 Toyotaka Sato1
Toyotaka Sato1 Shin-ichi Yokota2
Shin-ichi Yokota2 Ikuo Uchida3 Torahiko Okubo1
Ikuo Uchida3 Torahiko Okubo1 Masaru Usui1 Masahiro Kusumoto4 Masato Akiba4 Nobuhiro Fujii2
Masaru Usui1 Masahiro Kusumoto4 Masato Akiba4 Nobuhiro Fujii2 Yutaka Tamura1*
Yutaka Tamura1*- 1Laboratory of Food Microbiology and Food Safety, Department of Health and Environmental Sciences, School of Veterinary Medicine, Rakuno Gakuen University, Ebetsu, Japan
- 2Department of Microbiology, Sapporo Medical University School of Medicine, Sapporo, Japan
- 3Dairy Hygiene Research Division, Hokkaido Research Station, National Institute of Animal Health, Sapporo, Japan
- 4Bacterial and Parasitic Disease Research Division, Safety Research Team, National Institute of Animal Health, Ibaraki, Japan
Fluoroquinolone resistance can cause major clinical problems. Here, we investigated fluoroquinolone resistance mechanisms in a clinical Escherichia coli isolate, HUE1, which had no mutations quinolone resistance-determining regions (QRDRs) of DNA gyrase and topoisomerase IV. HUE1 demonstrated MICs that exceeded the breakpoints for ciprofloxacin, levofloxacin, and norfloxacin. HUE1 harbored oqxAB and qnrS1 on distinct plasmids. In addition, it exhibited lower intracellular ciprofloxacin concentrations and higher mRNA expression levels of efflux pumps and their global activators than did reference strains. The genes encoding AcrR (local AcrAB repressor) and MarR (MarA repressor) were disrupted by insertion of the transposon IS3-IS629 and a frameshift mutation, respectively. A series of mutants derived from HUE1 were obtained by plasmid curing and gene knockout using homologous recombination. Compared to the MICs of the parent strain HUE1, the fluoroquinolone MICs of these mutants indicated that qnrS1, oqxAB, acrAB, acrF, acrD, mdtK, mdfA, and tolC contributed to the reduced susceptibility to fluoroquinolone in HUE1. Therefore, fluoroquinolone resistance in HUE1 is caused by concomitant acquisition of QnrS1 and OqxAB and overexpression of AcrAB–TolC and other chromosome-encoded efflux pumps. Thus, we have demonstrated that QRDR mutations are not absolutely necessary for acquiring fluoroquinolone resistance in E. coli.
Introduction
Fluoroquinolones are widely used in the clinical treatment of various bacterial infections, such as urinary tract and blood stream infections caused by Escherichia coli. Many studies have reported the isolation of fluoroquinolone-resistant strains (Peña et al., 1995; Cizman et al., 2001; Sanchez et al., 2012). Fluoroquinolone resistance is mainly caused by point mutations in the quinolone resistance-determining regions (QRDRs) of the DNA gyrase (encoded by gyrA and gyrB) and topoisomerase IV (encoded by parC and parE) subunits (Yoshida et al., 1991; Conrad et al., 1996; Heisig, 1996; Breines et al., 1997). A slight decrease in susceptibility to fluoroquinolones is attributed to a single mutation in gyrA. Secondary mutations in gyrA and additional mutations in parC and/or parE are required to exceed the breakpoint of the fluoroquinolone MIC (Conrad et al., 1996; Heisig, 1996; Breines et al., 1997).
