



- 1Department of Microbial Ecology, Ecology Centre, University of Vienna, Vienna, Austria
- 2Department of Microbiology and Biotechnology, Biocenter Klein Flottbek, University of Hamburg, Hamburg, Germany
- 3Department of Computational Systems Biology, Ecology Centre, University of Vienna, Vienna, Austria
In marine systems, nitrate is the major reservoir of inorganic fixed nitrogen. The only known biological nitrate-forming reaction is nitrite oxidation, but despite its importance, our knowledge of the organisms catalyzing this key process in the marine N-cycle is very limited. The most frequently encountered marine NOB are related to Nitrospina gracilis, an aerobic chemolithoautotrophic bacterium isolated from ocean surface waters. To date, limited physiological and genomic data for this organism were available and its phylogenetic affiliation was uncertain. In this study, the draft genome sequence of N. gracilis strain 3/211 was obtained. Unexpectedly for an aerobic organism, N. gracilis lacks classical reactive oxygen defense mechanisms and uses the reductive tricarboxylic acid cycle for carbon fixation. These features indicate microaerophilic ancestry and are consistent with the presence of Nitrospina in marine oxygen minimum zones. Fixed carbon is stored intracellularly as glycogen, but genes for utilizing external organic carbon sources were not identified. N. gracilis also contains a full gene set for oxidative phosphorylation with oxygen as terminal electron acceptor and for reverse electron transport from nitrite to NADH. A novel variation of complex I may catalyze the required reverse electron flow to low-potential ferredoxin. Interestingly, comparative genomics indicated a strong evolutionary link between Nitrospina, the nitrite-oxidizing genus Nitrospira, and anaerobic ammonium oxidizers, apparently including the horizontal transfer of a periplasmically oriented nitrite oxidoreductase and other key genes for nitrite oxidation at an early evolutionary stage. Further, detailed phylogenetic analyses using concatenated marker genes provided evidence that Nitrospina forms a novel bacterial phylum, for which we propose the name Nitrospinae.
Introduction
Nitrite-oxidizing bacteria (NOB) are chemolithoautotrophic organisms catalyzing the oxidation of nitrite to nitrate, which is the second step of nitrification and a major biogeochemical process. Nitrite is substrate not only for nitrite oxidation but also for denitrification and anaerobic ammonium oxidation (anammox), both leading to nitrogen losses from ecosystems to the atmosphere. Therefore, the activity of NOB is a key factor controlling the retention of fixed nitrogen in the form of nitrate in the biosphere.
NOB are a phylogenetically diverse functional group. Known representatives belong to the genera Nitrobacter, Nitrotoga, and Nitrococcus within the Alpha-, Beta-, and Gammaproteobacteria, respectively (Winogradsky, 1890; Watson and Waterbury, 1971; Alawi et al., 2007), to the genus Nitrospira within the Nitrospirae (Daims et al., 2001), and to the genus Nitrolancetus within the Chloroflexi (Sorokin et al., 2012). The genus Nitrospina was provisionally assigned to the Deltaproteobacteria (Teske et al., 1994), but this affiliation has been questioned due to difficulties to classify this genus solely by 16S rRNA-based phylogenetic inference (Schloss and Handelsman, 2004). Most NOB lineages are highly versatile and can be found in terrestrial, limnic, and marine ecosystems as well as in man-made habitats such as wastewater treatment facilities. However, the dominant nitrite oxidizers in the oceans appear to be Nitrospina. The type species of this genus, Nitrospina gracilis, was isolated from ocean surface waters (Watson and Waterbury, 1971). It forms long, slender, Gram-negative rods, grows purely chemoautotrophically with nitrite and CO2 as the sole source of energy and carbon, respectively, and was described to be strictly aerobic using oxygen as terminal electron acceptor. No heterotrophic potential was observed and many organic substrates even inhibited the growth of this organism (Watson and Waterbury, 1971). Nitrospina-like bacteria were frequently detected by molecular methods in open ocean water (Fuchs et al., 2005; DeLong et al., 2006) as well as in marine sediments (Hunter et al., 2006; Jorgensen et al., 2012), but surprisingly also in marine oxygen minimum zones (OMZs; Labrenz et al., 2007; Fuchsman et al., 2011) and anoxic marine sediments (Davis et al., 2009; Jorgensen et al., 2012). Moreover, nitrite oxidation has been measured in OMZs (Füssel et al., 2012). Interestingly, the distribution profiles of Nitrospina and ammonia-oxidizing archaea (AOA) were found to correlate in some coastal and open ocean habitats (Mincer et al., 2007; Santoro et al., 2010). Prolonged coexistence of Nitrospina and AOA closely related to “Candidatus Nitrosopumilus maritimus” was even observed in an enrichment culture (Park et al., 2010), further strengthening the functional link between these nitrifiers. The only non-marine habitats Nitrospina-like 16S rRNA sequences were derived from are uranium mill tailings (Radeva and Selenska-Pobell, 2005) and an alpine subsurface radioactive thermal spring (Weidler et al., 2007). Thus, Nitrospina apparently are confined mainly to marine environments.
In marine ecosystems, nitrate accounts for as much as 88% of the fixed nitrogen, corresponding to 5.8 × 105 Tg (Gruber, 2008). While rapidly assimilated by phytoplankton in ocean surface waters, nitrate accumulates in the deep sea where it constitutes the largest pool of fixed inorganic nitrogen in the biosphere. The only biochemical reaction known to form nitrate is bacterial nitrite oxidation, which takes place during nitrification and, to a lesser extent, during anammox (Strous et al., 2006). Contrasting the obviously immense importance of Nitrospina as a major source of nitrate in marine environments, very little biochemical and genomic data is available for these enigmatic organisms, and even the phylogenetic affiliation of the genus is still unclear. This embarrassing lack of knowledge is mainly caused by the recalcitrance of Nitrospina to cultivation in the laboratory. Their slow growth rates with a minimum generation time of 24 h (Watson and Waterbury, 1971) further impede biochemical characterization. In this study, we employed a genomic approach to gain deeper insights into the metabolic potential of Nitrospina gracilis strain 3/211. The sequenced genome of this organism shed light on the core metabolism enabling Nitrospina of chemolithoautotrophic growth with nitrite. Comparative genomic analyses illuminated the evolutionary descent of Nitrospina and its nitrite-oxidizing capacity. By utilizing concatenated datasets of established marker proteins, the phylogenetic affiliation of Nitrospina could finally be assessed.
Materials and Methods
Cultivation
Cells of N. gracilis were grown in 5 l batch cultures in mineral salts medium prepared with 70% seawater containing 0.3 mM nitrite as the sole energy source. The marine medium (Watson and Waterbury, 1971) was composed of 700 ml filtered seawater and 300 ml distilled water, supplemented with 0.005 g l−1 CaCl2 × 2 H2O, 0.1 g l−1 MgSO4 × 7 H2O, 0.001 g l−1 FeSO4 × 7 H2O, 0.0017 g l−1 KH2PO4, 6 μg l−1 CuSO4 × 5 H2O, 25 μg l−1 Na2MoO4 × 2 H2O, 50 μg l−1 MnCl2 × 4 H2O, 0.5 μg l−1 CoCl2 × 6 H2O, and 25 μg l−1 ZnSO4 × 7 H2O. The pH was adjusted to 6.5–7.0 and changed to 7.4–7.6 within 2 days after autoclaving. Cultures were started with 1% inoculum and incubated at 28°C in the dark. When nitrite was consumed for the first time, the culture bottles were started to be stirred moderately. Nitrite consumption was measured and substrate was replenished regularly. To check for purity of the culture, aliquots were incubated in heterotrophic marine medium (0.15 g l−1 bactopeptone, 0.15 g l−1 yeast extract, 0.055 g l−1 sodium pyruvate in marine mineral salts medium) without nitrite.
DNA Extraction, Genome Sequencing, and Annotation
High-molecular weight genomic DNA was isolated following the hexadecyltrimethylammonium bromide (CTAB) protocol as provided by the DOE Joint Genome Institute (JGI, http://my.jgi.doe.gov/general/protocols.html) with minor modifications. Shortly, cells were resuspended in TE buffer and digested by the sequential addition of lysozyme and 10% (w/v) sodium dodecylsulfate (SDS) and Proteinase K. Following the subsequent incubation in CTAB/NaCl solution, the DNA-containing aqueous phase was extracted in Roti-Phenol/Chloroform/Isoamylalcohol (Carl Roth, Karlsruhe, Germany) and washed in Roti-C/I once to remove residual phenol. After DNA precipitation with isopropanol and washing with 70% ethanol, the DNA was suspended in TE buffer and stored at −20°C. Genome sequencing was performed at LGC Genomics (Berlin, Germany) using GS FLX Titanium sequencing technology. One 1/4 picotiter plate was run with an 8 kb paired-end large-insert library. Sequence read filtering and correction, contig assembly, and scaffold formation were done by LGC Genomics.
The draft genome of N. gracilis was integrated into the MicroScope annotation platform (Vallenet et al., 2009). After automated prediction and annotation of coding sequences (CDS), the annotation of all CDS in key pathways, including those for nitrite oxidation, respiration, and carbon fixation, was manually refined by using the respective tools of MaGe (Vallenet et al., 2006) as described in detail elsewhere (Lücker et al., 2010).
The draft genome sequence of Nitrospina gracilis strain 3/211 has been deposited at EMBL-EBI under project number PRJEB1269.
Amplification, Cloning, and Sequencing of nxrB Genes
As both copies of the operon containing the genes of nitrite oxidoreductase (NXR) were incomplete and lacked the nxrB gene coding for the beta subunit of NXR, these regions were specifically PCR-amplified from genomic DNA, cloned in E. coli by TOPO-TA Cloning (Life Technologies, Paisley, UK), and Sanger-sequenced (3130xl Genetic Analyzer, Applied Biosystems, Vienna, Austria). Operon-specific PCR was carried out using the newly designed primers Nspn_nxrA1_3064f (CACTCTTGCTGGACGTCA; forward) in combination with Nspn_nxrC1_111r (CATATCCACAACCACGTG; reverse) and Nspn_nxrA2_3064f (CATTCAGCATGGCAGAGC; forward) with Nspn_nxrC2_111r (CAAATCGATCACCACTCC; reverse). These primer pairs target the C-terminal ends of the alpha subunit genes nxrA and the N-terminal parts of the gamma subunits nxrC. Cycling conditions were as follows: an initial denaturation step (94°C, 5 min) was followed by 35 cycles of template denaturation (94°C, 30 s), primer annealing (52°C, 30 s), and product elongation (72°C, 1.5 min). Cycling was completed by a final elongation step (72°C, 20 min). The cloned amplicons were sequenced using M13 primers.
nxrB sequences obtained in this study have been deposited at GenBank under accession numbers KC262217 and KC262218.
Phylogenetic Analyses
An amino acid sequence database of heme-copper oxidase-family enzymes was established using the software ARB (Ludwig, 2004). Multiple sequence alignments were generated automatically using ClustalW2 (Larkin et al., 2007) and manually refined using the sequence editor included in ARB. Protein sequences of the NxrA and NxrB subunits of NXR were imported into existing datasets of type II DMSO reductase-family molybdopterin cofactor-binding proteins (Lücker et al., 2010). The sequences were aligned using the automatic aligner integrated in ARB, followed by manual refinement.
Amino acid sequences of 49 established phylogenetic marker proteins (Table A1 in Appendix) were downloaded from the Microbial Genome Database for Comparative Analysis (Uchiyama et al., 2010). Ortholog clusters were manually filtered to include bacterial sequences only. To avoid erroneous phylogenetic signals caused by horizontal gene transfer (HGT) or incorrect annotations, organisms with more than one entry for a query protein were excluded. This resulted in datasets with 443–502 entries for each protein. The single-protein datasets were automatically aligned with ClustalO (Sievers et al., 2011), running two iterations, followed by filtering for badly aligned regions or sites of rare insertions by Gblocks (Talavera and Castresana, 2007) with settings “Minimum Length of an Initial Block (−b0) = 2,” “Maximum Number of Contiguous Non-conserved Positions (−b3) = 50,” “Minimum Length of A Block (−b4) = 3,” and “Allowed Gap Positions (−b5) = all” to obtain less stringent filtering. The filtered alignment files were subsequently concatenated using an in-house generated C++ program and imported into ARB.
Phylogenetic analyses were performed by applying maximum-likelihood, maximum-parsimony, and Bayesian interference methods: RAxML [version 7.0.3 (Stamatakis et al., 2005); 100 rapid or 100 advanced bootstrap iterations], PhyML (Felsenstein, 2005), ProML and ProtPars [PHYLIP package, version 3.6 (Guindon and Gascuel, 2003); 100 bootstrap iterations], TreePuzzle [version 5.0 (Schmidt et al., 2002)], and MrBayes [version 3.1 (Ronquist and Huelsenbeck, 2003); 2–5 M generations] using the JTT, WAG, or Blosum62 substitution models. Where applicable, N-terminal signal peptide sequences were excluded from the analyses and manually created indel filters were used.
Phylome Construction
For the calculation of phylogenetic trees for each protein of the in silico deduced proteome of N. gracilis, the software PhyloGenie (Frickey and Lupas, 2004) was used as previously described (Lücker et al., 2010). The reference database for PhyloGenie was generated from the National Center for Biotechnology Information non-redundant protein database NCBI nr (Sayers et al., 2011), in which taxon names were edited to remove characters that would interfere with the export of phylogenetic trees in the Newick file format. The NCBI taxonomy database name file was adapted in a similar manner. PhyloGenie analysis was performed for each query protein using the default parameters with the following modification: −blammerparams = −taxid f. For the BLAST (Altschul et al., 1990) searches in PhyloGenie, NCBI BLAST (version 2.2.19) was used. Protein phylogenies were calculated on the basis of full or partial automatic alignments produced by the BLAMMER program included in PhyloGenie. Trees were calculated using RAxML (Stamatakis et al., 2005) with the JTT substitution model and 100 bootstrap iterations. All trees were subject of specific taxonomic queries using Phat (Frickey and Lupas, 2004). Furthermore the taxonomy of closest homologs was determined from these trees since this strategy is less prone to alignment artifacts than best hits from automatic multiple sequence alignments. An in-house Perl script determined the tree node closest to the respective query proteins from N. gracilis and consecutively sorted all operational taxonomic units behind this node by their distances to the query. The organism with the smallest distance was considered to contain the closest homologous protein.
