


- Department of Pathology, National Institute of Infectious Diseases, Tokyo, Japan
Retroelements comprise a large and successful family of transposable genetic elements that, through intensive infiltration, have shaped the genomes of humans and other mammals over millions of years. In fact, retrotransposons now account for approximately 45% of the human genome. Because of their genomic mobility called retrotransposition, some retroelements can cause genetic diseases; such retrotransposition events occur not only in germ cells but also in somatic cells, posing a threat to genomic stability throughout all cellular populations. In response, mammals have developed intrinsic immunity mechanisms that provide resistance against the deleterious effects of retrotransposition. Among these, seven members of the APOBEC3 (A3) family of cytidine deaminases serve as highly active, intrinsic, antiretroviral host factors. Certain A3 proteins effectively counteract infections of retroviruses such as HIV-1, as well as those of other virus families, while also blocking the transposition of retroelements. Based on their preferential expression in the germ cells, in which retrotransposons may be active, it is likely that A3 proteins were acquired through mammalian evolution primarily to inhibit retrotransposition and thereby maintain genomic stability in these cells. This review summarizes the recent advances in our understanding of the interplay between the retroelements currently active in the human genome and the anti-retroelement A3 proteins.
Introduction
The evolution of vertebrate genomes has been driven in part by the long history of their interaction with genetic transposable elements. These so-called retrotransposons, which replicate via RNA intermediates, can be divided into two groups depending on the presence or absence of long terminal repeats (LTRs). LTR retrotransposons are endogenous retroviruses that constitute nearly 10% of murine and human genomes, but they have been rendered mostly inactive due to the accumulation of mutations, although some murine intracisternal A-particles (IAP) and MusD sequences remain viable (Dewannieux et al., 2004; Ribet et al., 2004). Non-LTR retrotransposons comprise the majority of transposable elements; in fact, collectively, they account for more than one third of the human genome. They can be further subdivided into three types; long interspersed elements (LINEs), short interspersed elements (SINEs), and the composite hominid-specific retrotransposons, each of which contain the only transposable elements currently active in the human genome, i.e., LINE-1, Alu, and SINE-VNTR-Alu (SVA), respectively (Deininger and Batzer, 2002; Ostertag et al., 2003).
Retrotransposition, discussed in greater detail below, involves the reverse transcription of an RNA intermediate with subsequent genomic integration in a process driven by retrotransposon-encoded RNA-dependent DNA polymerase and endonuclease. The integration of these elements may have harmful consequences for the host, compromising genomic stability via insertions, deletions, and DNA rearrangements and thereby posing a threat to human health, as described in several reports of retrotransposition-induced genetic disorders (Kazazian et al., 1988; Wallace et al., 1991; Kobayashi et al., 1998). In response, eukaryotic organisms have evolved mechanisms to restrict uncontrolled retrotransposition. Anti-retroelement strategies include transcriptional silencing through DNA methylation (Walsh et al., 1998; Bourc'his and Bestor, 2004; Burden et al., 2005), post-transcriptional silencing via RNA interference (Soifer et al., 2005; Yang and Kazazian, 2006), and some cellular factors inhibiting retrotransposition at the post-translational level. Of these cellular factors, seven members of the apolipoprotein B mRNA-editing catalytic polypeptide-like 3 (APOBEC3; referred to hereafter as the A3) family of cytidine deaminases have been shown to act as potent inhibitors of a wide range of both exogenous retroviruses and endogenous retroelements (Sheehy et al., 2002; Esnault et al., 2005; Chen et al., 2006; Kinomoto et al., 2007). In this review, we focus on active endogenous retroelements, their deleterious effects on the human genome, and the anti-retroelement activity of A3 proteins.
Retrotransposons: an Overview
Unlike the murine LTR retrotransposons IAP and MusD, human versions, such as human endogenous retroviruses (HERV), have been mostly fossilized, and even those that are not are non-transposable. In contrast, many copies of human non-LTR retrotransposons can replicate through an RNA/protein complex intermediate and integrate into the host genome at a new site. The LINE retrotransposons, typified by LINE-1 (L1), account for approximately 17% of the human genome, corresponding to >500,000 copies (of which 100 copies are retrotransposition-competent). L1 retrotransposons are 6 kb in length and contain a 5′ untranslated region (UTR) that harbors a Pol II promoter; two ORFs necessary for their own replication; and a 3′ UTR containing a polyadenylation signal, followed by a poly(A) tail (Figure 1A, top). Briefly, L1 elements are first transcribed by RNA-polymerase II using a promoter located at the L1 5′ region (Ostertag and Kazazian, 2001). ORF1, encoding an RNA-binding protein, and ORF2, encoding a protein with reverse transcriptase and endonuclease activity, are then translated in the cytoplasm. The resulting proteins associate with L1 RNA to form a ribonucleoprotein (RNP) complex (Martin, 1991; Hohjoh and Singer, 1996; Figure 1B) that is transported back into the nucleus, where L1 is integrated into the host genome through a target-primed reverse transcription (Cost et al., 2002).
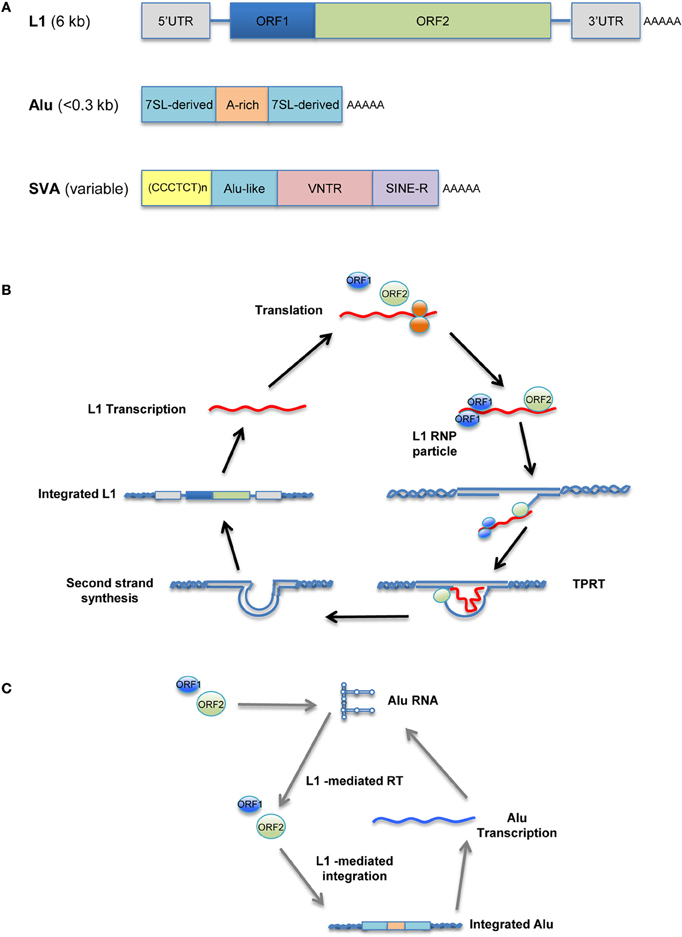
Figure 1. Retrotransposition cycle. Schematic representation of active human retrotransposons. (A) Top: L1 genomic organization, from the left: 5′ UTR, untranslated region; ORF-1, encoding an RNA-binding protein; linker region; ORF-2, encoding reverse transcriptase and endonuclease; 3′ UTR; AAA, poly(A) tail. Middle: Alu organization, from the left: 7SL-derived monomer; A-rich linker, A5TACA6; 7SL-derived monomer; AAA, poly(A) tail. Bottom: SVA organization, from the left: (CCCTCT)n, hexamer repeat; inverted Alu-like sequence; VNTR, variable number of tandem repeats; SINE-R, HERV-K-derived sequence; AAA, poly(A) tail. (B) Retrotransposition cycle: L1 elements are transcribed by RNA-polymerase II from an L1 promoter sequence. The L1 mRNA template is exported to the cytoplasm and translated. Retrotransposon-encoded proteins actively bind the L1 RNA transcript, forming a ribonucleoprotein particle (RNP) that is imported back into the nucleus. There, the L1-encoded endonuclease nicks an L1 target sequence (5′-TTTT/AA-3′) and the 3′-OH generated is used as a primer for target-primed reverse transcription (TPRT) by the L1-encoded reverse transcriptase, resulting in de novo integration into the host genome. (C) Alu as well as SVA elements are transcribed and hijack the L1-encoded enzymatic machinery to complete their respective retrotransposition cycles.
The human genome also contains more than 1 million copies of Alu elements; these are the most common SINE retrotransposons, representing 11% of our genome. The typical Alu element is approximately 300 bp in length and is formed by the fusion of two 7SL-RNA gene-derived monomers separated by an A-rich linker, followed by a poly(A) tail (Kriegs et al., 2007; Figure 1A, middle). Likewise, there are ~2700 copies of the composite SVA elements in the human genome. SVAs, which are approximately 2 kb long, are composed of CCCTCT hexameric repeats that are followed by an inverted Alu-like region, a region containing a variable number of tandem repeats (VNTRs), and a partial HERV-K env–LTR sequence termed SINE-R that ends with a polyadenylation signal, followed by a poly(A) tail (Ostertag et al., 2003; Figure 1A, bottom). Unlike L1, Alu and SVA elements are non-autonomous since they do not encode functional reverse transcriptase or endonuclease; instead, they use the enzymatic machinery of L1 for retrotransposition. Once Alu and SVA elements have been transcribed and exported to the cytoplasm, they hijack the L1-encoded enzymes in the vicinity of the ribosomes through mechanisms that are as-yet unclear (Figure 1C; Dewannieux et al., 2003; Ostertag et al., 2003).
