetivorans C2A, such as methanol and methylamine specific methyltransferases as well as cytoplasmic and membrane-bound heterosulfide reductases (Bose et al., 2009; Buan and Metcalf, 2010). The hydrogenases required for hydrogenotrophic methanogenesis are conserved in M. bakeri Fusaro and M. acetivorans C2A, cis-acting mutations were identified in the promoter regions of the respective genes of the latter that prevent gene expression and render the strain incapable of using H2/CO2 as growth substrates (Galagan et al., 2002; Guss et al., 2009). The ability to genetically manipulate Methanosarcina has not only contributed to a deeper understanding of methanogenesis, but has paved the way for the study of other cellular and metabolic functions such as transcriptional and post-transcriptional gene regulation (Bose et al., 2006, 2009; Bose and Metcalf, 2008; Opulencia et al., 2009) and mechanisms for synthesis and insertion of the 22nd genetically encoded amino acid, pyrrolysine, into methylamine methyltransferases of Methanosarcina (Mahapatra et al., 2006, 2007).
Ongoing and future research employing genetic methods in combination with the classical physiological and biochemical approaches will undoubtedly expand our understanding of methanogenic archaea even further. In addition, the ability to genetically manipulate Methanosarcina spp. opens up the possibility to metabolically engineer strains for the production of biogas from organic materials. Methane, one of the major end products of methanogenesis, represents a clean burning and renewable energy source. Methanosarcina spp. are among the most promising candidates for routine use in biofuel production, because among the methanogenic archaea Methanosarcina have the broadest range in substrates for methanogenesis and exhibit the highest tolerance to environmental stresses (De Vrieze et al., 2012). Engineering efforts to further enhance stress tolerance, optimize the efficiency of substrate usage and broaden the substrate range of Methanosarcina spp. could significantly improve their performance in industrial settings like anaerobic digesters, and optimize methane yields. For example, the introduction of a gene coding for a broad-specificity esterase from Pseudomonas veronii into M. acetivornas has resulted in a strain that efficiently converts methyl acetate and methyl propionate to methane (Lessner et al., 2010).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy (DE-FG02-02ER15296) and the National Science Foundation (MCB1022462) for support during the preparation of this review.
References
Ahmadi, M., and Yeow, J. T. (2011). Fabrication and characterization of a radiation sensor based on bacteriorhodopsin. Biosens. Bioelectron. 26, 2171–2176.
Anderson, I., Ulrich, L. E., Lupa, B., Susanti, D., Porat, I., Hooper, S. D., Lykidis, A., Sieprawska-Lupa, M., Dharmarajan, L., Goltsman, E., Lapidus, A., Saunders, E., Han, C., Land, M., Lucas, S., Mukhopadhyay, B., Whitman, W. B., Woese, C., Bristow, J., and Kyrpides, N. (2009). Genomic characterization of Methanomicrobiales reveals three classes of methanogens. PLoS ONE 4:e5797. doi: 10.1371/journal.pone.0005797
Balch, W. E., Fox, G. E., Magrum, L. J., Woese, C. R., and Wolfe, R. S. (1979). Methanogens: reevaluation of a unique biological group. Microbiol. Rev. 43, 260–296.
Beck, C. F., Mutzel, R., Barbe, J., and Muller, W. (1982). A multifunctional gene (tetR) controls Tn10-encoded tetracycline resistance. J. Bacteriol. 150, 633–642.
Bell, S. D. (2005). Archaeal transcriptional regulation–variation on a bacterial theme? Trends Microbiol. 13, 262–265.
Boccazzi, P., Zhang, J. K., and Metcalf, W. W. (2000). Generation of dominant selectable markers for resistance to pseudomonic acid by cloning and mutagenesis of the ileS gene from the archaeon Methanosarcina barkeri Fusaro. J. Bacteriol. 182, 2611–2618.
Bose, A., Kulkarni, G., and Metcalf, W. W. (2009). Regulation of putative methyl-sulphide methyltransferases in Methanosarcina acetivorans C2A. Mol. Microbiol. 74, 227–238.
Bose, A., and Metcalf, W. W. (2008). Distinct regulators control the expression of methanol methyltransferase isozymes in Methanosarcina acetivorans C2A. Mol. Microbiol. 67, 649–661.
Bose, A., Pritchett, M. A., Rother, M., and Metcalf, W. W. (2006). Differential regulation of the three methanol methyltransferase isozymes in Methanosarcina acetivorans C2A. J. Bacteriol. 188, 7274–7283.