Results
Genome Reconstruction
The obtained sequence reads were assembled into 109 contigs, which were arranged in four scaffolds. The largest scaffold encompassed 106 contigs, whereas the remaining three scaffolds consisted of single contigs whose location relative to the other contigs could not be determined unambiguously. The presence of frameshift mutations in some essential and highly conserved genes indicates that some pyrosequencing-associated errors could not be detected and corrected during read filtering and assembly. The obtained genome sequence of N. gracilis has a size of 3,067,213 bp and an average G + C content of 56.2%. It contains 3,147 predicted coding sequences (CDS; Table 1), but the large number of gene fragments (due to sequence gaps within genes and frameshifts) likely causes an overestimation of the CDS count. The genome sequence contains 1 complete rrn operon and 45 tRNA genes (1–5 for each of the 20 amino acids). The sequenced genome is near-complete based on the full set of tRNAs, the presence of all 127 clusters of orthologous groups (COGs) of proteins conserved in all 50 bacterial genomes currently in the COG database (Tatusov et al., 2003), and the estimated small gap sizes within the large scaffold.
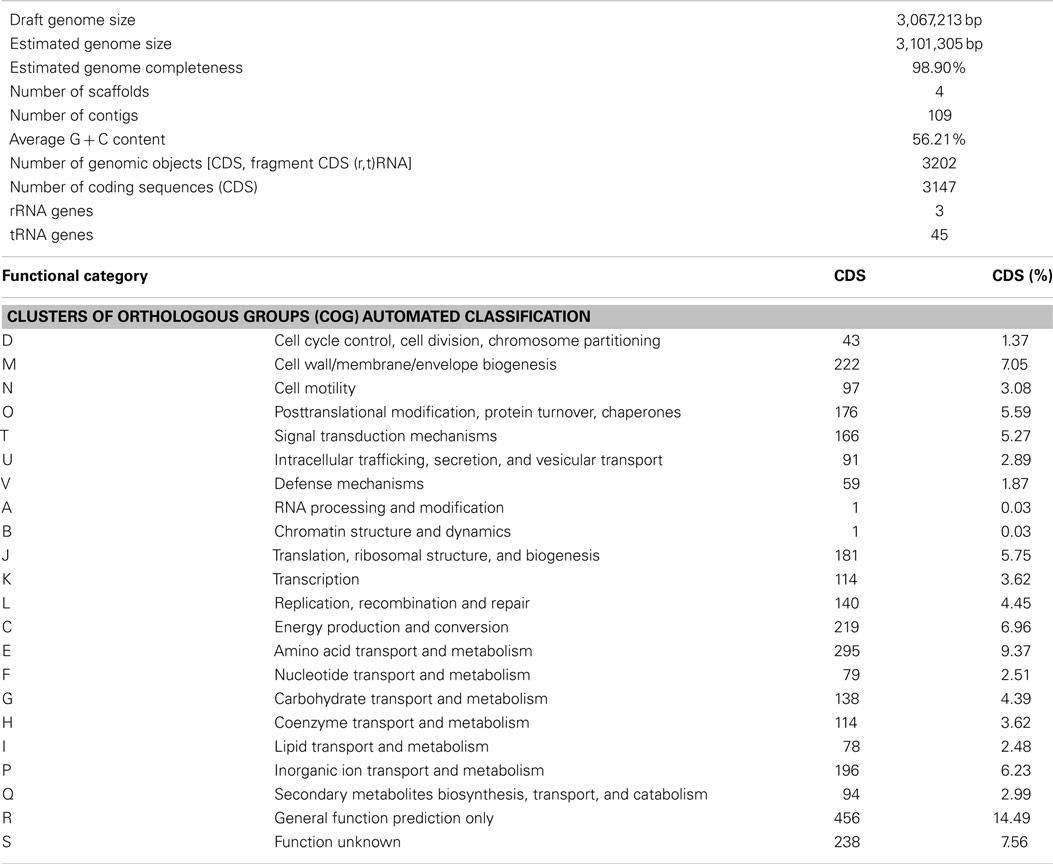
Table 1. Overview of key features of the Nitrospina gracili s strain 3/211 genome.
Notably, the smallest of the four scaffolds contained only an nxrB gene coding for the beta subunit of nitrite oxidoreductase (NXR). The NXR operon, which contains the genes of the alpha (nxrA), beta (nxrB), and gamma (nxrC) subunits, is duplicated in the N. gracilis genome. Since the sequences of the two nxrB genes are highly similar, the complete NXR operons could not be unambiguously assembled from the sequence reads. Therefore, operon-specific primers where designed and used to amplify the genomic regions between nxrA1 and nxrC1, and nxrA2 and nxrC2, respectively, in order to obtain by cloning and Sanger-sequencing the complete operon sequences including nxrB. When translated into their protein sequence, the two NxrB copies had a length of 425 amino acids and an identity of 100% (with five synonymous single-base substitutions on the nucleotide level).
Phylogeny
A set of 49 established phylogenetic marker proteins (Table A1 in Appendix) was extracted from the genome of N. gracilis and a range of other complete bacterial genomes covering all bacterial phyla for which genome sequences are available. The concatenated dataset of these proteins enabled a detailed phylogenetic analysis of the affiliation of Nitrospina within the Bacteria. Preliminary trees containing sequence information from 502 bacteria indicated deep phylogenetic branching of N. gracilis between the Proteobacteria and a group consisting of the Acidobacteria and Nitrospirae phyla (data not shown). Consecutively, a reduced dataset containing representatives of these and few other closely affiliated phyla was generated and used for more detailed analyses employing different tree calculation methods. These analyses clearly indicated a position of N. gracilis close to, but outside the Proteobacteria (Figure 1) and not within any other bacterial phylum either. Notably, the Epsilonproteobacteria did not cluster with the other Proteobacteria, a branching pattern that has been observed previously (Ludwig, 2010), and formed a separate branch together with the Aquificae. This grouping of Aquificae and Epsilonproteobacteria is consistent with other marker protein-based analyses that also found the Aquificae not to group as one of the most basal bacterial phyla (Korbel et al., 2002; Cavalier-Smith, 2006). The Acidobacteria and Nitrospirae formed a superphylum in the Bayesian interference analysis that grouped as the next most closely related phyla to Nitrospina (Figure 1A), followed by the aforementioned Aquificae/Epsilonproteobacteria group and the Deferribacteres. Maximum-likelihood analysis grouped Nitrospina as sister phylum of the Acidobacteria, with the Nitrospirae forming a separate branch (Figure 1B), and the maximum-parsimony approach indicated a superphylum of all three groups (Figure 1C). The separation of Nitrospina from the Proteobacteria thus is supported by all analyses, and the grouping with the Acidobacteria and Nitrospirae in some analyses strongly contradicts the possibility that Nitrospina forms a basal new class within the Proteobacteria.
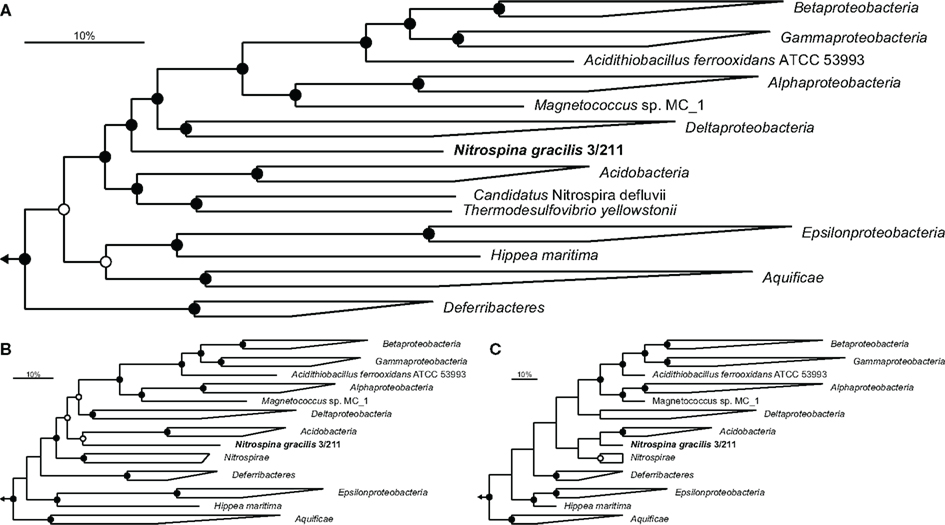
Figure 1. Phylogenetic affiliation of N. gracilis 3/211 (boldface). Trees were calculated using a concatenated dataset of 49 proteins (Table A1 in Appendix). Members of the phylum Planctomycetes were used as outgroup. Filled and open circles represent statistical support ≥90 and >75%, respectively. The scale bar represents 10% estimated sequence divergence. (A) Bayesian inference tree (SD = 0.009985) run for 8 M generations. In total, 6962 alignment positions were considered. (B,C) Maximum-likelihood and maximum-parsimony analysis, respectively, with 100 bootstrap iterations, using 7629 alignment positions.
To further assess the separate phylogenetic position of N. gracilis as member of a novel phylum, two additional methods were employed: comparison of shared conserved insertion and deletion signature sequences (indel analysis; Gupta and Griffiths, 2002) and gene transposition analysis (Kunisawa, 2006). Both analyses were consistent with the calculated phylogenetic trees by suggesting that N. gracilis belongs to a phylum related to, but separate from the Proteobacteria. N. gracilis carries the characteristic insertions in the transcription termination factor Rho and the alanine-tRNA ligase AlaS, which are common to all Proteobacteria and a range of phyla including the Chlamydiae and Aquificae (Gupta, 2000; Gupta and Griffiths, 2002), but lacks signature sequences in the inorganic phosphatase Ppi, molecular chaperone DnaK, and CTP synthetase PyrG that distinguish the Proteobacteria from the other phyla (Gupta, 2000, 2001). Analyses of gene transposition events (Kunisawa, 2010, 2011) also indicated a close relationship to the Proteobacteria, Nitrospirae, and to a lesser extent the Deferribacteres and Aquificae, but clearly distinguished N. gracilis from these established phyla based on unique differences in bordering genes.
Another way to look at ancestry and identify HGT events at the same time is by phylome reconstruction, which is the determination of the relationship of every protein encoded in a genome by phylogenetic inference (Frickey and Lupas, 2004). If the exact phylogenetic position of an organism is known this is an elegant method to determine the fraction of horizontally transferred genes and to identify potential donor organisms. For organisms of uncertain affiliation, the phylomic information may reveal ancestral relationships since most genes are inherited vertically. Therefore, the phylome of N. gracilis was reconstructed and the closest relative (based on the distance to the respective homolog in N. gracilis) was extracted from each single-protein tree. Of the 2,354 proteins of N. gracilis that have homologs in public databases, a large part clustered on the class level with Deltaproteobacteria (n = 514), followed by Gammaproteobacteria (n = 225) and Nitrospira (n = 211). On the genus level Nitrospira had most hits (n = 161), followed by the deltaproteobacterial Geobacter (n = 81), the recently described “Candidatus Methylomirabilis” (Ettwig et al., 2010; n = 68), and “Candidatus Kuenenia” (Strous et al., 2006; n = 51). The numerous closest homologs in Proteobacteria and Nitrospira are consistent with the phylogenetic trees as well as the indel and gene transposition analyses. The large number of proteins clustering with Nitrospira may also reflect a high degree of functional similarity in Nitrospina and Nitrospira. In contrast, the large number of proteins affiliated to homologs in Methylomirabilis and Kuenenia was unexpected and might indicate a large extent of HGT between these organisms and Nitrospina. One possible caveat of the applied tree query approach is that the next neighbors in a tree may not always have the shortest distance to each other. However, manual reassessment by using Phat within the PhyloGenie program (Frickey and Lupas, 2004) revealed the same trends for the relationship of Nitrospina proteins to the aforementioned groups (Figure 2). These results strongly corroborate the phylogenetic placement of Nitrospina in a distinct phylum with the Proteobacteria and Nitrospirae as most closely related known phyla.
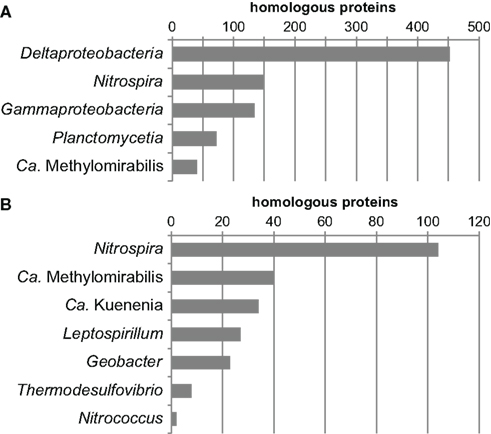
Figure 2. Summarized results of a phylogenetic analysis of the phylome of N. gracilis 3/211. The nearest phylogenetic neighbor (closest homolog) in other sequenced genomes was determined for each protein of N. gracilis. The resulting trees were queried for the closest homolog using Phat (Frickey and Lupas, 2004). The graphs depict the phylogenetic groups displaying the largest numbers of most closely related homologs on the (A) class and (B) genus level. In total, Phat could unambiguously assign closest homologs for 2,246 proteins.
