 Sayaka Mino1*
Sayaka Mino1* Hiroko Makita2
Hiroko Makita2 Tomohiro Toki3 Junichi Miyazaki2
Tomohiro Toki3 Junichi Miyazaki2 Shingo Kato4 Hiromi Watanabe5
Shingo Kato4 Hiromi Watanabe5 Hiroyuki Imachi2 Tomo-o Watsuji2
Hiroyuki Imachi2 Tomo-o Watsuji2 Takuro Nunoura2 Shigeaki Kojima6
Takuro Nunoura2 Shigeaki Kojima6 Tomoo Sawabe1
Tomoo Sawabe1 Ken Takai2
Ken Takai2 Satoshi Nakagawa1,2
Satoshi Nakagawa1,2- 1Laboratory of Microbiology, Faculty of Fisheries Sciences, Hokkaido University, Hakodate, Japan
- 2Subsurface Geobiology Advanced Research Project, Institute of Biogeosciences, Japan Agency for Marine-Earth Science and Technology, Yokosuka, Japan
- 3Department of Chemistry, Biology, and Marine Science, Faculty of Science, University of the Ryukyus, Nishihara, Okinawa, Japan
- 4Japan Collection of Microorganisms, RIKEN BioResource Center, Tsukuba, Japan
- 5Marine Biodiversity Research Program, Japan Agency for Marine-Earth Science and Technology, Yokosuka, Japan
- 6Department of Marine Ecosystem Dynamics, Atmosphere and Ocean Research Institute, The University of Tokyo, Kashiwa, Japan
Deep-sea hydrothermal vent fields are areas on the seafloor with high biological productivity fueled by microbial chemosynthesis. Members of the Aquificales genus Persephonella are obligately chemosynthetic bacteria, and appear to be key players in carbon, sulfur, and nitrogen cycles in high temperature habitats at deep-sea vents. Although this group of bacteria has cosmopolitan distribution in deep-sea hydrothermal ecosystem around the world, little is known about their population structure such as intraspecific genomic diversity, distribution pattern, and phenotypic diversity. We developed the multi-locus sequence analysis (MLSA) scheme for their genomic characterization. Sequence variation was determined in five housekeeping genes and one functional gene of 36 Persephonella hydrogeniphila strains originated from the Okinawa Trough and the South Mariana Trough (SNT). Although the strains share >98.7% similarities in 16S rRNA gene sequences, MLSA revealed 35 different sequence types (ST), indicating their extensive genomic diversity. A phylogenetic tree inferred from all concatenated gene sequences revealed the clustering of isolates according to the geographic origin. In addition, the phenotypic clustering pattern inferred from whole-cell matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF/MS) analysis can be correlated to their MLSA clustering pattern. This study represents the first MLSA combined with phenotypic analysis indicative of allopatric speciation of deep-sea hydrothermal vent bacteria.
Introduction
Mixing of hydrothermal fluids and ambient seawater at the seafloor creates physically and chemically dynamic habitats for microorganisms. Vent fluids physicochemistry is variable both spatially and temporally as a result of subsurface geological and geochemical processes (Edmond et al., 1979; Butterfield and Massoth, 1994; Butterfield et al., 2004). Diverse microorganisms including both Archaea and Bacteria have been isolated in pure cultures from various hydrothermal fields (Nakagawa and Takai, 2006). In addition, culture-independent studies revealed the dominance of yet-to-be cultured microorganisms in deep-sea hydrothermal environments (Haddad et al., 1995; Takai and Horikoshi, 1999; Reysenbach et al., 2000; Corre, 2001; Teske et al., 2002), and provided insight into the great heterogeneity of microbial communities between hydrothermal systems. The heterogeneity can be correlated to differences in the geological and chemical properties between different vents (Takai et al., 2004; Nakagawa et al., 2005a,b; Takai and Nakamura, 2011). On the other hand, there are also some cosmopolitan genera found in deep-sea hydrothermal systems occurring not only in the Mid-Ocean Ridge systems but in the Back-Arc Basin systems and the Volcanic Arc systems (Takai et al., 2006; Nakagawa and Takai, 2008; Kaye et al., 2011). Members of the genus Persephonella belonging to the order Aquificales, obligately sulfur- and/or hydrogen-oxidizing, chemoautotrophic, thermophilic bacteria, are widely distributed in deep-sea hydrothermal systems (Reysenbach et al., 2000, 2002; Takai et al., 2004; Nakagawa et al., 2005a,b; Ferrera et al., 2007; Takai et al., 2008). Although the widespread occurrence of this group suggests that they may play important role, many questions remained about their physiology, metabolism, and ecology within the environment because of the difficulty in isolating these strains. Some isolates have been characterized (Götz et al., 2002; Nakagawa et al., 2003), and implied their role in carbon, sulfur and nitrogen cycles in high temperature habitats at deep-sea vents (Reysenbach et al., 2002; Ferrera et al., 2007). However, little is known about the spatial or biogeographical pattern of Persephonella microdiversity and phenotypic heterogeneity.
Weak biogeographical signals in microbial communities are usually explained by the hypothesis of microbial cosmopolitanism formulated by Bass Becking (Wit and Bouvier, 2006). However, recent studies have explored the effects of dispersal limitation on microbial biogeography. Like macroorganisms, the genetic similarity negatively correlated with geographic distance, i.e., distance-decay relationship, have been reported for cyanobacteria, sulfate-reducing bacteria, marine planktonic bacteria, and hyperthermophilic archaea (Papke et al., 2003; Whitaker et al., 2003; Vergin et al., 2007; Oakley et al., 2010). In addition, the biogeographical diversity pattern was reported in detail for members of the “deep-sea hydrothermal vent euryarchaeota 2” (Flores et al., 2012). Microbial biogeographical studies have been usually based solely on genetic data. Microbial biogeography was recently studied at the phenotypic level (Rosselló-Mora et al., 2008), however, genetic and phenotypic correlation has not been explored. We investigated the spatial diversity pattern of Persephonella population by the combined use of comparative genetic and phenotypic characterizations.
