Abstract
The research on virion infectivity factor (Vif) protein had started in late 1980s right after HIV-1 was cloned, and the function of Vif had been a mystery for a long time. However, the research on Vif has finally lead to the identification of APOBEC3G, which opens up a new era in the research field of host restriction factors in HIV-1 infection followed by TRIM5α, Tetherin/BST-2, and SAMHD1. This suggests that continuation of basic research on fundamental questions is quite important. We still have many questions on Vif and APOBEC3 and should continue to work on these proteins in the future in order to better regulate HIV-1. We will discuss not only the history but also recent advances in Vif research.
INTRODUCTION
HIV-1 virion infectivity factor (Vif) was identified as an accessory gene right after the HIV-1 genome was sequenced. It is well conserved among lentiviruses except in equine infectious virus and plays a crucial role in the viral life cycle to facilitate viral infectivity as its name indicates (Desrosiers et al., 1998). In the early reports, Strebel and collegues described that the mutant virus deficient in the vif gene produces virion particles normally; however, the particles are ~1000 times less infectious than the wild type (Fisher et al., 1987; Strebel et al., 1987). The underlying mechanism of Vif function had been unsolved and a mystery for a long time.
EARLY OBSERVATIONS OF Vif FUNCTION LEAD TO IDENTIFICATION OF APOBEC3G
Virion infectivity factor exerts its function in a cell-type-specific manner. Vif is dispensable for producing infectious viral particles in permissive cells such as all known adherent cells (e.g., HeLa and 293T cells) and some T cell lines (e.g., CEM-SS and SupT1 cells); in contrast, Vif is indispensable in non-permissive cells such as physiologically relevant CD4+ T cells and macrophages, and other T cell lines (e.g., CEM and H9 cells; Gabuzda et al., 1992; Sakai et al., 1993; Simon et al., 1998b). These findings raise two possibilities; one is that permissive cells have a vif-like cellular factor which facilitates virion infectivity, another is that non-permissive cells possess an anti-HIV-1 host factor which is antagonized by Vif. Later studies using heterokaryon experiments have shown the latter possibility (Madani and Kabat, 1998; Simon et al., 1998a). In 2002, Malim’s group identified this factor using very sophisticated subtraction cloning methods between non-permissive CEM cells and its derivative subclone permissive CEM-SS cells, which was first called as CEM15 and is now known as APOBEC3G (Sheehy et al., 2002). Details of functions of APOBEC3G and other APOBEC3 family members are described and discussed in many reviews and other chapters of this issue (Goila-Gaur and Strebel, 2008; Wissing et al., 2010; Kitamura et al., 2011).
In addition to the above described main function, early studies also revealed several important Vif functions including dimerization (Yang et al., 2001), virion incorporation (Camaur and Trono, 1996; Simon et al., 1997), and phosphorylation (Yang et al., 1996; Yang and Gabuzda, 1998); however, the significances of these functions are not discussed much recently. Recently, a novel Vif function on cell cycle has been reported, which is discussed in more detail later.
Vif ANTAGONIZES APOBEC3G
As described above, the main function of Vif is to antagonize APOBEC3G. Right after identification of APOBEC3G, many studies have shown that Vif inhibits the virion incorporation of APOBEC3G, which is mainly attributable to degradation of cellular APOBEC3G via the proteasomal pathway (Marin et al., 2003; Sheehy et al., 2003; Stopak et al., 2003; Mehle et al., 2004b). However, some studies have also shown that Vif directly inhibits the virion incorporation of APOBEC3G (Opi et al., 2007) or that Vif inhibits translation of APOBEC3G (Mariani et al., 2003; Stopak et al., 2003).
Yu et al. (2003) have independently shown that Vif forms E3 ligase complexes with cellular proteins including Cullin 5, Elongin B, and C (Vif–Cul5–EloB/C complex) using mass-spectrometry techniques. They and others have also shown that this complex works as the E3 ligase for APOBEC3G to induce polyubiquitination of APOBEC3G and direct it to the 26S proteasome for degradation (Mehle et al., 2004a; Yu et al., 2004; Kobayashi et al., 2005). Iwatani et al. (2009) have identified four critical lysine residues (K297, K301, K303, and K334) in APOBEC3G which are required for Vif-mediated degradation, although others have reported that Vif can ubiquitinate and degrade a lysine-free APOBEC3G (Shao et al., 2010). Vif also antagonizes other APOBEC3 proteins from APOBEC3C to H by the same E3 ligase complex (Shirakawa et al., 2006).