Bowen, T. L., Lin, W. C., and Whitman, W. B. (1996). Characterization of guanine and hypoxanthine phosphoribosyltransferases in Methanococcus voltae. J. Bacteriol. 178, 2521–2526.
Bowen, T. L., and Whitman, W. B. (1987). Incorporation of exogenous purines and pyrimidines by Methanococcus voltae and isolation of analog-resistant mutants. Appl. Environ. Microbiol. 53, 1822–1826.
Brown, J. W., Daniels, C. J., and Reeve, J. N. (1989). Gene structure, organization, and expression in Archaebacteria. Crit. Rev. Microbiol. 16, 287–338.
Brown, J. W., and Reeve, J. N. (1985). Polyadenylated, noncapped RNA from the archaebacterium Methanococcus vannielii. J. Bacteriol. 162, 909–917.
Brown, J. W., and Reeve, J. N. (1986). Polyadenylated RNA isolated from the archaebacterium Halobacterium halobium. J. Bacteriol. 166, 686–688.
Buan, N., Kulkarni, G., and Metcalf, W. (2011). Genetic methods for Methanosarcina species. Methods Enzymol. 494, 23–42.
Buan, N. R., and Metcalf, W. W. (2010). Methanogenesis by Methanosarcina acetivorans involves two structurally and functionally distinct classes of heterodisulfide reductase. Mol. Microbiol. 75, 843–853.
Conway De Macario, E., Guerrini, M., Dugan, C. B., and Macario, A. J. (1996). Integration of foreign DNA in an intergenic region of the archaeon Methanosarcina mazei without effect on transcription of adjacent genes. J. Mol. Biol. 262, 12–20.
De Vrieze, J., Hennebel, T., Boon, N., and Verstraete, W. (2012). Methanosarcina: the rediscovered methanogen for heavy duty biomethanation. Bioresour. Technol. 112, 1–9.
Delong, E. F., and Pace, N. R. (2001). Environmental diversity of Bacteria and Archaea. Syst. Biol. 50, 470–478.
Ehlers, C., Weidenbach, K., Veit, K., Deppenmeier, U., Metcalf, W. W., and Schmitz, R. A. (2005). Development of genetic methods and construction of a chromosomal glnK1 mutant in Methanosarcina mazei strain Gö1. Mol. Genet. Genomics 273, 290–298.
Ellen, A. F., Zolghadr, B., Driessen, A. M., and Albers, S. V. (2010). Shaping the archaeal cell envelope. Archaea 2010, 608243.
Fox, G. E., Magrum, L. J., Balch, W. E., Wolfe, R. S., and Woese, C. R. (1977). Classification of methanogenic Bacteria by 16S ribosomal RNA characterization. Proc. Natl. Acad. Sci. U.S.A. 74, 4537–4541.
Galagan, J. E., Nusbaum, C., Roy, A., Endrizzi, M. G., Macdonald, P., Fitzhugh, W., Calvo, S., Engels, R., Smirnov, S., Atnoor, D., Brown, A., Allen, N., Naylor, J., Stange-Thomann, N., Dearellano, K., Johnson, R., Linton, L., McEwan, P., McKernan, K., Talamas, J., Tirrell, A., Ye, W., Zimmer, A., Barber, R. D., Cann, I., Graham, D. E., Grahame, D. A., Guss, A. M., Hedderich, R., Ingram-Smith, C., Kuettner, H. C., Krzycki, J. A., Leigh, J. A., Li, W., Liu, J., Mukhopadhyay, B., Reeve, J. N., Smith, K., Springer, T. A., Umayam, L. A., White, O., White, R. H., Conway De Macario, E., Ferry, J. G., Jarrell, K. F., Jing, H., Macario, A. J., Paulsen, I., Pritchett, M., Sowers, K. R., Swanson, R. V., Zinder, S. H., Lander, E., Metcalf, W. W., and Birren, B. (2002). The genome of M. acetivorans reveals extensive metabolic and physiological diversity. Genome Res. 12, 532–542.
Gernhardt, P., Possot, O., Foglino, M., Sibold, L., and Klein, A. (1990). Construction of an integration vector for use in the Archaebacterium Methanococcus voltae and expression of a eubacterial resistance gene. Mol. Gen. Genet. 221, 273–279.
Guss, A. M., Kulkarni, G., and Metcalf, W. W. (2009). Differences in hydrogenase gene expression between Methanosarcina acetivorans and Methanosarcina barkeri. J. Bacteriol. 191, 2826–2833.
Guss, A. M., Rother, M., Zhang, J. K., Kulkarni, G., and Metcalf, W. W. (2008). New methods for tightly regulated gene expression and highly efficient chromosomal integration of cloned genes for Methanosarcina species. Archaea 2, 193–203.