Materials and Methods
Field Site and Sampling
Samples, i.e., chimney structures, fluids, and sediments, were collected with R/V Natsushima and ROV Hyper-Dolphin or R/V Yokosuka and DSV Shinkai 6500 from the Okinawa Trough (OT) in 2007 and 2009, or the South Mariana Trough (SMT) in 2010 (Table 1). Vent fluids from the OT are characteristic in the high contents of methane and carbon dioxide (Kawagucci et al., 2011). Among the OT hydrothermal fields, this study focused on the Iheya North and Hatoma Knoll (Figure 1). In the SMT, four vent sites were studied (Figure 1). The Archaean site is located at a ridge flank, about 2 km apart from the backarc-spreading axis. Discharging fluids (Tmax = 318°C) was acidic and depleted in Cl− (Cl− = 401 mM) (Ishibashi et al., 2006). Pika site is located on an off-axis knoll, about 5 km from the axis. Fluid chemistry (Tmax = 330°C) of Pika site showed brine-rich signature (Cl− = 600 mM) (Ishibashi et al., 2006). Urashima site is newly discovered in 2010, and located at the northern foot of the western peak of the same knoll as Pika. Snail site is located on the active backarc-spreading axis. After retrieval on board, each of the chimney structures were sectioned immediately into the exterior surface and the inside parts, and slurried with 25 ml of sterilized seawater in the presence or absence of 0.05% (w/v) neutralized sodium sulfide in 100 ml glass bottles (Schott Glaswerke, Mainz, Germany). Bottles were then tightly sealed with butyl rubber caps under a gas phase of 100% N2 (0.2 MPa). Similarly, fluid, sediment, and biological samples were prepared anaerobically in 10 ml glass bottles. Samples were stored at 4°C until use.
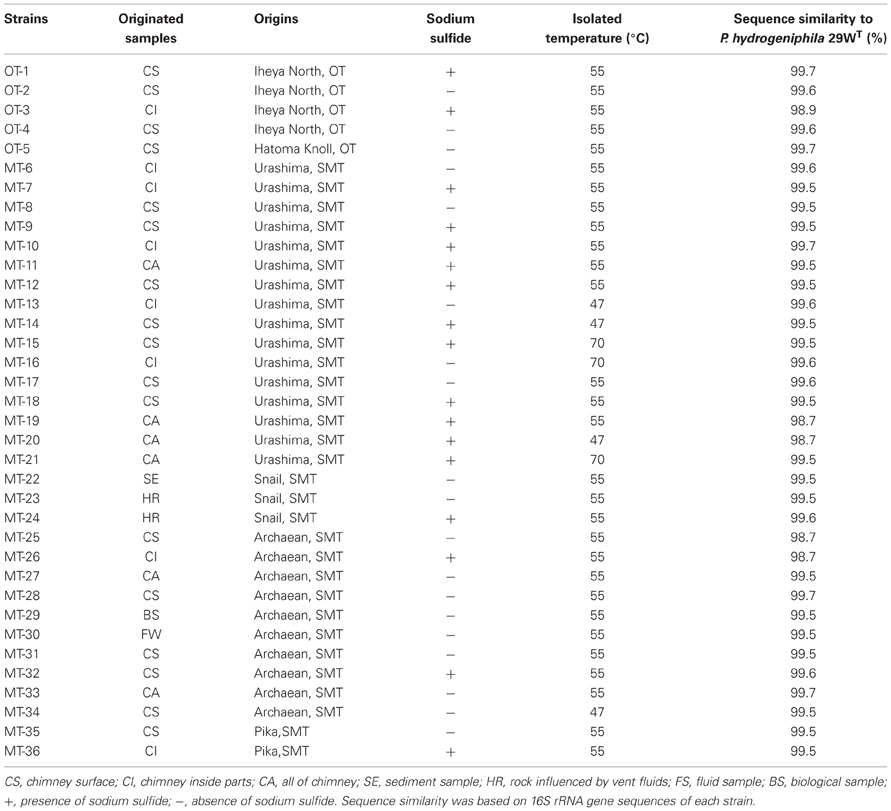
Table 1. Information of samples and cultivation temperature.
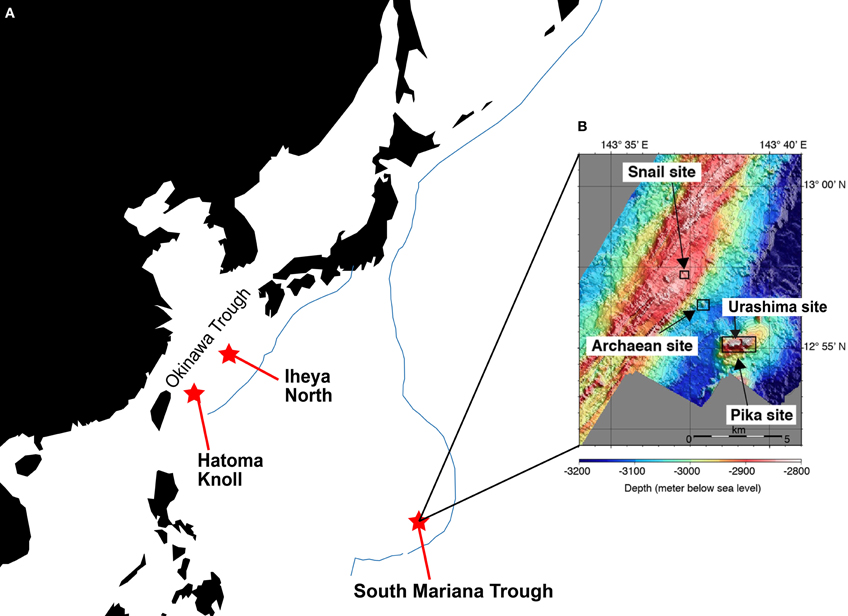
Figure 1. Sampling sites. (A) Location of the Iheya North field and Hatoma Knoll in the Okinawa Trough, and the South Mariana Trough. Blue line indicates subduction zone. (B) Location of four vent sites in the South Mariana Trough.
Enrichment, Isolation, and Phylogenetic Analysis
Serial dilution cultures were performed using the MMJHS medium (Takai et al., 2003) containing a mixture of electron donors and electron acceptors for hydrogen/sulfur-oxidizing chemoautotrophs at 47, 55 and 70°C. MMJHS medium included 1 g each of NaHCO3, Na2S2O3.5H2O, and NaNO3, 10 g of S0 and 10 ml vitamin solution (Balch et al., 1979) per liter of MJ synthetic seawater [gas phase: 80% H2 +20% CO2 (0.3 MPa)]. To obtain pure cultures, dilution-to-extinction was repeated at least 2 times (Baross, 1995). The purity was confirmed routinely by microscopic examination and by sequencing of the 16S rRNA gene using several PCR primers. Genomic DNA was extracted from isolates using the UltraClean Microbial DNA isolation Kit (MoBio Laboratories, Inc., Solana Beach, CA, USA) following the manufacturer's protocol. The 16S rRNA gene of each isolate was amplified by PCR using LA Taq polymerase (TaKaRa Bio, Otsu, Japan) as described previously (Takai et al., 2001). The primers used were Eubac 27F and 1492R (Weisburg et al., 1991). These amplicons were bidirectionally determined by the dideoxynucleotide chain-termination method. Almost complete sequences of the 16S rRNA gene were assembled using Sequencher ver 4.8 (Gene Codes Corporation, Ann Arbor, MI, USA). In order to determine the phylogenetic positions of isolates, the sequences were aligned using Greengenes NAST alignment tool (DeSantis et al., 2006), and compiled using ARB software version 03.08.22 (Ludwig et al., 2004).