Nitrite Oxidation and Nitrogen Metabolism
N. gracilis is a chemolithoautotrophic nitrite-oxidizing bacterium. It gains all energy required for growth from the oxidation of nitrite and fixes CO2 as sole carbon source, using electrons derived from nitrite oxidation. The key enzyme for nitrite oxidation is nitrite oxidoreductase, which shuttles two electrons per oxidized into the electron transport chain. The NXR belongs to the type II group of molybdopterin-binding enzymes within the DMSO reductase enzyme family (Jormakka et al., 2004). These enzymes are membrane-associated complexes of three subunits. The catalytic alpha subunit contains the molybdenum bis molybdopterin guanine dinucleotide (Mo-bis-MGD) cofactor and one [4Fe–4S] iron-sulfur (Fe-S) cluster. The beta subunit contains one [3Fe-4S] and three [4Fe-4S] cluster and transfers the electrons from the alpha subunit to the membrane-integral gamma subunit (Martinez-Espinosa et al., 2007). This membrane subunit functions as membrane anchor of the holoenzyme and channels the electrons to and from the electron transport chain via one or two hemes (Rothery et al., 2008).
N. gracilis contains two operons encoding putative NXR, which consist of the genes coding for all three subunits in an nxrABC order (Table S1 in Supplementary Material). The two operon copies are highly similar, with an overall identity of 94.6% on the nucleotide level. When translated into their protein sequences, the α (NxrA), β (NxrB), and γ (NxrC) subunits have identities of 94.9, 100, and 87.5%, respectively. NxrA1 and NxrA2 contain an N-terminal twin-arginine motif for export into the periplasmic space via the twin-arginine protein translocation (Tat) pathway. Both NxrB copies lack a signal peptide but may be co-translocated into the periplasm with the large subunit by a “hitchhiker” mechanism by the Tat pathway as proposed for “Candidatus Nitrospira defluvii” (Lücker et al., 2010) and observed for other members of the DMSO reductase-family of molybdopterin-binding enzymes (Martinez-Espinosa et al., 2007). The two γ subunits encoded in the NXR operons are predicted by Pfam (Finn et al., 2010) to contain a DMSO reductase-like heme b-binding domain and have a predicted N-terminal signal peptide that overlaps with a predicted single transmembrane helix. The Phobius (Käll et al., 2007) and SignalP 4.0 (Petersen et al., 2011) prediction methods favor secretion of NxrC into the periplasm via the Sec protein tanslocation pathway. This is puzzling, since NxrC of Ca. N. defluvii and the NarI homologs in NarG-like molybdopterin-binding enzymes are membrane-integral (Rothery et al., 2008; Lücker et al., 2010). It implies that NxrC of N. gracilis is a soluble heme b subunit as found in selenate (Ser) and chlorate (Clr) reductases (Bäcklund et al., 2009; Lowe et al., 2010) and ethylbenzene (Ebd) and dimethylsulfide (Ddh) dehydrogenases (Kniemeyer and Heider, 2001; McDevitt et al., 2002). These enzymes form soluble periplasmic protein complexes and interact with soluble cytochrome c proteins, which shuttle electrons to and from the membrane-integral respiratory chain (Lowe et al., 2010). In Nitrospina, however, the NXR complex is membrane-associated (Bartosch et al., 1999; Spieck and Bock, 2005b). Thus, an additional membrane anchor likely is required for the formation of active NXR in this organism. In addition to the two linked copies of NxrC, N. gracilis has genes of two other putative γ-subunits (Table S1 in Supplementary Material). NxrC3 is located in the direct vicinity of a TorD-like chaperone that most likely functions in Mo cofactor insertion into NxrA prior to Tat export, as has been proposed for other Mo-bis-MGD-containing enzymes such as selenate reductase (Guymer et al., 2009). This NxrC has one predicted membrane helix as well as the DMSO reductase-like heme b-binding domain. NxrC4 is encoded in a genetic context unrelated to nitrite oxidation and shows only low similarities to the other NxrC copies, but also contains one predicted transmembrane helix and the heme b-binding domain. Aside from these candidate NXR γ subunits, the genome of N. gracilis codes for at least one putative alternative NxrC (alt_NxrC1, Table S1 in Supplementary Material). This protein is remarkable since it contains not only two transmembrane helices, but also two cytochrome c-type heme c binding sites. The heme c groups might have an important function in nitrite oxidation, since cytochrome c is predicted to be the primary acceptor of the high-potential electrons derived from nitrite in NOB (; Sundermeyer-Klinger et al., 1984). Thus, electrons might be channeled through NxrB and possibly NxrC to the alt_NxrC subunit, where the c-type hemes could shuttle the electrons to acceptors downstream in the respiratory chain. The genome contains three additional copies of this putative alternative γ subunit (Table S1 in Supplementary Material), which all contain 1–3 cytochrome c binding sites and an additional DMSO reductase-like heme b-binding domain. However, alt_NxrC2 and 4 are predicted to be periplasmic rather than membrane-integral and might function as soluble electron transfer proteins as described for cytochrome c4 and selenate reductase (Lowe et al., 2010). Future studies may resolve the true subunit composition of the NXR complex in Nitrospina.
The N. gracilis genome encodes two copies of copper-containing nitrite reductase NirK (Figure 3; Table S1 in Supplementary Material). NirK1 contains all conserved amino acid residues required for binding of the two Cu(II) sites and of nitrite. In the second copy NirK2 the weak ligand of the type 1 copper center methionine is mutated to lysine. NirK1 thus is expected to be a functional nitrite reductase, while NirK2 might have reduced or altered activity (Wherland et al., 2005). Interestingly, all analyzed genomes of NOB except Nitrolancetus hollandicus (Sorokin et al., 2012) contain nirK genes, but the function of nitrite reduction in NOB has not clearly been determined yet. No known nitric oxide (NO) reductase is encoded in the N. gracilis genome and a role of NirK in denitrification therefore seems unlikely. NO stimulated NADH formation in Nitrobacter and was speculated to be an intermediate of nitrite oxidation in this organism (Freitag and Bock, 1990; Poughon et al., 2001). A later study proposed an alternative role of NO in the regulation of the cellular redox state in Nitrobacter (Starkenburg et al., 2008). In this model the terminal cytochrome c oxidase is reversibly inhibited by NO under low-oxygen conditions, leading to an increased flow of electrons through the reverse electron transport chain. This causes higher CO2 fixation rates and may increase the biosynthesis of the storage compound poly-β-hydroxybutyrate (Starkenburg et al., 2008). For NOB other than Nitrobacter no function of NirK has been proposed, and denitrification of nitrite to NO under anoxic conditions has not been observed yet.
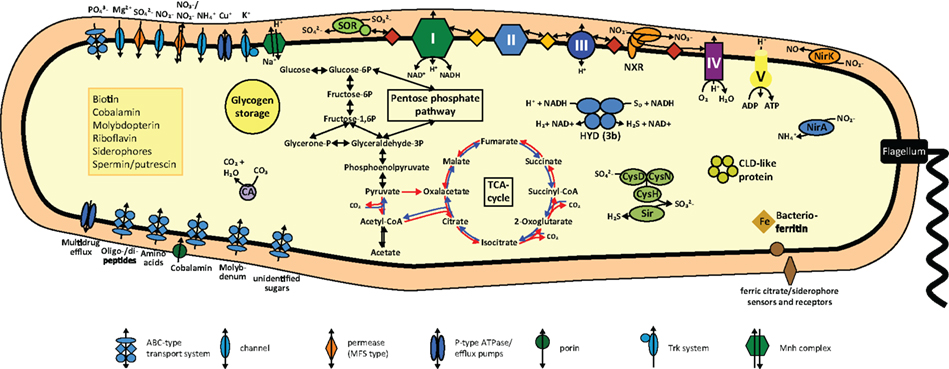
Figure 3. Cell metabolic cartoon based on the annotation of the N. gracilis 3/211 genome. CLD, chloride dismutase; CA, carbonic anhydrase; CysDNC, sulfate adenylyltransferase/adenylylsulfate kinase; HYD, hydrogenase; NirA, ferredoxin-nitrite reductase; NirK, Copper-containing nitrite reductase; NXR, nitrite oxidoreductase; Sir, ferredoxin-sulfite reductase; SOR, Sulfite:cytochrome c oxidoreductase. Enzyme complexes of the electron transport chain are labeled by Roman numerals. Red and orange diamonds represent cytochrome c proteins and quinones, red and blue arrows the oxidative and reductive TCA cycle, respectively.
N. gracilis grows on nitrite as sole source of both energy and nitrogen. Consistently, the genome contains the nirA gene for assimilatory ferredoxin-nitrite reductase, which is located directly next to nirC encoding a nitrite transporter of the formate/nitrite transporter family (Figure 3; Table S1 in Supplementary Material). A second nitrite transporter of the NarK nitrate/nitrite antiporter type, as well as two Amt-type ammonium transporters, likely is also involved in nitrogen uptake for assimilation (Table S1 in Supplementary Material).
Energy Metabolism and Reverse Electron Transport
N. gracilis encodes a terminal oxidase of the cytochrome cbb3-type (Figure 3; Table S1 in Supplementary Material). As the cbb3-type terminal oxidases have high affinities for O2 (Cosseau and Batut, 2004), its presence will allow Nitrospina to sustain respiration in low-oxygen environments. The electrons derived from nitrite oxidation most likely are shuttled via cytochrome c to this terminal oxidase for reduction of O2 as terminal electron acceptor, yielding the energy for the formation of a proton gradient across the cytoplasmic membrane. The proton motive force drives ATP formation by a canonical F1F0 ATPase (Figure 3; Table S1 in Supplementary Material). Interestingly, the three subunits of the cytochrome cbb3 oxidase of Nitrospina are fused into one gene encoding a protein of 954 aa. The N-terminal part of this protein corresponds to subunit I, has 13 predicted transmembrane helices, and contains most of the characteristic binding sites for two heme b and the low-potential copper (CuB) center. The C-terminal part corresponds to subunits II and III with one and two heme c binding sites, respectively. Interestingly, one amino acid replacement in the CuB center is unprecedented in heme-copper oxidases (Figure 4). In all members of this family the copper is complexed by three histidine residues, which are numbered His207, His257, and His258 in the cytochrome cbb3 oxidase of Pseudomonas stutzeri whose structure has been determined (Buschmann et al., 2010). At position 203 (P. stutzeri numbering) most of these enzymes contain a highly conserved tryptophan that is involved in passing electrons from the high-spin heme b3 to the CuB center where oxygen is reduced (Zaslavsky and Gennis, 2000; Pereira et al., 2001). This tryptophan is replaced by glycine in the N. gracilis enzyme, thus clearly impeding any charge transfer. Directly upstream, however, is an additional histidine residue found only in N. gracilis (His202; P. stutzeri numbering) whose positively charged side chain may adopt the role of the hydrophobic Trp203 in electron transfer. In addition, two uncommon tyrosine residues flanking the His207 at positions 205 and 208 (P. stutzeri numbering) might also function as electron carriers. Besides Nitrospina, the His202, Gly203, and Tyr208 residues are found only in subunit I of putative heme-copper oxidases from various Sinorhizobium genomes (e.g., Sinorhizobium meliloti SM11, Figure 4), where they occur in one of several homologous cytochrome cbb3 oxidases. Their presence in other organisms strongly suggests that these unusual amino acid substitutions are not sequencing errors in the N. gracilis genome.

Figure 4. Partial multiple sequence alignment of selected heme-copper oxidase-family members. Relationship of the enzymes is indicated by a cladogram. The oxidase type is indicated for each enzyme. Amino acid residues involved in the formation of the copper (CuB)-binding site are highlighted in color and by a symbol indicating function: dark blue, histidine residues involved in CuB-binding (c); purple, residues involved in electron transport from the high-spin heme to CuB(*), red, uncommon amino acids; orange, crosslinking tyrosine residue stabilizing one of the CuB-binding histidines (#), green and turquoise, alternative conserved residues in this position.
Nitrospina depends on the electrons derived from nitrite oxidation for the regeneration of reducing equivalents and for autotrophic CO2 fixation by the reductive tricarboxylic acid (rTCA) cycle. These electrons enter the respiratory chain at the level of cytochrome c due to the high redox potential of the / couple. Therefore, reverse electron transport to NAD+ () and to ferredoxin () is required and most likely powered by the proton motive force across the cytoplasmic membrane. Mechanistically, the cytochrome bc complex and NADH-quinone oxidoreductase (complexes III and I of the respiratory chain, respectively; Elbehti et al., 2000) are involved in electron transport from cytochrome c to NAD+, whereas a novel type of complex I may catalyze the reduction of ferredoxin. In addition, electrons can be shuttled from the quinone pool into the rTCA cycle by the succinate dehydrogenase/fumarate reductase (complex II; Lemos et al., 2002). Moreover, the use of organic storage compounds such as glycogen as energy source requires a respiratory chain for the forward transport of electrons. Genome-based hypotheses on forward and reverse electron transport in N. gracilis are outlined in the following paragraphs, in the order of electron flow from cytochrome c to NAD+ and ferredoxin.
The canonical cytochrome bc complex in bacteria is composed of three subunits, a Rieske-type [2Fe-2S] subunit, a cytochrome b subunit that contains the quinol binding site, and a c-type cytochrome. The cytochrome c subunit is least conserved among bacteria and usually is of a mono- or di-heme, sometimes also a tetraheme, type (Ilbert and Bonnefoy, 2012). N. gracilis has one operon that shows moderate similarity to complex III (Table S1 in Supplementary Material). The three proteins encoded in this cytochrome bc-like operon show highest similarity to the cyanobacterial cytochrome b6f rather than to the bc1 complex. They consist of a Fe-S subunit next to a split cytochrome b subunit. This fission of the cytochrome b into a cytochrome b6 and a so-called subunit IV is observed in many non-phototrophic bacteria that have this type of complex III (Sone et al., 1995; Schütz et al., 2000). Notably, Nitrospina carries elsewhere in the genome a second copy of the cytochrome b subunit that is not split, but also appears to be of the b/b6-type (Table S1 in Supplementary Material). The only putative cytochrome c subunit encoded next to the cytochrome bc-like operon is an atypical octaheme cytochrome c. Interestingly, the exactly same operon structure including a similar octaheme protein is conserved in the genome of the anammox organism “Candidatus Kuenenia stuttgartiensis” (de Almeida et al., 2011) and in “Candidatus Methylomirabilis oxyfera,” a noteworthy bacterium capable of nitrite-driven anaerobic methane oxidation (Ettwig et al., 2010).