Recently, we reported a fluoroquinolone-resistant E. coli isolate without QRDR mutations, named HUE1 (Sato et al., 2011). Its MICs for fluoroquinolones, such as ciprofloxacin (CIP) and levofloxacin (LVX), exceeded the breakpoints established by the Clinical and Laboratory Standards Institute (CLSI) (Clinical and Laboratory Standards Institute, 2011). HUE1 possesses two plasmid-mediated quinolone-resistant determinants (PMQRs), viz., oqxAB and qnrS. In this bacterium, OqxAB is a plasmid-encoded efflux pump; however, the gene is present on the chromosomal DNA in most Klebsiella pneumoniae and Enterobacter cloacae strains (Bin Kim et al., 2009). The presence of this pump confers resistance to several antimicrobial agents, such as olaquindox (OLA), trimethoprim (TMP), and chloramphenicol (CHL), and decreases bacterial susceptibility to fluoroquinolones (Hansen et al., 2007). QnrS, on the other hand, is a member of the pentapeptide-repeat protein family that protects DNA gyrase (and probably also topoisomerase IV) from binding to fluoroquinolones, thereby decreasing fluoroquinolone susceptibility (Jacoby, 2005). However, acquisition of these PMQRs alone results in only a low level of fluoroquinolone resistance, with MICs that do not exceed the breakpoints for fluoroquinolones (Jacoby, 2005; Hansen et al., 2007). E. coli isolates lacking QRDR mutations in gyrA and parC and showing concomitant acquisition of oqxAB and qnrS have previously been reported in China; however, these isolates did not exceed the breakpoint for CIP (Zhao et al., 2010).
Our previous findings suggested that the fluoroquinolone resistance of HUE1, which lacks QRDR mutations, is associated with not only with the presence of oqxAB and qnrS but also with other fluoroquinolone-resistance mechanism(s) (Sato et al., 2011). In the current study, we investigated the fluoroquinolone-resistance mechanisms of the HUE1 strain.
Methods
Bacterial Isolates
E. coli HUE1 had been isolated from the urinary catheter of a 77-year-old female patient at Hokkaido University Hospital (Sapporo, Japan) in 2007 (Sato et al., 2011). The somatic (O) serotype was determined by the slide agglutination test by using Escherichia coli O antisera (Denka Seiken, Tokyo, Japan), and the flagellar (H) serotype was determined using reference sera obtained from the Statens Serum Institut (Hillerød, Denmark).
Susceptibility Testing and Genetic Analysis
Norfloxacin (NOR) was purchased from Sigma-Aldrich (St Louis, MO). Other antibiotics were obtained as described previously (Sato et al., 2011). Susceptibility to fluoroquinolones [CIP, LVX, urifloxacin (URX), sitafloxacin (STX), and NOR], nalidixic acid (NAL), CHL, and TMP was determined by the agar plate dilution method, according to CLSI guidelines (Clinical and Laboratory Standards Institute, 2011). Phe-Arg-β-naphthylamide (PAβN; final concentration, 20 mg/L), which is an inhibitor of the resistance-nodulation-division (RND)-type efflux pump, was purchased from Sigma-Aldrich.
The presence of oqxA, oqxB, and qnrS was determined by PCR (Sorensen et al., 2003; Cattoir et al., 2007). Full-length nucleotide sequences of oqxAB, qnrS, acrA, acrB, acrR, acrE, acrF, acrS, tolC, soxS, soxR, and rob were determined by PCR and direct sequencing by using the primer pairs listed in Table 1. The nucleotide sequence of marR was determined as previously described (Lindgren et al., 2003). Nucleotide sequences were determined using a BigDye Terminator v3.1 Cycle Sequencing Kit (Life Technologies, Carlsbad, CA), and sequencing was performed in a 3130 Genetic Analyzer (Life Technologies). The oqxAB nucleotide sequence of HUE1 was submitted to GenBank (accession number AB601773). All gene sequences, except those of oqxAB and qnrS, were compared with those of Escherichia coli strain K12 substrain MG1655, which was deposited in GenBank (accession number U00096), as the reference strain.

Table 1. Sequences of primers used for PCR and DNA sequencing in this study.
Transformation of Plasmids Derived from HUE1 into DH5α
Plasmids were isolated from HUE1 as described previously (Kado and Liu, 1981). The plasmids were electroporated into E. coli DH5α (Takara, Shiga, Japan) by using an ECM600 (BTX, San Diego, CA, USA) under the following conditions: voltage, 1.8 kV; capacitance, 25 μF; and resistance, 200 ohms. oqxAB and/or qnrS transformants were screened using Muller-Hinton (MH) agar containing CIP, TMP, and CHL at concentrations ranging from 2- to 8-fold of the respective MIC values.