Virion infectivity factor binds to the E3 ligase complex through two interaction sites; it binds to Elongin C through its suppressors of cytokine signaling (SOCS) box motif (Mehle et al., 2004a; Yu et al., 2004), S144LQYLA149, and to Cullin 5 through a zinc-binding motif (Luo et al., 2005; Mehle et al., 2006), H108x5Cx17-18Cx3-5H139 (Figure 1). The SOCS box motif is well conserved among Vif proteins, indicating that this motif is crucial for Vif function, and mutation of S144, a presumed phosphorylation site in Vif, affects binding of Vif to Elongin C (Mehle et al., 2004a). The zinc-binding motif is also important for Vif function to form the E3 ligase complex. Therefore, a zinc chelating agent can inhibit Vif function in infectivity assays (Xiao et al., 2007).
FIGURE 1
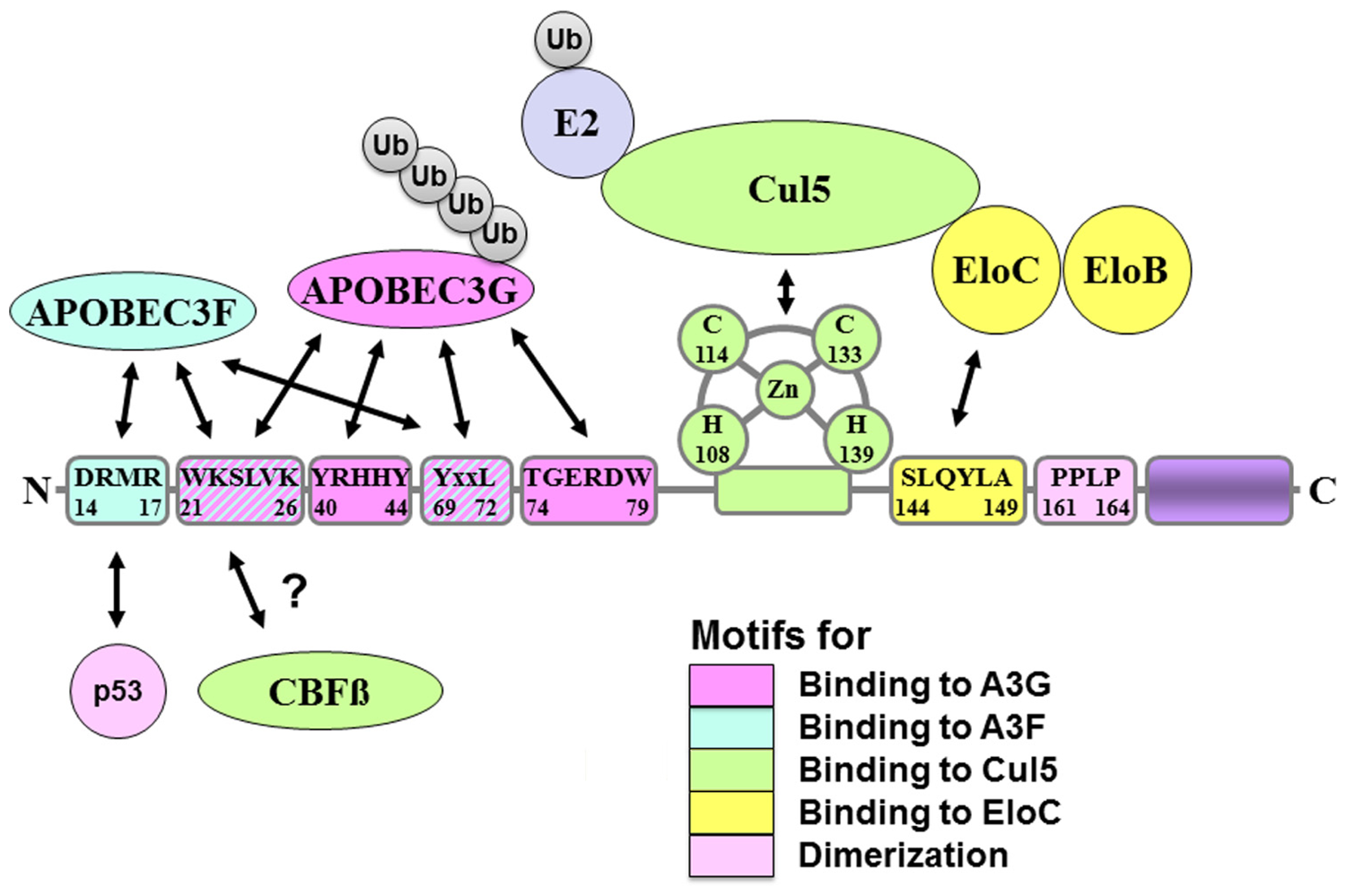
Schematic figure of the virion infectivity factor (Vif) protein and amino acid motifs for binding to Vif-interacting proteins. Pink indicates binding motifs for A3G; light blue indicates binding motifs for A3F; light green indicates binding motifs for Cul5; yellow indicates binding motifs for EloC; light pink indicates motifs for dimerization. Vif binds to p53 and CBFβ in its N-terminal regions, but binding motifs were not elucidated yet.
