 Matthew D. Woolard1*
Matthew D. Woolard1* Lydia M. Barrigan2,3
Lydia M. Barrigan2,3 James R. Fuller4
James R. Fuller4 Adam S. Buntzman2
Adam S. Buntzman2 Joshua Bryan1
Joshua Bryan1 Colin Manoil5
Colin Manoil5 Thomas H. Kawula3
Thomas H. Kawula3 Jeffrey A. Frelinger2
Jeffrey A. Frelinger2- 1Department of Microbiology and Immunology, Louisiana State University Health Sciences Center at Shreveport, Shreveport, LA, USA
- 2Department of Immunobiology, University of Arizona, Tucson, AZ, USA
- 3Department of Microbiology and Immunology, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
- 4DNA Identification Testing Division, Laboratory Corporation of America, Burlington, NC, USA
- 5Department of Genome Sciences, University of Washington, Seattle, WA, USA
Francisella tularensis is the causative agent of tularemia. We have previously shown that infection with F. tularensis Live Vaccine Strain (LVS) induces macrophages to synthesize prostaglandin E2 (PGE2). Synthesis of PGE2 by F. tularensis infected macrophages results in decreased T cell proliferation in vitro and increased bacterial survival in vivo. Although we understand some of the biological consequences of F. tularensis induced PGE2 synthesis by macrophages, we do not understand the cellular pathways (neither host nor bacterial) that result in up-regulation of the PGE2 biosynthetic pathway in F. tularensis infected macrophages. We took a genetic approach to begin to understand the molecular mechanisms of bacterial induction of PGE2 synthesis from infected macrophages. To identify F. tularensis genes necessary for the induction of PGE2 in primary macrophages, we infected cells with individual mutants from the closely related strain F. tularensis subspecies novicida U112 (U112) two allele mutant library. Twenty genes were identified that when disrupted resulted in U112 mutant strains unable to induce the synthesis of PGE2 by infected macrophages. Fourteen of the genes identified are located within the Francisella pathogenicity island (FPI). Genes in the FPI are required for F. tularensis to escape from the phagosome and replicate in the cytosol, which might account for the failure of U112 with transposon insertions within the FPI to induce PGE2. This implies that U112 mutant strains that do not grow intracellularly would also not induce PGE2. We found that U112 clpB::Tn grows within macrophages yet fails to induce PGE2, while U112 pdpA::Tn does not grow yet does induce PGE2. We also found that U112 iglC::Tn neither grows nor induces PGE2. These findings indicate that there is dissociation between intracellular growth and the ability of F. tularensis to induce PGE2 synthesis. These mutants provide a critical entrée into the pathways used in the host for PGE2 induction.
Introduction
Francisella tularensis is a facultative intracellular bacterium and the causative agent of tularemia. F. tularensis has a low infective dose, high morbidity, and can persist in the environment (Ellis et al., 2002). F. tularensis has also been produced as a bioweapon (Dennis et al., 2001), and is classified as a Category A Select Agent. There are four major subspecies of F. tularensis: F. tularensis subspecies tularensis, F. tularensis subspecies holarctica, F. tularensis subspecies mediasiatica, and F. tularensis subspecies novicida. F. tularensis, F. holarctica (including the live vaccine strain, LVS), and F. novicida all cause a fulminate disease in mice that is similar to tularemia in humans (Rick Lyons and Wu, 2007). There are clear differences in virulence between strains in mice. F. novicida, F. holarctica, and F. tularensis can have an LD50 of less than 10 organisms in intranasally inoculated mice, while F. holarctica LVS LD50 in mice is much higher (Pechous et al., 2009). Each strain varies in its capacity to cause disease in humans. F. novicida is highly attenuated in humans, only causing disease in immuno-compromised individuals (Hollis et al., 1989; Hand et al., 2012). F. holarctica is highly infectious in humans, but causes a milder form of tularemia compared to F. tularensis. F. holarctica LVS is highly attenuated for disease in humans but can cause disease in immunocompetent individuals (Tigertt, 1962; Hornick and Eigelsbach, 1966; Ellis et al., 2002). Though each strain has a different level of virulence in humans, they share high nucleotide sequence identity. F. novicida shares 95% nucleotide sequence identity with F. tularensis and F. holarctica (Rohmer et al., 2007), suggesting that homologous proteins function via similar mechanisms.
Key to F. tularensis’ virulence is its ability to escape the phagosome and replicate within the cytosol of host cells. Previous studies have identified over 200 genes that are necessary for intracellular growth of F. tularensis (Qin and Mann, 2006; Weiss et al., 2007; Kraemer et al., 2009; Asare and Abu Kwaik, 2010; Asare et al., 2010). Some of the genes required for escape from the phagosome and intracellular growth reside within the Francisella pathogenicity island (FPI; Barker et al., 2009). The FPI is a set of 16 genes that are highly conserved among all subspecies of F. tularensis (Barker et al., 2009). The FPI likely encodes a secretion system that is related to the recently discovered type VI secretion systems (T6SS; Nano and Schmerk, 2007; Ludu et al., 2008). The T6SS is involved in the virulence of several bacterial pathogens (Mougous et al., 2006; Pukatzki et al., 2006; Shalom et al., 2007; Ma and Mekalanos, 2010). Several regulators of FPI expression have been described. Two of the best studied are MglA and SspA, which positively regulate the transcription of FPI genes (Baron and Nano, 1998; Lauriano et al., 2004; Charity et al., 2007). The mechanisms by which FPI proteins promote F. tularensis escape and intra-macrophage growth are unknown. There is evidence that translocated products of T6SS in other bacteria are capable of modulating host immune responses (Pukatzki et al., 2007; Ma and Mekalanos, 2010; Suarez et al., 2010a,b). Though FPI gene products are clearly involved in phagosome escape and intracellular growth, the ability of these gene products to induce immunomodulatory responses has not been demonstrated to date.
Prostaglandin E2 (PGE2) synthesis induced by LVS from host cells alters both innate and adaptive immune responses. We demonstrated that F. tularensis LVS was capable of inducing macrophages to synthesize PGE2 and that this was independent of intracellular growth of F. tularensis (Woolard et al., 2007). In vitro, LVS-induced PGE2 synthesis inhibits T cell proliferation and skews their phenotypic development from IFN-γ+ T cells to IL-4+ T cells (Woolard et al., 2007). Through an indirect mechanism, PGE2 induces ubiquitin-mediated degradation of MHC II which results in decreased MHC II protein levels on the surface of macrophages (Wilson et al., 2009). Decreased MHC II surface expression would decrease the antigenic stimulatory capacity of these macrophages, likely making them less capable of activating F. tularensis-specific T cells. T cells are required for both clearance of F. tularensis and generation of long-term immune protection (Yee et al., 1996), thus the biological activity of PGE2 would be beneficial to F. tularensis survival in vivo. LVS-induced PGE2 synthesis during respiratory tularemia inhibits the generation of beneficial T cell response. The inhibition of PGE2 synthesis in vivo by indomethacin leads to increased number of IFN-γ+ T cells and decreased bacterial burden (Woolard et al., 2008). It is clear that induction of PGE2 synthesis is an important immune modulation mechanism utilized by F. holarctica to persist in the host.
Presently, none of the F. tularensis product(s) responsible for the induction of PGE2 synthesis in eukaryotic cells are known. Several bacterial products have been identified that are capable of inducing PGE2 synthesis. Bacterial peptidoglycan, LPS, and CpG DNA can up-regulate prostaglandin synthesis through interactions with TLR2, TLR4, and TLR9, respectively (Chen et al., 2001, 2004; Smith et al., 2002; Uematsu et al., 2002; Treffkorn et al., 2004). It is not known if F. tularensis is capable of inducing PGE2 through a similar mechanism. To date, few F. tularensis TLR ligands have been identified. F. tularensis LpnA and FTT1103 have been reported to be TLR2 ligands and DnaK a TLR4 ligand (Ashtekar et al., 2008; Forestal et al., 2008; Thakran et al., 2008). F. tularensis LPS fails to or only weakly stimulates a cytokine response by host cells (Kieffer et al., 2003; Hajjar et al., 2006). If F. tularensis LPS does stimulate host cells, it is likely in a TLR4 independent manner. Both TLR2 and TLR4 deficient macrophages produce PGE2 after infection (Woolard et al., unpublished data).
In this study we demonstrate that along with LVS, F. novicida U112 (U112), and F. tularensis subspecies tularensis Schu S4 (Schu S4) induce PGE2 synthesis by macrophages. We tested a F. novicida (U112) comprehensive transposon mutant library to identify genes necessary for induction of PGE2 synthesis by infected bone marrow-derived macrophages (BMDMs). This library allowed us to identify 20 genes that when disrupted result in U112 strains that are unable to induce the synthesis of PGE2 by infected macrophages. Identified genes included genes of the FPI and regulators of the FPI. All genes identified are highly conserved among all sequenced strains of F. tularensis (Charity et al., 2007, 2009; Nano and Schmerk, 2007; Meibom et al., 2008). We also demonstrate that the ability of F. novicida to induce PGE2 synthesis is likely not dependent on phagosome escape nor intracellular growth. This work likely suggests that the FPI is involved in immune modulation along with previously established mechanisms of phagosomal escape and intracellular growth.
Materials and Methods
Bacteria and Mouse Strains
The F. tularensis subspecies holarctica LVS was obtained from ATCC (29684; American Type Culture Collection, Manassas, VA, USA; Cowley and Elkins, 2003), the F. tularensis subspecies novicida U112 strain was previously published (Larson et al., 1955), and the F. tularensis subspecies tularensis Schu S4 strain (catalog no. NR-643) was obtained from the Biodefense Emerging Infections Research Resources Repository (Manassas, VA, USA). The two allele transposon library was previously described (Gallagher et al., 2007). For all studies, except for the original screen, F. novicida was propagated on tryptic soy agar supplemented with 0.1% cysteine while the F. novicida transposon mutants were propagated on the same agar with the addition of 20 μg/ml of kanamycin. F. holarctica LVS and F. tularensis Schu S4 were propagated on chocolate agar. Inocula were generated by collecting plate grown bacteria and diluting them in PBS to reach an OD600 of 1.00. Inocula were then diluted into appropriate cell culture medium for inoculation.