Hausner, W., Frey, G., and Thomm, M. (1991). Control regions of an archaeal gene. A TATA box and an initiator element promote cell-free transcription of the tRNA(Val) gene of Methanococcus vannielii. J. Mol. Biol. 222, 495–508.
Hennigan, A. N., and Reeve, J. N. (1994). mRNAs in the methanogenic Archaeon Methanococcus vannielii: numbers, half-lives and processing. Mol. Microbiol. 11, 655–670.
Huang, L. C., Wood, E. A., and Cox, M. M. (1997). Convenient and reversible site-specific targeting of exogenous DNA into a bacterial chromosome by use of the FLP recombinase: the FLIRT system. J. Bacteriol. 179, 6076–6083.
Jarrell, K. F., Walters, A. D., Bochiwal, C., Borgia, J. M., Dickinson, T., and Chong, J. P. (2011). Major players on the microbial stage: why Archaea are important. Microbiology 157, 919–936.
Jun, S. H., Reichlen, M. J., Tajiri, M., and Murakami, K. S. (2011). Archaeal RNA polymerase and transcription regulation. Crit. Rev. Biochem. Mol. Biol. 46, 27–40.
Keeling, P. J., Charlebois, R. L., and Doolittle, W. F. (1994). Archaebacterial genomes: eubacterial form and eukaryotic content. Curr. Opin. Genet. Dev. 4, 816–822.
Koonin, E. V., and Wolf, Y. I. (2008). Genomics of Bacteria and Archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 36, 6688–6719.
Kreisl, P., and Kandler, O. (1986). Chemical structure or the cell wall polymer of Methanosarcina. System Appl. Microbiol. 7, 293–299.
Kulkarni, G., Kridelbaugh, D. M., Guss, A. M., and Metcalf, W. W. (2009). Hydrogen is a preferred intermediate in the energy-conserving electron transport chain of Methanosarcina barkeri. Proc. Natl. Acad. Sci. U.S.A. 106, 15915–21590.
Lampe, D. J., Churchill, M. E., and Robertson, H. M. (1996). A purified mariner transposase is sufficient to mediate transposition in vitro. EMBO J. 15, 5470–5479.
Leigh, J. A., Albers, S. V., Atomi, H., and Allers, T. (2011). Model organisms for genetics in the domain Archaea: methanogens, halophiles, Thermococcales and Sulfolobales. FEMS Microbiol. Rev. 35, 577–608.
Lessner, D. J., Lhu, L., Wahal, C. S., and Ferry, J. G. (2010). An engineered methanogenic pathway derived from the domains Bacteria and Archaea. MBio 1, e00243–10.
Liu, Y., and Whitman, W. B. (2008). Metabolic, phylogenetic, and ecological diversity of the methanogenic Archaea. Ann. N.Y. Acad. Sci. 1125, 171–789.
Mahapatra, A., Patel, A., Soares, J. A., Larue, R. C., Zhang, J. K., Metcalf, W. W., and Krzycki, J. A. (2006). Characterization of a Methanosarcina acetivorans mutant unable to translate UAG as pyrrolysine. Mol. Microbiol. 59, 56–66.
Mahapatra, A., Srinivasan, G., Richter, K. B., Meyer, A., Lienard, T., Zhang, J. K., Zhao, G., Kang, P. T., Chan, M., Gottschalk, G., Metcalf, W. W., and Krzycki, J. A. (2007). Class I and class II lysyl-tRNA synthetase mutants and the genetic encoding of pyrrolysine in Methanosarcina spp. Mol. Microbiol. 64, 1306–1318.
Malys, N., and McCarthy, J. E. (2011). Translation initiation: variations in the mechanism can be anticipated. Cell. Mol. Life Sci. 68, 991–1003.
Metcalf, W. W., Zhang, J. K., Apolinario, E., Sowers, K. R., and Wolfe, R. S. (1997). A genetic system for Archaea of the genus Methanosarcina: liposome-mediated transformation and construction of shuttle vectors. Proc. Natl. Acad. Sci. U.S.A. 94, 2626–2631.
Metcalf, W. W., Zhang, J. K., and Wolfe, R. S. (1998). An anaerobic, intrachamber incubator for growth of Methanosarcina spp. on methanol-containing solid media. Appl. Environ. Microbiol. 64, 768–770.
Oelgeschlager, E., and Rother, M. (2009). In vivo role of three fused corrinoid/methyl transfer proteins in Methanosarcina acetivorans. Mol. Microbiol. 72, 1260–1272.