THE INTERACTION OF Vif WITH APOBEC3 PROTEINS
It is quite important to reveal the interaction sites between Vif and APOBEC3 proteins, because the regulation of this interaction may lead to the development of novel therapeutic strategies for HIV-1 infection. However, their structural information is not fully elucidated yet, because it is quite difficult to produce these proteins as soluble forms. Thus, the information described below is mainly obtained by many studies using site-directed mutagenesis, which sometimes shows different results.
First of all, the most important and confirmed evidence is that the interaction between Vif and APOBEC3G is critically dependent on D128PD130 in APOBEC3G (Huthoff and Malim, 2007). Many groups have simultaneously reported this evidence by comparing human and African green monkey (agm) APOBEC3G (Bogerd et al., 2004; Mangeat et al., 2004; Schrofelbauer et al., 2004; Xu et al., 2004). In detail, HIV-1 Vif binds and antagonizes human APOBEC3G, but not agm APOBEC3G. In contrast, SIVagm Vif antagonizes agm APOBEC3G, but not human APOBEC3G. By comparing amino acids residues and preparing chimeric APOBEC3G between human and agm APOBEC3G, they identified D128 as the determinant of the species-specific binding of Vif to APOBEC3G (Bogerd et al., 2004; Mangeat et al., 2004; Schrofelbauer et al., 2004; Xu et al., 2004). On the other hand, SIVmac and HIV-2 Vif can antagonize both human and agm APOBEC3G, indicating that the interaction between Vif and APOBEC3G is not restricted by D128, in other words, D128 is not the sole determinant for species-specific target by Vif (Gaur and Strebel, 2012). Furthermore, the interaction between Vif and APOBEC3G is regulated by phosphorylation of APOBEC3G at T32 by protein kinase A (Shirakawa et al., 2008).
The interaction sites in Vif are reported by many groups and are much more complicated. The binding site only for APOBEC3G is Y40RHHY44 (Russell and Pathak, 2007), while that only for APOBEC3F is D14RMR17 (Russell and Pathak, 2007), and T74GERxW79 (He et al., 2008). The binding sites for both APOBEC3G and F are W21KSLVK26 (Chen et al., 2009; Dang et al., 2009), V55xIPLx4-5LxΦx2YWxL72 (He et al., 2008), and Y69xxL72 (Pery et al., 2009; Figure 1). To identify the real interaction sites, we have to wait a little longer until we will get the structural information of these complexes.
Vif AND CBFβ
Recent mass-spectrometry screening of Vif-binding proteins has identified a T cell transcription factor, core-binding factor subunit beta (CBFβ), as an important Vif-binding protein (Jager et al., 2012; Zhang et al., 2012). CBFβ directly binds to Vif and plays a crucial role in forming a stable Vif–Cul5–EloB/C E3 ligase complex. Without CBFβ, the Vif–Cul5–EloB/C E3 ligase complex is not stable enough to polyubiquitinate APOBEC3G and its function is severely impaired. The binding sites of Vif with CBFβ are identified as W21 and W38 (Figure 1). However, the mechanisms by which CBFβ regulates the E3 ligase complex are still under investigation. Furthermore, since CBFβ is an important T cell transcription factor, it would be very interesting to determine whether Vif affects T cell differentiation.