Multi-Locus Sequence Analysis
Intraspecies diversity among isolates was evaluated using multi-locus sequence analysis [MLSA; formerly called multilocus sequence typing (MLST)] technique (Gevers et al., 2005). MLSA represents a universal and unambiguous method for strain genotyping, population genetics, and molecular evolutionary studies (Whitaker et al., 2005; Mazard et al., 2012). Genes selected for MLSA were tkt (transketolase), atpA (ATP synthase, A subunit), dnaK (Hsp 70 chaperon protein), napA (nitrate reductase, large subunit), metG (methionyl-tRNA synthetase), and gyrB (DNA gyrase, B subunit). Primers (Table 2) were designed according to the published complete genome sequences of Aquificales members, i.e., Persephonella marina EX-H1T (NC_012439) (Reysenbach et al., 2009), Sulfurihydrogenibium azorense (NC_012438) (Reysenbach et al., 2009), Sulfurihydrogenibium sp. YO3AOP1 (NC_010730) (Reysenbach et al., 2009), Hydrogenobaculum sp. Y04AAS1 (NC_011126) (Reysenbach et al., 2009), Aquifex aeolicus VF5 (NC_000918) (Deckert et al., 1998), and Hydrogenivirga sp. 128-5-R1-1 (NZ_ABHJ01000000) (Reysenbach et al., 2009). ClustalX version 2.0 was used for the alignment of nucleotide sequences (Larkin et al., 2007). PCR were performed under the following conditions: 96°C for 1 min, 35–38 cycles of 96°C for 20 s, annealing for 45 s at temperatures shown in Table 2, and 72°C for 2 min. The PCR products were confirmed by 1% agarose gel electrophoresis and purified with exonuclease I and shrimp alkaline phosphatase. If necessary, bands were excised and purified using Wizard® SV Gel and PCR Clean-up System (Promega, Madison, WI, USA). Purified PCR products were used as templates for Sanger sequencing reaction. The sequences were assembled and edited using Sequencher ver 4.8, and aligned with ClustalX. The sequences were translated into amino acid using Transeq program (EMBOSS; European Molecular Biology Open Software Suite).
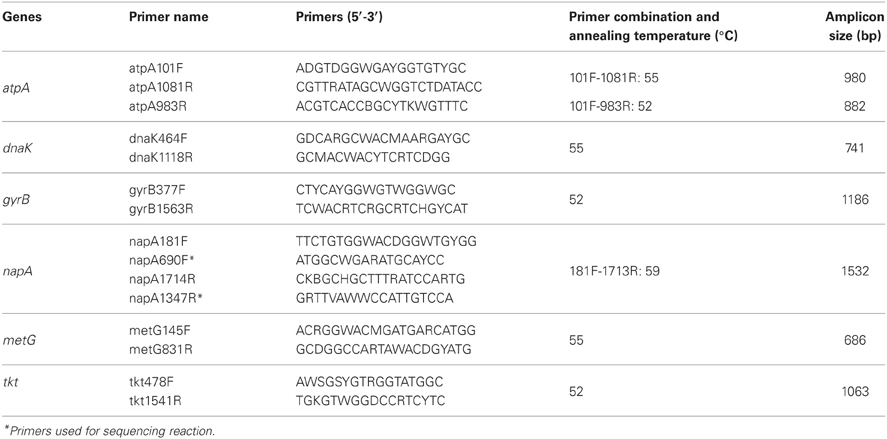
Table 2. Primers and PCR conditions for MLSA.
Z-test, non-synonymous (Ka)/synonymous (Ka) substitution, and Tajima's D test were performed as described elsewhere (Vergin et al., 2007). Briefly, values of Ka and Ks were determined using the software program SWAAP ver 1.0.3 (Pride, 2000), set to the Li method with a window size of 90 and step size of 18. Z-test was performed using MEGA ver 5.05 software (Tamura et al., 2011) with the following options: purifying selection, overall average, 1000 bootstrap replicates, pairwise deletion and the Pamilo–Bianchi–Li method. Tajima's D based on the total number of mutation were calculated using DnaSP ver 5 (Librado and Rozas, 2009). The combination of allele types for each isolate defined the sequence type (ST). Phylogenetic trees were constructed by the maximum likelihood (ML) method using MEGA ver 5.05. ML bootstrap support was computed after 100 reiterations. Split decomposition trees were constructed with SplitsTree ver 4 using the Neighbor-Net algorithm (Huson, 1998).
The levels of genetic variation within and between populations were calculated with the Arlequin ver 3.5 software (Excoffier and Lischer, 2010). FST values were estimated for groups of two or more strains and were tested for significance against 1000 randomized bootstrap resamplings. Average pairwise genetic distance and standard error based on 500 bootstrap resamplings of each population were estimated using MEGA ver 5.05. Mantel test was performed with XLSTAT software (www.xlstat.com). Sequences obtained in this study have been deposited in DDBJ/EMBL/GenBank under Accession No. AB773894-AB774147.
Geochemical Analysis
Chemical compositions listed in Table 6 were analyzed as previously described (Takai et al., 2008; Toki et al., 2008). End-member fluids compositions were estimated by the conventional method, that is extrapolation to Mg = 0 of linear relationship of concentration of each species to Mg among the obtained samples (Von Damm et al., 1985).
Preparation of Bacterial Samples for Whole-Cell MALDI-TOF/MS
Samples for whole-cell matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF/MS) were prepared as described in Hazen et al. (2009). Briefly, Persephonella strains were cultured in 3 ml of MMJHS medium at their isolated temperatures (Table 1). Following incubation, cells were washed once in 1 ml of 0.85% NaCl and twice in 1 ml of 50% ethanol at 4°C. Cell pellets were weighed and resuspended in 1% trifluoroacetic acid (TFA) to yield a final concentration of 0.2 mg cells/μl of 1% TFA. Equal volumes of the TFA bacterial suspension and the MALDI-TOF/MS matrix solution (10 mg/ml sinapinic acid in 50% acetonitrile, 50% water, and 0.1% TFA) were mixed in a microcentrifuge tube, and then 1.0 μl of this mixture was spotted in triplicate on a stainless steel MALDI-TOF/MS sample plate (corresponding to approximately 1.4 × 108 cells/spot). Samples were allowed to air dry before being loaded in the mass spectrometer.