Retrotransposons in Human Diseases
Approximately 100 examples of disease-causing retrotransposon insertions are currently reported in the literature. It is estimated that de novo insertions of L1, Alu, and SVA elements are responsible for approximately 0.3% of all disease-causing human mutations, corresponding to event rates of 1:100, 1:20, and 1:900 births, respectively (Cordaux and Batzer, 2009). L1-induced genetic diseases include the following: Duchenne muscular dystrophy and X-linked dilated cardiomyopathy, resulting from insertions in the dystrophin gene (Narita et al., 1993; Yoshida et al., 1998); progressive chorioretinal degeneration, caused by the CHM gene disruption (van Den Hurk et al., 2003); hemophilia A and B, due to insertions in the factor VIII and IX genes, respectively (Kazazian et al., 1988; Li et al., 2001; Mukherjee et al., 2004); and chronic granulomatous disease, the result of a mutation arising from an insertion in the CYBB gene (Meischl et al., 2000). Genetic diseases linked to Alu integration events include neurofibromatosis via an insertion in the NF1 gene (Wallace et al., 1991; Wimmer et al., 2011); Apert syndrome, a severe autosomal dominant disorder, due to integration of the element into the fibroblast growth-factor receptor 2 (FGFR2) gene (Oldridge et al., 1999); and progressive renal failure (Dent's disease) due to disruption of the renal chloride channel (CLCN5) gene (Claverie-Martin et al., 2005). The involvement of SVA retrotransposition in human diseases has also been documented; namely, an insertion in the ARH gene leads to autosomal recessive hypercholesterolemia (Wilund et al., 2002); disruption of the BTK gene causes X-linked agammaglobulinemia (XLA; Rohrer et al., 1999); and disruption of the fukutin gene results in Fukuyama-type congenital muscular dystrophy (Kobayashi et al., 1998). Importantly, ongoing retrotransposon insertions seem to occur not only in germ cells and early embryos but also in brain tissues (Coufal et al., 2009; Baillie et al., 2011), somatic cells in vitro (Kubo et al., 2006; Rangwala et al., 2009), and somatic malignant tissues (Economou-Pachnis and Tsichlis, 1985; Morse et al., 1988; Miki et al., 1992). Several reports have also shown retrotransposon-induced recombination in certain types of cancer (Schichman et al., 1994; Jeffs et al., 1998).
Cellular Mechanisms Limiting the Activity of Retroelements and Retroviruses
As noted above, since unrestricted retrotransposition would result in genome instability, eukaryotic organisms have developed several strategies to restrict these mobile elements. Firstly, retrotransposition can be regulated at the transcriptional level through several transcription factors. For example, L1 transcription is positively regulated by SOX11 (Tchenio et al., 2000), RUNX3 (Yang et al., 2003) and YY1 (Athanikar et al., 2004), and negatively regulated by SRY (Tchenio et al., 2000) and SOX2 (Muotri et al., 2005). DNA methylation by the methyl-CpG-binding protein MeCP2 results in the repression of L1 transcription in neurons (Walsh et al., 1998; Burden et al., 2005; Muotri et al., 2010). Secondly, retrotransposable elements are also susceptible to post-transcriptional regulation. For instance, endogenously encoded small interfering RNAs have been shown to reduce L1 retrotransposition in vitro (Soifer et al., 2005; Yang and Kazazian, 2006). Additionally, L1 transcripts that contain multiple polyadenylation signals lead to premature polyadenylation, resulting in the attenuation of L1 activity via truncation of its full-length transcripts (Perepelitsa-Belancio and Deininger, 2003). Thirdly, some cellular factors regulate retrotransposition at the post-translational level. In mice, the 3′–5′ exonuclease Trex1 digests retroelement-derived DNA to suppress the autoimmune response (Stetson et al., 2008), Consistent with this, mutations in human Trex1 cause autoimmune diseases like familial chilblain lupus and Aicardi-Goutieres syndrome (Crow et al., 2006). Likewise, HIV-1 restriction factors such as the cytidine deaminases, the focus of this review, can inhibit L1 and Alu retrotransposition through a mechanism that is still unknown.
In humans, the cellular cytidine deaminase family comprises several members, including activation-induced cytidine deaminase (AID), APOBEC1, APOBEC2, the A3 family, and APOBEC4 (Harris and Liddament, 2004; Conticello, 2008; Smith et al., 2012). APOBEC1 is the catalytic subunit of an RNA-editing complex that deaminates C6666→U in the mRNA of the lipid-transport protein apolipoprotein B, thereby creating a premature stop codon that leads to a truncated protein in gastrointestinal tissues (Teng et al., 1993). APOBEC1 proteins from multiple small-animal species exhibit inhibitory activity against not only exogenous retroviruses (Ikeda et al., 2008) but also endogenous retroviruses, such as murine IAP and MusD sequences, as well as L1 elements (Ikeda et al., 2011). AID plays a role in the adaptive humoral immune system by inducing somatic hypermutations and class switch recombination, which allows affinity maturation and memory development; however, its precise mechanism of action remains to be determined (Honjo et al., 2005). As described in detail in a subsequent section, members of the A3 family are potent inhibitors of both exogenous retroviruses and endogenous retroelements. A3G, the most extensively studied member of the A3 family, was the first cytidine deaminase shown to restrict infection by Vif-deficient HIV-1 viruses. Briefly, as depicted in Figure 2, A3G is incorporated into budding virions and thus exerts its antiviral effect at the post-entry step in target cells, either by mediating extensive deamination of the minus-strand of viral DNA during reverse transcription, which results in G → A hypermutations in the proviral DNA plus strand (deaminase-dependent mechanism) (Harris et al., 2003; Mangeat et al., 2003; Zhang et al., 2003), or by binding to HIV-1 RNA, leading to physical impairment of reverse transcription (deaminase-independent mechanism; Newman et al., 2005; Bishop et al., 2006; Iwatani et al., 2007). Consequently, primate lentiviruses have evolved to counteract the antiretroviral activity of A3G by acquiring Vif. This accessory protein prevents A3G incorporation into virions through its proteasomal degradation (Marin et al., 2003; Sheehy et al., 2003; Stopak et al., 2003). We and others have shown that Vif proteins derived from different HIV-1 subtypes differ in their potency of A3G inhibition, suggesting differential levels of viral fitness among clades (Iwabu et al., 2010; Binka et al., 2012). APOBEC2, a cardiac- and skeletal muscle-specific cytidine deaminase, is required for muscle development and early embryogenesis (Etard et al., 2010; Sato et al., 2010; Vonica et al., 2011). The physiological role of APOBEC4 remains to be determined.
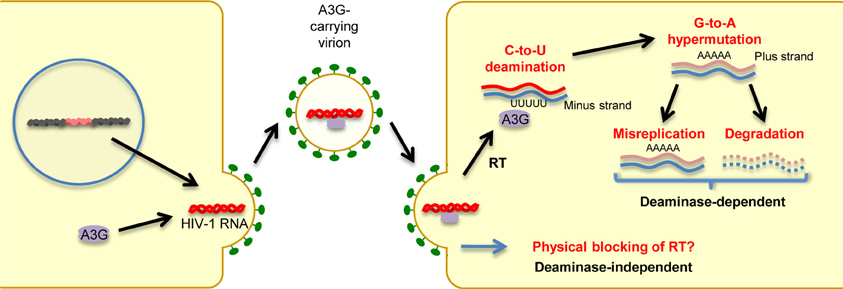
Figure 2. Mechanisms of antiretroviral and anti-retroelement activity of A3G protein. A3G potently restricts Vif-deficient HIV-1 viruses: A3G is incorporated into virus particles in virus-producing cells. Following the infection of target cells, A3G inhibits viral replication either by binding to HIV-1 RNA, leading to physical blocking of reverse transcription (deaminase-independent mechanism), or by deaminating the viral minus-strand DNA during reverse transcription, thus generating G → A hypermutations in the proviral DNA plus strand.