Vif IS ALSO UBIQUITINATED
Fujita et al. (2004) have reported that expression of the Vif protein in virus-producing cells is maintained at very low levels, which is regulated by the ubiquitin–proteasome pathway. It is because its high expression inhibits viral infectivity by affecting proteolytic processing of Gag protein (Akari et al., 2004). We have identified the E3 ligase for Vif as mouse double minute 2 homolog (MDM2; Izumi et al., 2009; Figure 2). Since Vif is a component of a Cul5–EloB/C complex, one report showed that this complex ubiquitinated Vif (Mehle et al., 2004a). Another report showed that other E3 ligases such as neural precursor cell expressed developmentally down-regulated protein 4 (Nedd4) and atrophin-interacting protein 4 (AIP4) bound to Vif, however, it didn’t show the direct evidence of Vif ubiquitination by these ligases (Dussart et al., 2004). The identification of the E3 ligase has lead to elucidation of the mechanisms of Vif-induced G2 cell cycle arrest described below.
FIGURE 2
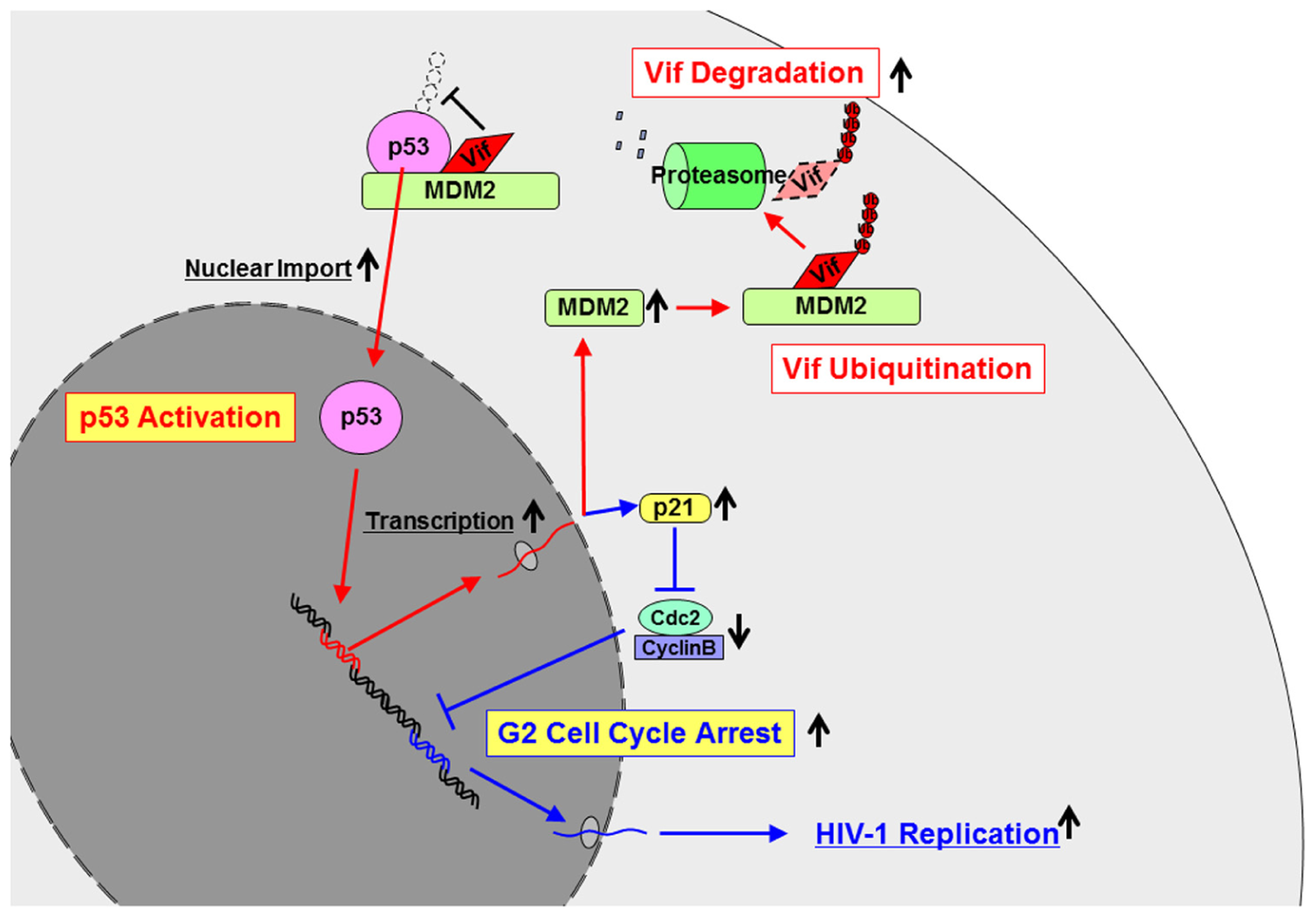
The mechanisms how Vif is ubiquitinated and degraded and how Vif induces G2 cell cycle arrest. Vif is ubiquitinated and degraded by MDM2. On the contrary, Vif inhibits ubiquitination of p53 by MDM2 to induce activation and nuclear import of p53. Activated p53 induces transcription of several genes including MDM2 and p21. Enhanced expression of MDM2 may lead to more Vif ubiquitination and degradation, which forms the autoregulatory circuit of Vif expression. On the other hand, activation of p21 leads to G2 cell cycle arrest, resulting in more HIV-1 replication.