MALDI-TOF/MS and Data Processing
All mass spectra were acquired using the MALDI-TOF/MS spectrometer (4700 proteomics analyzer; Applied Biosystems, Foster City, CA, USA) in the linear and positive-ion modes. The laser (N2, 337 nm) intensity was set above the ion generation threshold. Mass spectra were recorded in the m/z range of 2000–14,000. The acceptance criteria, based on 1000 laser shots per spot, were signal intensities between 2000 and 55,000 counts and a signal/noise ratio of 10 or greater.
Raw mass spectra from three spots were normalized using Data Explorer software (Applied Biosystems, Foster City, CA, USA) by baseline correction and combined to generate an averaged peak list. The peaks around 2000 m/z were excluded as noise.
The peaks were ranked according to their signal intensities, and the top 15 most intense peaks were chosen for further analysis. The relative intensity ratio was calculated for the 15 peaks. Squared distance was estimated based on the presence or absence of peaks by Ward's minimum variance method using MVSP software ver 3.21 (Kovach Computing Services, Wales, UK). The presence or absence of peaks was determined within a tolerance of 14 Da.
Results
Isolation of Persephonella Strains
We investigated a total of 36 Persephonella strains originating from various hydrothermal samples from the OT (4 strains from Iheya North, and 1 strain from Hatoma Knoll) and the SMT (16 strains from Urashima site, 10 strains from Archaean site, 3 strains from Snail site, and 2 strains from Pika site) (Figure 2 and Table 1). All of the 36 strains shared >98.7% 16S rRNA gene similarities with one another and with P. hydrogeniphila 29WT.

Figure 2. Phylogenetic relationship of isolates and representative Persephonella species as determined by neighbor-joining analysis of 16S rRNA gene sequences. Tree was constructed by using 1119 sites that could be unambiguously aligned. Origins of isolates and DDBJ accession numbers are shown in parentheses. Branch points conserved with bootstrap values of >75% (filled circle) and bootstrap value of 50–74% (open circles) are indicated.
Genetic Diversity of Persephonella Population
We developed a MLSA scheme for the Persephonella population based on five housekeeping genes and a functional gene. The gene fragments sequenced varied from 501 to 882 bp in length (Table 1), and nucleotide sequence similarity at MLSA loci varied from 94.6 to 96.3% (average 95.8%). We obtained concatenated sequences of 4254 bp and identified a total of 702 variable positions. Ratios of non-synonymous to synonymous substitutions (Ka/Ks) were much smaller than 1 for all loci (Table 3), indicating the genes were subject to purifying selection, conforming to the general requirements for MLSA loci (Maiden, 2006). This was statistically supported by the high values from the Z-test (Table 3).
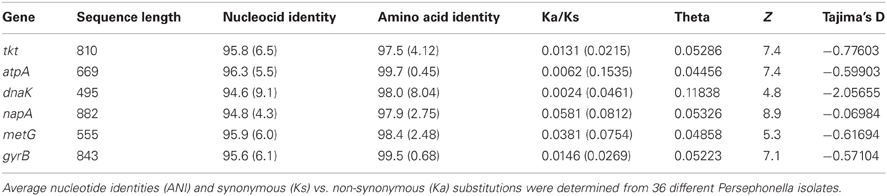
Table 3. Genetic features at the six MLSA loci.
Population Genetic Structure
Typing based on sequences of six protein-coding gene fragments revealed 35 different STs among 36 isolates, indicating the high genetic diversity of Persephonella population (Table 4). The number of different alleles per locus varied between 16 for metG and 24 for tkt. Strains MT-17 and −18 had identical sequences for all MLSA loci. These strains were isolated from the same chimney sample, but the slurries were prepared in the presence (used for strain MT-18) or absence (used for strain MT-17) of 0.05% (w/v) sodium sulfide. In other cases, the presence of sodium sulfide in slurries resulted in the isolation of strains classified into different STs (Table 1).
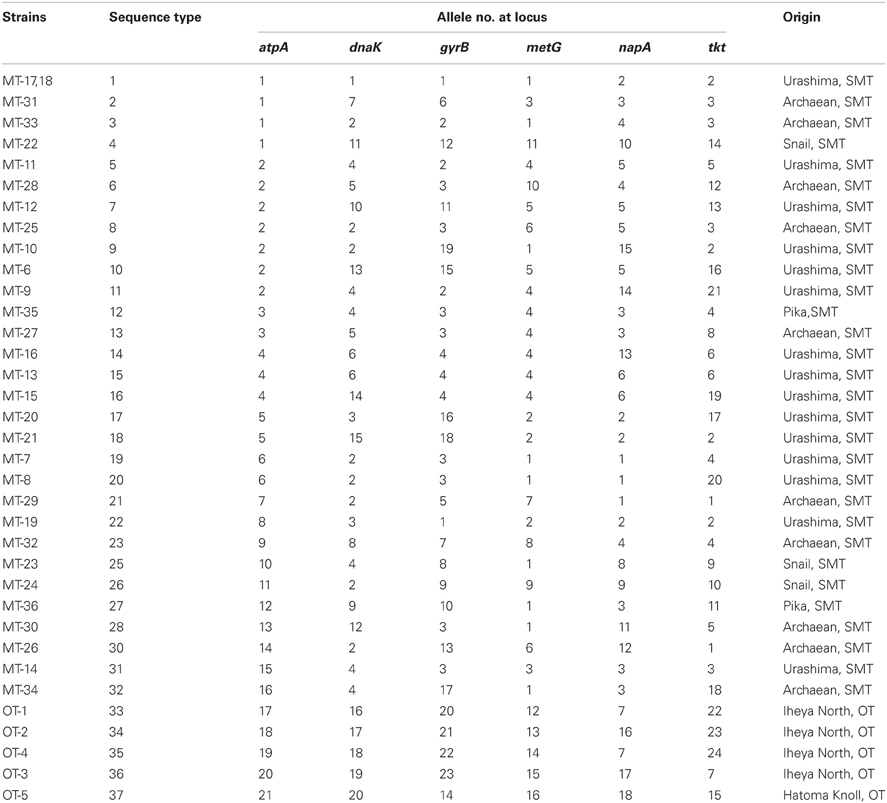
Table 4. Allelic properties at six loci of Persephonella isolates analyzed in this study.
The split graph obtained from the concatenated sequence data displayed bushy network structures with complex parallelogram formation indicative of extensive homologous recombination (Figure 3). The result of PHI test (Bruen et al., 2006) for the concatenated sequences also showed the presence of the past recombination events during the evolution of Persephonella (p < 0.05).

Figure 3. Neighbor-Net graph based on the concatenated sequences of 6 protein-coding genes of Persephonella isolates showing a bushy network structure indicative of homologous recombination. Scale bar represent 0.1 substitutions per nucleotide position. Origins of isolates are indicated as follows: light blue, Iheya North; blue, Hatoma Knoll; red, Archaean; orange, Snail; green, Pika; brown, Urashima.