The genome of N. gracilis contains the genes for all 14 subunits of NADH:quinone oxidoreductase (NUO; complex I) needed for the forward transport of electrons from NADH to quinone (Figure 3; Table S1 in Supplementary Material). All Fe-S clusters required for electron channeling between NADH and quinone are preserved, as are the NADH and flavin mononucleotide (FMN)-binding patterns within subunit NuoF. The nuoA-M genes are clustered in one operon on the lagging strand, whereas nuoN is located on the opposite strand five genes downstream of nuoA. Besides the canonical NADH:quinone oxidoreductase (NUO-1), a second copy is present, but the genes coding for this NUO-2 are distributed across the genome (Table S1 in Supplementary Material). While nuoB2 and nuoF2 are not organized in an operon structure, the other genes of NUO-2 form two clusters consisting of nuoACHIJKLMN and nuoEGD, respectively. Intriguingly, this complex shows substantial differences to NUO-1 in the binding patterns for cofactors in the catalytic subunits. NADH in canonical NUOs (including NUO-1 of N. gracilis) binds to NuoF, where two electrons are transferred onto the primary electron acceptor FMN that serves as a two-to-one-electron converter (Brandt, 2006). Consecutively, FMN feeds single electrons into a chain of seven conserved Fe-S clusters distributed throughout subunits NuoE, G, I, and B in the hydrophilic arm of the complex, at the end of which quinone reduction occurs (Sazanov and Hinchliffe, 2006). The N. gracilis NuoF2 lacks the FMN binding site, but contains one [2Fe-2S] and two [4Fe-4S] ferredoxin-like clusters in addition to the Fe-S cluster N3 and the NADH binding domain. Similarly, NuoG2 has the characteristic cysteine binding patterns for Fe-S clusters N1b, N4, N5, and N7, along with one additional [4Fe-4S] cluster. Without the FMN cofactor a function as NUO is unlikely, but a possible alternative role for NUO-2 might be in the reverse electron transport from quinol to low-potential ferredoxin. Two enzymes of the rTCA cycle (see below) belong to the 2-oxoacid:ferredoxin oxidoreductase family and require reduced low-potential ferredoxin as electron donor for the reductive carboxylation of their substrates. Common mechanisms to reduce low-potential ferredoxin with electrons from NADH (Rnf complex; Biegel and Müller, 2010) or H2 (Ech complex; Welte et al., 2010) employ membrane-integral complexes that use the proton motive force for reverse electron transport to ferredoxin ( to -420 mV). These complexes evolved from a NUO-like ancestor (Buckel and Thauer, 2013). Nitrospina lacks these known pathways, but the NUO-2 complex is an interesting candidate to perform ferredoxin reduction with quinol as electron donor. Due to the lack of the FMN binding site, the NuoF2 subunit cannot interact with a two-electron carrier such as NAD+/NADH. However, as NADH and ferredoxin binding sites display homology in some proteins (Hanke et al., 2011), the predicted NADH binding site of NUO-2 might interact with the single-electron carrier ferredoxin. Powered by proton motive force and with the help of the four additional Fe-S clusters in NuoG2 and NuoF2, two of which are of the bacterial [4Fe-4S] ferredoxin type, it might be possible to elevate electrons from the quinol pool not only to the redox potential of NADH, but further to the potential of ferredoxin. Indeed, the ferredoxin-like [4Fe-4S] clusters 6a and 6b of NuoI display a negative shift of their redox midpoint potential of <600 mV when expressed heterologously without incorporation into the NUO complex (Yano et al., 1999), and the NuoI homolog in Ech is responsible for ferredoxin reduction (Forzi et al., 2005). The extra [4Fe-4S] clusters in NuoF2 could thus be sufficiently electronegative to interact with ferredoxin. Interestingly, in Ca. K. stuttgartiensis a highly similar complex is present that also contains all additional Fe-S clusters in the NuoGF subunits and apparently lacks a FMN binding site. The organization of all corresponding genes in one operon in Kuenenia strongly suggests that their products are subunits of the same enzyme complex. The reduction of ferredoxin by reverse electron transport is important also in Kuenenia, which employs the acetyl-coenzyme A pathway for CO2 fixation (Strous et al., 2006). The NUO-2 complex of Kuenenia has been tentatively annotated as formate:quinone oxidoreductase (Strous et al., 2006), but biochemical data endorsing this function is lacking. Future experimental work may reveal whether the NUO-2 complex indeed is a novel mechanism for reverse electron transport spanning the large difference in redox potential between quinol and ferredoxin.
Complex II, the succinate:quinone oxidoreductase (SQR), links the TCA cycle to the quinone pool. This complex can work in both directions. It catalyzes either the oxidation of succinate in the course of the oxidative TCA cycle and oxidative phosphorylation, or the reduction of fumarate to channel electrons into the rTCA cycle (Cecchini et al., 2002). SQRs are classified in five types (A–E; Lemos et al., 2002) and consist of three (type B and E) or four (type A, C, and D) subunits. Many organisms encode a separate succinate dehydrogenase (SDH) and fumarate reductase (FRD), which are expressed differentially under oxic and anoxic conditions, but can also replace each other functionally (Maklashina et al., 1998). SDH and FRD in the same organism usually are similar in sequence and subunit composition, or only one enzyme is present. In contrast, N. gracilis contains two highly different versions of SQRs. One enzyme is of type B that is found also in bacilli and Epsilonproteobacteria, whereas the other one belongs to type E found mainly in Archaea (Lemos et al., 2002). Both types consist of three subunits (Table S1 in Supplementary Material). The type B SQR-1 of N. gracilis consists of a FAD-binding flavoprotein, a Fe-S protein containing all conserved cysteine residues for binding one [2Fe-2S], one [4Fe-4S], and one [3Fe-4S] cluster, and a diheme cytochrome b membrane subunit with five predicted transmembrane helices (Lancaster and Simon, 2002). This SQR type is a good candidate for a fumarate reductase, because it interacts with menaquinone (MQ) rather than ubiquinone (Schirawski and Unden, 1998) and MQ has a lower redox potential () than the succinate/fumarate couple (). In Bacillus subtilis fumarate reduction with menaquinol catalyzed by a type B SQR is electrogenic, so that the reaction contributes to the proton gradient over the cytoplasmic membrane (Schnorpfeil et al., 2001; Lancaster et al., 2008). However, the quinone types present in Nitrospina are not known yet. The type E SQR-2 has some fundamental differences to the type B SQR-1 in its subunit composition and predicted binding sites for prosthetic groups. The large subunit also is a FAD-binding flavoprotein, but it has only low similarity to the corresponding subunit of SQR-1. The Fe-S subunit contains a second [4Fe-4S] cluster instead of the [3Fe-4S] cluster (Gomes et al., 1999), resulting in a slightly lower redox potential of the binuclear center (Lemos et al., 2001). Further, type E SQRs contain an unusual membrane subunit (SdhE) that is unrelated to the membrane anchors of the SQR types A–D. The respective gene is split in the N. gracilis genome sequence, but this may be caused by an uncorrected sequencing error. SdhE does not bind heme b, has only one small putative membrane-spanning segment at the C-terminus, and contains two tandem repeats of the conserved cysteine motif CX31CCGX34CX2C (Janssen et al., 1997). These motifs bind an additional high-potential [2Fe-2S] cluster that functionally replaces the missing heme b (Iwasaki et al., 2002). Despite these differences, SdhE is essential for membrane attachment of the SQR complex, possibly mediated by amphipathic helices, and (mena-)quinone interaction (Lemos et al., 2002). Although type E SQRs can work reversibly, they appear to preferentially catalyze succinate oxidation in most organisms carrying these enzymes (Gomes et al., 1999). The slightly lower redox potential of the active center allows this SQR to efficiently donate electrons to quinone or directly to Rieske-type Fe-S proteins as found in complex III (Janssen et al., 1997). Hence, the SQR-2 of N. gracilis is the more likely candidate than SQR-1 for a SDH that is active during the degradation of carbon storage compounds.
Interestingly, N. gracilis contains genes for a cytoplasmic NiFe-hydrogenase (Figure 3; Table S1 in Supplementary Material). These genes are organized in a single operon and encode the four subunits of the holoenzyme and six accessory proteins involved in hydrogenase assembly and maturation. Sequence analysis revealed binding sites for nickel in the α subunit and for one and three [4Fe-4S] clusters in the β and δ subunit, respectively. The γ subunit contains predicted binding sites for one [2Fe-2S] cluster, FAD, and NAD(P). Characteristic amino acid patterns in the large subunit clearly classify the enzyme as a type 3b bidirectional (NADP) hydrogenase (Vignais and Billoud, 2007). Members of this group catalyze the reversible oxidation of H2 with NAD(P)+ (Silva et al., 2000). Furthermore, most type 3b hydrogenases can reduce elemental sulfur (S0) or polysulfide to H2S. Since they display higher affinities for polysulfides than for H+, they are also referred to as sulfhydrogenases (Ma et al., 2000). The physiological role of this enzyme in N. gracilis is unclear, but three functions appear to be possible: (i) When present, H2 may be oxidized to provide reduced NAD(P)H for biosynthetic processes and energy generation. (ii) Polysulfide (or H+) can be used as terminal electron acceptor during fermentation of intracellular glycogen deposits in absence of more favorable electron acceptors. (iii) Polysulfide (or S0) can be reduced to H2S for sulfur assimilation during protein biosynthesis if neither sulfate nor sulfite is available. None of these reactions has been reported for N. gracilis so far and all three possibilities seem unlikely in the aerobic marine habitat. However, the hydrogenase may be advantageous under hypoxic or even anoxic conditions (see also below).
N. gracilis encodes a periplasmic sulfite:cytochrome c oxidoreductase that may couple sulfite oxidation to sulfate with the reduction of two cytochromes (Figure 3; Table S1 in Supplementary Material). In eukaryotes this reaction mainly serves as a detoxification mechanism for sulfite formed during the degradation of sulfur-containing amino acids, but it can be used for energy conservation by sulfur-oxidizing prokaryotes (Kappler et al., 2000). Since N. gracilis also has all genes needed for sulfate assimilation via 3′-phosphoadenylyl sulfate and the subsequent reduction to sulfide, the sulfite:cytochrome c oxidoreductase is probably not involved in sulfur assimilation. Hence, N. gracilis might be able to use sulfite as alternative energy and/or electron source, but physiological data showing this metabolic capacity are lacking.
CO2 Fixation and Carbon Metabolism
According to cultivation-based experiments, N. gracilis is an obligate autotroph that meets all carbon demands by CO2 fixation (Watson and Waterbury, 1971). Surprisingly, Nitrospina employs the rTCA cycle for this task. This CO2 fixation pathway is unexpected in an aerobic organism, because two of its key enzymes belong to the 2-oxoacid oxidoreductase (OR) family whose members normally are highly oxygen-sensitive (Berg, 2011). However, Hydrogenobacter thermophilus possesses unusual five-subunit types of 2-oxoglutarate:ferredoxin (OGOR) and pyruvate:ferredoxin oxidoreductase (POR) that are functional under oxic conditions (Yoon et al., 1996, 1997). Moreover, the aerobic nitrite oxidizer Ca. N. defluvii also utilizes this pathway using such five-subunit OGOR and POR (Lücker et al., 2010). N. gracilis encodes a five-subunit POR with high identity of the subunits (60–76%) to the Nitrospira holoenzyme. The OGOR of N. gracilis lacks the δ subunit, but the remaining subunits also share high identities with their homologs in Nitrospira (53–68%; Table S1 in Supplementary Material). The δ subunit was required for heterologous expression of the H. thermophilus holoenzyme in E. coli, but it does not contain any known binding motifs for prosthetic groups (Ikeda et al., 2006). A function of this subunit in the expression or assembly of the active OGOR complex might be fulfilled by the δ subunit of POR or by a yet unidentified chaperone. The reduced ferredoxins required by the two ORs could either be provided by the alternative NUO-2 complex (see above) or by a complex involving electron transfer flavoproteins (ETFs). Clostridium kluyveri can couple the reduction of ferredoxin with NADH to crotonyl-CoA reduction, similar to the reaction in the ETF:quinone reductase in mammals (Herrmann et al., 2008). In this electron bifurcation reaction, the energy derived from the electron transfer from NADH to crotonyl-CoA is used to simultaneously catalyze the unfavorable reduction of a [4Fe-4S] ferredoxin (Buckel and Thauer, 2013). The two FAD-containing ETFs as well as a putative FAD-containing acyl-CoA dehydrogenase necessary are encoded in the N. gracilis genome (Table S1 in Supplementary Material), but it appears counterintuitive that Nitrospina should rely on the fatty acid degradation pathway for the reduction of ferredoxin during autotrophic growth. Alternatively, the ETF proteins might interact with an unidentified flavin-containing ETF:quinone reductase, that couples the ferredoxin reduction by NADH to the reduction of quinone.
Besides OGOR and POR, the presence of ATP citrate lyase (ACL) is generally considered to be a prerequisite of the rTCA cycle (Hügler et al., 2007). The N. gracilis genome contains two CDS with very low similarities to the large and small subunits of known ACLs (Table S1 in Supplementary Material). They contain the characteristic succinyl-CoA synthase-like NAD(P)- and CoA-binding domains and the ATP-grasp fold (Kanao et al., 2001) in the large and small subunit, respectively. The alternative pathway of ATP cleavage in Aquificaceae and Leptospirillum, which involves citryl-CoA synthase and citryl-CoA lyase (Hügler et al., 2007; Levican et al., 2008), could not be identified in N. gracilis. Therefore, we assume that the predicted ACL is functional despite its low similarity to validated ACLs from other organisms.