A NOVEL Vif FUNCTION: G2 CELL CYCLE ARREST
In early 1990s, viral protein R (Vpr) had been shown to induce G2 cell cycle arrest in HIV-1-infected cells (He et al., 1995; Re et al., 1995; Roshal et al., 2003; Nakai-Murakami et al., 2006). Many groups have extensively worked on Vpr-induced G2 arrest in terms of its molecular mechanisms and published many papers. Although only one paper reported the virological significance of G2 arrest induced by Vpr (Goh et al., 1998), the basic and fundamental questions of why the virus needs to induce G2 arrest still remain unsolved. More than 10 years had passed since then, and two recent reports came out, describing that Vif as well as Vpr-induce G2 arrest in HIV-1-infected cells (Sakai et al., 2006; Wang et al., 2007). We have recently shown the molecular mechanisms by which Vif induces G2 arrest (Izumi et al., 2010; Figure 2). Vif activates p53, which is well known as a tumor suppressor gene and the regulator of cell cycle as “a guardian of the genome.” Vif binds and activates p53 by stabilizing and sequestering it to the nucleus. Activation of p53 induces its downstream cascade such as activation of p21 and inactivation of Cdc2/CyclinB, resulting in G2 arrest. Furthermore, we identified the amino acid residues in Vif responsible for its interaction with p53 and a Vif mutant which does not induce G2 arrest. Using a mutant virus which possesses the vif mutant, we have demonstrated that Vif-induced G2 arrest facilitates viral replication (Izumi et al., 2010; Figure 2). Thus, HIV-1 needs to have G2 cell cycle arrest to efficiently replicate so that it possesses two accessory genes such as vif and vpr. Vif induces G2 arrest in a p53-dependent manner, while Vpr accomplishes the same goal in a p53-independent manner.
CONCLUSION
HIV Vif is an intriguing viral protein, not only because it opens up a new era in the research field of host restriction factors, but also because it has a variety of functions for the viral life cycle by interacting several cellular proteins. It suggests that it might be a good target for control of HIV-1 infection.
Statements
Acknowledgments
This work was partly supported by grants-in-aid from the Ministry of Education, Culture, Sports, Science, and Technology and from the Ministry of Health, Labour and Welfare in Japan. This work was also partly supported by grants from SENSHIN Medical Research Foundation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AkariH.FujitaM.KaoS.KhanM. A.Shehu-XhilagaM.AdachiA.et al (2004). High level expression of human immunodeficiency virus type-1 Vif inhibits viral infectivity by modulating proteolytic processing of the Gag precursor at the p2/nucleocapsid processing site.J. Biol. Chem.27912355–12362.
2
BogerdH. P.DoehleB. P.WiegandH. L.CullenB. R. (2004). A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV type 1 virion infectivity factor.Proc. Natl. Acad. Sci. U.S.A.1013770–3774.
3
CamaurD.TronoD. (1996). Characterization of human immunodeficiency virus type 1 Vif particle incorporation.J. Virol.706106–6111.
4
ChenG.HeZ.WangT.XuR.YuX. F. (2009). A patch of positively charged amino acids surrounding the human immunodeficiency virus type 1 Vif SLVx4Yx9Y motif influences its interaction with APOBEC3G.J. Virol.838674–8682.