The F. novicida two allele transposon library was previously described (Gallagher et al., 2007). The LVS ∆mglA, LVS ∆sspA, LVS ∆mglA pmglA, and LVS ∆sspA psspA were previously published (Fuller et al., 2009). The dotU deletion construct was made by splice overlap extension PCR retaining the start and stop codons of dotU and fusing the first four and last two codons in frame and retaining 0.8 kb of flanking sequence. The constructs were cloned into the suicide vector pMP590 and sequenced to confirm the integrity of the DNA sequence. The LVS dotU mutant was generated by allelic exchange, selection for plasmid co-integrates, and counter selection on sucrose containing media to identify plasmid and dotU allele resolution as described (Fuller et al., 2008). The following primers were utilized to generated the SOE fragment; FTL0119 5′ ext 5′-GAGTTTTTTCCACCTCTGAGGATGTTTC, FTL0119 5′ int 5′-GAAAGACTTTAAAGAGATAGAATAATAAGGGTAAGAGGAGATTTATATGAGTCAGATAATATC, FTL0119 3′ int 5′-CTCCTCTTACCCTTATTATTCTATCTCTTTAAAGTCTTTCATTTATAATATCCTTTATATAGAG, FTL0119 3′ ext 5′-CATACATATTTAACCAAGTATTAGAAGATAATGGCTCAG. Loss of the wild-type and retention of the deletion dotU alleles were confirmed by PCR. Since dotU is duplicated in the LVS genome, a second round of mutagenesis was performed on the single dotU mutant strain to create an LVS dotU double-deletion strain. Plasmids for complementation were created by ligating cloned region of dotU into the PKK MCS plasmid. dotU expression from the PKK MCS plasmid was regulated by the putative PI promoter. The following primers were used; FTL0119 forward 5′-CTTAATTAAATGAAAGACTTTAAAGAGATAGAAATTATTCTAGATATTATAAAAAC, FTL0119 reverse 5′-TGTCGACCCAGCTTAATAAAATTAGTAAGCTTAAAAGAAACAGTC.
C57Bl/6J (B6) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). All animals used in this study were maintained under specific pathogen-free conditions in the American Association of Laboratory Animal Care-accredited University of North Carolina Department of Laboratory Animal Medicine Facilities or American Association of Laboratory Animal Care-accredited Louisiana State University Health Science Center at Shreveport Animal Medicine Facilities. All work was approved by each facility’s Animal Care and Use Committee (UNC #04-200, LSUHSC P10-010).
Generation of Bone Marrow-Derived Macrophages
Bone marrow cells from B6 mouse femurs were cultured in 30% L cell-conditioned medium as previously described (Woolard et al., 2007). Briefly bone marrow cells were flushed from B6 mouse femurs and incubated for 7 days on non-tissue culture-treated 15-cm2 dishes with L cell-conditioned medium as a source of GM-CSF. Following differentiation, non-adherent cells were removed by multiple washes with PBS and BMDMs were removed from plates by incubation with 10 mM EDTA in PBS. Since L cell-condition media and FBS batches can affect the amount of PGE2 induction by infected macrophages we utilized the same L cell-conditioned media and FBS batches for each series of experiments to minimize variability in PGE2 synthesis between experiments.
Bone Marrow-Derived Macrophage Infections
Bone marrow-derived macrophages were plated in 96-well flat bottom plates (105/well). Macrophages were allowed to adhere for 2 h. Macrophages were mock infected or infected with LVS, Schu S4, U112, or U112 transposon insertion strains at different multiplicity of infections (MOIs) as indicated. Bacteria were centrifuged onto the macrophage monolayer at 300 g for 5 min to allow closer contact and more efficient infection. Two hours after inoculation, extracellular bacteria were killed by the addition of 50 μg/ml of gentamicin for 45 min. Supernatants were removed and cells were washed with antibiotic-free complete medium. Fresh antibiotic-free complete medium was added and cells were incubated for 24 h. Supernatants were then collected and spun at 300 g for 10 min to remove eukaryotic cells. Supernatants were sterilized by UV. Representative supernatants were plated onto chocolate agar after UV treatment to ensure complete killing of F. tularensis. Supernatant was then stored at -80°C until needed.
Identification of Transposon Insertion Strains
The transposon library has previously been described (Gallagher et al., 2007). In brief, the 3,050-member library includes two insertion alleles in 1488 genes (the majority of total Francisella ORFs). The alleles chosen were primarily insertions positioned between 5 and 70% within the ORF and are thus likely to represent null mutations. After single-colony purification, the mutants were arrayed in 96-well format and sequence-mapped to confirm their identities (see Table 2 of Gallagher et al., 2007 for the summary of this information).
The two allele mutant library was screened in a 96-well format. Transposon insertion strains were grown up in 96-well deep well plates containing 1 ml of Tryptic Soy broth containing 15 μg/ml carbenicillin, 20 μg/ml kanamycin, and supplemented with 0.1% l-cysteine-HCl. After over-night growth an aliquot of supernatants from each transposon insertion strain was taken and OD600 was determined. MOI were normalized by average plate OD600. B6 BMDM was inoculated at an MOI of 500:1 to guarantee sufficient inocula in each well to induce PGE2 synthesis. In our experience increasing MOI inocula increases the number of macrophages infected. Twenty-four hours after inoculation supernatants were collected and then stored at -80°C until needed.
PGE2 Assay
Prostaglandin E2 in cell culture supernatants was measured using a commercial PGE2 enzyme immunoassay kit (Assay Design, Ann Arbor, MI, USA) as per manufacturer’s instructions. Transposon insertion strains were deemed defective in the ability to induce PGE2 when the levels of PGE2 by any transposon insertion strain were three standard deviations (SD) below the mean of the entire plate.
Bacterial Growth Assay
Macrophages were mock infected or infected with U112, U112 clpB::Tn, U112 pdpA::Tn, or U112 iglC::Tn strains at an MOI of 100:1. At 4 and 24 h post-inoculation, supernatants were removed. 100 μl of 0.05% sodium dodecyl sulfate in PBS was used to lyse the BMDM. Samples were transferred to tubes containing 900 μl PBS and vortexed on high setting for 1 min. Samples were serially diluted and plated on chocolate agar to determine bacterial numbers.
Confocal and Transmission Electron Microscopy
J774.1 macrophages (from ATCC #TIB-67) were seeded on coverslips at a density of 6 × 105 cells/well. Prior to infection, bacteria were carboxyfluorescein succinimidyl ester (CFSE) labeled as previously described (Bosio and Dow, 2005) with the following modifications: CFSE was added to bacteria at a final concentration of 5 μM and incubated for 20 min at 37°C. Macrophages were inoculated with CFSE-labeled U112, U112 clpB::Tn, U112 pdpA::Tn, or U112 iglC::Tn mutants at an MOI of 200:1. Bacteria were centrifuged onto the macrophages at 300 g for 5 min. Two hours after inoculation, extracellular bacteria were killed by the addition of 25 μg/ml of gentamicin for 45 min and then media was replaced with antibiotic-free media. At 4 h post-inoculation, LAMP-1 association with bacteria was determined as previously described (Schmerk et al., 2009a). Briefly, wells were washed with PBS and fixed for 20 min at room temperature with 2% (w/v) formaldehyde and 1% (w/v) sucrose in PBS. Cells were permeabilized using methanol. Coverslips were blocked with 5% bovine serum albumin, incubated overnight at 4°C with anti-mouse LAMP-1 (1D4B eBioscience), washed three times with PBS, and stained with donkey anti-rat IgG Alexafluor594 secondary antibody (Invitrogen) for 2 h at room temperature. After three PBS washes, the coverslips were mounted in DAPI-containing mounting media (Vector Laboratories, Inc.) to label the DNA. Cells were imaged using a Leica SP2 Laser Scanning Confocal Microscope using a 63× oil immersion lens. A minimum of 20 cells per strain were captured. To remove subjectivity in determining co-localization of bacteria with LAMP-1 images were analyzed using Volocity software (Improvision/Perkin Elmer) to determine bacterial association with LAMP-1. Co-localization was determined by the shared of red and green pixels at the same location. To determine whether a bacterium resided in a LAMP-1 positive vesicle, the voxel spy tool was used to closely examine whether the LAMP-1 red pixels surrounded the CFSE green pixels that labeled the bacterium. If the red pixels surrounded >50% of the green pixels, the bacterium was categorized as residing within a LAMP-1+ vesicle.
B6 BMDMs were inoculated with U112, U112 clpB::Tn, U112 iglC::Tn, or U112 pdpA::Tn at an MOI of 500:1 to maximize the number of infected BMDMs. 2 h after inoculation, the media was removed and replaced with media containing 50 μg/ml gentamicin (Sigma-Aldrich, St. Louis, MO, USA). Gentamicin-containing media was removed 1 h after treatment and replaced with antibiotic-free media. Four hours post-inoculation, the BMDM monolayer was fixed using gluteraldehyde and post-fixed with osmium tetroxide. Images were obtained using a Phillips CM-12 transmission electron microscope using 25,000× magnification.
Statistical Analysis
Student’s t-tests were used for statistical analysis between two group experiments. Multi group comparisons were done by ANOVA followed by Dunnett’s Multiple Comparison Test. When appropriate, data were logarithmically transformed before statistical analysis and confirmed by a demonstrated increase in power of the test after transformation of the data. Data analysis on the rescreen (Figure 2) was accomplished by one-way ANOVA analysis followed by Student’s t-test. A p-value ≤ 0.05 was considered statistically significant.
Results
F. tularensis Subspecies novicida and tularensis Induced the Synthesis of PGE2 by Infected Macrophages
We have previously demonstrated that F. tularensis subspecies holarctica LVS induces PGE2 synthesis in infected macrophages. To enable the use of the two allele transposon mutant library we needed to determine if the ability to induce PGE2 synthesis by infected macrophages is shared among Francisella subspecies. We tested both F. novicida U112 and F. tularensis Schu S4 for their ability to induce B6 BMDMs to synthesize PGE2 upon infection. We inoculated BMDM with LVS, U112, or Schu S4 at an MOI of 200:1. All strains tested were capable of inducing synthesis of PGE2 by infected macrophages (Figure 1). This demonstrates that the ability to induce PGE2 synthesis is conserved among F. tularensis strains.