Population Difference Between the OT and the SMT
A ML phylogenetic tree derived from the concatenated alignment of six loci showed two different clades with high bootstrap support (Figure 4). The two clades corresponded to the two geographic regions, showing that the SMT strains share a common evolutionary history distinct from the OT strains. The FST value confirmed that the OT and the SMT populations were significantly different (FST = 0.8711, p < 0.05) (Table 5).

Figure 4. Maximum likelihood tree based on concatenated sequences of all MLSA loci. Origins of isolates are indicated as follows: light blue, Iheya North; blue, Hatoma Knoll; red, Archaean; orange, Snail; green, Pika; brown, Urashima.
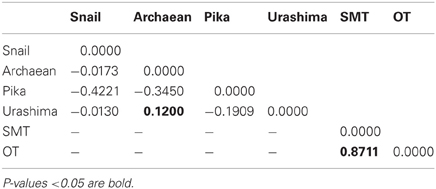
Table 5. FST values between each population.
Correlation Between Chemistry and Genetic Diversity
Geochemical analysis revealed that different vent fluids had distinctive end-member chemical compositions (Table 6). Although the vent fluids from Archaean and Pika were respectively Cl−-depleted and -enriched in 2004 and 2005 (Ishibashi et al., 2006), no significant difference was found between them in this study. We assessed the relative contributions of environmental factors (such as pH and maximum temperature of vent fluids) and geographic distance to Persephonella genetic structure using the Mantel test. The pH of SMT vent fluids (pH 3.0–3.5) were significantly lower than those (pH 5.0–5.2) of OT (Table 6). However, we found no significant correlation between the genetic distance and the absolute difference in vent fluid pH and temperature (Mantel r = −0.28, p = 0.4). In contrast, a large, significant correlation coefficient (Mantel r = 0.993, p < 0.0001) was found in a Mantel test of all pairwise comparisons of the genetic and the geographic distance between strains (Figure 5).
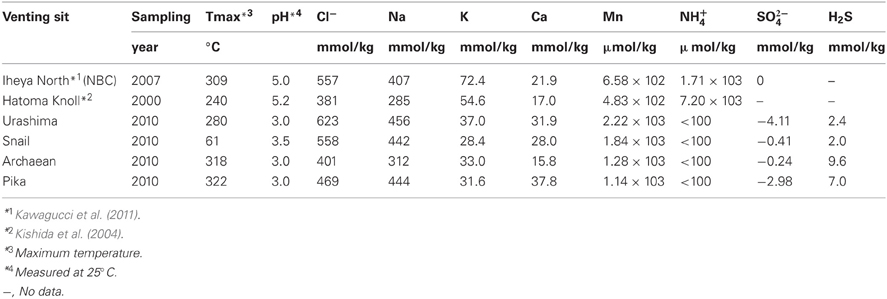
Table 6. End-member compositions of vent fluids from the OT and the SMT.
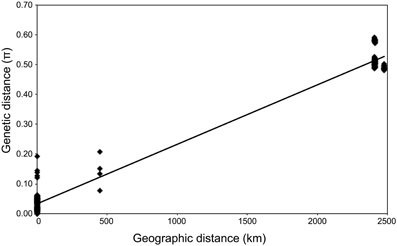
Figure 5. Relationship between genetic and geographic distance (R2 = 0.98). π, number of base substitutions per site from between sequences.
Whole-Cell MALDI-TOF/MS Analysis
MALDI-TOF/MS fingerprinting of whole microbial cells was highly reproducible. A peak at m/z 9678 in the MALDI-TOF/MS spectra was detected in all strains despite their geographical origin (Figure 6). Some peaks were detected in some strains with relatively low intensities. Cluster analysis based on the presence or absence of peaks identified two clusters that would correspond to the geographical regions of isolation (Figure 7). Two Persephonella trees, one generated from the whole-cell MALDI-TOF/MS data and a ML tree from concatenated MLSA sequences, show similar topologies (Figure 7).
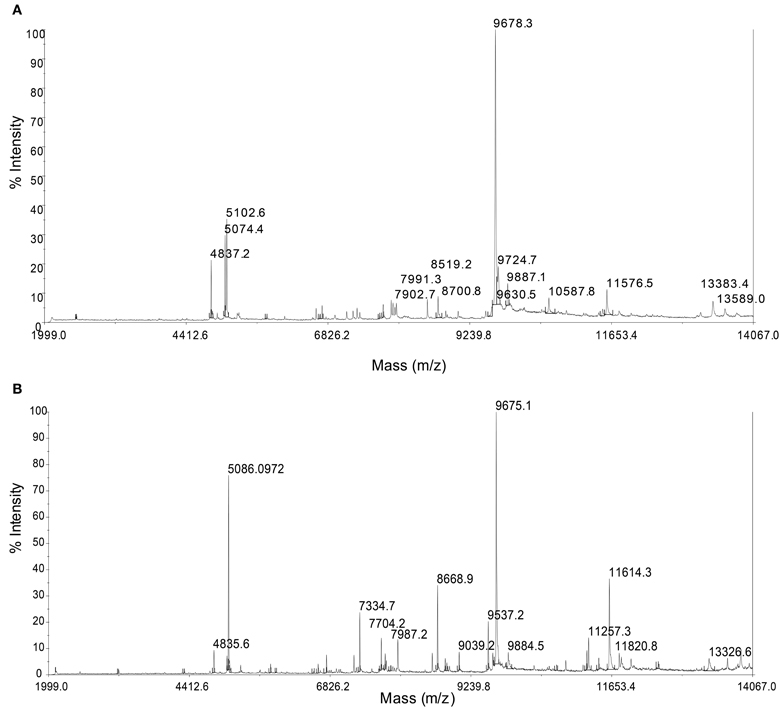
Figure 6. Representative whole-cell MALDI-TOF/MS spectra from the OT strain (OT-2, A) and the SMT strain (MT-26, B).
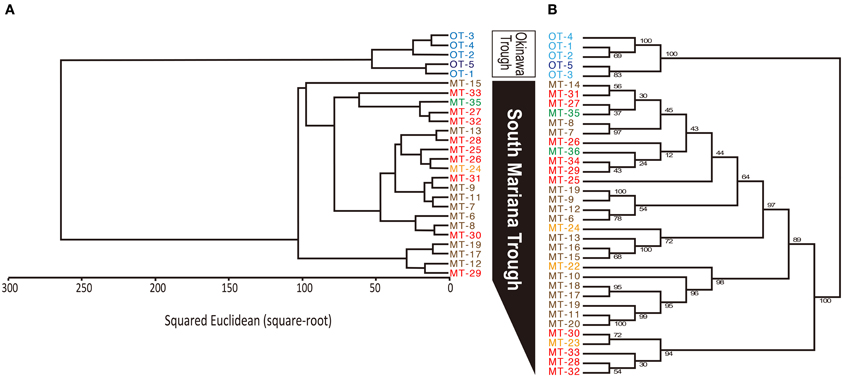
Figure 7. Correlation between genotype and phenotype inferred from MALDI-TOF/MS analysis (A) and MLSA (B). Origins of isolates are indicated as follows: light blue, Iheya North; blue, Hatoma Knoll; red, Archaean; orange, Snail; green, Pika; brown, Urashima.