5
DangY.WangX.ZhouT.YorkI. A.ZhengY. H. (2009). Identification of a novel WxSLVK motif in the N terminus of human immunodeficiency virus and simian immunodeficiency virus Vif that is critical for APOBEC3G and APOBEC3F neutralization.J. Virol.838544–8552.
6
DesrosiersR. C.LifsonJ. D.GibbsJ. S.CzajakS. C.HoweA. Y.ArthurL. O.et al (1998). Identification of highly attenuated mutants of simian immunodeficiency virus.J. Virol.721431–1437.
7
DussartS.CourcoulM.BessouG.DouaisiM.DuvergerY.VigneR.et al (2004). The Vif protein of human immunodeficiency virus type 1 is posttranslationally modified by ubiquitin.Biochem. Biophys. Res. Commun.31566–72.
8
FisherA. G.EnsoliB.IvanoffL.ChamberlainM.PettewayS.RatnerL.et al (1987). The sor gene of HIV-1 is required for efficient virus transmission in vitro.Science237888–893.
9
FujitaM.AkariH.SakuraiA.YoshidaA.ChibaT.TanakaK.et al (2004). Expression of HIV-1 accessory protein Vif is controlled uniquely to be low and optimal by proteasome degradation.Microbes Infect.6791–798.
10
GabuzdaD. H.LawrenceK.LanghoffE.TerwilligerE.DorfmanT.HaseltineW. A.et al (1992). Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes.J. Virol.666489–6495.
11
GaurR.StrebelK. (2012). Insights into the dual activity of SIVmac239 Vif against human and African green monkey APOBEC3G.PLoS ONE 7:e48850. 10.1371/journal.pone.0048850
12
GohW. C.RogelM. E.KinseyC. M.MichaelS. F.FultzP. N.NowakM. A.et al (1998). HIV-1 Vpr increases viral expression by manipulation of the cell cycle: a mechanism for selection of Vpr in vivo.Nat. Med.465–71.
13
Goila-GaurR.StrebelK. (2008). HIV-1 Vif, APOBEC, and intrinsic immunity.Retrovirology551.
14
HeJ.ChoeS.WalkerR.Di MarzioP.MorganD. O.LandauN. R. (1995). Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity.J. Virol.696705–6711.
15
HeZ.ZhangW.ChenG.XuR.YuX. F. (2008). Characterization of conserved motifs in HIV-1 Vif required for APOBEC3G and APOBEC3F interaction.J. Mol. Biol.3811000–1011.
16
HuthoffH.MalimM. H. (2007). Identification of amino acid residues in APOBEC3G required for regulation by human immunodeficiency virus type 1 Vif and Virion encapsidation.J. Virol.813807–3815.
17
IwataniY.ChanD. S.LiuL.YoshiiH.ShibataJ.YamamotoN.et al (2009). HIV-1 Vif-mediated ubiquitination/degradation of APOBEC3G involves four critical lysine residues in its C-terminal domain.Proc. Natl. Acad. Sci. U.S.A.10619539–19544.
18
IzumiT.IoK.MatsuiM.ShirakawaK.ShinoharaM.NagaiY.et al (2010). HIV-1 viral infectivity factor interacts with TP53 to induce G2 cell cycle arrest and positively regulate viral replication.Proc. Natl. Acad. Sci. U.S.A.10720798–20803.
19
IzumiT.Takaori-KondoA.ShirakawaK.HigashitsujiH.ItohK.IoK.et al (2009). MDM2 is a novel E3 ligase for HIV-1 Vif.Retrovirology61.
20
JagerS.KimD. Y.HultquistJ. F.ShindoK.LarueR. S.KwonE.et al (2012). Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection.Nature481371–375.
21
KitamuraS.OdeH.IwataniY. (2011). Structural features of antiviral APOBEC3 proteins are linked to their functional activities.Front. Microbiol. 2:258. 10.3389/fmicb.2011.00258
22
KobayashiM.Takaori-KondoA.MiyauchiY.IwaiK.UchiyamaT. (2005). Ubiquitination of APOBEC3G by an HIV-1 Vif–Cullin5–Elongin B–Elongin C complex is essential for Vif function.J. Biol. Chem.28018573–18578.