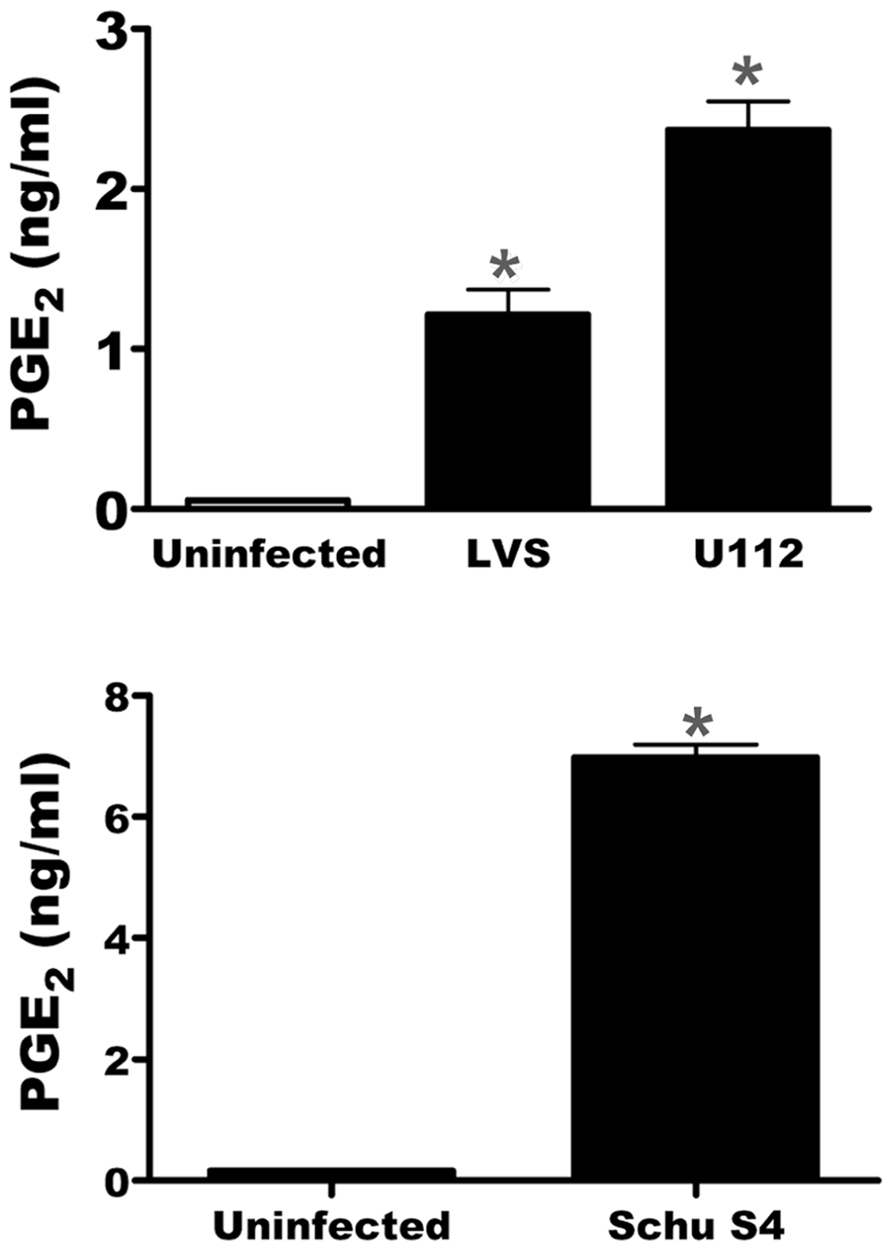
FIGURE 1. U112 and Schu S4 induces the synthesis of PGE2 from bone marrow-derived macrophages (BMDMs). BMDMs were either mock inoculated or inoculated with LVS, U112, or Schu S4 at an MOI of 200:1. Twenty-four hours after inoculation supernatants were collected and PGE2 concentration was determined. Data represents three independent experiments and expressed as the mean ± SEM. Asterisk “*”denotes statistical difference (p ≤ 0.05) from uninfected BMDM (n = 3).
Screening the Two Allele Mutant Library Identifies Several Genes Necessary for the Francisella Induction of PGE2 by Infected Macrophages
Since we demonstrated that U112 induced macrophage synthesis of PGE2, we used the F. novicida two allele transposon mutant library (Gallagher et al., 2007) to identify mutants that were unable to induce PGE2 synthesis. During the initial testing of the 3050 F. novicida U112 transposon mutants, we defined a F. novicida U112 transposon mutant as defective in induction of PGE2 synthesis by infected BMDM when BMDM produced relative PGE2 amounts that were three SD lower than the plate average amount of PGE2. The use of the three SD rule allowed us to minimize the likelihood (0.3%) of identifying false positives. The initial screen identified 33 genes that when disrupted made F. novicida unable to induce PGE2 synthesis by infected macrophages (Table S1 in Supplementary Material). This included 10 genes located in the FPI. We retested all U112 transposon insertion mutants with transposon insertions in the identified 33 genes. Furthermore, since the initial screen identified 10 genes of the FPI, we included all FPI transposon mutants within the two allele transposon mutant library in this rescreen to ensure these genes important in pathogenesis were carefully evaluated. BMDMs were inoculated with individual transposon insertion strains (89 mutants representing the original 33 genes identified and 10 genes from the FPI not originally identified) at an MOI of 200:1 and PGE2 levels were measured 24 h post-inoculation (Figure 2). We utilized an MOI of 200:1 since we have previously demonstrated this MOI results in a reproducible significant increase in detectable PGE2 from infected macrophages (Woolard et al., 2007). Each U112 transposon mutant was tested a minimum of four times. No difference was noted between strains with insertions in the same gene; as such the values were combined for representation in Figure 2. We were able to confirm 20 genes that when disrupted resulted in F. novicida strains that did not induce the synthesis of PGE2 by infected BMDM (Figure 2). With the exception of mglA and rpoB, which were only represented once, each gene identified encodes a product involved in the induction of PGE2 that was represented at least twice in the U112 two allele transposon mutant library. The genes identified in the screen of the two allele mutant library are summarized in Table 1. The identified genes were located in the FPI or were genes that encode some of the previously identified regulators of the FPI (sspA, mglA, mglB, and trmE) with the exception of rpoB and clpB (Baron and Nano, 1998; Charity et al., 2007, 2009; Guina et al., 2007; Nano and Schmerk, 2007). These genes are highly conserved in all F. tularensis subspecies sequenced to date (Charity et al., 2007, 2009; Nano and Schmerk, 2007; Meibom et al., 2008). Of note, not all genes encoded within the FPI are necessary for U112-induced PGE2 synthesis as pdpA::Tn, pdpD::Tn, and pdpE::Tn were able to induce PGE2 synthesis similarly to wild-type U112. Thus, the screen identified 20 F. novicida genes that are necessary for the induction of PGE2.
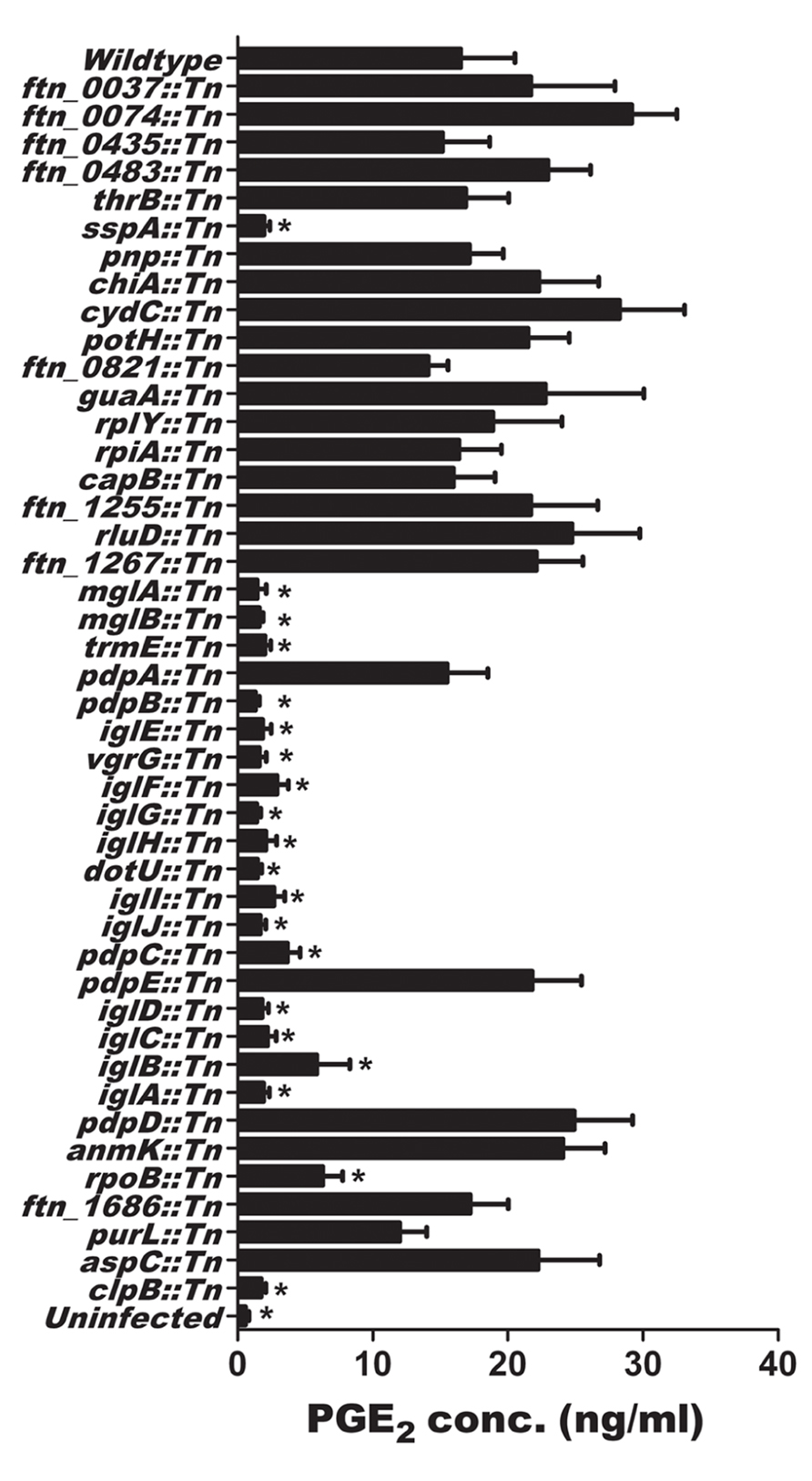
FIGURE 2. Identification of U112 genes necessary for the induction of PGE02 from bone marrow-derived macrophages (BMDMs). BMDMs were inoculated with F. novicida U112 or individual transposon insertion strains at an MOI of 200:1. Twenty-four hours after inoculation, supernatants were collected and PGE2 concentration was determined. Each transposon insertion mutant strain was tested four times. Bars represents the mean of all independent transposon insertions mutants within the same gene ± SEM, Asterisk “*” denotes statistical difference (p ≤ 0.05) from U112 inoculated BMDM (n ≥ 4).