Discussion
Here we investigated the microdiversity and phenotypic heterogeneity of extremely thermophilic chemolithoautotrophic bacteria in deep-sea hydrothermal vents. Genetic and phenotypic differences corresponding to the geographic origins were discovered by the combined use of MLSA and whole-cell MALDI-TOF/MS fingerprinting. The biogeography of hydrothermal vent-associated microbial community has been well studied (Takai et al., 2004; Nakagawa et al., 2005a,b; Kato et al., 2010). Members of the genus Persephonella have been found in global hydrothermal vent fields, however, their genetic and phenotypic heterogeneities were poorly understood.
Genetic Difference Between OT and SMT Populations
We identified 35 STs among 36 Persephonella strains by MLSA based on 6 protein-coding genes, indicating high genetic diversity of Persephonella population. The same ST is rarely shared among Persephonella strains, however, all SMT strains have the same alleles with other SMT strains but not with OT strains in one or more MLSA loci, suggesting that OT and SMT populations are significantly different. Likewise, two OT strains, i.e., OT-1 and -4, have the same allele (no. 7) at napA (Table 4), although the number of OT strains obtained in this study is small.
The split decomposition tree showed the evidence of recombination (Figure 3), which might contribute to increased STs. Previous studies showed that recombination generated the large number of unique combinations of alleles in some archaea and bacteria (Suerbaum et al., 2001; Whitaker et al., 2005; Doroghazi and Buckley, 2010).
Biogeography of Persephonella
The phylogenetic analysis based on concatenated gene sequences separated the strains into two clusters according to their geographic origins (Figure 4). The FST value supported significant biogeographical isolation between SMT and OT populations. These results indicate that ubiquitous occurrence of Persephonella in deep-sea vents has not resulted from widespread contemporary dispersal but is an ancient historical legacy.
The microbial distribution seems to be not only influenced by local environmental conditions (Martiny et al., 2006). In this study, we observed clear correlation between the genetic distance and the geographic distance of isolates (Figure 5) as described in thermophilic archaea (Whitaker et al., 2003; Flores et al., 2012). On the contrary, genetic distance has no significant correlation with the difference in vent fluid pH and temperature. We cannot rule out the possibility that other factors not determined in this study, including grazing pressure and virus activity, may be correlated with the genetic difference of Persephonella. Recently, H2 concentration in vent fluids was shown to have an impact on the formation of microbial community structures in deep-sea vents (Takai and Nakamura, 2011).
Correlation Between Genotypic and Phenotypic Heterogeneity
Some peaks in the MALDI-TOF/MS spectra were shared among some Persephonella strains. Major peaks of whole-cell MALDI-TOF/MS analysis are considered to reflect ribosomal proteins (Fenselau and Plamen, 2001; Ryzhov and Fenselau, 2001) and thus are independent of growth conditions (Bernardo et al., 2002). There were also some minor peaks that were specific to SMT or OT strains. Likewise the concatenated nucleotide alignment of MLSA loci, MALDI-TOF/MS data clustered the strains into two distinct groups corresponding to the geographic regions (Figure 7), suggesting that protein expression of Persephonella is tuned to function optimally in their original habitats. The genotypic and phenotypic correlation found among Persephonella isolates indicates the occurrence of allopatric speciation.
Conclusion
By using both comparative genetic and phenotypic population characterizations, this study for the first time indicated the Persephonella populations were geographically distinct. Since the Persephonella members are extremely thermophilic chemoautotrophs endemic to deep-sea vents, considerable dispersal barriers for the migration to spatially distinct niches should exist. Focal points raised by this study for future research include the effects of cold, oxic deep-sea conditions on the viability of deep-sea vent (hyper) thermophiles during the dispersal, the biogeographical comparison with other ubiquitous thermophiles with different metabolic traits (e.g., heterotrophic fermenters and methanogens), and the comparison with moderately thermophiles or mesophiles with similar energy/carbon metabolisms (e.g., Epsilonproteobacteria) in deep-sea vents.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the captain and crew of the R/V Yokosuka and R/V Natsushima and Shinkai 6500 and Hyper Dolphin operation team on cruises JAMSTEC YK10-10, YK10-11, NT07-13, and NT09-11. Sayaka Mino was supported by the Research Fellowship of the Japan Society for the Promotion of Science.
References
Balch, W. E., Fox, G. E., Magrum, L. J., Woese, C. R., and Wolfe, R. S. (1979). Methanogens: reevaluation of a unique biological group. Microbiol. Rev. 43, 260–296.
Baross, J. A. (1995). “Isolation, growth and maintenance of hyperthermophiles,” in Archaea: A Laboratory Manual, Thermophiles, eds F. T. Robb and A. R. Place (New York, NY: Cold Spring Harbor Laboratory), 15–23.
Bernardo, K., Fleer, S., Pakulat, N., Krut, O., Hünger, F., and Krönke, M. (2002). Identification of Staphylococcus aureus exotoxins by combined sodium dodecyl sulfate gel electrophoresis and matrix-assisted laser desorption/ ionization-time of flight mass spectrometry. Proteomics 2, 740–746.
Bruen, T. C., Philippe, H., and Bryant, D. (2006). A simple and robust statistical test for detecting the presence of recombination. Genetics 172, 2665–2681.
Butterfield, D. A., and Massoth, G. J. (1994). Geochemistry of north cleft segment vent fluids: temporal changes in chlorinity and their possible relation to recent volcanism. J. Geophys. Res. 99, 4951–4968.
Butterfield, D. A., Roe, K. K., Lilley, M. D., Huber, J., Baross, J. A., Embley, R. W., et al. (2004). “Mixing, reaction and microbial activity in the sub-seafloor revealed by temporal and spatial variation in diffuse flow vents at Axial Volcano,” in The Subseafloor Biosphere at Mid-Ocean Ridges, Geophysical Monograph Series. Vol. 144, eds W. S. D. Wilcock, E. F. DeLong, D. S. Kelley, J. A. Baross, and S. C. Cary (Washington, DC: American Geophysical Union), 269–289.
Corre, E. (2001). ε-Proteobacterial diversity from a deep-sea hydrothermal vent on the Mid-Atlantic Ridge. FEMS Microbiol. Lett. 205, 329–335.