Each of the other enzymes involved in the reductive and/or oxidative TCA cycles (Berg, 2011) has one homolog in the N. gracilis genome (Table S1 in Supplementary Material). There are, however, notable differences to the typical enzyme set for the anaplerotic reactions replenishing the oxaloacetate pool: usually, oxaloacetate is formed from pyruvate either in one single step by pyruvate carboxylase or via phosphoenolpyruvate (PEP) by the enzymes pyruvate, water dikinase, and PEP carboxylase. Intriguingly, N. gracilis lacks these enzymes, but alternatives for these routes are present. Firstly, instead of pyruvate carboxylase Nitrospina appears to employ a membrane-bound oxaloacetate decarboxylase. This enzyme complex usually performs the oxidative decarboxylation of oxaloacetate and directly uses the released energy for the extrusion of sodium ions across the membrane. In the presence of a sodium gradient, however, the reaction can be reversed to carboxylate pyruvate (Dimroth and Hilpert, 1984). Such dependency on a sodium gradient to drive this reaction might also explain why Nitrospina is restricted to marine habitat types. Secondly, the two-step route via PEP can be mediated by the enzymes pyruvate, phosphate dikinase, and the ATP-utilizing PEP carboxykinase. Pyruvate, phosphate dikinase replaces pyruvate, water dikinase in many phototrophic organisms, but usually functions in ATP synthesis from PEP in chemoautotrophic bacteria (Lim et al., 2007). The second enzyme, PEP carboxykinase, is mainly involved in gluconeogenesis by replenishing PEP from oxaloacetate, but can also catalyze the carboxylation of PEP (Zamboni et al., 2004).
Observation by electron microscopy of N. gracilis cells revealed glycogen deposits as intracellular storage compounds (Watson and Waterbury, 1971). Accordingly, automatic genome annotation predicted all genes required for gluconeogenesis and glycogen formation (Figure 3; Table S1 in Supplementary Material). The stored glycogen can be catabolized and used as energy source via the Embden–Meyerhof–Parnas pathway and the oxidative (oTCA). Organotrophic growth of N. gracilis was not observed previously (Watson and Waterbury, 1971). Consistently, no transport systems for the import of organic substrates were identified in the genome except one, merely putative, ABC-type sugar transporter of unknown specificity.
Resistance and Defense
Although N. gracilis is an aerobic organism, it lacks classical defense mechanisms against reactive oxygen species (ROS) usually found in aerobic bacteria. No homolog of superoxide dismutase (SOD) or reductase (SOR) could be identified in the genome, and catalase appears to be absent, too. Hydrogen peroxide might be degraded by a cytochrome c peroxidase and peroxiredoxins, possibly assisted by the thioredoxin and glutaredoxin systems, which all are encoded in the genome. The protection mechanism against superoxide radicals is more puzzling. Ca. N. defluvii also lacks SOD and catalase, and the presence of a manganese import system has led to speculations that some resistance of this organism against ROS may be conferred by manganese (Lücker et al., 2010). Although the N. gracilis genome contains no known specific manganese import system, protection from ROS in the periplasm might be mediated by a periplasmic multicopper protein that is highly conserved in the Ca. N. defluvii genome, too. This putative multicopper oxidase shows similarity to an enzyme of Bacillus sp. strain SG-1 that is capable of manganese oxidation to MnO2 (van Waasbergen et al., 1996). Since multicopper oxidases are one-electron oxidants, the oxidation of Mn(II) to Mn(IV) proceeds via a Mn(III) intermediate (Spiro et al., 2009). Mn(III) can be stabilized in the cell by binding to siderophores (Parker et al., 2004) or pyrophosphate (Tebo et al., 2004), and Mn(III) pyrophosphates contribute up to 100% of the total dissolved manganese pool in marine suboxic zones (Trouwborst et al., 2006). Mn(III) can oxidize to harmless O2, becoming reduced back to Mn(II) (Barnese et al., 2008; Batinic-Haberle et al., 2011). Interestingly, N. gracilis also has genes for siderophores that may function in Mn(III) stabilization. One could speculate that siderophores loaded with Mn(III) may also be transported into the cell, extending ROS protection from the periplasmic space into the cytoplasm. Here, pyrophosphate for further Mn(III) stabilization could be provided by inorganic phosphatase. Thus, Mn(III) produced by the multicopper oxidase could be stabilized and may contribute to ROS protection. Aside from manganese, polyamines can confer protection against oxidative stress and scavenge free radicals (Chattopadhyay et al., 2003; Tkachenko et al., 2012). As the N. gracilis genome contains the genes needed for producing the polyamines putrescine and spermidine, the organism might also employ this ROS defense mechanism.
N. gracilis appears to be well protected against a plethora of toxic compounds (Table S1 in Supplementary Material). Arsenate reductase (Ji and Silver, 1992) and mercuric reductase (Laddaga et al., 1987) were identified in the genome. Thiosulfate sulfurtransferase and cyanate hydratase convert thiocyanate to cyanate and then to ammonia and CO2 for cyanate detoxification (Kamennaya et al., 2008). This mechanism may also allow N. gracilis to use (thio)cyanate as source of ammonium for assimilation. A range of transporters for monovalent copper, diverse mono- and divalent cations, acriflavin, as well as multidrug export systems complete the diverse defense mechanisms of N. gracilis. Multiple Na+ export systems, including an putative eight subunit Mnh-type secondary Na+/H+antiporter (Swartz et al., 2007), likely contribute to salt tolerance necessary for growth in the marine environment.
Discussion
Phylogeny and Diversity of Nitrospina
As each gene is shaped by a unique set of functional constraints through evolution, phylogenetic trees based on different single genes, which are calculated for the same set of organisms, may show conflicting topologies (Rokas et al., 2003). Therefore, comparative phylogenetic analyses using different individual marker genes often suffer from incongruent trees that hamper firm conclusions on the affiliation of an organism or even a whole lineage. This problem can be ameliorated by using concatenated datasets of multiple marker genes or proteins, as analyses based on such datasets are not biased toward the evolutionary history of a single marker (Rokas et al., 2003; Strous et al., 2006). This approach and the now available genome sequence of N. gracilis allowed us to assess the phylogenetic affiliation of Nitrospina with an unprecedented high degree of confidence. Contrasting previous phylogenies, which were based on 16S rRNA gene sequences only (Teske et al., 1994), our analyses strongly suggest that N. gracilis does not belong to the Deltaproteobacteria but represents a distinct, deep-branching line of descent within the Bacteria. This conclusion gains further support from analyses of conserved insertions and deletions in highly conserved genes (Gupta and Griffiths, 2002) and of gene transposition events (Kunisawa, 2006), which are not affected by the quality of sequence alignments and other possible biases of tree calculation methods. For the novel phylum we therefore propose the tentative name Nitrospinae phyl. nov. and the reclassification of the family Nitrospinaceae (Garrity et al., 2005) in the Nitrospinia class. nov., and Nitrospinales ord. nov., with N. gracilis as the type species. The species description remains as published by Watson and Waterbury (1971). However, the exact position of the Nitrospinae in the bacterial tree could not be resolved unambiguously. Some trees suggested a position between the Proteobacteria and a group consisting of Nitrospirae and Acidobacteria, whereas others indicated a superphylum comprising the Nitrospirae, Acidobacteria, and Nitrospinae (Figure 1). Ambiguous branching patterns of underrepresented lineages in phylogenetic trees can sometimes be resolved by adding more reference sequences related to these lineages (Peplies et al., 2008). However, this is currently not possible for Nitrospina since no closely related organisms are known.
Evolutionary History of Nitrite Oxidation
Two main groups of NXR are known which can be distinguished based on their subcellular localization and phylogenetic affiliation within the type II DMSO reductase-family molybdopterin cofactor-binding enzymes. In Nitrobacter, Nitrococcus, and Nitrolancetus NXR faces the cytoplasmic side of the cytoplasmic membrane. This type of NXR is closely related to the respiratory nitrate reductase (NAR) of E. coli and many denitrifying bacteria (Lücker et al., 2010; Sorokin et al., 2012). Furthermore, the proteobacterial NOB Nitrobacter and Nitrococcus contain stacks of intracytoplasmic membranes (ICM), which are densely packed with NXR complexes also facing the cytoplasm (Spieck and Bock, 2005a). Since these ICM resemble those of related phototrophic proteobacteria (purple bacteria), Teske et al. (1994) hypothesized that Nitrobacter and Nitrococcus evolved from phototrophic ancestors where nitrite was used as electron donor for anoxygenic photosynthesis. Indeed, phototrophic nitrite oxidizers from the genera Rhodopseudomonas and Thiocapsa, which are closely related to Nitrobacter and Nitrococcus, were discovered recently (Schott et al., 2010). In contrast, Nitrospira lack ICM. Their NXR is also attached to the cytoplasmic membrane, but it is oriented toward the periplasm (Spieck et al., 1998; Lücker et al., 2010). Previously, we showed that this periplasmically oriented NXR is phylogenetically distinct from the cytoplasmically oriented enzyme and clusters together with the NXR of the anammox organism Ca. K. stuttgartiensis (Lücker et al., 2010). Anammox bacteria oxidize nitrite not for energy generation but to replenish the respiratory chain with electrons needed for CO2 fixation (Jetten et al., 2009). Interestingly, the different subcellular orientation and phylogenetic separation of the two NXR types mirror differences in the nitrite requirements of the respective NOB, which were determined experimentally (Schramm et al., 1999; Bartosch et al., 2002; Freitag et al., 2005; Sorokin et al., 2012). All known NOB with the periplasmic NXR achieve growth with lower nitrite concentrations than NOB with the cytoplasmic enzyme (Sorokin et al., 2012). A likely explanation is the higher efficiency of periplasmic NXR, which conserves more energy per oxidized nitrite. Since the additional O atom in nitrate stems from water, two protons are released in the periplasmic space if nitrite is oxidized by a periplasmic NXR. An additional two protons are consumed in the cytoplasm during O2 reduction to water by the terminal oxidase. Thus, the activity of a periplasmic NXR directly contributes to the proton motive force. This is not the case with a cytoplasmically oriented NXR, where the protons are released and consumed at the same side of the membrane (Lücker et al., 2010). N. gracilis lacks ICM and is adapted to low nitrite concentrations (Watson and Waterbury, 1971). Consistently, its two copies of the NxrA subunit are highly similar to the periplasmic NxrA of Ca. N. defluvii (54–55% identity). Surprisingly, however, their sequence identity to the NxrA of Ca. K. stuttgartiensis is even higher (62%), indicating a closer evolutionary relationship between the NXRs of Nitrospina and anammox bacteria. Indeed, phylogenetic analysis showed that the NxrA of Nitrospina clusters together with the marine “Ca. Scalindula profunda” within the anammox group, whereas Nitrospira branches off at the ancestral node of the NXR branch (Figure 5A). This branching pattern implies that the genes coding for the periplasmic NXR were horizontally transferred from an ancestor of Nitrospira to an ancestral anammox organism (or vice versa) and from a (putatively marine) anammox bacterium to Nitrospina. Vertical inheritance can be excluded due to the large phylogenetic distance between these organisms at the inter-phylum level. The first HGT between ancestral Nitrospira and anammox requires the coexistence of these organisms in an anoxic or hypoxic environment, which was already proposed based on the first analysis of a Nitrospira genome (Lücker et al., 2010). Since Nitrospina-like bacteria often co-occur with anammox organisms in low-oxygen marine environments (Füssel et al., 2012), the second HGT from anammox to Nitrospina appears possible as well. The use of the rTCA cycle for carbon fixation also is fully consistent with a microaerophilic or even anaerobic evolutionary origin of Nitrospina. The periplasmic NXR might originally have evolved in ancestral Nitrospira or anammox bacteria, or it was received by one of these lineages from an unknown donor. As the closest known branch contains bacterial and archaeal nitrate reductases (Figure 5A), one may speculate that such a donor was a nitrate-reducing or nitrite-oxidizing bacterium or archaeon. In this context it appears striking that the reaction of recent periplasmic NXR is reversible and the enzyme still catalyzes both nitrite oxidation and nitrate reduction (Ehrich et al., 1995).
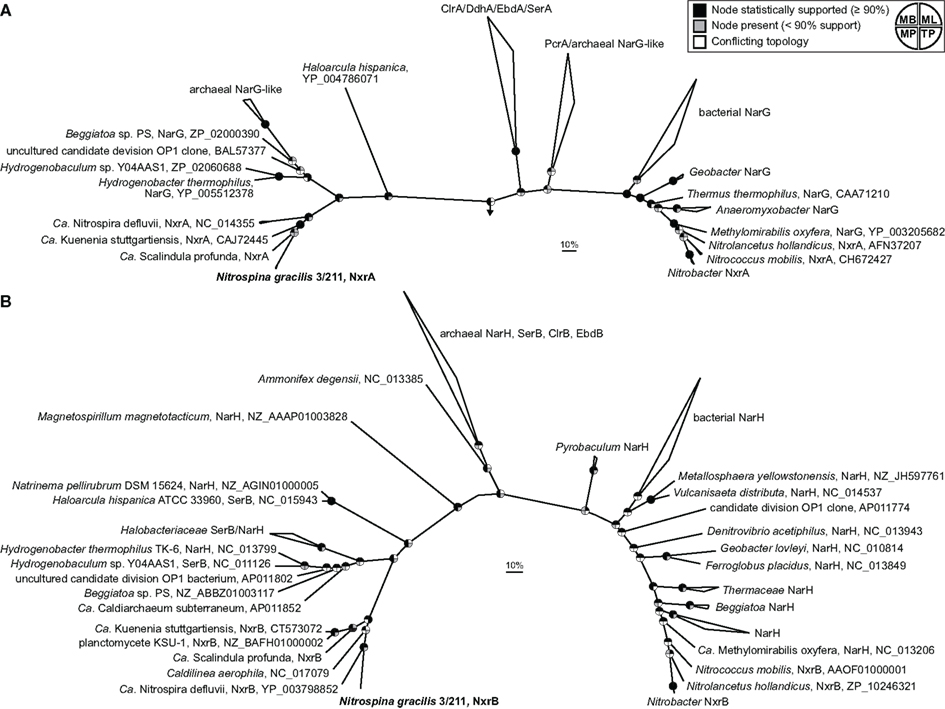
Figure 5. Phylogeny of N. gracilis 3/211 NXR (boldface) and related enzymes. Both trees show selected enzymes from the DMSO reductase type II family. Names of validated enzymes are indicated (Clr, chlorate reductase; Ddh, dimethylsulfide dehydrogenase; Ebd, ethylbenzene dehydrogenase; Nar, Nitrate reductase; Nxr, nitrite oxidoreductase; Pcr, perchlorate reductase; Ser, selenate reductase). More distantly related molybdoenzymes were used as outgroup. The scale bar represents 10% estimated sequence divergence. Pie charts indicate statistical support of nodes based on Bayesian inference, bootstrap analysis, or treepuzzle support. MB, Bayesian inference; ML, maximum-likelihood (RAxML); MP, maximum-parsimony (ProtPars); TP, maximum-likelihood (TreePuzzle). (A) Bayesian interference tree (SD = 0.006924) of the large (α) subunit. In total, 1,475 alignment positions were considered. (B) Bayesian interference tree (SD = 0.015471) of the small (β) subunit, obtained by using 532 alignment positions.