23
LuoK.XiaoZ.EhrlichE.YuY.LiuB.ZhengS.et al (2005). Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G.Proc. Natl. Acad. Sci. U.S.A.10211444–11449.
24
MadaniN.KabatD. (1998). An endogenous inhibitor of human immunodeficiency virus in human lymphocytes is overcome by the viral Vif protein.J. Virol.7210251–10255.
25
MangeatB.TurelliP.LiaoS.TronoD. (2004). A single amino acid determinant governs the species-specific sensitivity of APOBEC3G to Vif action.J. Biol. Chem.27914481–14483.
26
MarianiR.ChenD.SchrofelbauerB.NavarroF.KonigR.BollmanB.et al (2003). Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif.Cell11421–31.
27
MarinM.RoseK. M.KozakS. L.KabatD. (2003). HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation.Nat. Med.91398–1403.
28
MehleA.GoncalvesJ.Santa-MartaM.McpikeM.GabuzdaD. (2004a). Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif–Cul5 complex that promotes APOBEC3G degradation.Genes Dev.182861–2866.
29
MehleA.StrackB.AncutaP.ZhangC.McpikeM.GabuzdaD. (2004b). Vif Overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin–proteasome pathway.J. Biol. Chem.2797792–7798.
30
MehleA.ThomasE. R.RajendranK. S.GabuzdaD. (2006). A zinc-binding region in Vif binds Cul5 and determines cullin selection.J. Biol. Chem.28117259–17265.
31
Nakai-MurakamiC.ShimuraM.KinomotoM.TakizawaY.TokunagaK.TaguchiT.et al (2006). HIV-1 Vpr induces ATM-dependent cellular signal with enhanced homologous recombination.Oncogene26477–486.
32
OpiS.KaoS.Goila-GaurR.KhanM. A.MiyagiE.TakeuchiH.et al (2007). Human immunodeficiency virus type 1 Vif inhibits packaging and antiviral activity of a degradation-resistant APOBEC3G variant.J. Virol.818236–8246.
33
PeryE.RajendranK. S.BrazierA. J.GabuzdaD. (2009). Regulation of APOBEC3 proteins by a novel YXXL motif in human immunodeficiency virus type 1 Vif and simian immunodeficiency virus SIVagm Vif.J. Virol.832374–2381.
34
ReF.BraatenD.FrankeE. K.LubanJ. (1995). Human immunodeficiency virus type 1 Vpr arrests the cell cycle in G2 by inhibiting the activation of p34cdc2-cyclin B.J. Virol.696859–6864.
35
RoshalM.KimB.ZhuY.NghiemP.PlanellesV. (2003). Activation of the ATR-mediated DNA Damage Response by the HIV-1 viral protein R.J. Biol. Chem.27825879–25886.
36
RussellR. A.PathakV. K. (2007). Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F.J. Virol.818201–8210.
37
SakaiH.ShibataR.SakuragiJ.SakuragiS.KawamuraM.AdachiA. (1993). Cell-dependent requirement of human immunodeficiency virus type 1 Vif protein for maturation of virus particles.J. Virol.671663–1666.
38
SakaiK.DimasJ.LenardoM. J. (2006). The Vif and Vpr accessory proteins independently cause HIV-1-induced T cell cytopathicity and cell cycle arrest.Proc. Natl. Acad. Sci. U.S.A.1033369–3374.
39
SchrofelbauerB.ChenD.LandauN. R. (2004). A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif).Proc. Natl. Acad. Sci. U.S.A.1013927–3932.
40
ShaoQ.WangY.HildrethJ. E.LiuB. (2010). Polyubiquitination of APOBEC3G is essential for its degradation by HIV-1 Vif.J. Virol.844840–4844.
41
SheehyA. M.GaddisN. C.ChoiJ. D.MalimM. H. (2002). Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein.Nature418646–650.
42
SheehyA. M.GaddisN. C.MalimM. H. (2003). The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif.Nat. Med.91404–1407.
43
ShirakawaK.Takaori-KondoA.KobayashiM.TomonagaM.IzumiT.FukunagaK.et al (2006). Ubiquitination of APOBEC3 proteins by the Vif–Cullin5–ElonginB–ElonginC complex.Virology344263–266.
44
ShirakawaK.Takaori-KondoA.YokoyamaM.IzumiT.MatsuiM.IoK.et al (2008). Phosphorylation of APOBEC3G by protein kinase A regulates its interaction with HIV-1 Vif.Nat. Struct. Mol. Biol.151184–1191.