Deckert, G., Warren, P. V., Gaasterland, T., Young, W. G., Lenox, A. L., and Graham, D. E. (1998). The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature 392, 353–358.
DeSantis, T. Z., Hugenholtz, P., Keller, K., Brodie, E. L., Larsen, N., and Piceno, Y. M. (2006). NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 34, W394–W399.
Doroghazi, J. R., and Buckley, D. H. (2010). Widespread homologous recombination within and between Streptomyces species. ISME J. 4, 1136–1143.
Edmond, J. M., McDuff, R. E., and Chan, L. H. (1979). Ridge crest hydrothermal activity and the balances of the major and minor elements in the ocean: the Galapagos data. Earth Planet. Sci. Lett. 46, 1–18.
Excoffier, L., and Lischer, H. E. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567.
Fenselau, C., and Plamen, A. D. (2001). Characterization of intact microorganisms by MALDI mass spectrometory. Mass Spectrom. Rev. 20, 157–171.
Ferrera, I., Longhorn, S., Banta, A. B., Liu, Y., Preston, D., and Reysenbach, A. L. (2007). Diversity of 16S rRNA gene, ITS region and aclB gene of the Aquificales. Extremophiles 11, 57–64.
Flores, G. E., Wagner, I. D., Liu, Y., and Reysenbach, A. L. (2012). Distribution, abundance, and diversity patterns of the thermoacidophilic “deep-sea hydrothermal vent euryarchaeota 2”. Front. Microbiol. 3:47. doi: 10.3389/fmicb.2012.00047
Gevers, D., Cohan, F. M., Lawrence, J. G., Spratt, B. G., Coenye, T., and Feil, E. J. (2005). Re-evaluating prokaryotic species. Nat. Rev. Microbiol. 3, 733–739.
Götz, D., Banta, A., Beveridge, T. J., Rushdi, A. I., Simoneit, B. R., and Reysenbach, A. L. (2002). Persephonella marina gen. nov., sp. nov. and Persephonella guaymasensis sp. nov., two novel, thermophilic, hydrogen-oxidizing microaerophiles from deep-sea hydrothermal vents. Int. J. Syst. Evol. Microbiol. 52, 1349–1359.
Haddad, A., Camacho, F., Durand, P., and Cary, S. C. (1995). Phylogenetic characterization of the epibiotic bacteria associated with the hydrothermal vent polychaete Alvinella pompejana. Appl. Environ. Microbiol. 61, 1679–1687.
Hazen, T. H., Martinez, R. J., Chen, Y., Lafon, C., Garrett, N. M., and Parsons, M. B. (2009). Rapid identification of Vibrio parahaemolyticus by whole-cell matrix-assisted laser desorption ionization-time of flight mass spectrometry. Appl. Environ. Microbiol. 75, 6745–6756.
Huson, D. H. (1998). SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics 14, 68–73.
Ishibashi, J., Suzuki, R., Yamanaka, T., Toki, T., Kimura, H., Noguchi, T., et al. (2006). Seafloor hydrothermal activity at off-axial seamounts of backarc spreading in southern Mariana Trough. Geochim. Cosmochim. Acta 70, A279.
Kato, S., Takano, Y., Kakegawa, T., Oba, H., Inoue, K., Kobayashi, C., et al. (2010). Biogeography and biodiversity in sulfide structures of active and inactive vents at deep-sea hydrothermal fields of the southern mariana trough. Appl. Environ. Microbiol. 76, 2968–2979.
Kawagucci, S., Chiba, H., Ishibashi, J., Yamanaka, T., Toki, T., Muramatsu, Y., et al. (2011). Hydrothermal fluid geochemistry at the Iheya North field in the mid-Okinawa Trough: Implication for origin of methane in subseafloor fluid circulation systems. Geochem. J. 45, 109–124.
Kaye, J. Z., Sylvan, J. B., Edwards, K. J., and Baross, J. A. (2011). Halomonas and Marinobacter ecotypes from hydrothermal vent, subseafloor and deep-sea environments. FEMS Microbiol. Ecol. 75, 123–133.
Kishida, K., Sohrin, Y., Okamura, K., and Ishibachi, J. (2004). Tungsten enriched in submarine hydrothermal fluids. Earth Planet. Sci. Lett. 222, 819–827.
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948.
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452.
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar. et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371.
Martiny, J. B., Bohannan, B. J., Brown, J. H., Colwell, R. K., Fuhrman, J. A., Green, J. L., et al. (2006). Microbial biogeography: putting microorganisms on the map. Nat. Rev. Microbiol. 4, 102–112.
Mazard, S., Ostrowski, M., Partensky, F., and Scanlan, D. J. (2012). Multi-locus sequence analysis, taxonomic resolution and biogeography of marine Synechococcus. Environ. Microbiol. 14, 372–386.
Nakagawa, S., and Takai, K. (2006). The isolation of thermophiles from deep-sea hydrothermal environments. Methods Microbiol. 35, 55–91.
Nakagawa, S., and Takai, K. (2008). Deep-sea vent chemoautotrophs: diversity, biochemistry and ecological significance. FEMS microbiol. Ecol. 65, 1–14.
Nakagawa, S., Takai, K., Horikoshi, K., and Sako, Y. (2003). Persephonella hydrogeniphila sp. nov., a novel thermophilic, hydrogen-oxidizing bacterium from a deep-sea hydrothermal vent chimney. Int. J. Syst. Evol. Microbiol. 53, 863–869.
Nakagawa, S., Takai, K., Inagaki, F., Hirayama, H., Nunoura, T., Horikoshi, K., et al. (2005a). Distribution, phylogenetic diversity and physiological characteristics of epsilon-Proteobacteria in a deep-sea hydrothermal field. Environ. Microbiol. 7, 1619–1632.
Nakagawa, S., Takai, K., Inagaki, F., Chiba, H., Ishibashi, J., Kataoka, S., et al. (2005b). Variability in microbial community and venting chemistry in a sediment-hosted backarc hydrothermal system: impacts of subseafloor phase-separation. FEMS Microbiol. Ecol. 54, 141–155.
Oakley, B. B., Carbonero, F., Van der Gast, C. J., Hawkins, R. J., and Purdy, K. J. (2010). Evolutionary divergence and biogeography of sympatric niche-differentiated bacterial populations. ISME J. 4, 488–497.
Papke, R. T., Ramsing, N. B., Bateson, M. M., and Ward, D. M. (2003). Geographical isolation in hot spring cyanobacteria. Environ. Microbiol. 5, 650–659.
Pride, D. T. (2000). SWAAP Version 1.0.0 - Sliding Windows Alignment Analysis Program: A Tool for Analyzing Patterns of Substitutions and Similarity in Multiple Alignments. Distributed by the author.