In phylogenetic trees based on the smaller and highly conserved NxrB subunit, N. gracilis and Nitrospira cluster together and the anammox lineage branches off first (Figure 5B). This topology is unclear with respect to the aforementioned order of HGT events, because the common ancestral node may represent a member of the anammox lineage, Nitrospirae, Nitrospinae, or an unknown donor. Additional NxrB sequences from each lineage, once available, might disambiguate the branching order. However, the clustering of the NxrB of Nitrospira and Nitrospina may also reflect similar functions in the respiratory chain. NxrB is more downstream than NxrA and channels electrons to the gamma subunit, NxrC. In both aerobic NOB, the electrons from nitrite are shuttled either toward a terminal oxidase or into a reverse electron transport chain. This bifurcated electron flow from nitrite may require different forms of NxrC, which interact with the respective downstream acceptors. Consistently, the genomes of both Ca. N. defluvii and N. gracilis contain several different NxrC-like CDS (Lücker et al., 2010 and this study). NxrB must then interact with different types of NxrC. This flexibility may require adaptations in the sequence of NxrB not found in anammox bacteria, where nitrite oxidation is always followed by reverse electron flow. Alternative explanations for the differences between the NxrA and NxrB trees include the possibility that these NXR subunits do not share the same evolutionary history but were subject to different HGT events. Interestingly, a member of the Chloroflexi, Caldilinea aerophila, also encodes an NxrB-like protein that branches between Nitrospina/Nitrospira and the anammox group (Figure 5B). A closer inspection of its publicly available genome sequence revealed that C. aerophila also carries genes for the TorD-like chaperone and NxrC, but the catalytic NxrA is lacking. Thus, the functions of its NXR-like proteins remain to be investigated.
Phylogenomics (Frickey and Lupas, 2004), further illuminated the extent of HGT between Nitrospina, Nitrospira, and the anammox lineage (represented by Kuenenia; Figure 2). A set of genes is highly conserved among all three organisms (Figure 6). In Ca. K. stuttgartiensis these genes form one cluster that codes for the NXR subunits and other proteins that may form a membrane-bound complex involved in nitrite oxidation and electron transfer into the respiratory chain (de Almeida et al., 2011). Exactly these genes are conserved between Kuenenia and Nitrospira (Lücker et al., 2010), and their conservation in Nitrospina strongly supports their importance for nitrite oxidation in these bacteria. Besides the NXR α, β, and γ subunits, these genes encode an alternative NxrC subunit, the TorD-like chaperone, one mono- and one diheme cytochrome c, one putative zinc finger- and two cupredoxin-like copper-binding proteins, and one conspicuous cytochrome bd-like oxidase. The two cytochromes c likely are involved in electron transport from NXR to the terminal cytochrome c oxidase for energy generation and to the cytochrome bc complex for reverse electron transport. Alternatively, the interaction with the terminal oxidase or the bc complex might involve copper instead of heme-based electron carriers. This would explain the presence of genes coding for cupredoxin-like proteins in the cluster (see also de Almeida et al., 2011). The function of the bd-like oxidase is more enigmatic. The predicted protein contains 14 transmembrane helices and shows partial similarity to subunit I of canonical cytochrome bd terminal oxidases. In comparative analyses, the N-terminal parts of the canonical bd and bd-like oxidases align well and the ligands for heme b-binding (Borisov et al., 2011) are conserved. In place of the periplasmically oriented Q-loop found in canonical bd oxidases, however, the bd-like oxidases have three predicted membrane helices, and all residues responsible for quinol binding in the canonical enzyme (Borisov et al., 2011) are lacking. Thus, unless the bd-like oxidases have evolved an alternative quinol binding site, a role comparable to canonical cytochrome bd terminal oxidases is unlikely. However, if the enzymes can interact with quinone they might function like an alternative complex III and enable reverse electron transport from nitrite to the quinone pool, either through direct interaction with NXR, or mediated by the copper- or heme-based electron carriers also encoded in the gene cluster. Interestingly, the gene cluster also contains the gene for the aforementioned multicopper oxidase that might be involved in ROS defense in Nitrospina and Nitrospira (Figure 6). The lack of this gene in Ca. K. stuttgartiensis would be in line with a function in ROS defense, which would be largely obsolete in strictly anaerobic anammox bacteria.
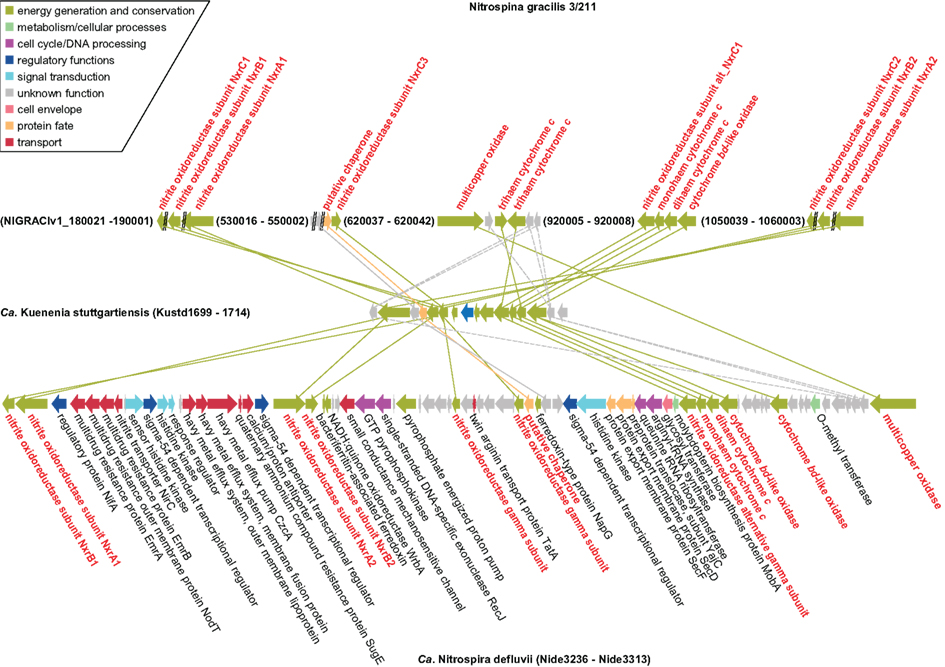
Figure 6. Schematic illustration of the genomic regions in N. gracilis 3/211, Ca. K. stuttgartiensis, and Ca. N. defluvii that contain genes coding for NXR, chaperones, electron carriers, cytochrome bd-like oxidases, multicopper oxidases, and conserved proteins of unknown function. Solid lines indicate proteins that are closest homologs based on protein phylogeny. Dashed lines connect homologous proteins that are not the closest relatives in the respective phylogenetic trees. Proteins and connecting lines are color-coded according to functional classes. CDS and intergenic regions are drawn to scale.
Conclusion
N. gracilis is the most abundant and widespread nitrite oxidizer in marine systems and is of central importance to the oceanic nitrogen cycle. The extended phylogenetic analyses performed in this study have shown that N. gracilis represents a novel bacterial phylum. Intriguingly, N. gracilis extends an evolutionary model of nitrite oxidation that is based on lateral gene transfer across phylum boundaries, and it strengthens the previously proposed evolutionary link between anammox bacteria and NOB with a periplasmic NXR (Lücker et al., 2010; Sorokin et al., 2012). Like with Nitrospira, the use of an oxygen-sensitive CO2 fixation pathway and the lack of canonical ROS defense mechanisms suggest an evolutionary origin of Nitrospina from microaerophilic or even anaerobic ancestors. Consistently, Nitrospina-like organisms are frequently encountered in hypoxic marine habitats. However, nitrite accumulation in the truly anoxic core regions of OMZs (Thamdrup et al., 2012) indicates that the observed nitrite oxidation across OMZs, and also in marine sediments, still requires oxygen and probably is caused by the activity of microaerophilic NOB including Nitrospina.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Christiane Dorninger and Jasmin Schwarz for help with DNA extraction and for cloning of the NXR operons and Ilias Lagkouvardos for providing the Perl script for phylome sorting. Martina Grill and Florian Goldenberg are acknowledged for technical support, as is the LABGeM team at Genoscope. This work was supported by the Austrian Science Fund (FWF; grant P24101-B22) and the Vienna Science and Technology Fund (WWTF; grant LS09-40).
Author Note
Data deposition: The genome sequence reported in this paper has been deposited in the EMBL-EBI SRA archive (project number PRJEB1269), the partial nxrABC operon sequences at GenBank (accession numbers KC262217 and KC262218).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Evolutionary_and_Genomic_Microbiology/10.3389/fmicb.2013.00027/abstract
Table S1. Nitrospina gracilis strain 3/211 proteins with predicted functions in key metabolic pathways.
References
Alawi, M., Lipski, A., Sanders, T., Pfeiffer, E. M., and Spieck, E. (2007). Cultivation of a novel cold-adapted nitrite oxidizing betaproteobacterium from the Siberian Arctic. ISME J. 1, 256–264.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Bäcklund, A. S., Bohlin, J., Gustavsson, N., and Nilsson, T. (2009). Periplasmic c cytochromes and chlorate reduction in Ideonella dechloratans. Appl. Environ. Microbiol. 75, 2439–2445.
Barnese, K., Gralla, E. B., Cabelli, D. E., and Selverstone Valentine, J. (2008). Manganous phosphate acts as a superoxide dismutase. J. Am. Chem. Soc. 130, 4604–4606.
Bartosch, S., Hartwig, C., Spieck, E., and Bock, E. (2002). Immunological detection of Nitrospira-like bacteria in various soils. Microb. Ecol. 43, 26–33.
Bartosch, S., Wolgast, I., Spieck, E., and Bock, E. (1999). Identification of Nitrite-oxidizing bacteria with monoclonal antibodies recognizing the nitrite oxidoreductase. Appl. Environ. Microbiol. 65, 4126–4133.
Batinic-Haberle, I., Rajic, Z., Tovmasyan, A., Reboucas, J. S., Ye, X., Leong, K. W., et al. (2011). Diverse functions of cationic Mn(III) N-substituted pyridylporphyrins, recognized as SOD mimics. Free Radic. Biol. Med. 51, 1035–1053.
Berg, I. A. (2011). Ecological aspects of the distribution of different autotrophic CO2 fixation pathways. Appl. Environ. Microbiol. 77, 1925–1936.
Biegel, E., and Müller, V. (2010). Bacterial Na+-translocating ferredoxin:NAD+ oxidoreductase. Proc. Natl. Acad. Sci. U.S.A. 107, 18138–18142.
Borisov, V. B., Gennis, R. B., Hemp, J., and Verkhovsky, M. I. (2011). The cytochrome bd respiratory oxygen reductases. Biochim. Biophys. Acta 1807, 1398–1413.
Brandt, U. (2006). Energy converting NADH:quinone oxidoreductase (complex I). Annu. Rev. Biochem. 75, 69–92.
Buckel, W., and Thauer, R. K. (2013). Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochim. Biophys. Acta 1827, 94–113.
Buschmann, S., Warkentin, E., Xie, H., Langer, J. D., Ermler, U., and Michel, H. (2010). The structure of cbb3 cytochrome oxidase provides insights into proton pumping. Science 329, 327–330.
Cecchini, G., Schröder, I., Gunsalus, R. P., and Maklashina, E. (2002). Succinate dehydrogenase and fumarate reductase from Escherichia coli. Biochim. Biophys. Acta 1553, 140–157.
Chattopadhyay, M. K., Tabor, C. W., and Tabor, H. (2003). Polyamines protect Escherichia coli cells from the toxic effect of oxygen. Proc. Natl. Acad. Sci. U.S.A. 100, 2261–2265.
Cosseau, C., and Batut, J. (2004). Genomics of the ccoNOQP-encoded cbb3 oxidase complex in bacteria. Arch. Microbiol. 181, 89–96.
Daims, H., Nielsen, J. L., Nielsen, P. H., Schleifer, K. H., and Wagner, M. (2001). In situ characterization of Nitrospira-like nitrite-oxidizing bacteria active in wastewater treatment plants. Appl. Environ. Microbiol. 67, 5273–5284.
Davis, R. E., Stakes, D. S., Wheat, C. G., and Moyer, C. L. (2009). Bacterial variability within an iron-silica-manganese-rich hydrothermal mound located off-axis at the cleft segment, Juan de Fuca Ridge. Geomicrobiol. J. 26, 570–580.
de Almeida, N. M., Maalcke, W. J., Keltjens, J. T., Jetten, M. S. M., and Kartal, B. (2011). Proteins and protein complexes involved in the biochemical reactions of anaerobic ammonium-oxidizing bacteria. Biochem. Soc. Trans. 39, 303.