Plasmid Curing, Plasmid Re-Introduction, and Southern Hybridization
Curing of plasmids and generation of spontaneous mutants were performed as previously described, with slight modifications (Deane and Rawlings, 2004). Briefly, strains were grown in 4 mL LB broth at 40°C and incubated for periods ranging from a week to a month. Then, clones were picked and grown in MH agar containing sub-MIC concentrations of CIP, TMP, and CHL or no antimicrobials, in order to obtain oqxAB- and/or qnrS-cured clones. Re-introduction of plasmids harboring oqxAB and/or qnrS into plasmid-cured mutants was performed using electroporation as described above. The presence of oqxA, oqxB, and qnrS was detected by PCR and Southern hybridization of plasmids, as previously described (Tamamura et al., 2011). Probes for oqxB and qnrS1 were prepared by PCR by using specific primers, as described previously (Cattoir et al., 2007; Bin Kim et al., 2009). Probe labeling was carried out using a PCR DIG labeling mix (Roche Diagnostics, Tokyo, Japan).
Real-Time Reverse-Transcription (RT) PCR
Overnight cultures were diluted 1:100 in LB broth and grown to the mid-logarithmic phase. RNA was isolated using an RNeasy Mini kit (Qiagen, Hilden, Germany). Gene expression was estimated by quantitative real-time reverse-transcription (RT)-PCR by using the probe and primer pairs shown in Table 2. RT-PCR was performed using a QuantiTect Probe RT-PCR kit (Qiagen) in 20 μL reactions containing 2.5 ng of purified RNA, 0.2 μM of probe, and 0.5 μM of each of the forward and reverse primers. The cycling conditions included reverse transcription at 50°C for 20 min and PCR involving initial activation at 95°C for 15 min and 45 cycles each consisting of 1 min at 55°C and 30 s at 60°C, in a LightCycler 480 system (Roche, Mannheim, Germany). The E. coli strain AG100 (K-12 argE3 thi-1 rpsL xyl mtl D(gal-uvrB) supE44) (Okusu et al., 1996), which was