Differential Antiviral and Anti-Retroelement Activities of A3 Cytidine Deaminases
Members of the A3 family contain either single (A3A, A3C, A3H) or double (A3B, A3DE, A3F, and A3G) cytidine deaminase domains (CDA). In A3G and A3F, the N-terminal CDA is responsible for RNA-dependent oligomerization, while the C-terminal CDA mainly mediates the deamination of single-stranded DNA (Hache et al., 2005; Newman et al., 2005). Some A3 family members strongly inhibit a wide range of exogenous retroviruses, as well as other viral pathogens, including herpesviruses, parvoviruses, papillomaviruses, and hepadnaviruses (Baumert et al., 2007; Vartanian et al., 2008; Narvaiza et al., 2009; Suspène et al., 2011b). The importance of A3 proteins in vivo has been demonstrated in murine studies in which mice lacking the A3 gene were shown to be more susceptible to viral infection than their wild-type counterparts (Okeoma et al., 2007, 2009; Takeda et al., 2008). A3 proteins also inhibit the mobilization of endogenous retroviruses, such as MusD, IAP, and the yeast LTR-retrotransposon Ty1 (Esnault et al., 2005; Schumacher et al., 2008), in addition to their inhibitory activity on L1 and Alu retrotransposition. The gene copy number of A3 family members is species-specific in mammals, in which except for primates, one, two, or three A3 proteins are encoded, whereas in humans and in non-human primates, seven A3 proteins have been recognized (A3A, A3B, A3C, A3DE, A3F, A3G, and A3H; Sawyer et al., 2004; OhAinle et al., 2006). Of note, expansion of the A3 gene cluster in primate genomes correlates with a sharp reduction in retrotransposition activity, suggesting that these restriction factors have evolved to protect mammalian hosts from retroelements (Sawyer et al., 2004; Schumann, 2007). Antiretroviral and anti-retroelement potencies were shown to differ in the seven members of A3 family, independently of their subcellular localization (Kinomoto et al., 2007). However, the exact mechanism by which A3 proteins inhibit retrotransposition is unclear. The current findings on antiviral and anti-retroelement activities of A3 members are summarized below and in Table 1.
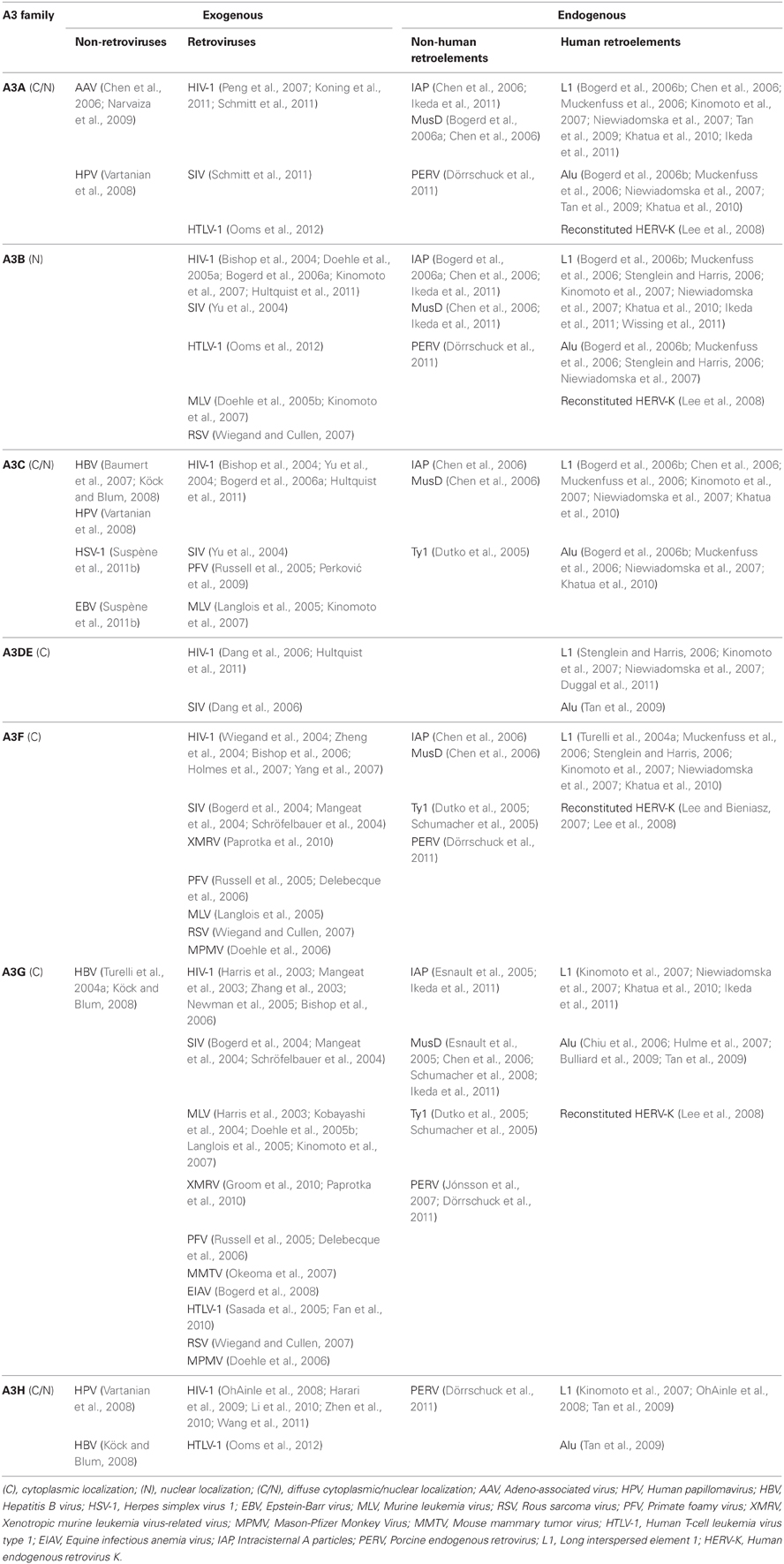
Table 1. Antiviral and anti-retroelement spectrum of A3 family members.
A3A
Human A3A (hA3A) lacks inhibitory activity against HIV-1 produced from 293T cells overexpressing this protein (Bishop et al., 2004; Bogerd et al., 2006a; Kinomoto et al., 2007; Hultquist et al., 2011), since it is not incorporated into virions (Goila-Gaur et al., 2007; Aguiar et al., 2008). In human monocytic cells as targets, however, hA3A blocks the early phase of HIV-1 infection (Peng et al., 2007; Koning et al., 2011) but is counteracted by the HIV-2/SIV (simian immunodeficiency virus) accessory protein Vpx (Berger et al., 2010, 2011). Also, hA3A can inhibit infections by adeno-associated virus (Chen et al., 2006; Narvaiza et al., 2009), human papillomavirus (HPV; Vartanian et al., 2008), porcine endogenous retrovirus (PERV; Dörrschuck et al., 2011), and human T-cell leukemia virus type 1 (HTLV-1; Ooms et al., 2012). Importantly, in vitro overexpression experiments have demonstrated that hA3A effectively inhibits the retrotranspositions of L1, Alu (Bogerd et al., 2006b; Chen et al., 2006; Muckenfuss et al., 2006; Kinomoto et al., 2007; Niewiadomska et al., 2007; Tan et al., 2009; Khatua et al., 2010; Ikeda et al., 2011), IAP, and MusD (Bogerd et al., 2006a; Chen et al., 2006; Ikeda et al., 2011) through a deaminase-independent mechanism. hA3A is intrinsically able to restrict infection of the genetically reconstituted HERV-K in a deaminase-dependent manner (Lee et al., 2008). In a recent report, hA3A was shown to induce somatic hypermutation in human mitochondrial and nuclear DNA; in the latter, this included genes associated with the development of cancer (Suspène et al., 2011a).
A3B
Human A3B (hA3B) is the sole member of the A3 family with an exclusive nuclear localization (Bogerd et al., 2006a; Muckenfuss et al., 2006; Stenglein and Harris, 2006; Kinomoto et al., 2007; Pak et al., 2011), sharing a common nuclear import mechanism with AID (Lackey et al., 2012). hA3B inhibits infections of HIV-1 and SIV, independently of the presence of Vif (Bishop et al., 2004; Yu et al., 2004; Doehle et al., 2005a; Hultquist et al., 2011). Since hA3B expression is extremely low in target CD4+ T-cells (Bishop et al., 2004; Doehle et al., 2005a; Koning et al., 2009; Refsland et al., 2010), Vif might have failed to evolve the mechanism to antagonize this antiretroviral factor. Also, hA3B restricts infections by murine leukemia virus (MLV; Doehle et al., 2005b; Kinomoto et al., 2007), PERV (Dörrschuck et al., 2011), HTLV-1 (Ooms et al., 2012), and Rous sarcoma virus (RSV; Wiegand and Cullen, 2007). Like hA3A, in vitro overexpression of hA3B inhibits the retrotranspositions of L1, Alu (Bogerd et al., 2006b; Muckenfuss et al., 2006; Stenglein and Harris, 2006; Kinomoto et al., 2007; Niewiadomska et al., 2007; Khatua et al., 2010; Ikeda et al., 2011), IAP, and MusD (Bogerd et al., 2006a; Chen et al., 2006; Ikeda et al., 2011) in a deaminase-independent manner, while inhibiting the reconstituted HERV-K infection through a deaminase-dependent mechanism (Lee et al., 2008). In a recent study, endogenously expressed hA3B effectively restricted L1 retrotransposition in both transformed cells and human embryonic stem cells (Wissing et al., 2011).