TABLE 1. Genes required for Francisella induction of PGE2 synthesis in Francisella-infected macrophages.
F. tularensis LVS Mutant Strains with Deletions of mglA, sspA, or dotU do not Induce PGE2 Synthesis from Infected Macrophages
To begin to address if the genes identified in U112 also encode products that contribute to LVS to induce PGE2 synthesis by infected macrophages we utilized clean deletion mutants. Two of the genes identified in the screen of the two allele library, U112 mglA::Tn and U112 sspA::Tn, encode positive transcriptional regulators (Baron and Nano, 1998; Lauriano et al., 2004; Charity et al., 2007). We also identified several genes of the FPI, including dotU. DotU is necessary for stabilization of the FPI secretion apparatus, and mutants lacking dotU do not have a functional FPI secretion system (Broms et al., 2012). To examine the possibility that LVS mutants lacking mglA, sspA, or dotU do not to induce PGE2 synthesis, we tested these mutant strains for induction of PGE2 synthesis by BMDMs. BMDMs were inoculated with LVS, LVS∆mglA, LVS∆mglA (pmglA), LVS∆sspA, LVS∆sspA (psspA), LVS∆dotU, or LVS∆dotU (pdotU) at an MOI of 200:1. Twenty-four hours after inoculation the levels of PGE2 were determined. Neither LVS∆mglA, LVS∆sspA nor LVS∆dotU mutant strains induce significant PGE2 synthesis from infected macrophages (Figure 3). This phenotype was reversed by trans complementation with the appropriate plasmid (Figure 3), suggesting that U112 and LVS induce PGE2 synthesis through similar mechanisms.
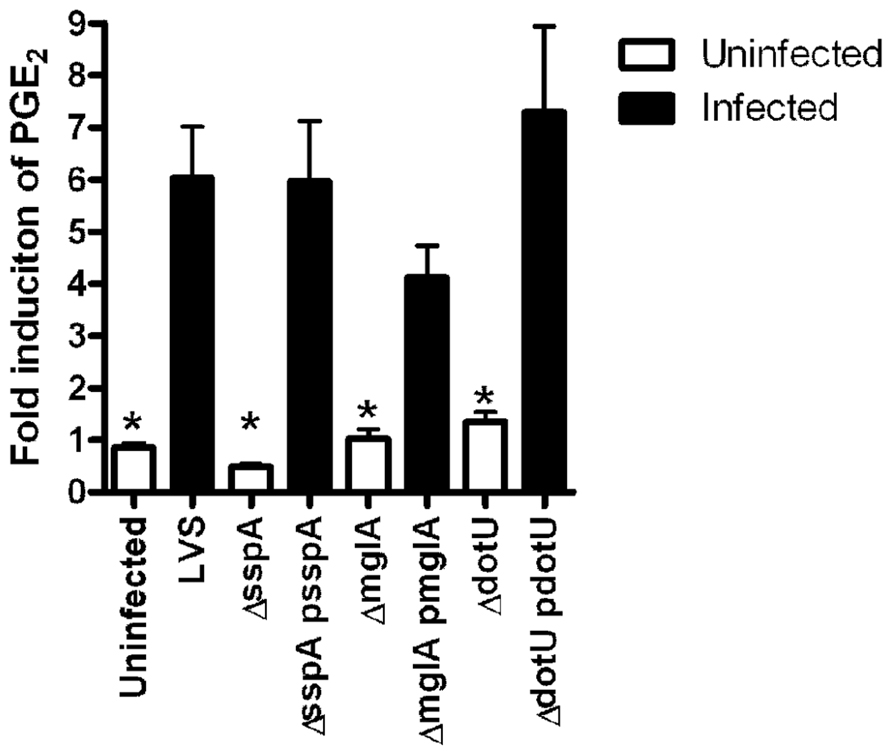
FIGURE 3.mglA, sspA, and dotU are necessary for LVS induction of PGE2 synthesis. Bone marrow-derived macrophages were inoculated with LVS, LVS∆mglA, LVS∆mglA (pmglA), LVS∆sspA, LVS∆sspA (psspA), LVS∆dotU, LVS∆dotU (pdotU), or LVS at an MOI of 200:1. Twenty-four hours after inoculation the levels of PGE2 were determined. Experiments were done in triplicate; error bars represent SEM (n = 3).
Dissociation of Intracellular Growth and Induction of PGE2 by Francisella
Escape from the phagosome and replication in the cytosol of host cells are critical for F. tularensis survival. All of the genes identified in this screen have been identified in other screens examining disease pathogenesis and intracellular growth (Maier et al., 2007; Su et al., 2007; Weiss et al., 2007; Kraemer et al., 2009; Asare and Abu Kwaik, 2010; Asare et al., 2010). Thus, it may be that failure to either escape the phagosome or replicate explains why these F. novicida mutants did not to induce PGE2 synthesis. Previous studies that examined infection of macrophages by F. novicida pdpA::Tn and ∆pdpA strains demonstrated that PdpA is required for escape from the phagosome (Schmerk et al., 2009a,b). Similarly, IglC has been shown to be required for F. novicida and F. holarctica phagosomal escape (Lindgren et al., 2004; Bonquist et al., 2008). In contrast, F. holarctica mutants with a transposon insertion in clpB escape the phagosome and replicate (Meibom et al., 2008). The characterization of the trafficking phenotypes of Francisella strains with mutations in clpB, pdpA, and iglC suggested we could use the two allele mutant library clpB::Tn, pdpA::Tn, and iglC::Tn mutant strains as tools to investigate the requirement of escape and intracellular growth for PGE2 induction. We understand these experiments do not prove that these genes are necessarily involved in the induction of PGE2, but rather eliminate or confirm if either the act of escaping the phagosome or replicating in the cytosol is what is necessary and sufficient to induce PGE2 synthesis in macrophages.
To determine the intracellular localization of these strains, we inoculated the J774.1 macrophage cell line, as we and others have successfully used this cell line in the past to examine intracellular localization of F. tularensis (Tempel et al., 2006; Fuller et al., 2008), at an MOI of 500:1 with CFSE labeled U112, U112 clpB::Tn, U112 iglC::Tn, and U112 pdpA::Tn and examined their association with LAMP-1 using Confocal microscopy (Figure 4). A high MOI was used to ensure our ability to identify intracellular bacteria and their respective intracellular localization. We confirmed that U112 infected J774 cells synthesize increased amounts of PGE2 upon both U112 and LVS infection compared to uninfected samples (data not shown). We analyzed the associations of bacteria and LAMP-1 using Volocity image software and showed the percentage of bacteria associated with LAMP-1 by pixel association (Figure 4A). We found only 34% of U112 remained associated with LAMP-1 4 h post-inoculation. Similarly, 34% of U112 clpB::Tn remained associated with LAMP-1 4 h post-inoculation. In contrast, U112 iglC::Tn and U112 pdpA::Tn resided mainly in the phagosome 4 h post-inoculation displaying 71% and 70% LAMP-1 association, respectively. We confirmed the intracellular localization of U112 clpB::Tn, U112 iglC::Tn, and U112 pdpA::Tn by transmission electron microscopy (Figure 4B). These data indicate U112 clpB::Tn, U112 iglC::Tn, and U112 pdpA::Tn have intracellular trafficking patterns that are similar to those of previously published clpB, iglC, and pdpA transposon insertion strains (Lindgren et al., 2004; Bonquist et al., 2008; Meibom et al., 2008; Schmerk et al., 2009a,b). As noted above, these data show that the U112 pdpA::Tn mutant is able to induce PGE2 even though it was diminished in its ability to escape the phagosome. If PGE2 synthesis induction required phagosomal escape, we would expect U112 pdpA::Tn would not induce PGE2 synthesis, as seen with U112 iglC::Tn. However, pdpA::Tn does induce PGE2 at similar levels to wild-type U112 (Figure 2). Thus, our data suggest PGE2 induction is unaffected by intracellular trafficking/localization.