gifted by Dr. Helen I. Zgurskaya (University of Oklahoma, USA), was used as a control. Expression levels of gapA were used to normalize expression ratios. Data, except for those of oqxB and qnrS1, were calibrated against expression levels in AG100, which were set as 1, to determine fold changes in expression. Data for oqxB and qnrS1 were calibrated to the respective levels in HUE1, which were set as 1. The data shown represent the mean values of three independent experiments.
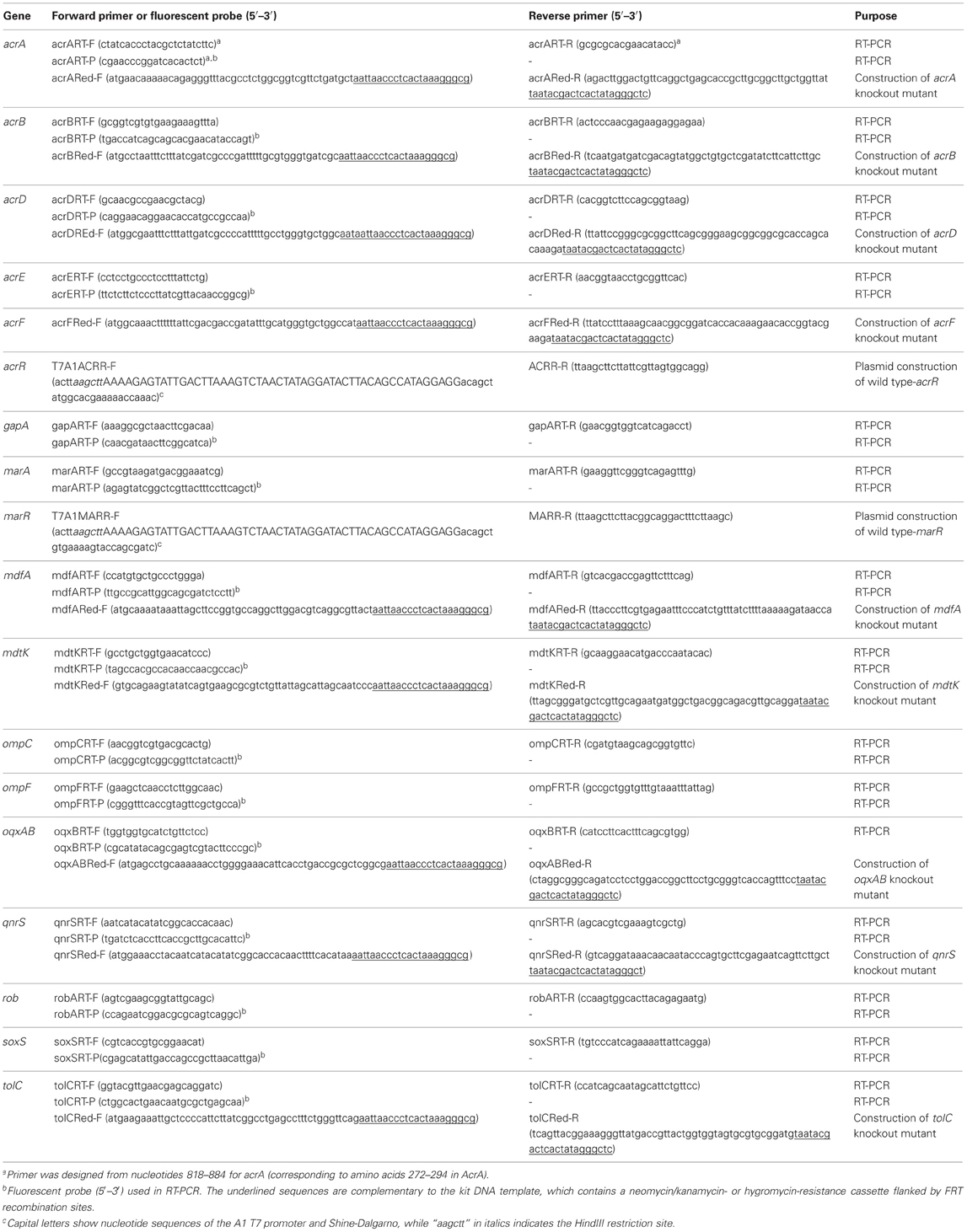
Table 2. Sequences of real-time RT-PCR probes and primers for RT-PCR or construction of knockout mutants designed in this study..
Construction of Gene Deletion Mutants
Gene disruption was performed by Red/ET recombination by using the Quick and Easy Gene Deletion kit (Gene Bridges GmbH, Heidelberg, Germany) according to the manufacturer's protocol. The relevant forward and reverse primers are shown in Table 2. Briefly, target gene-specific minigenes, containing a neomycin/kanamycin- or hygromycin-resistance cassette, were constructed by PCR with primers containing 46 bp of upstream and downstream sequences that contained the start codon or the stop codon (or the last codon before the stop codon) of the target genes, respectively (Table 2). The target genes were replaced with minigenes containing the drug-resistance cassette by using Red/ET recombination. The integration of interrupted genes was verified by PCR and DNA sequencing.
Plasmid Construction
Each of the wild-type acrR and wild-type marR DNA segments were amplified by PCR using DH5α genomic DNA as a template and primers containing the A1 T7 promoter, consensus Shine-Dalgarno, and HindIII restriction sites (Table 2), which were designed according to a previous report (Edgar et al., 2012). These PCR products were cloned into the HindIII site of pUC19 (wt-acrR or wt-marR), and the plasmids were then transformed into HUE1.