DeLong, E. F., Preston, C. M., Mincer, T., Rich, V., Hallam, S. J., Frigaard, N.-U., et al. (2006). Community genomics among stratified microbial assemblages in the ocean’s interior. Science 311, 496–503.
Dimroth, P., and Hilpert, W. (1984). Carboxylation of pyruvate and acetyl coenzyme A by reversal of the sodium pumps oxaloacetate decarboxylase and methylmalonyl-CoA decarboxylase. Biochemistry 23, 5360–5366.
Ehrich, S., Behrens, D., Lebedeva, E., Ludwig, W., and Bock, E. (1995). A new obligately chemolithoautotrophic, nitrite-oxidizing bacterium, Nitrospira moscoviensis sp. nov. and its phylogenetic relationship. Arch. Microbiol. 164, 16–23.
Elbehti, A., Brasseur, G., and Lemesle-Meunier, D. (2000). First evidence for existence of an uphill electron transfer through the bc1 and NADH-Q oxidoreductase complexes of the acidophilic obligate chemolithotrophic ferrous ion-oxidizing bacterium Thiobacillus ferrooxidans. J. Bacteriol. 182, 3602–3606.
Ettwig, K. F., Butler, M. K., Le Paslier, D., Pelletier, E., Mangenot, S., Kuypers, M. M., et al. (2010). Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464, 543–548.
Felsenstein, J. (2005). PHYLIP (Phylogeny Inference Package) version 3.6. Seattle: Department of Genome Sciences, University of Washington.
Finn, R. D., Mistry, J., Tate, J., Coggill, P., Heger, A., Pollington, J. E., et al. (2010). The Pfam protein families database. Nucleic Acids Res. 38, D211–D222.
Forzi, L., Koch, J., Guss, A. M., Radosevich, C. G., Metcalf, W. W., and Hedderich, R. (2005). Assignment of the [4Fe-4S] clusters of Ech hydrogenase from Methanosarcina barkeri to individual subunits via the characterization of site-directed mutants. FEBS J. 272, 4741–4753.
Freitag, A., and Bock, E. (1990). Energy conservation in Nitrobacter. FEMS Microbiol. Lett. 66, 157–162.
Freitag, T. E., Chang, L., Clegg, C. D., and Prosser, J. I. (2005). Influence of inorganic nitrogen management regime on the diversity of nitrite-oxidizing bacteria in agricultural grassland soils. Appl. Environ. Microbiol. 71, 8323–8334.
Frickey, T., and Lupas, A. N. (2004). PhyloGenie: automated phylome generation and analysis. Nucleic Acids Res. 32, 5231–5238.
Fuchs, B. M., Woebken, D., Zubkov, M. V., Burkill, P., and Amann, R. (2005). Molecular identification of picoplankton populations in contrasting waters of the Arabian Sea. Aquat. Microb. Ecol. 39, 145–157.
Fuchsman, C. A., Kirkpatrick, J. B., Brazelton, W. J., Murray, J. W., and Staley, J. T. (2011). Metabolic strategies of free-living and aggregate-associated bacterial communities inferred from biologic and chemical profiles in the Black Sea suboxic zone. FEMS Microbiol. Ecol. 78, 586–603.
Füssel, J., Lam, P., Lavik, G., Jensen, M. M., Holtappels, M., Gunter, M., et al. (2012). Nitrite oxidation in the Namibian oxygen minimum zone. ISME J. 6, 1200–1209.
Garrity, G. M., Bell, J. A., and Lilburn, T. (2005). “Nitrospinaceae fam. nov.,” in Bergey’s Manual® of Systematic Bacteriology, eds D. J. Brenner, N. R. Krieg, G. M. Garrity, J. T. Staley, D. R. Boone, P. Vos, M. Goodfellow, F. A. Rainey, and K.-H. Schleifer (New York: Springer), 999.
Gomes, C. M., Lemos, R. S., Teixeira, M., Kletzin, A., Huber, H., Stetter, K. O., et al. (1999). The unusual iron sulfur composition of the Acidianus ambivalens succinate dehydrogenase complex. Biochim. Biophys. Acta 1411, 134–141.
Gruber, N. (2008). “The Marine Nitrogen Cycle: Overview and Challenges,” in Nitrogen in the Marine Environment, 2nd Edn, eds D. G. Capone, D. A. Bronk, M. R. Mulholland, and E. J. Carpenter (San Diego: Academic Press), 1–50.
Guindon, S., and Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704.
Gupta, R. S. (2000). The phylogeny of Proteobacteria: relationships to other eubacterial phyla and eukaryotes. FEMS Microbiol. Rev. 24, 367–402.
Gupta, R. S. (2001). The branching order and phylogenetic placement of species from completed bacterial genomes, based on conserved indels found in various proteins. Int. Microbiol. 4, 187–202.
Gupta, R. S., and Griffiths, E. (2002). Critical issues in bacterial phylogeny. Theor. Popul. Biol. 61, 423–434.
Guymer, D., Maillard, J., and Sargent, F. (2009). A genetic analysis of in vivo selenate reduction by Salmonella enterica serovar Typhimurium LT2 and Escherichia coli K12. Arch. Microbiol. 191, 519–528.
Hanke, G. T., Satomi, Y., Shinmura, K., Takao, T., and Hase, T. (2011). A screen for potential ferredoxin electron transfer partners uncovers new, redox dependent interactions. Biochim. Biophys. Acta 1814, 366–374.
Herrmann, G., Jayamani, E., Mai, G., and Buckel, W. (2008). Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J. Bacteriol. 190, 784–791.
Hügler, M., Huber, H., Molyneaux, S. J., Vetriani, C., and Sievert, S. M. (2007). Autotrophic CO2 fixation via the reductive tricarboxylic acid cycle in different lineages within the phylum Aquificae: evidence for two ways of citrate cleavage. Environ. Microbiol. 9, 81–92.
Hunter, E. M., Mills, H. J., and Kostka, J. E. (2006). Microbial community diversity associated with carbon and nitrogen cycling in permeable shelf sediments. Appl. Environ. Microbiol. 72, 5689–5701.
Ikeda, T., Ochiai, T., Morita, S., Nishiyama, A., Yamada, E., Arai, H., et al. (2006). Anabolic five subunit-type pyruvate:ferredoxin oxidoreductase from Hydrogenobacter thermophilus TK-6. Biochem. Biophys. Res. Commun. 340, 76–82.
Ilbert, M., and Bonnefoy, V. (2012). Insight into the evolution of the iron oxidation pathways. Biochim. Biophys. Acta. doi:10.1016/j.bbabio.2012.1010.1001. [Epub ahead of print].
Iwasaki, T., Kounosu, A., Aoshima, M., Ohmori, D., Imai, T., Urushiyama, A., et al. (2002). Novel [2Fe-2S]-type redox center C in SdhC of archaeal respiratory complex II from Sulfolobus tokodaii strain 7. J. Biol. Chem. 277, 39642–39648.
Janssen, S., Schafer, G., Anemuller, S., and Moll, R. (1997). A succinate dehydrogenase with novel structure and properties from the hyperthermophilic archaeon Sulfolobus acidocaldarius: genetic and biophysical characterization. J. Bacteriol. 179, 5560–5569.
Jetten, M. S., Niftrik, L., Strous, M., Kartal, B., Keltjens, J. T., and Op Den Camp, H. J. (2009). Biochemistry and molecular biology of anammox bacteria. Crit. Rev. Biochem. Mol. Biol. 44, 65–84.
Ji, G., and Silver, S. (1992). Reduction of arsenate to arsenite by the ArsC protein of the arsenic resistance operon of Staphylococcus aureus plasmid pI258. Proc. Natl. Acad. Sci. U.S.A. 89, 9474–9478.
Jorgensen, S. L., Hannisdal, B., Lanzen, A., Baumberger, T., Flesland, K., Fonseca, R., et al. (2012). Correlating microbial community profiles with geochemical data in highly stratified sediments from the Arctic Mid-Ocean Ridge. Proc. Natl. Acad. Sci. U.S.A. 109, E2846–E2855.
Jormakka, M., Richardson, D., Byrne, B., and Iwata, S. (2004). Architecture of NarGH reveals a structural classification of Mo-bisMGD enzymes. Structure 12, 95–104.
Käll, L., Krogh, A., and Sonnhammer, E. L. L. (2007). Advantages of combined transmembrane topology and signal peptide prediction–the Phobius web server. Nucleic Acids Res. 35, W429–W432.
Kamennaya, N. A., Chernihovsky, M., and Anton, F. P. (2008). The cyanate utilization capacity of marine unicellular cyanobacteria. Limnol. Oceanogr. 53, 2485–2494.
Kanao, T., Fukui, T., Atomi, H., and Imanaka, T. (2001). ATP-citrate lyase from the green sulfur bacterium Chlorobium limicola is a heteromeric enzyme composed of two distinct gene products. Eur. J. Biochem. 268, 1670–1678.
Kappler, U., Bennett, B., Rethmeier, J., Schwarz, G., Deutzmann, R., McEwan, A. G., et al. (2000). Sulfite:cytochrome c Oxidoreductase from Thiobacillus novellus: purification, characterization, and molecular biology of a heterodimeric member of the sulfite oxidase family. J. Biol. Chem. 275, 13202–13212.
Kniemeyer, O., and Heider, J. (2001). Ethylbenzene dehydrogenase, a novel hydrocarbon-oxidizing molybdenum/iron-sulfur/heme enzyme. J. Biol. Chem. 276, 21381–21386.
Korbel, J. O., Snel, B., Huynen, M. A., and Bork, P. (2002). SHOT: a web server for the construction of genome phylogenies. Trends Genet. 18, 158–162.
Kunisawa, T. (2006). Dichotomy of major bacterial phyla inferred from gene arrangement comparisons. J. Theor. Biol. 239, 367–375.
Kunisawa, T. (2010). Evaluation of the phylogenetic position of the sulfate-reducing bacterium Thermodesulfovibrio yellowstonii (phylum Nitrospirae) by means of gene order data from completely sequenced genomes. Int. J. Syst. Evol. Microbiol. 60, 1090–1102.
Kunisawa, T. (2011). Inference of the phylogenetic position of the phylum Deferribacteres from gene order comparison. Antonie Van Leeuwenhoek 99, 417–422.
Labrenz, M., Jost, G., and Jurgens, K. (2007). Distribution of abundant prokaryotic organisms in the water column of the central Baltic Sea with an oxic-anoxic interface. Aquat. Microb. Ecol. 46, 177–190.
Laddaga, R. A., Chu, L., Misra, T. K., and Silver, S. (1987). Nucleotide sequence and expression of the mercurial-resistance operon from Staphylococcus aureus plasmid pI258. Proc. Natl. Acad. Sci. U.S.A. 84, 5106–5110.
Lancaster, C. R. D., Herzog, E., Juhnke, H. D., Madej, M. G., Müller, F. G., Paul, R., et al. (2008). Electroneutral and electrogenic catalysis by dihaem-containing succinate:quinone oxidoreductases. Biochem. Soc. Trans. 36, 996–1000.
Lancaster, C. R. D., and Simon, J. (2002). Succinate:quinone oxidoreductases from Epsilonproteobacteria. Biochim. Biophys. Acta 1553, 84–101.
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0 . Bioinformatics 23, 2947–2948.
Lemos, R. S., Fernandes, A. S., Pereira, M. M., Gomes, C. M., and Teixeira, M. (2002). Quinol:fumarate oxidoreductases and succinate:quinone oxidoreductases: phylogenetic relationships, metal centres and membrane attachment. Biochim. Biophys. Acta 1553, 158–170.
Lemos, R. S., Gomes, C. M., and Teixeira, M. (2001). Acidianus ambivalens complex II typifies a novel family of succinate dehydrogenases. Biochem. Biophys. Res. Commun. 281, 141–150.
Levican, G., Ugalde, J. A., Ehrenfeld, N., Maass, A., and Parada, P. (2008). Comparative genomic analysis of carbon and nitrogen assimilation mechanisms in three indigenous bioleaching bacteria: predictions and validations. BMC Genomics 9:581. doi:10.1186/1471-2164-9-581
Lim, K., Read, R. J., Chen, C. C. H., Tempczyk, A., Wei, M., Ye, D., et al. (2007). Swiveling domain mechanism in pyruvate phosphate dikinase. Biochemistry 46, 14845–14853.
Lowe, E. C., Bydder, S., Hartshorne, R. S., Tape, H. L. U., Dridge, E. J., Debieux, C. M., et al. (2010). Quinol-cytochrome c oxidoreductase and cytochrome c4 mediate electron transfer during selenate respiration in Thauera selenatis. J. Biol. Chem. 285, 18433–18442.
Lücker, S., Wagner, M., Maixner, F., Pelletier, E., Koch, H., Vacherie, B., et al. (2010). A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. Proc. Natl. Acad. Sci. U.S.A. 107, 13479–13484.
Ludwig, W. (2010). “Molecular phylogeny of microorganisms: is rRNA still a useful marker?” in Molecular Phylogeny of Microorganisms, eds A. Oren and R. T. Papke (Norfolk: Caister Academic Press), 65–84.
Ma, K., Weiss, R., and Adams, M. W. (2000). Characterization of hydrogenase II from the hyperthermophilic archaeon Pyrococcus furiosus and assessment of its role in sulfur reduction. J. Bacteriol. 182, 1864–1871.
Maklashina, E., Berthold, D. A., and Cecchini, G. (1998). Anaerobic expression of Escherichia coli succinate dehydrogenase: functional replacement of fumarate reductase in the respiratory chain during anaerobic growth. J. Bacteriol. 180, 5989–5996.
Martinez-Espinosa, R. M., Dridge, E. J., Bonete, M. J., Butt, J. N., Butler, C. S., Sargent, F., et al. (2007). Look on the positive side! The orientation, identification and bioenergetics of ‘Archaeal’ membrane-bound nitrate reductases. FEMS Microbiol. Lett. 276, 129–139.