A3C
Human A3C (hA3C) is abundantly expressed in numerous tissues and cell types (Jarmuz et al., 2002), and its expression is unresponsive to interferon-α (Koning et al., 2009). Although hA3C is efficiently incorporated into retroviral particles, it exhibits only partial antiviral activity against HIV-1, with or without Vif (Bishop et al., 2004; Yu et al., 2004; Bogerd et al., 2006a; Hultquist et al., 2011). By contrast, hA3C is able to efficiently block the replication of SIV, which also encapsidates this protein but is readily antagonized by SIV Vif (Yu et al., 2004). hA3C can inhibit infection of primate foamy virus (PFV), which carries an hA3 antagonistic Bet protein (Russell et al., 2005; Perković et al., 2009). The overexpression of hA3C results in a moderate inhibition of L1 and Alu retrotranspositions (Bogerd et al., 2006b; Chen et al., 2006; Muckenfuss et al., 2006; Kinomoto et al., 2007; Niewiadomska et al., 2007; Khatua et al., 2010) but effectively inhibits those of IAP, MusD, and Ty1 (Dutko et al., 2005; Chen et al., 2006). In a recent study, hA3C was shown to restrict infections by MLV (Langlois et al., 2005; Kinomoto et al., 2007), hepatitis B virus (HBV; Baumert et al., 2007; Köck and Blum, 2008), HPV (Vartanian et al., 2008), herpes simplex virus 1 and Epstein-Barr virus (Suspène et al., 2011b).
A3DE
Human A3DE (hA3DE) overexpression has moderate effects on L1 and Alu retrotransposition (Stenglein and Harris, 2006; Kinomoto et al., 2007; Niewiadomska et al., 2007; Tan et al., 2009; Duggal et al., 2011). Similarly, hA3DE exhibits low levels of anti-HIV-1 and anti-SIV activities, both of which are antagonized by the respective Vif proteins (Dang et al., 2006; Hultquist et al., 2011). The reduced activity is determined by a cysteine residue located at amino acid position 320 of hA3DE. Substitution with the corresponding tyrosine present in A3F resulted in a 20-fold increase of A3DE activity (Dang et al., 2011). Indeed, the chimpanzee version of A3DE, carrying a tyrosine residue at this position, shows much higher antiretroviral activity, while both human and chimpanzee A3DEs exhibit similar levels of inhibition against retroelements, suggesting that the host defense activity of A3DE against retroelements has been evolutionarily conserved (Duggal et al., 2011).
A3F/A3G
With regard to the antiretroviral potencies of human A3G (hA3G) and A3F (hA3F) proteins, overwhelming amount of information is well-summarized elsewhere (Harris and Liddament, 2004; Huthoff and Towers, 2008; Malim, 2009). Similar to hA3G, as introduced in the previous section, hA3F has been shown to potently restrict the replication of Vif-deficient HIV-1 viruses in target cells after its incorporation into budding virions through both deaminase-dependent and -independent mechanisms (Bishop et al., 2004; Wiegand et al., 2004; Zheng et al., 2004; Holmes et al., 2007; Yang et al., 2007). The in vitro overexpression of hA3G inhibits not only retroviral infections, such as those by HTLV-1 (Sasada et al., 2005; Fan et al., 2010), SIV (Bogerd et al., 2004; Mangeat et al., 2004; Schröfelbauer et al., 2004), PFV (Russell et al., 2005; Delebecque et al., 2006), equine infectious anemia virus (Bogerd et al., 2008), MLV (Harris et al., 2003; Kobayashi et al., 2004; Doehle et al., 2005b; Langlois et al., 2005; Kinomoto et al., 2007), Mason-Pfizer monkey virus (MPMV; Doehle et al., 2006), xenotropic murine leukemia virus-related virus (XMRV; Groom et al., 2010;, Paprotka et al., 2010), PERV (Jónsson et al., 2007; Dörrschuck et al., 2011), and RSV (Wiegand and Cullen, 2007), but also retrotranspositions of non-human LTR retroelements, such as IAP, MusD, and Ty1 (Dutko et al., 2005; Esnault et al., 2005; Chen et al., 2006; Schumacher et al., 2008; Ikeda et al., 2011). This cytidine deaminase is also effective against HBV (Turelli et al., 2004a; Köck and Blum, 2008). With regard to potential to inhibit L1 retrotransposition, conflicting results have been reported; some lines of evidence suggest that hA3G has anti-Alu activity (Chiu et al., 2006; Hulme et al., 2007; Bulliard et al., 2009; Tan et al., 2009) but little or no anti-L1 activity (Turelli et al., 2004b; Bogerd et al., 2006b; Muckenfuss et al., 2006; Stenglein and Harris, 2006). We and others, however, have shown that hA3G is also able to restrict L1 retrotransposition, albeit less potently than hA3A or hA3B, through deaminase-independent mechanisms (Kinomoto et al., 2007; Niewiadomska et al., 2007; Khatua et al., 2010; Ikeda et al., 2011). These discrepancies might be due to cell-type differences in hA3 protein expression levels, as we have described previously (Kinomoto et al., 2007). A putative mechanism of L1 inhibition by hA3G is as follows: When L1 forms the RNP complex in the cytoplasm (Figure 1B, right half), cytoplasmic hA3G protein might be able to access the complex through the interaction with L1 RNA, and then enter the nucleus together with the complex. This could result in the effective inhibition of L1 reverse transcription, by physically blocking the access to the chromosomal DNA, or by impeding the movement of the reverse transcriptase on a template L1 RNA. In the case of the infection by reconstituted HERV-K, hA3G carries out deamination showing only marginal inhibition of infectivity (Lee et al., 2008). Among the viruses and retroelements described above, hA3F is known to inhibit infections of PFV (Russell et al., 2005; Delebecque et al., 2006), MLV (Langlois et al., 2005), XMRV (Paprotka et al., 2010), MPMV (Doehle et al., 2006), PERV (Dörrschuck et al., 2011), RSV (Wiegand and Cullen, 2007), and reconstituted HERV-K (Lee and Bieniasz, 2007; Lee et al., 2008), as well as the retrotransposons; L1 (Chen et al., 2006; Muckenfuss et al., 2006; Stenglein and Harris, 2006; Kinomoto et al., 2007; Niewiadomska et al., 2007; Khatua et al., 2010), Ty1 (Dutko et al., 2005; Schumacher et al., 2005), MusD and IAP (Chen et al., 2006).
A3H
Human A3H (hA3H) is the most distantly related of the hA3 members and is known for its functional polymorphisms. Currently, four major haplotypes (I–IV) have been identified in human populations, among which haplotype I has the highest allelic frequencies (OhAinle et al., 2008). Haplotypes I, III, and IV generate unstable proteins with very little, if any, antiretroviral and anti-retroelement activity. Haplotype II, however, expresses a stable protein with relatively high inhibitory activity on HIV-1 (OhAinle et al., 2008; Harari et al., 2009; Li et al., 2010; Zhen et al., 2010; Wang et al., 2011) and HTLV-1 (Ooms et al., 2012), and its overexpression effectively restricts L1 retrotransposition (OhAinle et al., 2008; Tan et al., 2009). These observations suggest that the relative lack of anti-retroviral and anti-retroelement potencies in hA3H is not due to insufficient enzymatic activity but to the instability of the protein. It should be noted that hA3H haplotype II is mainly localized to the cytoplasm, while the haplotype I protein passively diffuses into the nucleus (Li and Emerman, 2011). The ability of hA3H to block infections of HPV (Vartanian et al., 2008), HBV (Köck and Blum, 2008), and PERV (Dörrschuck et al., 2011) has also been reported, although the responsible haplotypes have not been described.
Concluding Remarks
Retrotransposable elements have successfully proliferated over tens of millions of years of mammalian evolution, such that they now constitute 45% of the human genome. Retrotransposition spreads DNA fragments to different genomic sites and is thus considered to be one of the driving forces in genome evolution by contributing to the formation of new genes. On the other hand, the price to pay for such genomic innovation, in which retrotransposons integrate in their host genomes, is the potential disruption of essential genes, resulting in deleterious effects, some of which are clearly associated with genetic diseases and tumorigenesis. Consequently, to prevent uncontrolled retrotransposition, host organisms have evolved several defense mechanisms. Among these, the seven members of A3 family have the ability to restrict not only a broad range of exogenous retroviruses but also endogenous retroelements, as described herein. Interestingly, high-level A3 expression is seen in the testis and ovary and in embryonic stem cells (Jarmuz et al., 2002; Bogerd et al., 2006b; OhAinle et al., 2006), in which the retroelements are hypomethylated and therefore active (Bourc'his and Bestor, 2004; Dupressoir and Heidmann, 1996). These findings support the evolutionary acquisition of A3 proteins to protect these cells primarily from the genomic instability caused by the disruptive effect of endogenous retroelements. Further investigations of A3-mediated intrinsic immunity are likely to provide insights into the molecular mechanisms of the host defenses that do not allow retrotransposons to escape from the seven members of A3.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the Ministry of Health, Labor and Welfare of Japan (Research on HIV/AIDS; H24-005 and H24-008), and from the Ministry of Education, Science, Technology, Sports and Culture of Japan.