Reysenbach, A. L., Dorothée, G., Banta, A., Jeanthon, C., and Fouqut, Y. (2002). Expanding the distribution of the Aquificales to the deep-sea vents on Mid-Atlantic Ridge and Central Indian Ridge. Cah. Biol. Mar. 43, 425–428.
Reysenbach, A. L., Hamamura, N., Podar, M., Griffiths, E., Ferreira, S., Hochstein, R., et al. (2009). Complete and draft genome sequences of six members of the Aquificales. J. Bacteriol. 191, 1992–1993.
Reysenbach, A. L., Longnecker, K., and Kirshtein, J. (2000). Novel bacterial and archaeal lineages from an in situ growth chamber deployed at a Mid-Atlantic Ridge hydrothermal vent. Appl. Environ. Microbiol. 66, 3798–3806.
Rosselló-Mora, R., Lucio, M., Peña, A., Brito-Echeverría, J., López-López, A., Valens-Vadell, M., et al. (2008). Metabolic evidence for biogeographic isolation of the extremophilic bacterium Salinibacter ruber. ISME J. 2, 242–253.
Ryzhov, V., and Fenselau, C. (2001). Characterization of the protein subset desorbed by MALDI from whole bacterial cells. Anal. Chem. 73, 746–750.
Suerbaum, S., Lohrengel, M., Sonnevend, A., Ruberg, F., and Kist, M. (2001). Allelic diversity and recombination in Campylobacter jejuni. J. Bacteriol. 183, 2553–2559.
Takai, K., Gamo, T., Tsunogai, U., Nakayama, N., Hirayama, H., Nealson, K. H., et al. (2004). Geochemical and microbiological evidence for a hydrogen-based, hyperthermophilic subsurface lithoautotrophic microbial ecosystem (HyperSLiME) beneath an active deep-sea hydrothermal field. Extremophiles 8, 269–282.
Takai, K., and Horikoshi, K. (1999). Genetic diversity of Archaea in deep-sea hydrothermal vent environments. Genetics 152, 1285–1297.
Takai, K., Inagaki, F., Nakagawa, S., Hirayama, H., Nunoura, T., Sako, Y., et al. (2003). Isolation and phylogenetic diversity of members of previously uncultivated epsilon-Proteobacteria in deep-sea hydrothermal fields. FEMS Microbiol. Lett. 218, 167–174.
Takai, K., Komatsu, T., Inagaki, F., and Horikoshi, K. (2001). Distribution of archaea in a black smoker chimney structure. Appl. Environ. Microbiol. 67, 3618–3629.
Takai, K., Nakagawa, S., Reysenbach, A. L., and Hoek, J. (2006). “Microbial ecology of mid-ocean ridges and back-arc basins,” in Back-arc Spreading Systems: Geological, Biological, Chemical, and Physical Interactions, Geophysical Monograph Series. Vol. 166, eds D. M. Christie, C. R. Fisher, S. M. Lee, and S. Givens (Washington, DC: American Geophysical Union), 185–213.
Takai, K., and Nakamura, K. (2011). Archaeal diversity and community development in deep-sea hydrothermal vents. Curr. Opin. Microbiol. 14, 282–291.
Takai, K., Nunoura, T., Ishibashi, J., Lupton, J., Suzuki, R., Hamasaki, H., et al. (2008). Variability in the microbial communities and hydrothermal fluid chemistry at the newly discovered Mariner hydrothermal field, southern Lau Basin. J. Geophys. Res. 113:G02031. doi: 10.1029/2007JG000636
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739.
Teske, A., Hinrichs, K. U., Edgcomb, V., de Vera Gomez, A., Kysela, D., Sylva, S. P., et al. (2002). Microbial diversity of hydrothermal sediments in the Guaymas Basin: evidence for anaerobic methanotrophic communities. Appl. Environ. Microbiol. 68, 1994–2007.
Toki, T., Tsunogai, U., Ishibashi, J., Utsumi, M., and Gamo, T. (2008). Methane enrichment in low-temperature hydrothermal fluids from the Suiyo Seamount in the Izu-Bonin Arc of the western Pacific Ocean. J. Geophys. Res. 113:B08S13. doi: 10.1029/2007JB005476
Vergin, K. L., Tripp, H. J., Wilhelm, L. J., Denver, D. R., Rappé, M. S., and Giovannoni, S. J. (2007). High intraspecific recombination rate in a native population of Candidatus pelagibacter ubique (SAR11). Environ. Microbiol. 9, 2430–2440.
Von Damm, K. L. V., Edmond, J. M., and Grant, B. (1985). Chemistry of submarine hydrothermal solutions at 21°N, East Pacific Rise. Geochem. Cosmochim. Acta 49, 2197–2220.
Weisburg, W. G., Barns, S. M., Pelletier, D. A., and Lane, D. J. (1991). 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173, 697–703.
Whitaker, R. J., Grogan, D. W., and Taylor, J. W. (2003). Geographic barriers isolate endemic populations of hyperthermophilic archaea. Science 301, 976–978.
Whitaker, R. J., Grogan, D. W., and Taylor, J. W. (2005). Recombination shapes the natural population structure of the hyperthermophilic archaeon Sulfolobus islandicus. Mol. Biol. Evol. 22, 2354–2361.
Keywords: population structure, biogeography, deep-sea hydrothermal vent, Persephonella, Aquificales, MLSA, MALDI-TOF/MS, chemolithoautotroph
Citation: Mino S, Makita H, Toki T, Miyazaki J, Kato S, Watanabe H, Imachi H, Watsuji T, Nunoura T, Kojima S, Sawabe T, Takai K and Nakagawa S (2013) Biogeography of Persephonella in deep-sea hydrothermal vents of the Western Pacific. Front. Microbiol. 4:107. doi: 10.3389/fmicb.2013.00107
Received: 30 December 2012; Paper pending published: 28 February 2013;
Accepted: 13 April 2013; Published online: 25 April 2013.
Edited by:
Anna-Louise Reysenbach, Portland State University, USAReviewed by:
Barbara J. Campbell, University of Delaware, USAElizaveta Bonch-Osmolovskyaya, Winogradsky Institute of Microbiology, Russian Academy of Sciences, Russia
Copyright © 2013 Mino, Makita, Toki, Miyazaki, Kato, Watanabe, Imachi, Watsuji, Nunoura, Kojima, Sawabe, Takai and Nakagawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Sayaka Mino, Laboratory of Microbiology, Faculty of Fisheries Sciences, Hokkaido University, 3-1-1, Minato-cho, Hakodate 041-8611, Japan. e-mail:bWlub0BlYy5ob2t1ZGFpLmFjLmpw