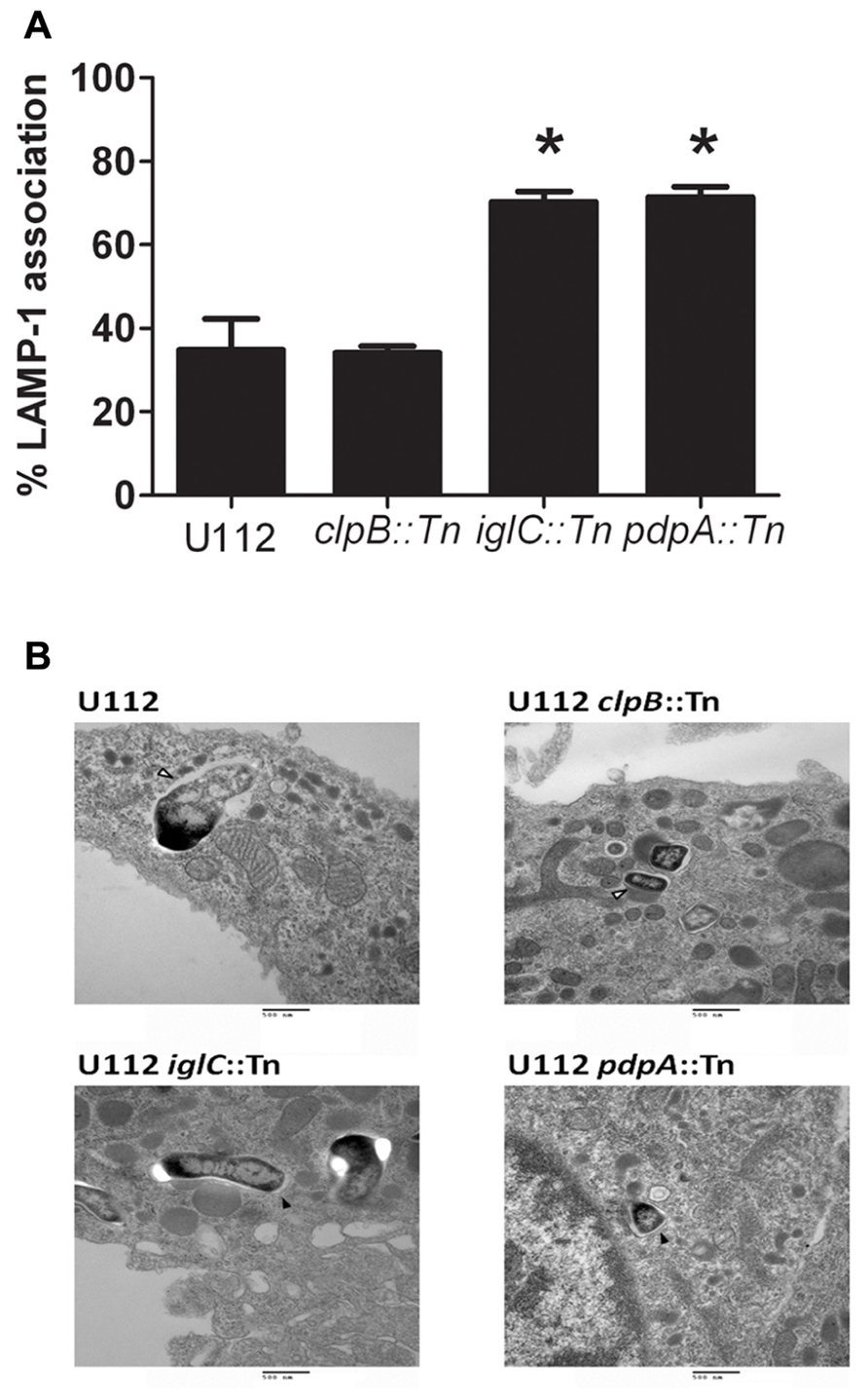
FIGURE 4. Induction of PGE2 does not require full escape from the phagosome. (A) Bacterial association with LAMP-1 was scored using Volocity (n ≥ 20 imaged cells per strain with an average of one bacterium per macrophage). Co-localization was determined by the shared of red and green pixels at the same location. Data represents three independent experiments and expressed as the mean ± SEM. Asterisk “*” denotes statistical difference (p ≤ 0.05) from U112-infected cells. (B) BMDMs were inoculated with U112, U112 clpB::Tn, U112 iglC::Tn, or U112 pdpA::Tn at an MOI of 500:1. Association of the bacterium with the phagosomal membrane was determined 4 h post-inoculation using transmission electron microscopy. Open arrowheads denote bacteria no longer surrounded by an intact phagosomal membrane. Filled arrowheads denote bacteria surrounded by a phagosomal membrane.
To determine if intracellular growth was required for F. novicida induction of PGE2 synthesis we inoculated BMDM at an MOI of 100:1 with U112, U112 clpB::Tn, U112 pdpA::Tn, and U112 iglC::Tn and counted intracellular CFUs over time. We used an MOI 100:1 to maximize differences in intracellular CFUs at 4 and 24 h post-inoculation. At higher MOIs extensive cell death of BMDMs by 24 h post-inoculation made it difficult to measure intracellular growth (data not shown). The number of intracellular bacteria was determined at 4 and 24 h post-inoculation, while the concentration of PGE2 in supernatants was determined at 24 h post-inoculation. The U112 clpB::Tn strain grew within BMDM similarly to wild-type U112, while the U112 pdpA::Tn and U112 iglC::Tn strains failed to grow in BMDM (Figure 5). In fact, there were fewer intra-macrophage U112 pdpA::Tn and U112 iglC::Tn bacteria at 24 h post-inoculation than at 4 h post-inoculation. Wild-type U112 and U112 pdpA::Tn were able to induce PGE2 synthesis, while U112 clpB::Tn and U112 iglC::Tn did not. The fact that pdpA::Tn induced PGE2 synthesis without intra-macrophage growth and clpB::Tn did not induce PGE2 synthesis while still able to grow in the macrophage demonstrates dissociation between intracellular growth and the ability of F. novicida to induce infected BMDM to synthesize PGE2.
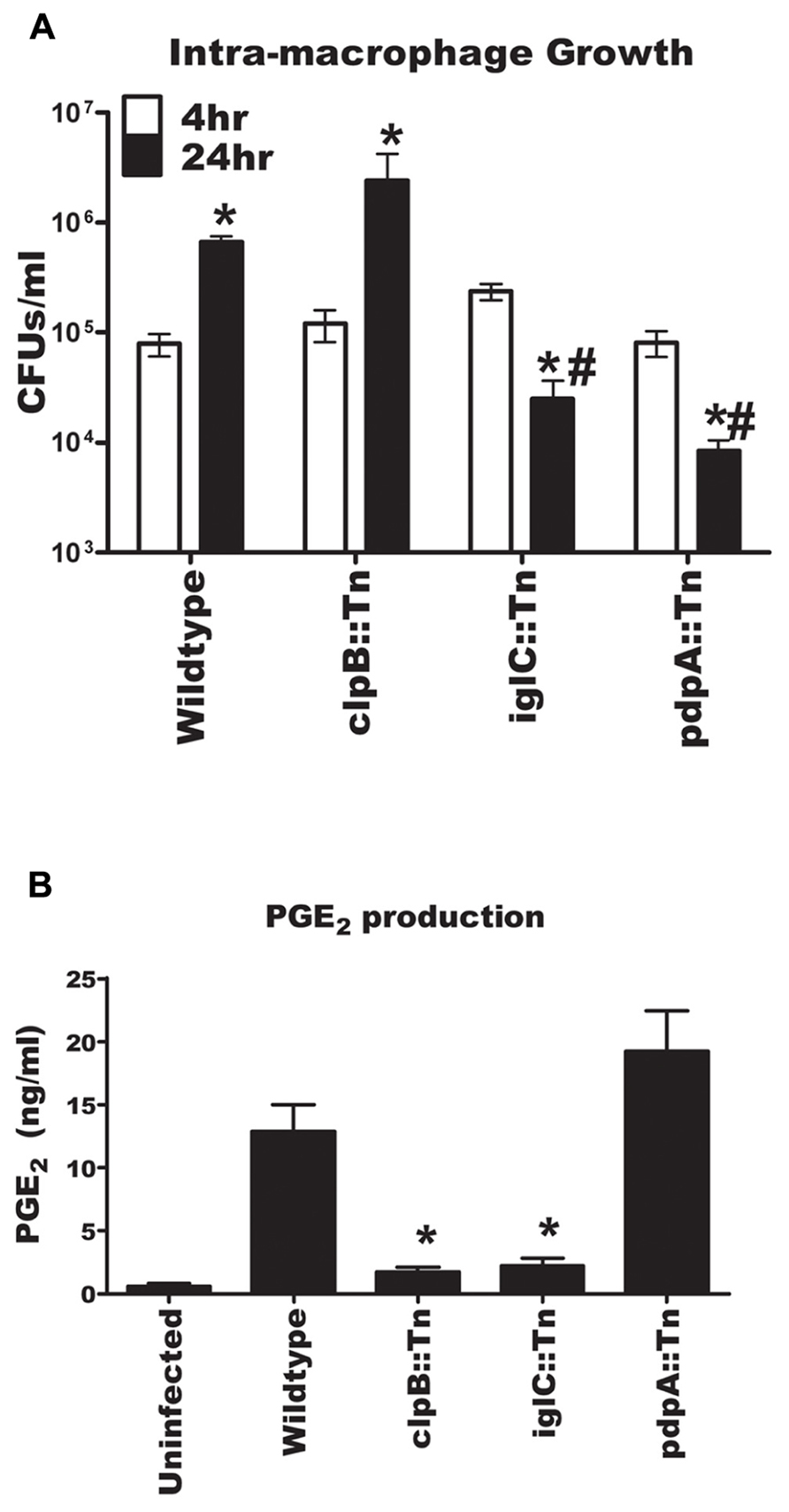
FIGURE 5. Dissociation of intracellular growth and the induction of PGE2 from bone marrow-derived macrophages (BMDMs). (A) BMDMs were inoculated with U112, U112 clpB::Tn, U112 iglC::Tn, or U112 pdpA::Tn at an MOI of 100:1. CFU were determined at 4 and 24 h post-inoculation. Data represents three independent experiments and expressed as the means ± SEM. Asterisk “*” denotes statistical difference (p ≤ 0.05) from corresponding 4 h sample. #BMDM denotes statistical difference (p ≤ 0.05) from 24 h U112-infected BMDM (n = 3). (B) BMDMs were inoculated with U112, U112 clpB::Tn, U112 iglC::Tn, or U112 pdpA::Tn at an MOI of 100:1. Twenty-four hours after inoculation supernatants were collected and PGE2 concentration was determined. Data represents three independent experiments and expressed as the mean ± SEM. Asterisk “*” denotes statistical difference (p ≤ 0.05) from U112-infected BMDM.
Discussion
The induction of PGE2 synthesis by LVS-infected macrophages disrupts T cell responses allowing LVS to persist in the host (Woolard et al., 2007, 2008). We demonstrate here that induction of PGE2 synthesis by infected BMDMs is conserved among F. novicida, F. holarctica, and F. tularensis. Synthesis of PGE2 by U112-infected macrophages allowed us to screen the comprehensive U112 two allele transposon mutant library to identify Francisella genes that are potentially involved in the induction of PGE2 synthesis by Francisella-infected macrophages. Our screen identified 20 genes that when disrupted resulted in strains that failed to induce PGE2 synthesis by F. novicida-infected BMDM. These 20 genes are highly conserved in all sequenced Francisella subspecies (Charity et al., 2007, 2009; Nano and Schmerk, 2007; Meibom et al., 2008). Eighteen of the genes identified in this study either mapped to the FPI or represent positive transcriptional regulators of the FPI (Nano and Schmerk, 2007). Seventeen of the 20 identified genes have been demonstrated to be involved in mouse virulence (Su et al., 2007; Weiss et al., 2007). Most, but not all of these genes, encode proteins that have been implicated in escape from the phagosome and intracellular growth (Su et al., 2007; Weiss et al., 2007). The data presented here suggest these gene products may be responsible for the induction of PGE2 biosynthesis in infected BMDM independent of their role in phagosomal escape and intracellular growth.