References
Aguiar, R. S., Lovsin, N., Tanuri, A., and Peterlin, B. M. (2008). Vpr.A3A chimera inhibits HIV replication. J. Biol. Chem. 283, 2518–2525.
Athanikar, J. N., Badge, R. M., and Moran, J. V. (2004). A YY1-binding site is required for accurate human LINE-1 transcription initiation. Nucleic Acids Res. 32, 3846–3855.
Baillie, J. K., Barnett, M. W., Upton, K. R., Gerhardt, D. J., Richmond, T. A., De Sapio, F., Brennan, P. M., Rizzu, P., Smith, S., Fell, M., Talbot, R. T., Gustincich, S., Freeman, T. C., Mattick, J. S., Hume, D. A., Heutink, P., Carninci, P., Jeddeloh, J. A., and Faulkner, G. J. (2011). Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479, 534–537.
Baumert, T. F., Rösler, C., Malim, M. H., and von Weizsäcker, F. (2007). Hepatitis B virus DNA is subject to extensive editing by the human deaminase APOBEC3C. Hepatology 46, 682–689.
Berger, A., Münk, C., Schweizer, M., Cichutek, K., Schüle, S., and Flory, E. (2010). Interaction of Vpx and apolipoprotein B mRNA-editing catalytic polypeptide 3 family member, A (APOBEC3A) correlates with efficient lentivirus infection of monocytes. J. Biol. Chem. 285, 12248–12254.
Berger, G., Durand, S., Fargier, G., Nguyen, X.-N., Cordeil, S., Bouaziz, S., Muriaux, D., Darlix, J.-L., and Cimarelli, A. (2011). APOBEC3A is a specific inhibitor of the early phases of HIV-1 infection in myeloid cells. PLoS Pathog. 7:e1002221. doi: 10.1371/journal.ppat.1002221
Binka, M., Ooms, M., Steward, M., and Simon, V. (2012). The activity spectrum of Vif from multiple HIV-1 subtypes against APOBEC3G, APOBEC3F, and APOBEC3H. J. Virol. 86, 49–59.
Bishop, K. N., Holmes, R. K., and Malim, M. H. (2006). Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 80, 8450–8458.
Bishop, K. N., Holmes, R. K., Sheehy, A. M., Davidson, N. O., Cho, S.-J., and Malim, M. H. (2004). Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 14, 1392–1396.
Bogerd, H. P., Doehle, B. P., Wiegand, H. L., and Cullen, B. R. (2004). A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV type 1 virion infectivity factor. Proc. Natl. Acad. Sci. U.S.A. 101, 3770–3774.
Bogerd, H. P., Tallmadge, R. L., Oaks, J. L., Carpenter, S., and Cullen, B. R. (2008). Equine infectious anemia virus resists the antiretroviral activity of equine APOBEC3 proteins through a packaging-independent mechanism. J. Virol. 82, 11889–11901.
Bogerd, H. P., Wiegand, H. L., Doehle, B. P., Lueders, K. K., and Cullen, B. R. (2006a). APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 34, 89–95.
Bogerd, H. P., Wiegand, H. L., Hulme, A. E., Garcia-Perez, J. L., O'Shea, K. S., Moran, J. V., and Cullen, B. R. (2006b). Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc. Natl. Acad. Sci. U.S.A. 103, 8780–8785.
Bourc'his, D., and Bestor, T. H. (2004). Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 431, 96–99.
Bulliard, Y., Turelli, P., Röhrig, U. F., Zoete, V., Mangeat, B., Michielin, O., and Trono, D. (2009). Functional analysis and structural modeling of human APOBEC3G reveal the role of evolutionarily conserved elements in the inhibition of human immunodeficiency virus type 1 infection and Alu transposition. J. Virol. 83, 12611–12621.
Burden, A. F., Manley, N. C., Clark, A. D., Gartler, S. M., Laird, C. D., and Hansen, R. S. (2005). Hemimethylation and non-CpG methylation levels in a promoter region of human LINE-1 (L1) repeated elements. J. Biol. Chem. 280, 14413–14419.
Chen, H., Lilley, C. E., Yu, Q., Lee, D. V., Chou, J., Narvaiza, I., Landau, N. R., and Weitzman, M. D. (2006). APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol. 16, 480–485.
Chiu, Y.-L., Witkowska, H. E., Hall, S. C., Santiago, M., Soros, V. B., Esnault, C., Heidmann, T., and Greene, W. C. (2006). High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc. Natl. Acad. Sci. U.S.A. 103, 15588–15593.
Claverie-Martin, F., Flores, C., Anton-Gamero, M., Gonzalez-Acosta, H., and Garcia-Nieto, V. (2005). The Alu insertion in the CLCN5 gene of a patient with Dent's disease leads to exon 11 skipping. J. Hum. Genet. 50, 370–374.
Cordaux, R., and Batzer, M. A. (2009). The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 10, 691–703.
Cost, G. J., Feng, Q., Jacquier, A., and Boeke, J. D. (2002). Human L1 element target-primed reverse transcription in vitro. EMBO J. 21, 5899–5910.
Coufal, N. G., Garcia-Perez, J. L., Peng, G. E., Yeo, G. W., Mu, Y., Lovci, M. T., Morell, M., O'Shea, K. S., Moran, J. V., and Gage, F. H. (2009). L1 retrotransposition in human neural progenitor cells. Nature 460, 1127–1131.
Crow, Y. J., Hayward, B. E., Parmar, R., Robins, P., Leitch, A., Ali, M., Black, D. N., van Bokhoven, H., Brunner, H. G., Hamel, B. C., Corry, P. C., Cowan, F. M., Frints, S. G., Klepper, J., Livingston, J. H., Lynch, S. A., Massey, R. F., Meritet, J. F., Michaud, J. L., Ponsot, G., Voit, T., Lebon, P., Bonthron, D. T., Jackson, A. P., Barnes, D. E., and Lindahl, T. (2006). Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat. Genet. 38, 917–920.
Dörrschuck, E., Fischer, N., Bravo, I. G., Hanschmann, K.-M., Kuiper, H., Spötter, A., Möller, R., Cichutek, K., Münk, C., and Tönjes, R. R. (2011). Restriction of porcine endogenous retrovirus by porcine APOBEC3 cytidine deaminases. J. Virol. 85, 3842–3857.
Dang, Y., Abudu, A., Son, S., Harjes, E., Spearman, P., Matsuo, H., and Zheng, Y.-H. (2011). Identification of a single amino acid required for APOBEC3 antiretroviral cytidine deaminase activity. J. Virol. 85, 5691–5695.
Dang, Y., Wang, X., Esselman, W. J., and Zheng, Y.-H. (2006). Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J. Virol. 80, 10522–10533.
Delebecque, F., Suspene, R., Calattini, S., Casartelli, N., Saib, A., Froment, A., Wain-Hobson, S., Gessain, A., Vartanian, J. P., and Schwartz, O. (2006). Restriction of foamy viruses by APOBEC cytidine deaminases. J. Virol. 80, 605–614.
Dewannieux, M., Dupressoir, A., Harper, F., Pierron, G., and Heidmann, T. (2004). Identification of autonomous IAP LTR retrotransposons mobile in mammalian cells. Nat. Genet. 36, 534–539.
Dewannieux, M., Esnault, C., and Heidmann, T. (2003). LINE-mediated retrotransposition of marked Alu sequences. Nat. Genet. 35, 41–48.
Doehle, B. P., Bogerd, H. P., Wiegand, H. L., Jouvenet, N., Bieniasz, P. D., Hunter, E., and Cullen, B. R. (2006). The betaretrovirus Mason-Pfizer monkey virus selectively excludes simian APOBEC3G from virion particles. J. Virol. 80, 12102–12108.
Doehle, B. P., Schäfer, A., and Cullen, B. R. (2005a). Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology 339, 281–288.
Doehle, B. P., Schäfer, A., Wiegand, H. L., Bogerd, H. P., and Cullen, B. R. (2005b). Differential sensitivity of murine leukemia virus to APOBEC3-mediated inhibition is governed by virion exclusion. J. Virol. 79, 8201–8207.
Duggal, N. K., Malik, H. S., and Emerman, M. (2011). The breadth of antiviral activity of Apobec3DE in chimpanzees has been driven by positive selection. J. Virol. 85, 11361–11371.
Dupressoir, A., and Heidmann, T. (1996). Germ line-specific expression of intracisternal A-particle retrotransposons in transgenic mice. Mol. Cell. Biol. 16, 4495–4503.
Dutko, J. A., Schafer, A., Kenny, A. E., Cullen, B. R., and Curcio, M. J. (2005). Inhibition of a yeast LTR retrotransposon by human APOBEC3 cytidine deaminases. Curr. Biol. 15, 661–666.
Economou-Pachnis, A., and Tsichlis, P. N. (1985). Insertion of an Alu SINE in the human homologue of the Mlvi-2 locus. Nucleic Acids Res. 13, 8379–8387.
Esnault, C., Heidmann, O., Delebecque, F., Dewannieux, M., Ribet, D., Hance, A. J., Heidmann, T., and Schwartz, O. (2005). APOBEC3G cytidine deaminase inhibits retrotransposition of endogenous retroviruses. Nature 433, 430–433.