McDevitt, C. A., Hugenholtz, P., Hanson, G. R., and McEwan, A. G. (2002). Molecular analysis of dimethyl sulphide dehydrogenase from Rhodovulum sulfidophilum: its place in the dimethyl sulphoxide reductase family of microbial molybdopterin-containing enzymes. Mol. Microbiol. 44, 1575–1587.
Mincer, T. J., Church, M. J., Taylor, L. T., Preston, C., Karl, D. M., and Delong, E. F. (2007). Quantitative distribution of presumptive archaeal and bacterial nitrifiers in Monterey Bay and the North Pacific Subtropical Gyre. Environ. Microbiol. 9, 1162–1175.
Park, B.-J., Park, S.-J., Yoon, D.-N., Schouten, S., Sinninghe Damste, J. S., and Rhee, S.-K. (2010). Cultivation of autotrophic ammonia-oxidizing archaea from marine sediments in coculture with sulfur-oxidizing bacteria. Appl. Environ. Microbiol. 76, 7575–7587.
Parker, D. L., Sposito, G., and Tebo, B. M. (2004). Manganese(III) binding to a pyoverdine siderophore produced by a manganese(II)-oxidizing bacterium. Geochim. Cosmochim. Acta 68, 4809–4820.
Peplies, J., Kottmann, R., Ludwig, W., and Glöckner, F. O. (2008). A standard operating procedure for phylogenetic inference (SOPPI) using (rRNA) marker genes. Syst. Appl. Microbiol. 31, 251–257.
Pereira, M. M., Santana, M., and Teixeira, M. (2001). A novel scenario for the evolution of haem-copper oxygen reductases. Biochim. Biophys. Acta 1505, 185–208.
Petersen, T. N., Brunak, S., Von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786.
Poughon, L., Dussap, C. G., and Gros, J. B. (2001). Energy model and metabolic flux analysis for autotrophic nitrifiers. Biotechnol. Bioeng. 72, 416–433.
Radeva, G., and Selenska-Pobell, S. (2005). Bacterial diversity in water samples from uranium wastes as demonstrated by 16S rDNA and ribosomal intergenic spacer amplification retrievals. Can. J. Microbiol. 51, 910–923.
Rokas, A., Williams, B. L., King, N., and Carroll, S. B. (2003). Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 425, 798–804.
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574.
Rothery, R. A., Workun, G. J., and Weiner, J. H. (2008). The prokaryotic complex iron-sulfur molybdoenzyme family. Biochim. Biophys. Acta 1778, 1897–1929.
Santoro, A. E., Casciotti, K. L., and Francis, C. A. (2010). Activity, abundance and diversity of nitrifying archaea and bacteria in the central California Current. Environ. Microbiol. 12, 1989–2006.
Sayers, E. W., Barrett, T., Benson, D. A., Bolton, E., Bryant, S. H., Canese, K., et al. (2011). Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 39, D38–D51.
Sazanov, L. A., and Hinchliffe, P. (2006). Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science 311, 1430–1436.
Schirawski, J., and Unden, G. (1998). Menaquinone-dependent succinate dehydrogenase of bacteria catalyzes reversed electron transport driven by the proton potential. Eur. J. Biochem. 257, 210–215.
Schloss, P. D., and Handelsman, J. (2004). Status of the microbial census. Microbiol. Mol. Biol. Rev. 68, 686–691.
Schmidt, H. A., Strimmer, K., Vingron, M., and Von Haeseler, A. (2002). TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18, 502–504.
Schnorpfeil, M., Janausch, I. G., Biel, S., Kröger, A., and Unden, G. (2001). Generation of a proton potential by succinate dehydrogenase of Bacillus subtilis functioning as a fumarate reductase. Eur. J. Biochem. 268, 3069–3074.
Schott, J., Griffin, B. M., and Schink, B. (2010). Anaerobic phototrophic nitrite oxidation by Thiocapsa sp. strain KS1 and Rhodopseudomonas sp. strain LQ17. Microbiology 156, 2428–2437.
Schramm, A., De Beer, D., Van Den Heuvel, J. C., Ottengraf, S., and Amann, R. (1999). Microscale distribution of populations and activities of Nitrosospira and Nitrospira spp. along a macroscale gradient in a nitrifying bioreactor: quantification by in situ hybridization and the use of microsensors. Appl. Environ. Microbiol. 65, 3690–3696.
Schütz, M., Brugna, M., Lebrun, E., Baymann, F., Huber, R., Stetter, K.-O., et al. (2000). Early evolution of cytochrome bc complexes. J. Mol. Biol. 300, 663–675.
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi:10.1038/msb.2011.75
Silva, P. J., Van Den Ban, E. C., Wassink, H., Haaker, H., De Castro, B., Robb, F. T., et al. (2000). Enzymes of hydrogen metabolism in Pyrococcus furiosus. Eur. J. Biochem. 267, 6541–6551.
Sone, N., Sawa, G., Sone, T., and Noguchi, S. (1995). Thermophilic Bacilli have split cytochrome b genes for cytochrome b6 and subunit IV. J. Biol. Chem. 270, 10612–10617.
Sorokin, D. Y., Lücker, S., Vejmelkova, D., Kostrikina, N. A., Kleerebezem, R., Rijpstra, W. I., et al. (2012). Nitrification expanded: discovery, physiology and genomics of a nitrite-oxidizing bacterium from the phylum Chloroflexi. ISME J. 6, 2245–2256.
Spieck, E., and Bock, E. (2005a). “The lithoautotrophic nitrite-oxidizing bacteria,” in Bergey’s Manual® of Systematic Bacteriology, eds D. J. Brenner, N. R. Krieg, J. T. Staley, and G. M. Garrity (New York: Springer), 149–153.
Spieck, E., and Bock, E. (2005b). “Nitrospina Watson and Waterbury 1971, 225AL,” in Bergey’s Manual® of Systematic Bacteriology, eds D. J. Brenner, N. R. Krieg, G. M. Garrity, J. T. Staley, D. R. Boone, P. Vos, M. Goodfellow, F. A. Rainey, and K.-H. Schleifer (New York: Springer), 999–1003.
Spieck, E., Ehrich, S., Aamand, J., and Bock, E. (1998). Isolation and immunocytochemical location of the nitrite-oxidizing system in Nitrospira moscoviensis. Arch. Microbiol. 169, 225–230.
Spiro, T. G., Bargar, J. R., Sposito, G., and Tebo, B. M. (2009). Bacteriogenic manganese oxides. Acc. Chem. Res. 43, 2–9.
Stamatakis, A., Ludwig, T., and Meier, H. (2005). RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21, 456–463.
Starkenburg, S. R., Arp, D. J., and Bottomley, P. J. (2008). Expression of a putative nitrite reductase and the reversible inhibition of nitrite-dependent respiration by nitric oxide in Nitrobacter winogradskyi Nb-255. Environ. Microbiol. 10, 3036–3042.
Strous, M., Pelletier, E., Mangenot, S., Rattei, T., Lehner, A., Taylor, M. W., et al. (2006). Deciphering the evolution and metabolism of an anammox bacterium from a community genome. Nature 440, 790–794.
Sundermeyer-Klinger, H., Meyer, W., Warninghoff, B., and Bock, E. (1984). Membrane-bound nitrite oxidoreductase of Nitrobacter: evidence for a nitrate reductase system. Arch. Microbiol. 140, 153–158.
Swartz, T. H., Ito, M., Ohira, T., Natsui, S., Hicks, D. B., and Krulwich, T. A. (2007). Catalytic properties of Staphylococcus aureus and Bacillus members of the secondary cation/proton antiporter-3 (Mrp) family are revealed by an optimized assay in an Escherichia coli host. J. Bacteriol. 189, 3081–3090.
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577.
Tatusov, R., Fedorova, N., Jackson, J., Jacobs, A., Kiryutin, B., Koonin, E., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. doi:10.1186/1471-2105-4-41
Tebo, B. M., Bargar, J. R., Clement, B. G., Dick, G. J., Murray, K. J., Parker, D., et al. (2004). Biogenic manganese oxides: properties and mechanisms of formation. Annu. Rev. Earth Planet. Sci. 32, 287–328.
Teske, A., Alm, E., Regan, J. M., Toze, S., Rittmann, B. E., and Stahl, D. A. (1994). Evolutionary relationships among ammonia- and nitrite-oxidizing bacteria. J. Bacteriol. 176, 6623–6630.
Thamdrup, B., Dalsgaard, T., and Revsbech, N. P. (2012). Widespread functional anoxia in the oxygen minimum zone of the Eastern South Pacific. Deep Sea Res. Part 1 Oceanogr. Res. Pap. 65, 36–45.
Tkachenko, A. G., Akhova, A. V., Shumkov, M. S., and Nesterova, L. Y. (2012). Polyamines reduce oxidative stress in Escherichia coli cells exposed to bactericidal antibiotics. Res. Microbiol. 163, 83–91.
Trouwborst, R. E., Clement, B. G., Tebo, B. M., Glazer, B. T., and Luther, G. W. (2006). Soluble Mn(III) in suboxic zones. Science 313, 1955–1957.
Uchiyama, I., Higuchi, T., and Kawai, M. (2010). MBGD update 2010: toward a comprehensive resource for exploring microbial genome diversity. Nucleic Acids Res. 38, D361–D365.
Vallenet, D., Engelen, S., Mornico, D., Cruveiller, S., Fleury, L., Lajus, A., et al. (2009). MicroScope: a platform for microbial genome annotation and comparative genomics. Database 2009, bap021.
Vallenet, D., Labarre, L., Rouy, Z., Barbe, V., Bocs, S., Cruveiller, S., et al. (2006). MaGe: a microbial genome annotation system supported by synteny results. Nucleic Acids Res. 34, 53–65.
van Waasbergen, L. G., Hildebrand, M., and Tebo, B. M. (1996). Identification and characterization of a gene cluster involved in manganese oxidation by spores of the marine Bacillus sp. strain SG-1. J. Bacteriol. 178, 3517–3530.
Vignais, P. M., and Billoud, B. (2007). Occurrence, classification, and biological function of hydrogenases: an overview. Chem. Rev. 107, 4206–4272.
Watson, S. W., and Waterbury, J. B. (1971). Characteristics of two marine nitrite oxidizing bacteria, Nitrospina gracilis nov. gen. nov. sp. and Nitrococcus mobilis nov. gen. nov. sp. Arch. Microbiol. 77, 203–230.
Weidler, G. W., Dornmayr-Pfaffenhuemer, M., Gerbl, F. W., Heinen, W., and Stan-Lotter, H. (2007). Communities of Archaea and Bacteria in a subsurface radioactive thermal spring in the Austrian Central Alps, and evidence of ammonia-oxidizing Crenarchaeota. Appl. Environ. Microbiol. 73, 259–270.
Welte, C., Kallnik, V., Grapp, M., Bender, G., Ragsdale, S., and Deppenmeier, U. (2010). Function of Ech hydrogenase in ferredoxin-dependent, membrane-bound electron transport in Methanosarcina mazei. J. Bacteriol. 192, 674–678.
Wherland, S., Farver, O., and Pecht, I. (2005). Intramolecular electron transfer in nitrite reductases. Chemphyschem 6, 805–812.
Winogradsky, S. (1890). Recherches sur les organismes de la nitrification. Ann. Inst. Pasteur 4, 213–231.
Yano, T., Magnitsky, S., Sled, V. D., Ohnishi, T., and Yagi, T. (1999). Characterization of the putative 2x[4Fe-4S]-binding NQO9 subunit of the proton-translocating NADH-quinone oxidoreductase (NDH-1) of Paracoccus denitrificans. Expression, reconstitution, and EPR characterization. J. Biol. Chem. 274, 28598–28605.
Yoon, K. S., Ishii, M., Igarashi, Y., and Kodama, T. (1996). Purification and characterization of 2-oxoglutarate:ferredoxin oxidoreductase from a thermophilic, obligately chemolithoautotrophic bacterium, Hydrogenobacter thermophilus TK-6. J. Bacteriol. 178, 3365–3368.
Yoon, K. S., Ishii, M., Kodama, T., and Igarashi, Y. (1997). Purification and characterization of pyruvate:ferredoxin oxidoreductase from Hydrogenobacter thermophilus TK-6. Arch. Microbiol. 167, 275–279.
Zamboni, N., Maaheimo, H., Szyperski, T., Hohmann, H.-P., and Sauer, U. (2004). The phosphoenolpyruvate carboxykinase also catalyzes C3 carboxylation at the interface of glycolysis and the TCA cycle of Bacillus subtilis. Metab. Eng. 6, 277–284.
Zaslavsky, D., and Gennis, R. B. (2000). Proton pumping by cytochrome oxidase: progress, problems and postulates. Biochim. Biophys. Acta 1458, 164–179.
Appendix

Table A1. Proteins used for phylogeny.
Keywords: Nitrospina, nitrite oxidation, nitrification, nitrite-oxidizing bacteria, marine nitrogen cycle, nitrite oxidoreductase
Citation: Lücker S, Nowka B, Rattei T, Spieck E and Daims H (2013) The genome of Nitrospina gracilis illuminates the metabolism and evolution of the major marine nitrite oxidizer. Front. Microbio. 4:27. doi: 10.3389/fmicb.2013.00027
Received: 05 December 2012; Paper pending published: 26 December 2012;
Accepted: 02 February 2013; Published online: 21 February 2013.
Edited by:
Boran Kartal, Radboud University, Nijmegen, NetherlandsReviewed by:
Peter Bottomley, Oregon State University, USAJan T. Keltjens, Radboud University Nijmegen, Netherlands
Copyright: © 2013 Lücker, Nowka, Rattei, Spieck and Daims. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Sebastian Lücker and Holger Daims, Department of Microbial Ecology, University of Vienna, Althanstr. 14, 1090 Vienna, Austria. e-mail:bHVlY2tlckBtaWNyb2JpYWwtZWNvbG9neS5uZXQ=;ZGFpbXNAbWljcm9iaWFsLWVjb2xvZ3kubmV0