45
SimonJ. H.FouchierR. A.SoutherlingT. E.GuerraC. B.GrantC. K.MalimM. H. (1997). The Vif and Gag proteins of human immunodeficiency virus type 1 colocalize in infected human T cells.J. Virol.715259–5267.
46
SimonJ. H.GaddisN. C.FouchierR. A.MalimM. H. (1998a). Evidence for a newly discovered cellular anti-HIV-1 phenotype.Nat. Med.41397–1400.
47
SimonJ. H.MillerD. L.FouchierR. A.SoaresM. A.PedenK. W.MalimM. H. (1998b). The regulation of primate immunodeficiency virus infectivity by Vif is cell species restricted: a role for Vif in determining virus host range and cross-species transmission.EMBO J.171259–1267.
48
StopakK.De NoronhaC.YonemotoW.GreeneW. C. (2003). HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability.Mol. Cell12591–601.
49
StrebelK.DaughertyD.ClouseK.CohenD.FolksT.MartinM. A. (1987). The HIV ‘A’ (sor) gene product is essential for virus infectivity.Nature328728–730.
50
WangJ.ShackelfordJ. M.CasellaC. R.ShiversD. K.RapaportE. L.LiuB.et al (2007). The Vif accessory protein alters the cell cycle of human immunodeficiency virus type 1 infected cells.Virology359243–252.
51
WissingS.GallowayN. L.GreeneW. C. (2010). HIV-1 Vif versus the APOBEC3 cytidine deaminases: an intracellular duel between pathogen and host restriction factors.Mol. Aspects Med.31383–397.
52
XiaoZ.EhrlichE.LuoK.XiongY.YuX. F. (2007). Zinc chelation inhibits HIV Vif activity and liberates antiviral function of the cytidine deaminase APOBEC3G.FASEB J.21217–222.
53
XuH.SvarovskaiaE. S.BarrR.ZhangY.KhanM. A.StrebelK.et al (2004). A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion.Proc. Natl. Acad. Sci. U.S.A.1015652–5657.
54
YangS.SunY.ZhangH. (2001). The multimerization of human immunodeficiency virus type I Vif protein: a requirement for Vif function in the viral life cycle.J. Biol. Chem.2764889–4893.
55
YangX.GabuzdaD. (1998). Mitogen-activated protein kinase phosphorylates and regulates the HIV-1 Vif protein.J. Biol. Chem.27329879–29887.
56
YangX.GoncalvesJ.GabuzdaD. (1996). Phosphorylation of Vif and its role in HIV-1 replication.J. Biol. Chem.27110121–10129.
57
YuX.YuY.LiuB.LuoK.KongW.MaoP.et al (2003). Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif–Cul5–SCF complex.Science3021056–1060.
58
YuY.XiaoZ.EhrlichE. S.YuX.YuX.-F. (2004). Selective assembly of HIV-1 Vif–Cul5–ElonginB–ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines.Genes Dev.182867–2872.
59
ZhangW.DuJ.EvansS. L.YuY.YuX. F. (2012). T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction.Nature481376–379.
Summary
Keywords
HIV-1 Vif, restriction factor, ubiquitin ligase, cell cycle arrest, p53, MDM2
Citation
Takaori-Kondo A and Shindo K (2013) HIV-1 Vif: a guardian of the virus that opens up a new era in the research field of restriction factors. Front. Microbio. 4:34. doi: 10.3389/fmicb.2013.00034
Received
28 November 2012
Accepted
07 February 2013
Published
20 February 2013
Volume
4 - 2013
Edited by
Atsushi Koito, Kumamoto University, Japan
Reviewed by
Mikako Fujita, Kumamoto University, Japan; Klaus Strebel, National Institutes of Health, USA; Vinay K. Pathak, National Cancer Institute, USA
Copyright
© Takaori-Kondo and Shindo.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Akifumi Takaori-Kondo, Department of Hematology and Oncology, Graduate School of Medicine, Kyoto University, Shogoin-Kawaracho 54, Sakyo-ku, Kyoto 606-8507, Japan. e-mail: atakaori@kuhp.kyoto-u.ac.jp
This article was submitted to Frontiers in Virology, a specialty of Frontiers in Microbiology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.