Etard, C., Roostalu, U., and Strahle, U. (2010). Lack of Apobec2-related proteins causes a dystrophic muscle phenotype in zebrafish embryos. J. Cell Biol. 189, 527–539.
Fan, J., Ma, G., Nosaka, K., Tanabe, J., Satou, Y., Koito, A., Wain-Hobson, S., Vartanian, J.-P., and Matsuoka, M. (2010). APOBEC3G generates nonsense mutations in human T-cell leukemia virus type 1 proviral genomes in vivo. J. Virol. 84, 7278–7287.
Goila-Gaur, R., Khan, M. A., Miyagi, E., Kao, S., and Strebel, K. (2007). Targeting APOBEC3A to the viral nucleoprotein complex confers antiviral activity. Retrovirology 4, 61.
Groom, H. C. T., Yap, M. W., Galão, R. P., Neil, S. J. D., and Bishop, K. N. (2010). Susceptibility of xenotropic murine leukemia virus-related virus (XMRV) to retroviral restriction factors. Proc. Natl. Acad. Sci. U.S.A. 107, 5166–5171.
Hache, G., Liddament, M. T., and Harris, R. S. (2005). The retroviral hypermutation specificity of APOBEC3F and APOBEC3G is governed by the C-terminal DNA cytosine deaminase domain. J. Biol. Chem. 280, 10920–10924.
Harari, A., Ooms, M., Mulder, L. C. F., and Simon, V. (2009). Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J. Virol. 83, 295–303.
Harris, R. S., Bishop, K. N., Sheehy, A. M., Craig, H. M., Petersen-Mahrt, S. K., Watt, I. N., Neuberger, M. S., and Malim, M. H. (2003). DNA deamination mediates innate immunity to retroviral infection. Cell 113, 803–809.
Harris, R. S., and Liddament, M. T. (2004). Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 4, 868–877.
Hohjoh, H., and Singer, M. F. (1996). Cytoplasmic ribonucleoprotein complexes containing human LINE-1 protein and RNA. EMBO J. 15, 630–639.
Holmes, R. K., Koning, F. A., Bishop, K. N., and Malim, M. H. (2007). APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. J. Biol. Chem. 282, 2587–2595.
Honjo, T., Nagaoka, H., Shinkura, R., and Muramatsu, M. (2005). AID to overcome the limitations of genomic information. Nat. Immunol. 6, 655–661.
Hulme, A. E., Bogerd, H. P., Cullen, B. R., and Moran, J. V. (2007). Selective inhibition of Alu retrotransposition by APOBEC3G. Gene 390, 199–205.
Hultquist, J. F., Lengyel, J. A., Refsland, E. W., Larue, R. S., Lackey, L., Brown, W. L., and Harris, R. S. (2011). Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 85, 11220–11234.
Huthoff, H., and Towers, G. J. (2008). Restriction of retroviral replication by APOBEC3G/F and TRIM5alpha. Trends Microbiol. 16, 612–619.
Ikeda, T., Abd El Galil, K. H., Tokunaga, K., Maeda, K., Sata, T., Sakaguchi, N., Heidmann, T., and Koito, A. (2011). Intrinsic restriction activity by apolipoprotein B mRNA editing enzyme APOBEC1 against the mobility of autonomous retrotransposons. Nucleic Acids Res. 39, 5538–5554.
Ikeda, T., Ohsugi, T., Kimura, T., Matsushita, S., Maeda, Y., Harada, S., and Koito, A. (2008). The antiretroviral potency of APOBEC1 deaminase from small animal species. Nucleic Acids Res. 36, 6859–6871.
Iwabu, Y., Kinomoto, M., Tatsumi, M., Fujita, H., Shimura, M., Tanaka, Y., Ishizaka, Y., Nolan, D., Mallal, S., Sata, T., and Tokunaga, K. (2010). Differential anti-APOBEC3G activity of HIV-1 Vif proteins derived from different subtypes. J. Biol. Chem. 285, 35350–35358.
Iwatani, Y., Chan, D. S., Wang, F., Maynard, K. S., Sugiura, W., Gronenborn, A. M., Rouzina, I., Williams, M. C., Musier-Forsyth, K., and Levin, J. G. (2007). Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 35, 7096–7108.
Jarmuz, A., Chester, A., Bayliss, J., Gisbourne, J., Dunham, I., Scott, J., and Navaratnam, N. (2002). An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 79, 285–296.
Jeffs, A. R., Benjes, S. M., Smith, T. L., Sowerby, S. J., and Morris, C. M. (1998). The BCR gene recombines preferentially with Alu elements in complex BCR-ABL translocations of chronic myeloid leukaemia. Hum. Mol. Genet. 7, 767–776.
Jónsson, S. R., Larue, R. S., Stenglein, M. D., Fahrenkrug, S. C., Andrésdóttir, V., and Harris, R. S. (2007). The restriction of zoonotic PERV transmission by human APOBEC3G. PLoS ONE 2:e893. doi: 10.1371/journal.pone.0000893
Köck, J., and Blum, H. E. (2008). Hypermutation of hepatitis B virus genomes by APOBEC3G, APOBEC3C and APOBEC3H. J. Gen. Virol. 89, 1184–1191.
Kazazian, H. H., Wong, C., Youssoufian, H., Scott, A. F., Phillips, D. G., and Antonarakis, S. E. (1988). Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature 332, 164–166.
Khatua, A. K., Taylor, H. E., Hildreth, J. E. K., and Popik, W. (2010). Inhibition of LINE-1 and Alu retrotransposition by exosomes encapsidating APOBEC3G and APOBEC3F. Virology 400, 68–75.
Kinomoto, M., Kanno, T., Shimura, M., Ishizaka, Y., Kojima, A., Kurata, T., Sata, T., and Tokunaga, K. (2007). All APOBEC3 family proteins differentially inhibit LINE-1 retrotransposition. Nucleic Acids Res. 35, 2955–2964.
Kobayashi, K., Nakahori, Y., Miyake, M., Matsumura, K., Kondo-Iida, E., Nomura, Y., Segawa, M., Yoshioka, M., Saito, K., Osawa, M., Hamano, K., Sakakihara, Y., Nonaka, I., Nakagome, Y., Kanazawa, I., Nakamura, Y., Tokunaga, K., and Toda, T. (1998). An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 394, 388–392.
Kobayashi, M., Takaori-Kondo, A., Shindo, K., Abudu, A., Fukunaga, K., and Uchiyama, T. (2004). APOBEC3G targets specific virus species. J. Virol. 78, 8238–8244.
Koning, F. A., Goujon, C., Bauby, H., and Malim, M. H. (2011). Target cell-mediated editing of HIV-1 cDNA by APOBEC3 proteins in human macrophages. J. Virol. 85, 13448–13452.
Koning, F. A., Newman, E. N., Kim, E. Y., Kunstman, K. J., Wolinsky, S. M., and Malim, M. H. (2009). Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 83, 9474–9485.
Kriegs, J. O., Churakov, G., Jurka, J., Brosius, J., and Schmitz, J. (2007). Evolutionary history of 7SL RNA-derived SINEs in Supraprimates. Trends Genet. 23, 158–161.
Kubo, S., Seleme, M. D. C., Soifer, H. S., Perez, J. L. G., Moran, J. V., Kazazian, H. H., and Kasahara, N. (2006). L1 retrotransposition in nondividing and primary human somatic cells. Proc. Natl. Acad. Sci. U.S.A. 103, 8036–8041.
Lackey, L., Demorest, Z. L., Land, A. M., Hultquist, J. F., Brown, W. L., and Harris, R. S. (2012). APOBEC3B and AID have similar nuclear import mechanisms. J. Mol. Biol. 419, 301–314.
Langlois, M.-A., Beale, R. C. L., Conticello, S. G., and Neuberger, M. S. (2005). Mutational comparison of the single-domained APOBEC3C and double-domained APOBEC3F/G anti-retroviral cytidine deaminases provides insight into their DNA target site specificities. Nucleic Acids Res. 33, 1913–1923.
Lee, Y. N., and Bieniasz, P. D. (2007). Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog. 3:e10. doi: 10.1371/journal.ppat.0030010
Lee, Y. N., Malim, M. H., and Bieniasz, P. D. (2008). Hypermutation of an ancient human retrovirus by APOBEC3G. J. Virol. 82, 8762–8770.
Li, M. M. H., and Emerman, M. (2011). Polymorphism in human APOBEC3H affects a phenotype dominant for subcellular localization and antiviral activity. J. Virol. 85, 8197–8207.
Li, M. M., Wu, L. I., and Emerman, M. (2010). The range of human APOBEC3H sensitivity to lentiviral Vif proteins. J. Virol. 84, 88–95.
Li, X., Scaringe, W. A., Hill, K. A., Roberts, S., Mengos, A., Careri, D., Pinto, M. T., Kasper, C. K., and Sommer, S. S. (2001). Frequency of recent retrotransposition events in the human factor IX gene. Hum. Mutat. 17, 511–519.
Malim, M. H. (2009). APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 675–687.