Accumulation Assays
Intracellular CIP concentrations were assayed using a fluorometric uptake assay (Usui et al., 2009). Forty milligrams of wet cells was treated with CIP (final concentration, 10 mg/L) in the presence or absence of carbonyl cyanide m-chlorophenylhydrazone (CCCP, Sigma-Aldrich; final concentration, 150 μM). The fluorescence of CIP was measured at excitation and emission wavelengths of 277 and 445 nm, respectively, by using an RF-5000 fluorescence spectrophotometer (Shimadzu, Kyoto, Japan) (Nakaminami et al., 2010). The data shown represent the mean value ± standard deviation values calculated from at least three independent experiments.
Statistical Analysis
Statistical significance was determined by the Student's t-test. Differences among more than three groups were determined using the Mann–Whitney U-test. A P-value of 0.05 or less was considered statistically significant.
Results
Characteristics of HUE1 and Genetic Analysis of OqxAB and QnrS
HUE1 was identified as ST48 (according to the Max-Planck-Institut für Infektionsbiologie database), phylogenetic group A (Sato et al., 2011), and O125:H37. Its NOR exceeded the breakpoints established by the CLSI, similarly with CIP and LVX previously tested (Sato et al., 2011). A 4421-bp DNA segment containing oqxAB and a 647-bp DNA segment containing qnrS derived from HUE1 were sequenced. The sequence of oqxAB was 100% identical to that of plasmid pOLA52 (accession number EU370913) in an E. coli isolate obtained from swine in Sweden (Hansen et al., 2004). The sequence of qnrS was 100% identical to that of qnrS1 in Shigella flexneri (accession number AB187515) (Hata et al., 2005). Plasmid profiling and Southern blotting analysis showed that oqxAB and qnrS1 were located on 2 independent plasmids, pHFQ1 (>165 kb) and pHFQ2 (>100 kb), respectively (Figure 1, lane 1).
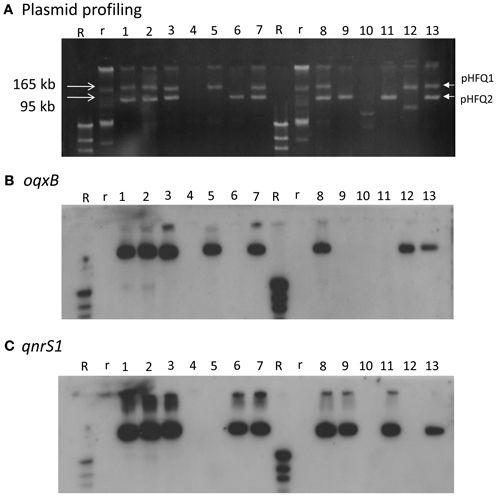
Figure 1. Plasmid profile and Southern hybridization of oqxB and qnrS1 in HUE1, its oqxAB- and/or qnrS1-cured strains, and oqxAB− and/or qnrS1−re-introduced strains. (A) Plasmid profile. Electrophoresis was performed at 100 V for 70 min with a 0.8% agarose gel. (B) Southern hybridization with the oqxB probe. (C) Southern hybridization with the qnrS probe. Lane 1, HUE1; lane 2, HUE1A2; lane 3, HUE1A; lane 4, HUE1A-Curoqxqnr; lane 5, HUE1A-Reoqx; lane 6, HUE1A-Reqnr; lane 7, HUE1A-Reoqxqnr; lane 8, HUE1; lane 9, HUE1B-Curoqx; lane 10, HUE1B-Curoqxqnr; lane 11, HUE1B-Reqnr; lane 12, HUE1B-Reoqx; lane 13, HUE1B-Reoqxqnr; R, DNA Molecular Weight MarkII, DIG-labeled: r, BAC-Tracker Supercoiled DNA Ladder.
Introduction of pHFQ1 (carrying oqxAB) into DH5α slightly increased the MICs for fluoroquinolones (except for LVX) and NAL, as compared with those for the host strain DH5α (Table 3). Introduction of pHFQ2 (carrying qnrS1) resulted in higher MICs for fluoroquinolones and NAL than those seen for DH5α/pHFQ1. However, introduction of both pHFQ1 and pHFQ2 into DH5α did not increase the MICs beyond the breakpoints for fluoroquinolone. Deletion of oqxAB or qnrS from the transformants by Red/ET recombination reverted MICs for fluoroquinolones to levels similar to those of DH5α.