The FPI likely encodes a secretion system. The FPI proteins PdpB, VgrG, DotU, IglA, and IglB are homologous to T6SS proteins from other bacterial pathogens (Ludu et al., 2008; de Bruin et al., 2011; Broms et al., 2012; Robb et al., 2012). The FPI was initially identified in F. novicida via mutations in iglA and iglC that resulted in F. novicida strains that no longer replicated within macrophages (Gray et al., 2002). Recent work has identified the FPI genes that encode proteins required for intracellular growth and include pdpA, pdpB, dotU, vgrG, iglABCDEFHJ, and potentially iglG and iglI (Nano et al., 2004; Santic et al., 2005, 2007; de Bruin et al., 2007, 2011; Bonquist et al., 2008; Broms et al., 2011). The genes pdpC, pdpD, pdpE, and anmK are not required for intracellular growth (de Bruin et al., 2011). Our screen demonstrated that disruptions in FPI genes dotU, vgrG, pdpBC, and iglABCGEDFGHIJ resulted in U112 strains unable to induce PGE2 synthesis by infected macrophages. At this time we are unsure whether all gene products are necessary, or whether some mutants where identified due to polar effects of transposon insertions. This is possible as the FPI is believed to be organized in two operons (Nano and Schmerk, 2007). Future work will be necessary to define which FPI gene products are truly necessary for induction of PGE2 synthesis from infected macrophages. Disruptions in pdpADE and anmK did not impair the bacteria’s ability to induce synthesis of PGE2. We were not surprised that pdpD and anmK mutants are not impaired, as we believe the mechanism of induction of PGE2 synthesis is conserved between Francisella strains. The anmK gene is not present in LVS while the pdpD is truncated in LVS and presumably non-functional (Ludu et al., 2008). The deletion of pdpE from F. novicida had no effect on the bacteria’s ability to grow in macrophages or cause disease. At this time the role of PdpE in FPI function is unknown (de Bruin et al., 2011). PdpA is involved in both intracellular growth and virulence. However, PdpA is not believed to be a component of the FPI secretion system (Schmerk et al., 2009a,b) which may explain why the pdpA::Tn mutant is still capable of inducing PGE2 synthesis. Some of the transposon mutants (pdpE::TN, pdpD::Tn, and anmK::Tn) were capable of inducing enhanced PGE2 secretion from infected macrophages. The mechanism behind this is unknown and future work will be done to examine this phenomenon. Regardless, it is clear that disruption of F. novicida’s genes in the FPI diminishes its ability to induce PGE2 synthesis from infected macrophages.
There are six genes located outside of the FPI that when disrupted resulted in strains unable to induce the synthesis of PGE2 from U112 infected macrophages. Four of those (trmE, sspA, mglA, and mglB) have previously been identified to encode positive transcriptional regulators of genes found both in the FPI and outside the FPI (Baron and Nano, 1998; Lauriano et al., 2004; Charity et al., 2007, 2009; Schmerk et al., 2009a,b). The work of the Dove laboratory has clearly identified other transcriptional regulators which include CaiC, CphA, PigR, and SpoT in F. tularensis LVS (Charity et al., 2009). The two allele mutant library lacks transposon insertional mutants in spoT and pigR, while the caiC and the cphA transposon mutant strain induced PGE2 synthesis from macrophages. This result suggests differential transcriptional regulation of the FPI between U112 and LVS; however future work would be required to corroborate this observation. RpoB is a component of the RNAP catalytic core responsible for the transcription of genes (Allison et al., 1985). The U112 rpoB::Tn was likely identified due to a general disruption of transcription. The fact the rpoB::Tn mutant failed to induce PGE2 synthesis would predict finding other components of the RNAP catalytic core. However, the two allele library did not contain mutants with transposon insertions in either rpoA or rpoD, while rpoC::Tn and rpoZ::Tn mutant strains induced PGE2 synthesis. ClpB, a stress response protein, has been previously demonstrated to be important in Francisella disease pathogenesis. A U112 clpB mutant was identified due to a delay in intra-macrophage growth, while a disruption of clpB in F. holarctica LVS resulted in a strain that could grow in vitro in macrophages, but failed to effectively multiply in mice (Gray et al., 2002; Meibom et al., 2008). We did not observe a intra-macrophage growth defect of the two allele clpB::Tn. In Listeria monocytogenes and Porphyromonas gingivalis, ClpB homologs are necessary for virulence during animal infections (Chastanet et al., 2004; Yuan et al., 2007). ClpB/ClpV homologs have been identified in other T6SS where their AAA+ ATPase activity supply energy for the protein secretion process (Mougous et al., 2006; Shalom et al., 2007; Shrivastava and Mande, 2008; Bonemann et al., 2009). ClpB regulates the protein levels of DnaK, FTL_0525, FTL_0311, FTL_0588, and FTL_0207 (Meibom et al., 2008). Since none of these genes were identified as necessary for induction of PGE2 synthesis, it would suggest ClpB may have other unidentified functions. It has not been demonstrated to regulate the protein levels of the FPI. Future work will be needed to define the mechanism of ClpB-mediated induction of PGE2 synthesis from infected macrophages, and whether this is through regulation of FPI genes, function of FPI gene products, or through a FPI-independent mechanism.
Infection of macrophages with U112, LVS, or Schu S4 results in the induction of PGE2. This demonstrates that the ability to induce PGE2 synthesis from infected macrophages is conserved among F. tularensis subspecies. In fact, U112 and Schu S4 induce more PGE2 than LVS at similar doses. This difference in PGE2 induction may be partially responsible for difference in virulence in these different subspecies. While we have previously demonstrated differences in innate immune responses to Schu S4, LVS, and U112 in intranasally inoculated mice, it is unknown if these different responses are due to differences in PGE2 induction (Hall et al., 2008). Further work will address this difference in PGE2 induction and the potential effect of PGE2 on disease pathogenesis. The demonstration that inactivation of FPI genes in F. novicida results in the inability to induce PGE2 biosynthesis and the fact that the FPI is highly conserved among all subspecies of F. tularensis would suggest that the mechanism of PGE2 induction is conserved among these subspecies. The fact that LVS mglA, sspA, and dotU mutant strains did not induce PGE2 synthesis further suggests the likelihood that F. tularensis subspecies tularensis, F. tularensis subspecies holarctica, and F. tularensis subspecies novicida have conserved mechanisms of induction of PGE2 synthesis. However, we cannot discount the possibility that F. tularensis subspecies tularensis may possess additional mechanisms for induction of PGE2 synthesis that F. novicida or F. holarctica do not.
The FPI is necessary for the organism to escape the phagosome and replicate in the cytosol (Nano et al., 2004; Santic et al., 2005, 2007; de Bruin et al., 2011). The reason the transposon insertion mutants we identified in U112 failed to induce PGE2 synthesis may be due to their failure to escape the phagosome and subsequently replicate. Failure to escape the phagosome may create a physiologic barrier between F. tularensis and the eukaryotic molecule that is responsible for sensing and responding to F. tularensis. There are many intracellular receptors that can recognize bacterial products (Franchi et al., 2009). ASC, a component of the inflammasome, and AIM2 (which recognizes F. tularensis DNA) are crucial for control of Francisella intra-macrophage growth in vitro and infection in vivo (Mariathasan et al., 2005; Fernandes-Alnemri et al., 2010; Jones et al., 2010). Inflammasome activation is also capable of inducing eicosanoid production (von Moltke et al., 2012). However, we believe that failure to escape into the cytosol is not the reason the transposon insertion mutant strains we identified in this study failed to induce PGE2 synthesis by infected macrophages. In other studies, pdpA::Tn and ∆pdpA F. novicida mutants fail to fully escape the phagosome (Mariathasan et al., 2005; Schmerk et al., 2009a,b). The U112 pdpA::Tn strain in the U112 two allele mutant library does not escape the phagosome to the same level as wild-type U112. Recently, 92 transposon mutant strains from the two allele mutant library were identified that did not escape the phagosome (Asare and Abu Kwaik, 2010). We showed all of these strains were able to induce PGE2. Thus, it is unlikely that the mutants we did identify failed to induce PGE2 solely because they failed to escape from the phagosome. Future work that identifies both the F. tularensis effector molecule and the corresponding eukaryotic binding partner will allow us to more definitively dissociate F. tularensis trafficking and induction of PGE2 synthesis from Francisella-infected macrophages.
Previous studies have identified 201 genes outside the FPI that are required for Francisella intra-macrophage growth (Qin and Mann, 2006; Maier et al., 2007; Asare and Abu Kwaik, 2010; Asare et al., 2010). U112 strains with insertions in any one of these 201 genes were all capable of inducing PGE2 synthesis from infected macrophages. We did not identify known F. tularensis auxotrophs as being defective in the ability to induce PGE2. Transposon insertions in purA, purF, carA, carB, and pyrB produce strains that have a defect in intracellular growth yet are able to induce macrophage synthesis of PGE2 (Maier et al., 2007; Quarry et al., 2007; Schulert et al., 2009). Our studies using U112, U112 clpB::Tn, U112 pdpA::Tn, and U112 iglC::Tn strains demonstrate dissociation between intra-macrophage growth, the ability of F. tularensis to fully escape the phagosome, and the ability to induce PGE2. These data also confirm our earlier report that UV inactivation of LVS, which inhibits replication, did not impact LVS’s ability to induce PGE2 synthesis from infected macrophages (Woolard et al., 2007). Further characterization and understanding of the molecular interactions between F. tularensis and eukaryotic cells that lead to the induction of PGE2 will provide new insight into tularemia pathogenesis.
Author Contributions
Matthew D. Woolard carried out all experiments except confocal and TEM microscopy. Lydia M. Barrigan and Adam S. Buntzman designed and carried out all confocal and TEM experiments. James R. Fuller and Joshua Bryan designed and generated strains used in study. Matthew D. Woolard drafted the manuscript. Colin Manoil aided in the design and use of the transposon library. Matthew D. Woolard, Thomas H. Kawula, Jeffrey A. Frelinger, and Colin Manoil designed and coordinated experiments and analyzed data. All authors read and approved the final manuscript.
Conflict of Interest Statement
James R. Fuller is currently employed by Laboratory Corporations of America. All work conducted by James R. Fuller was prior to his employment to Laboratory Corporations of America, as such LaboratoryCorporations of America have no proprietary claim to any work presented in this publication.
Acknowledgments
The authors wish to thank Lucinda L. Hensley and Kenneth Peterson, PhD for valuable discussion and thoughtful review of the manuscript. We also thank William Day and the University of Arizona’s University Spectroscopy and Imaging Facilities for assistance with the electron microscopy. This study was funded by NIH grant #U54 AI057157 from Southeastern Regional Center of Excellence for Emerging Infections and Biodefense, NIH Grant R01-AI 078345, and NIH Grant K22-AI083373.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Microbial Immunology/10.3389/fmicb.2013.00016/abstract
References
Allison, L. A., Moyle, M., Shales, M., and Ingles, C. J. (1985). Extensive homology among the largest subunits of eukaryotic and prokaryotic RNA polymerases. Cell 42, 599–610.