Mangeat, B., Turelli, P., Liao, S., and Trono, D. (2004). A single amino acid determinant governs the species-specific sensitivity of APOBEC3G to Vif action. J. Biol. Chem. 279, 14481–14483.
Mangeat, B., Turelli, P., Caron, G., Friedli, M., Perrin, L., and Trono, D. (2003). Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424, 99–103.
Marin, M., Rose, K. M., Kozak, S. L., and Kabat, D. (2003). HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 9, 1398–1403.
Martin, S. L. (1991). Ribonucleoprotein particles with LINE-1 RNA in mouse embryonal carcinoma cells. Mol. Cell. Biol. 11, 4804–4807.
Meischl, C., Boer, M., Ahlin, A., and Roos, D. (2000). A new exon created by intronic insertion of a rearranged LINE-1 element as the cause of chronic granulomatous disease. Eur. J. Hum. Genet. 8, 697–703.
Miki, Y., Nishisho, I., Horii, A., Miyoshi, Y., Utsunomiya, J., Kinzler, K. W., Vogelstein, B., and Nakamura, Y. (1992). Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 52, 643–645.
Morse, B., Rotherg, P. G., South, V. J., Spandorfer, J. M., and Astrin, S. M. (1988). Insertional mutagenesis of the myc locus by a LINE-1 sequence in a human breast carcinoma. Nature 333, 87–90.
Muckenfuss, H., Hamdorf, M., Held, U., Perković, M., Löwer, J., Cichutek, K., Flory, E., Schumann, G. G., and Münk, C. (2006). APOBEC3 proteins inhibit human LINE-1 retrotransposition. J. Biol. Chem. 281, 22161–22172.
Mukherjee, S., Mukhopadhyay, A., Banerjee, D., Chandak, G. R., and Ray, K. (2004). Molecular pathology of haemophilia B: identification of five novel mutations including a LINE 1 insertion in Indian patients. Haemophilia 10, 259–263.
Muotri, A. R., Chu, V. T., Marchetto, M. C. N., Deng, W., Moran, J. V., and Gage, F. H. (2005). Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435, 903–910.
Muotri, A. R., Marchetto, M. C., Coufal, N. G., Oefner, R., Yeo, G., Nakashima, K., and Gage, F. H. (2010). L1 retrotransposition in neurons is modulated by MeCP2. Nature 468, 443–446.
Narita, N., Nishio, H., Kitoh, Y., Ishikawa, Y., Minami, R., Nakamura, H., and Matsuo, M. (1993). Insertion of a 5′ truncated L1 element into the 3′ end of exon 44 of the dystrophin gene resulted in skipping of the exon during splicing in a case of Duchenne muscular dystrophy. J. Clin. Invest. 91, 1862–1867.
Narvaiza, I., Linfesty, D. C., Greener, B. N., Hakata, Y., Pintel, D. J., Logue, E., Landau, N. R., and Weitzman, M. D. (2009). Deaminase-independent inhibition of parvoviruses by the APOBEC3A cytidine deaminase. PLoS Pathog. 5:e1000439. doi: 10.1371/journal.ppat.1000439
Newman, E. N. C., Holmes, R. K., Craig, H. M., Klein, K. C., Lingappa, J. R., Malim, M. H., and Sheehy, A. M. (2005). Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 15, 166–170.
Niewiadomska, A. M., Tian, C., Tan, L., Wang, T., Sarkis, P. T. N., and Yu, X.-F. (2007). Differential inhibition of long interspersed element 1 by APOBEC3 does not correlate with high-molecular-mass-complex formation or P-body association. J. Virol. 81, 9577–9583.
OhAinle, M., Kerns, J. A., Li, M. M. H., Malik, H. S., and Emerman, M. (2008). Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 4, 249–259.
OhAinle, M., Kerns, J. A., Malik, H. S., and Emerman, M. (2006). Adaptive evolution and antiviral activity of the conserved mammalian cytidine deaminase APOBEC3H. J. Virol. 80, 3853–3862.
Okeoma, C. M., Lovsin, N., Peterlin, B. M., and Ross, S. R. (2007). APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature 445, 927–930.
Okeoma, C. M., Low, A., Bailis, W., Fan, H. Y., Peterlin, B. M., and Ross, S. R. (2009). Induction of APOBEC3 in vivo causes increased restriction of retrovirus infection. J. Virol. 83, 3486–3495.
Oldridge, M., Zackai, E. H., McDonald-McGinn, D. M., Iseki, S., Morriss-Kay, G. M., Twigg, S. R., Johnson, D., Wall, S. A., Jiang, W., Theda, C., Jabs, E. W., and Wilkie, A. O. (1999). De novo alu-element insertions in FGFR2 identify a distinct pathological basis for Apert syndrome. Am. J. Hum. Genet. 64, 446–461.
Ooms, M., Krikoni, A., Kress, A. K., Simon, V., and Münk, C. (2012). APOBEC3A, APOBEC3B and APOBEC3H haplotype 2 restrict human T-lymphotropic virus type I (HTLV-1). J. Virol. 86, 6097–6108.
Ostertag, E. M., Goodier, J. L., Zhang, Y., and Kazazian, H. H. Jr. (2003). SVA elements are nonautonomous retrotransposons that cause disease in humans. Am. J. Hum. Genet. 73, 1444–1451.
Ostertag, E. M., and Kazazian, H. H. Jr. (2001). Biology of mammalian L1 retrotransposons. Annu. Rev. Genet. 35, 501–538.
Pak, V., Heidecker, G., Pathak, V. K., and Derse, D. (2011). The role of amino-terminal sequences in cellular localization and antiviral activity of APOBEC3B. J. Virol. 85, 8538–8547.
Paprotka, T., Venkatachari, N. J., Chaipan, C., Burdick, R., Delviks-Frankenberry, K. A., Hu, W.-S., and Pathak, V. K. (2010). Inhibition of xenotropic murine leukemia virus-related virus by APOBEC3 proteins and antiviral drugs. J. Virol. 84, 5719–5729.
Peng, G., Greenwell-Wild, T., Nares, S., Jin, W., Lei, K. J., Rangel, Z. G., Munson, P. J., and Wahl, S. M. (2007). Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood 110, 393–400.
Perepelitsa-Belancio, V., and Deininger, P. (2003). RNA truncation by premature polyadenylation attenuates human mobile element activity. Nat. Genet. 35, 363–366.
Perković, M., Schmidt, S., Marino, D., Russell, R. A., Stauch, B., Hofmann, H., Kopietz, F., Kloke, B.-P., Zielonka, J., Ströver, H., Hermle, J., Lindemann, D., Pathak, V. K., Schneider, G., Löchelt, M., Cichutek, K., and Münk, C. (2009). Species-specific inhibition of APOBEC3C by the prototype foamy virus protein bet. J. Biol. Chem. 284, 5819–5826.
Rangwala, S., Zhang, L., and Kazazian, H. (2009). Many LINE1 elements contribute to the transcriptome of human somatic cells. Genome Biol. 10, R100.
Refsland, E. W., Stenglein, M. D., Shindo, K., Albin, J. S., Brown, W. L., and Harris, R. S. (2010). Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 38, 4274–4284.
Ribet, D., Dewannieux, M., and Heidmann, T. (2004). An active murine transposon family pair: retrotransposition of “master” MusD copies and ETn trans-mobilization. Genome Res. 14, 2261–2267.
Rohrer, J., Minegishi, Y., Richter, D., Eguiguren, J., and Conley, M. E. (1999). Unusual mutations in Btk: an insertion, a duplication, an inversion, and four large deletions. Clin. Immunol. 90, 28–37.
Russell, R. A., Wiegand, H. L., Moore, M. D., Schafer, A., McClure, M. O., and Cullen, B. R. (2005). Foamy virus Bet proteins function as novel inhibitors of the APOBEC3 family of innate antiretroviral defense factors. J. Virol. 79, 8724–8731.
Sasada, A., Takaori-Kondo, A., Shirakawa, K., Kobayashi, M., Abudu, A., Hishizawa, M., Imada, K., Tanaka, Y., and Uchiyama, T. (2005). APOBEC3G targets human T-cell leukemia virus type 1. Retrovirology 2, 32.
Sato, Y., Probst, H. C., Tatsumi, R., Ikeuchi, Y., Neuberger, M. S., and Rada, C. (2010). Deficiency in APOBEC2 leads to a shift in muscle fiber type, diminished body mass, and myopathy. J. Biol. Chem. 285, 7111–7118.
Sawyer, S. L., Emerman, M., and Malik, H. S. (2004). Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2:e275. doi: 10.1371/journal.pbio.0020275
Schichman, S. A., Caligiuri, M. A., Strout, M. P., Carter, S. L., Gu, Y., Canaani, E., Bloomfield, C. D., and Croce, C. M. (1994). ALL-1 tandem duplication in acute myeloid leukemia with a normal karyotype involves homologous recombination between Alu elements. Cancer Res. 54, 4277–4280.
Schmitt, K., Guo, K., Algaier, M., Ruiz, A., Cheng, F., Qiu, J., Wissing, S., Santiago, M. L., and Stephens, E. B. (2011). Differential virus restriction patterns of rhesus macaque and human APOBEC3A: implications for lentivirus evolution. Virology 419, 24–42.