Opulencia, R. B., Bose, A., and Metcalf, W. W. (2009). Physiology and posttranscriptional regulation of methanol:coenzyme M methyltransferase isozymes in Methanosarcina acetivorans C2A. J. Bacteriol. 191, 6928–6935.
Plasterk, R. H. (1996). The Tc1/mariner transposon family. Curr. Top. Microbiol. Immunol. 204, 125–143.
Pritchett, M. A., Zhang, J. K., and Metcalf, W. W. (2004). Development of a markerless genetic exchange method for Methanosarcina acetivorans C2A and its use in construction of new genetic tools for methanogenic Archaea. Appl. Environ. Microbiol. 70, 1425–1433.
Rother, M., Boccazzi, P., Bose, A., Pritchett, M. A., and Metcalf, W. W. (2005). Methanol-dependent gene expression demonstrates that methyl-coenzyme M reductase is essential in Methanosarcina acetivorans C2A and allows isolation of mutants with defects in regulation of the methanol utilization pathway. J. Bacteriol. 187, 5552–5559.
Rother, M., and Metcalf, W. W. (2005). Genetic technologies for Archaea. Curr. Opin. Microbiol. 8, 745–751.
Schweizer, H. P. (2003). Applications of the Saccharomyces cerevisiae Flp-FRT system in bacterial genetics. J. Mol. Microbiol. Biotechnol. 5, 67–77.
Sowers, K. R., Boone, J. E., and Gunsalus, R. P. (1993). Disaggregation of Methanosarcina spp. and growth as single cells at elevated osmolarity. Appl. Environ. Microbiol. 59, 3832–3839.
Sowers, K. R., and Gunsalus, R. P. (1988). Plasmid DNA from the acetotrophic methanogen Methanosarcina acetivorans. J. Bacteriol. 170, 4979–4982.
Thauer, R. K., Kaster, A. K., Seedorf, H., Buckel, W., and Hedderich, R. (2008). Methanogenic Archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6, 579–591.
Thavasi, V., Lazarova, T., Filipek, S., Kolinski, M., Querol, E., Kumar, A., Ramakrishna, S., Padros, E., and Renugopalakrishnan, V. (2009). Study on the feasibility of bacteriorhodopsin as bio-photosensitizer in excitonic solar cell: a first report. J. Nanosci. Nanotechnol. 9, 1679–1687.
Thorpe, H. M., and Smith, M. C. (1998). In vitro site-specific integration of bacteriophage DNA catalyzed by a recombinase of the resolvase/invertase family. Proc. Natl. Acad. Sci. U.S.A. 95, 5505–5510.
Tosi, L. R., and Beverley, S. M. (2000). cis and trans factors affecting Mos1 mariner evolution and transposition in vitro, and its potential for functional genomics. Nucleic Acids Res. 28, 784–790.
Welander, P. V., and Metcalf, W. W. (2008). Mutagenesis of the C1 oxidation pathway in Methanosarcina barkeri: new insights into the Mtr/Mer bypass pathway. J. Bacteriol. 190, 1928–1936.
White, M. F. (2011). Homologous recombination in the Archaea: the means justify the ends. Biochem. Soc. Trans. 39, 15–19.
Woese, C. R., Kandler, O., and Wheelis, M. L. (1990). Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. U.S.A. 87, 4576–4579.
Zhang, J. K., Pritchett, M. A., Lampe, D. J., Robertson, H. M., and Metcalf, W. W. (2000). In vivo transposon mutagenesis of the methanogenic archaeon Methanosarcina acetivorans C2A using a modified version of the insect mariner-family transposable element Himar1. Proc. Natl. Acad. Sci. U.S.A. 97, 9665–9670.
Keywords: Methanosarcina, genetic manipulation, mutagenesis, markerless deletion, genetypic complementation, gene expression
Citation: Kohler PRA and Metcalf WW (2012) Genetic manipulation of Methanosarcina spp.. Front. Microbio. 3:259. doi: 10.3389/fmicb.2012.00259
Received: 03 May 2012; Paper pending published: 21 May 2012;
Accepted: 03 July 2012; Published online: 24 July 2012.
Edited by:
Frank T. Robb, University of California, USAReviewed by:
Jonathan H. Badger, J. Craig Venter Institute, USAPaul Blum, University of Nebraska, USA
Isaac Cann, University of Illinois, USA
Copyright © 2012 Kohler and Metcalf. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: William W. Metcalf, Department of Microbiology, University of Illinois at Urbana-Champaign, B103 C&LSL, 601 S. Goodwin, Urbana, IL 61801-3763, USA. e-mail:bWV0Y2FsZkBpbGxpbm9pcy5lZHU=