Asare, R., and Abu Kwaik, Y. (2010). Molecular complexity orchestrates modulation of phagosome biogenesis and escape to the cytosol of macrophages by Francisella tularensis. Environ. Microbiol. 12, 2559–2586.
Asare, R., Akimana, C., Jones, S., and Abu Kwaik, Y. (2010). Molecular bases of proliferation of Francisella tularensis in arthropod vectors. Environ. Microbiol. 12, 2587–2612.
Ashtekar, A. R., Zhang, P., Katz, J., Deivanayagam, C. C., Rallabhandi, P., Vogel, S. N., et al. (2008). TLR4-mediated activation of dendritic cells by the heat shock protein DnaK from Francisella tularensis. J. Leukoc. Biol. 84, 1434–1446.
Barker, J. R., Chong, A., Wehrly, T. D., Yu, J. J., Rodriguez, S. A., Liu, J., et al. (2009). The Francisella tularensis pathogenicity island encodes a secretion system that is required for phagosome escape and virulence. Mol. Microbiol. 74, 1459–1470.
Baron, G. S., and Nano, F. E. (1998). MglA and MglB are required for the intramacrophage growth of Francisella novicida. Mol. Microbiol. 29, 247–259.
Bonemann, G., Pietrosiuk, A., Diemand, A., Zentgraf, H., and Mogk, A. (2009). Remodelling of VipA/VipB tubules by ClpV-mediated threading is crucial for type VI protein secretion. EMBO J. 28, 315–325.
Bonquist, L., Lindgren, H., Golovliov, I., Guina, T., and Sjostedt, A. (2008). MglA and Igl proteins contribute to the modulation of Francisella tularensis live vaccine strain-containing phagosomes in murine macrophages. Infect. Immun. 76, 3502–3510.
Bosio, C. M., and Dow, S. W. (2005). Francisella tularensis induces aberrant activation of pulmonary dendritic cells. J. Immunol. 175, 6792–6801.
Broms, J. E., Lavander, M., Meyer, L., and Sjostedt, A. (2011). IglG and IglI of the Francisella pathogenicity island are important virulence determinants of Francisella tularensis LVS. Infect. Immun. 79, 3683–3696.
Broms, J. E., Meyer, L., Lavander, M., Larsson, P., and Sjostedt, A. (2012). DotU and VgrG, core components of type VI secretion systems, are essential for Francisella LVS pathogenicity. PLoS ONE 7:e34639. doi: 10.1371/journal.pone.0034639
Charity, J. C., Blalock, L. T., Costante-Hamm, M. M., Kasper, D. L., and Dove, S. L. (2009). Small molecule control of virulence gene expression in Francisella tularensis. PLoS Pathog. 5:e1000641. doi: 10.1371/journal.ppat.1000641
Charity, J. C., Costante-Hamm, M. M., Balon, E. L., Boyd, D. H., Rubin, E. J., and Dove, S. L. (2007). Twin RNA polymerase-associated proteins control virulence gene expression in Francisella tularensis. PLoS Pathog. 3:e84. doi: 10.1371/journal.ppat.0030084
Chastanet, A., Derre, I., Nair, S., and Msadek, T. (2004). clpB, a novel member of the Listeria monocytogenes CtsR regulon, is involved in virulence but not in general stress tolerance. J. Bacteriol. 186, 1165–1174.
Chen, B. C., Chang, Y. S., Kang, J. C., Hsu, M. J., Sheu, J. R., Chen, T. L., et al. (2004). Peptidoglycan induces nuclear factor-kappaB activation and cyclooxygenase-2 expression via Ras, Raf-1, and ERK in RAW 264.7 macrophages. J. Biol. Chem. 279, 20889–20897.
Chen, Y., Zhang, J., Moore, S. A., Ballas, Z. K., Portanova, J. P., Krieg, A. M., et al. (2001). CpG DNA induces cyclooxygenase-2 expression and prostaglandin production. Int. Immunol. 13, 1013–1020.
Cowley, S. C., and Elkins, K. L. (2003). Multiple T cell subsets control Francisella tularensis LVS intracellular growth without stimulation through macrophage interferon gamma receptors. J. Exp. Med. 198, 379–389.
de Bruin, O. M., Duplantis, B. N., Ludu, J. S., Hare, R. F., Nix, E. B., Schmerk, C. L., et al. (2011). The biochemical properties of the Francisella pathogenicity island (FPI)-encoded proteins IglA, IglB, IglC, PdpB and DotU suggest roles in type VI secretion. Microbiology 157, 3483–3491.
de Bruin, O. M., Ludu, J. S., and Nano, F. E. (2007). The Francisella pathogenicity island protein IglA localizes to the bacterial cytoplasm and is needed for intracellular growth. BMC Microbiol. 7:1. doi: 10.1186/1471-2180-7-1
Dennis, D. T., Inglesby, T. V., Henderson, D. A., Bartlett, J. G., Ascher, M. S., Eitzen, E., et al. (2001). Tularemia as a biological weapon: medical and public health management. JAMA 285, 2763–2773.
Ellis, J., Oyston, P. C., Green, M., and Titball, R. W. (2002). Tularemia. Clin. Microbiol. Rev. 15, 631–646.
Fernandes-Alnemri, T., Yu, J. W., Juliana, C., Solorzano, L., Kang, S., Wu, J., et al. (2010). The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393.
Forestal, C. A., Gil, H., Monfett, M., Noah, C. E., Platz, G. J., Thanassi, D. G., et al. (2008). A conserved and immunodominant lipoprotein of Francisella tularensis is proinflammatory but not essential for virulence. Microb. Pathog. 44, 512–523.
Franchi, L., Warner, N., Viani, K., and Nunez, G. (2009). Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 227, 106–128.
Fuller, J.R., Craven, R.R., Hall, J.D., Kijek, T.M., Taft-Benz, S., and Kawula, T.H. (2008). RipA, a cytoplasmic membrane protein conserved among Francisella species, is required for intracellular survival. Infect. Immun. 76, 4934–4943.
Fuller, J. R., Kijek, T.M., Taft-Benz, S., and Kawula, T.H. (2009). Environmental and intracellular regulation of Francisella tularensis ripA. BMC Microbiol. 9:216. doi: 10.1186/1471-2180-9-216
Gallagher, L. A., Ramage, E., Jacobs, M. A., Kaul, R., Brittnacher, M., and Manoil, C. (2007). A comprehensive transposon mutant library of Francisella novicida, a bioweapon surrogate. Proc. Natl. Acad. Sci. U.S.A. 104, 1009–1014.
Gray, C. G., Cowley, S. C., Cheung, K. K., and Nano, F. E. (2002). The identification of five genetic loci of Francisella novicida associated with intracellular growth. FEMS Microbiol. Lett. 215, 53–56.
Guina, T., Radulovic, D., Bahrami, A. J., Bolton, D. L., Rohmer, L., Jones-Isaac, K. A., et al. (2007). MglA regulates Francisella tularensis subsp. novicida (Francisella novicida) response to starvation and oxidative stress. J. Bacteriol. 189, 6580–6586.
Hajjar, A. M., Harvey, M. D., Shaffer, S. A., Goodlett, D. R., Sjostedt, A., Edebro, H., et al. (2006). Lack of in vitro and in vivo recognition of Francisella tularensis subspecies lipopolysaccharide by Toll-like receptors. Infect. Immun. 74, 6730–6738.
Hall, J. D., Woolard, M. D., Gunn, B. M., Craven, R. R., Taft-Benz, S., Frelinger, J. A., et al. (2008). Infected-host-cell repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4, LVS, or U112. Infect. Immun. 76, 5843–5852.
Hand, J., Scott-Waldron, C., and Balsamo, G. (2012). Outbreak of Francisella novicida infections among occupants at a long-term residential facility – Louisiana, April–July, 2011. LA Morbidity Rep. 23, 1 and 6.
Hollis, D. G., Weaver, R. E., Steigerwalt, A. G., Wenger, J. D., Moss, C. W., and Brenner, D. J. (1989). Francisella philomiragia comb. nov. (formerly Yersinia philomiragia) and Francisella tularensis biogroup novicida (formerly Francisella novicida) associated with human disease. J. Clin. Microbiol. 27, 1601–1608.
Hornick, R. B., and Eigelsbach, H. T. (1966). Aerogenic immunization of man with live Tularemia vaccine. Bacteriol. Rev. 30, 532–538.
Jones, J. W., Kayagaki, N., Broz, P., Henry, T., Newton, K., O’Rourke, K., et al. (2010). Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. U.S.A. 107, 9771–9776.
Kieffer, T. L., Cowley, S., Nano, F. E., and Elkins, K. L. (2003). Francisella novicida LPS has greater immunobiological activity in mice than F. tularensis LPS, and contributes to F. novicida murine pathogenesis. Microbes Infect. 5, 397–403.
Kraemer, P. S., Mitchell, A., Pelletier, M. R., Gallagher, L. A., Wasnick, M., Rohmer, L., et al. (2009). Genome-wide screen in Francisella novicida for genes required for pulmonary and systemic infection in mice. Infect. Immun. 77, 232–244.
Larson, C. L., Wicht, W., and Jellison, W. L. (1955). A new organism resembling P. tularensis isolated from water. Public Health Rep. 70, 253–258.
Lauriano, C. M., Barker, J. R., Yoon, S. S., Nano, F. E., Arulanandam, B. P., Hassett, D. J., et al. (2004). MglA regulates transcription of virulence factors necessary for Francisella tularensis intraamoebae and intramacrophage survival. Proc. Natl. Acad. Sci. U.S.A. 101, 4246–4249.
Lindgren, H., Golovliov, I., Baranov, V., Ernst, R. K., Telepnev, M., and Sjostedt, A. (2004). Factors affecting the escape of Francisella tularensis from the phagolysosome. J. Med. Microbiol. 53, 953–958.
Ludu, J. S., de Bruin, O. M., Duplantis, B. N., Schmerk, C. L., Chou, A. Y., Elkins, K. L., et al. (2008). The Francisella pathogenicity island protein PdpD is required for full virulence and associates with homologues of the type VI secretion system. J. Bacteriol. 190, 4584–4595.