Schröfelbauer, B., Chen, D., and Landau, N. R. (2004). A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif). Proc. Natl. Acad. Sci. U.S.A. 101, 3927–3932.
Schumacher, A. J., Haché, G., Macduff, D. A., Brown, W. L., and Harris, R. S. (2008). The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. J. Virol. 82, 2652–2660.
Schumacher, A. J., Nissley, D. V., and Harris, R. S. (2005). APOBEC3G hypermutates genomic DNA and inhibits Ty1 retrotransposition in yeast. Proc. Natl. Acad. Sci. U.S.A. 102, 9854–9859.
Schumann, G. G. (2007). APOBEC3 proteins: major players in intracellular defence against LINE-1-mediated retrotransposition. Biochem. Soc. Trans. 35, 637–642.
Sheehy, A. M., Gaddis, N. C., Choi, J. D., and Malim, M. H. (2002). Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418, 646–650.
Sheehy, A. M., Gaddis, N. C., and Malim, M. H. (2003). The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9, 1404–1407.
Smith, H. C., Bennett, R. P., Kizilyer, A., McDougall, W. M., and Prohaska, K. M. (2012). Functions and regulation of the APOBEC family of proteins. Semin. Cell Dev. Biol. 23, 258–268.
Soifer, H. S., Zaragoza, A., Peyvan, M., Behlke, M. A., and Rossi, J. J. (2005). A potential role for RNA interference in controlling the activity of the human LINE-1 retrotransposon. Nucleic Acids Res. 33, 846–856.
Stenglein, M. D., and Harris, R. S. (2006). APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J. Biol. Chem. 281, 16837–16841.
Stetson, D. B., Ko, J. S., Heidmann, T., and Medzhitov, R. (2008). Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598.
Stopak, K., De Noronha, C., Yonemoto, W., and Greene, W. C. (2003). HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 12, 591–601.
Suspène, R., Aynaud, M.-M., Guétard, D., Henry, M., Eckhoff, G., Marchio, A., Pineau, P., Dejean, A., Vartanian, J.-P., and Wain-Hobson, S. (2011a). Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proc. Natl. Acad. Sci. U.S.A. 108, 4858–4863.
Suspène, R., Aynaud, M.-M., Koch, S., Pasdeloup, D., Labetoulle, M., Gaertner, B., Vartanian, J.-P., Meyerhans, A., and Wain-Hobson, S. (2011b). Genetic editing of herpes simplex virus 1 and Epstein-Barr herpesvirus genomes by human APOBEC3 cytidine deaminases in culture and in vivo. J. Virol. 85, 7594–7602.
Takeda, E., Tsuji-Kawahara, S., Sakamoto, M., Langlois, M. A., Neuberger, M. S., Rada, C., and Miyazawa, M. (2008). Mouse APOBEC3 restricts friend leukemia virus infection and pathogenesis in vivo. J. Virol. 82, 10998–11008.
Tan, L., Sarkis, P. T., Wang, T., Tian, C., and Yu, X. F. (2009). Sole copy of Z2-type human cytidine deaminase APOBEC3H has inhibitory activity against retrotransposons and HIV-1. FASEB J. 23, 279–287.
Tchenio, T., Casella, J. F., and Heidmann, T. (2000). Members of the SRY family regulate the human LINE retrotransposons. Nucleic Acids Res. 28, 411–415.
Teng, B., Burant, C. F., and Davidson, N. O. (1993). Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science 260, 1816–1819.
Turelli, P., Mangeat, B., Jost, S., Vianin, S., and Trono, D. (2004a). Inhibition of hepatitis B virus replication by APOBEC3G. Science 303, 1829.
Turelli, P., Vianin, S., and Trono, D. (2004b). The innate antiretroviral factor APOBEC3G does not affect human LINE-1 retrotransposition in a cell culture assay. J. Biol. Chem. 279, 43371–43373.
van Den Hurk, J. a. J. M., van de Pol, D. J. R., Wissinger, B., van Driel, M. A., Hoefsloot, L. H., De Wijs, I. J., van den Born, L. I., Heckenlively, J. R., Brunner, H. G., Zrenner, E., Ropers, H.-H., and Cremers, F. P. M. (2003). Novel types of mutation in the choroideremia (CHM) gene: a full-length L1 insertion and an intronic mutation activating a cryptic exon. Hum. Genet. 113, 268–275.
Vartanian, J.-P., Guétard, D., Henry, M., and Wain-Hobson, S. (2008). Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 320, 230–233.
Vonica, A., Rosa, A., Arduini, B. L., and Brivanlou, A. H. (2011). APOBEC2, a selective inhibitor of TGFbeta signaling, regulates left-right axis specification during early embryogenesis. Dev. Biol. 350, 13–23.
Wallace, M. R., Andersen, L. B., Saulino, A. M., Gregory, P. E., Glover, T. W., and Collins, F. S. (1991). A de novo Alu insertion results in neurofibromatosis type 1. Nature 353, 864–866.
Walsh, C. P., Chaillet, J. R., and Bestor, T. H. (1998). Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 20, 116–117.
Wang, X., Abudu, A., Son, S., Dang, Y., Venta, P. J., and Zheng, Y.-H. (2011). Analysis of human APOBEC3H haplotypes and anti-human immunodeficiency virus type 1 activity. J. Virol. 85, 3142–3152.
Wiegand, H. L., Doehle, B. P., Bogerd, H. P., and Cullen, B. R. (2004). A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 23, 2451–2458.
Wiegand, H. L., and Cullen, B. R. (2007). Inhibition of alpharetrovirus replication by a range of human APOBEC3 proteins. J. Virol. 81, 13694–13699.
Wilund, K. R., Yi, M., Campagna, F., Arca, M., Zuliani, G., Fellin, R., Ho, Y.-K., Garcia, J. V., Hobbs, H. H., and Cohen, J. C. (2002). Molecular mechanisms of autosomal recessive hypercholesterolemia. Hum. Mol. Genet. 11, 3019–3030.
Wimmer, K., Callens, T., Wernstedt, A., and Messiaen, L. (2011). The NF1 gene contains hotspots for L1 endonuclease-dependent de novo insertion. PLoS Genet. 7:e1002371. doi: 10.1371/journal.pgen.1002371
Wissing, S., Montano, M., Garcia-Perez, J. L., Moran, J. V., and Greene, W. C. (2011). Endogenous APOBEC3B restricts LINE-1 retrotransposition in transformed cells and human embryonic stem cells. J. Biol. Chem. 286, 36427–36437.
Yang, N., and Kazazian, H. H. Jr. (2006). L1 retrotransposition is suppressed by endogenously encoded small interfering RNAs in human cultured cells. Nat. Struct. Mol. Biol. 13, 763–771.
Yang, N., Zhang, L., Zhang, Y., and Kazazian, H. H. Jr. (2003). An important role for RUNX3 in human L1 transcription and retrotransposition. Nucleic Acids Res. 31, 4929–4940.
Yang, Y., Guo, F., Cen, S., and Kleiman, L. (2007). Inhibition of initiation of reverse transcription in HIV-1 by human APOBEC3F. Virology 365, 92–100.
Yoshida, K., Nakamura, A., Yazaki, M., Ikeda, S., and Takeda, S. (1998). Insertional mutation by transposable element, L1, in the DMD gene results in X-linked dilated cardiomyopathy. Hum. Mol. Genet. 7, 1129–1132.
Yu, Q., Chen, D., Konig, R., Mariani, R., Unutmaz, D., and Landau, N. R. (2004). APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J. Biol. Chem. 279, 53379–53386.
Zhang, H., Yang, B., Pomerantz, R. J., Zhang, C., Arunachalam, S. C., and Gao, L. (2003). The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424, 94–98.
Zhen, A., Wang, T., Zhao, K., Xiong, Y., and Yu, X. F. (2010). A single amino acid difference in human APOBEC3H variants determines HIV-1 Vif sensitivity. J. Virol. 84, 1902–1911.
Keywords: retroelements, retrotransposition, LINE-1, Alu, APOBEC3, HIV-1, Vif, restriction factors
Citation: Arias JF, Koyama T, Kinomoto M and Tokunaga K (2012) Retroelements versus APOBEC3 family members: No great escape from the magnificent seven. Front. Microbio. 3:275. doi: 10.3389/fmicb.2012.00275
Received: 26 June 2012; Paper pending published: 11 July 2012;
Accepted: 13 July 2012; Published online: 14 August 2012.
Edited by:
Yukihito Ishizaka, National Center for Global Health and Medicine, JapanReviewed by:
Yasumasa Iwatani, National Hospital Organization Nagoya Medical Center, JapanYoshitaka Sato, Kobe University School of Medicine, Japan
Copyright © 2012 Arias, Koyama, Kinomoto and Tokunaga. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Kenzo Tokunaga, Department of Pathology, National Institute of Infectious Diseases, Shinjuku-ku, Tokyo 162-8640, Japan. e-mail:dG9rdW5hZ2FAbmloLmdvLmpw