Ma, A..T., and Mekalanos, J..J. (2010). In vivo actin cross-linking induced by Vibrio cholerae type VI secretion system is associated with intestinal inflammation. Proc. Natl. Acad. Sci. U.S.A. 107, 4365–4370.
Maier, T. M., Casey, M. S., Becker, R. H., Dorsey, C. W., Glass, E. M., Maltsev, N., et al. (2007). Identification of Francisella tularensis Himar1-based transposon mutants defective for replication in macrophages. Infect. Immun. 75, 5376–5389.
Mariathasan, S., Weiss, D. S., Dixit, V. M., and Monack, D. M. (2005). Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J. Exp. Med. 202, 1043–1049.
Meibom, K. L., Dubail, I., Dupuis, M., Barel, M., Lenco, J., Stulik, J., et al. (2008). The heat-shock protein ClpB of Francisella tularensis is involved in stress tolerance and is required for multiplication in target organs of infected mice. Mol. Microbiol. 67, 1384–1401.
Mougous, J. D., Cuff, M. E., Raunser, S., Shen, A., Zhou, M., Gifford, C. A., et al. (2006). A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science 312, 1526–1530.
Nano, F. E., and Schmerk, C. (2007). The Francisella pathogenicity island. Ann. N. Y. Acad. Sci. 1105, 122–137.
Nano, F. E., Zhang, N., Cowley, S. C., Klose, K. E., Cheung, K. K., Roberts, M. J., et al. (2004). A Francisella tularensis pathogenicity island required for intramacrophage growth. J. Bacteriol. 186, 6430–6436.
Pechous, R. D., Mccarthy, T. R., and Zahrt, T. C. (2009). Working toward the future: insights into Francisella tularensis pathogenesis and vaccine development. Microbiol. Mol. Biol. Rev. 73, 684–711.
Pukatzki, S., Ma, A. T., Revel, A. T., Sturtevant, D., and Mekalanos, J. J. (2007). Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc. Natl. Acad. Sci. U.S.A. 104, 15508–15513.
Pukatzki, S., Ma, A. T., Sturtevant, D., Krastins, B., Sarracino, D., Nelson, W. C., et al. (2006). Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc. Natl. Acad. Sci. U.S.A. 103, 1528–1533.
Qin, A., and Mann, B. J. (2006). Identification of transposon insertion mutants of Francisella tularensis tularensis strain Schu S4 deficient in intracellular replication in the hepatic cell line HepG2. BMC Microbiol. 6:69. doi: 10.1186/1471-2180-6-69
Quarry, J. E., Isherwood, K. E., Michell, S. L., Diaper, H., Titball, R. W., and Oyston, P. C. (2007). A Francisella tularensis subspecies novicida purF mutant, but not a purA mutant, induces protective immunity to tularemia in mice. Vaccine 25, 2011–2018.
Rick Lyons, C., and Wu, T. H. (2007). Animal models of Francisella tularensis infection. Ann. N. Y. Acad. Sci. 1105, 238–265.
Robb, C. S., Nano, F. E., and Boraston, A. B. (2012). The structure of the conserved type six secretion protein TssL (DotU) from Francisella novicida. J. Mol. Biol. 419,277–283.
Rohmer, L., Fong, C., Abmayr, S., Wasnick, M., Larson Freeman, T. J., Radey, M., et al. (2007). Comparison of Francisella tularensis genomes reveals evolutionary events associated with the emergence of human pathogenic strains. Genome Biol. 8, R102.
Santic, M., Molmeret, M., Barker, J. R., Klose, K. E., Dekanic, A., Doric, M., et al. (2007). A Francisella tularensis pathogenicity island protein essential for bacterial proliferation within the host cell cytosol. Cell Microbiol. 9, 2391–2403.
Santic, M., Molmeret, M., Klose, K. E., Jones, S., and Kwaik, Y. A. (2005). The Francisella tularensis pathogenicity island protein IglC and its regulator MglA are essential for modulating phagosome biogenesis and subsequent bacterial escape into the cytoplasm. Cell Microbiol. 7, 969–979.
Schmerk, C. L., Duplantis, B. N., Howard, P. L., and Nano, F. E. (2009a). A Francisella novicida pdpA mutant exhibits limited intracellular replication and remains associated with the lysosomal marker LAMP-1. Microbiology 155, 1498–1504.
Schmerk, C. L., Duplantis, B. N., Wang, D., Burke, R. D., Chou, A. Y., Elkins, K. L., et al. (2009b). Characterization of the pathogenicity island protein PdpA and its role in the virulence of Francisella novicida. Microbiology 155, 1489–1497.
Schulert, G. S., Mccaffrey, R. L., Buchan, B. W., Lindemann, S. R., Hollenback, C., Jones, B. D., et al. (2009). Francisella tularensis genes required for inhibition of the neutrophil respiratory burst and intramacrophage growth identified by random transposon mutagenesis of strain LVS. Infect. Immun. 77, 1324–1336.
Shalom, G., Shaw, J. G., and Thomas, M. S. (2007). In vivo expression technology identifies a type VI secretion system locus in Burkholderia pseudomallei that is induced upon invasion of macrophages. Microbiology 153, 2689–2699.
Shrivastava, S., and Mande, S. S. (2008). Identification and functional characterization of gene components of type VI secretion system in bacterial genomes. PLoS ONE 3:e2955. doi: 10.1371/journal.pone.0002955
Smith, R. S., Kelly, R., Iglewski, B. H., and Phipps, R. P. (2002). The Pseudomonas autoinducer N-(3-oxododecanoyl) homoserine lactone induces cyclooxygenase-2 and prostaglandin E2 production in human lung fibroblasts: implications for inflammation. J. Immunol. 169, 2636–2642.
Su, J., Yang, J., Zhao, D., Kawula, T. H., Banas, J. A., and Zhang, J. R. (2007). Genome-wide identification of Francisella tularensis virulence determinants. Infect. Immun. 75, 3089–3101.
Suarez, G., Sierra, J. C., Erova, T. E., Sha, J., Horneman, A. J., and Chopra, A. K. (2010a). A type VI secretion system effector protein, VgrG1, from Aeromonas hydrophila that induces host cell toxicity by ADP ribosylation of actin. J. Bacteriol. 192, 155–168.
Suarez, G., Sierra, J. C., Kirtley, M. L., and Chopra, A. K. (2010b). Role of Hcp, a type 6 secretion system effector, of Aeromonas hydrophila in modulating activation of host immune cells. Microbiology 156, 3678–3688.
Tempel, R., Lai, X. H., Crosa, L., Kozlowicz, B., and Heffron, F. (2006). Attenuated Francisella novicida transposon mutants protect mice against wild-type challenge. Infect. Immun. 74, 5095–5105.
Thakran, S., Li, H., Lavine, C. L., Miller, M. A., Bina, J. E., Bina, X. R., et al. (2008). Identification of Francisella tularensis lipoproteins that stimulate the Toll-like receptor (TLR) 2/TLR1 heterodimer. J. Biol. Chem. 283, 3751–3760.
Tigertt, W. D. (1962). Soviet viable Pasteurella tularensis vaccines. A review of selected articles. Bacteriol. Rev. 26, 354–373.
Treffkorn, L., Scheibe, R., Maruyama, T., and Dieter, P. (2004). PGE2 exerts its effect on the LPS-induced release of TNF-alpha, ET-1, IL-1alpha, IL-6 and IL-10 via the EP2 and EP4 receptor in rat liver macrophages. Prostaglandins Other Lipid Mediat. 74, 113–123.
Uematsu, S., Matsumoto, M., Takeda, K., and Akira, S. (2002). Lipopolysaccharide-dependent prostaglandin E(2) production is regulated by the glutathione-dependent prostaglandin E(2) synthase gene induced by the Toll-like receptor 4/MyD88/NF-IL6 pathway. J. Immunol. 168, 5811–5816.
von Moltke, J., Trinidad, N. J., Moayeri, M., Kintzer, A. F., Wang, S. B., Van Rooijen, N., et al. (2012). Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 490, 107–111.
Weiss, D. S., Brotcke, A., Henry, T., Margolis, J. J., Chan, K., and Monack, D. M. (2007). In vivo negative selection screen identifies genes required for Francisella virulence. Proc. Natl. Acad. Sci. U.S.A. 104, 6037–6042.
Wilson, J. E., Katkere, B., and Drake, J. R. (2009). Francisella tularensis induces ubiquitin-dependent major histocompatibility complex class II degradation in activated macrophages. Infect. Immun. 77, 4953–4965.
Woolard, M. D., Hensley, L. L., Kawula, T. H., and Frelinger, J. A. (2008). Respiratory Francisella tularensis live vaccine strain infection induces Th17 cells and prostaglandin E2, which inhibits generation of gamma interferon-positive T cells. Infect. Immun. 76, 2651–2659.
Woolard, M. D., Wilson, J. E., Hensley, L. L., Jania, L. A., Kawula, T. H., Drake, J. R., et al. (2007). Francisella tularensis-infected macrophages release prostaglandin E2 that blocks T cell proliferation and promotes a Th2-like response. J. Immunol. 178, 2065–2074.
Yee, D., Rhinehart-Jones, T. R., and Elkins, K. L. (1996). Loss of either CD4+ or CD8+ T cells does not affect the magnitude of protective immunity to an intracellular pathogen, Francisella tularensis strain LVS. J. Immunol. 157, 5042–5048.
Keywords: Francisella, prostaglandin E2
Citation: Woolard MD, Barrigan LM, Fuller JR, Buntzman AS, Bryan J, Manoil C, Kawula TH and Frelinger JA (2013) Identification of Francisella novicida mutants that fail to induce prostaglandin E2 synthesis by infected macrophages. Front. Microbio. 4:16. doi: 10.3389/fmicb.2013.00016
Received: 20 November 2012; Accepted: 24 January 2013;
Published online: 11 February 2013.
Edited by:
Gregoire S. Lauvau, Albert Einstein College of Medicine, USAReviewed by:
Anders Sjostedt, Umeå University, SwedenThomas Henry, Institut National de la Santé et de la Recherche Médicale, France
David Weiss, Emory University, USA
Copyright: © 2013 Woolard, Barrigan, Fuller, Buntzman, Bryan, Manoil, Kawula and Frelinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Matthew D. Woolard, Department of Microbiology and Immunology, Louisiana State University Health Sciences Center at Shreveport, 1501 Kings Highway, Shreveport, LA 71130, USA. e-mail:bXdvb2xhQGxzdWhzYy5lZHU=