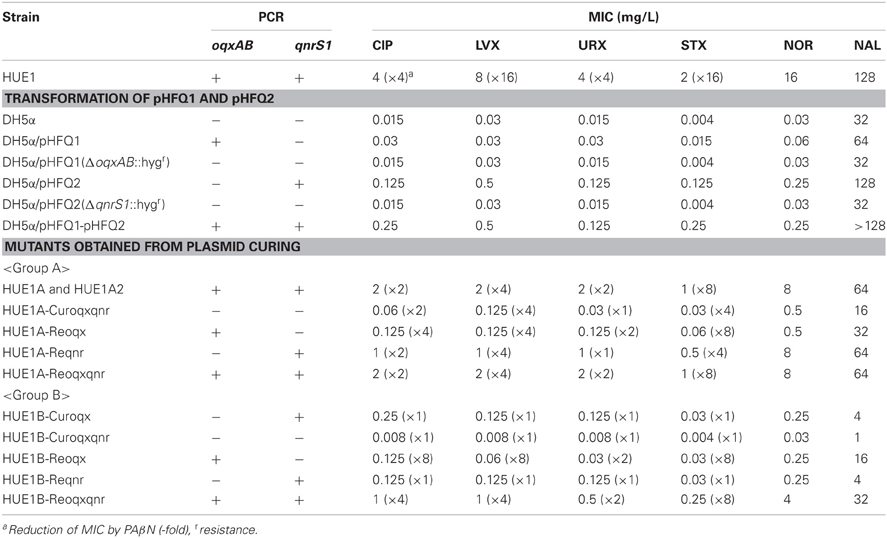
Table 3. Fluoroquinolones and NAL susceptibilities of HUE1, transformants derived from DH5α, and HUE1mutants derived from plasmid curing and reintroduction.
Characterization of Mutants Derived from Plasmid Curing and Plasmid Re-Introduction
Eleven mutants obtained by curing and re-introduction of the plasmids showed altered fluoroquinolone MICs (Figure 1 and Table 3). These mutants were grouped into two types (groups A and B). In the case of group A, we first obtained two strains, named HUE1A and HUE1A2. Although these two mutants still harbored pHFQ1 (carrying oqxAB) and pHFQ2 (carrying qnrS1; Figure 1, lanes 2 and 3), the MICs of NAL and fluoroquinolones were 2- or 4-fold lower than those of the parental strain, HUE1. Secondary screening of mutants by using HUE1A yielded a mutant, HUE1A-Curoqxqnr, which had lost pHFQ1 and pHFQ2 (Figure 1, lane 4). This mutant had 16- to 64-fold lower MICs for fluoroquinolones than the parental strain, HUE1A. Re-introduction of pHFQ1 and pHFQ2 into HUE1A-Curoqxqnr yielded HUE1A-Reoqxqnr, in which fluoroquinolone MICs recovered to the levels seen for HUE1A, but not to the MICs of the parent strain, HUE1.
In the case of group B, we first obtained a mutant, HUE1B-Curoqx, which had lost pHFQ1; a second screening using HUE1B-Curoqx yielded HUE1B-Curoqxqnr, which had lost both pHFQ1 and pHFQ2 (Figure 1, lanes 9 and 10). Interestingly, the fluoroquinolone MICs of HUE1B-Curoqxqnr were 512- and 1280-fold lower than those of HUE1 and were 4- to 16-fold lower than those of HUE1A-Curoqxqnr (Table 3).
Similar differences with respect to the MICs of groups A and B were also observed for the mutant series in which pHFQ2 (carrying qnrS1) had been re-introduced. However, most fluoroquinolone MICs of group B strains in which pHFQ1 (carrying oqxAB) had been re-introduced were only 2- to 4-fold lower than those of mutants in group A.
Effects of Efflux Pump Inhibitors and Genetic Analysis of Efflux Pump Components in the HUE1 Strain and its Mutants
The efflux pump inhibitor PAβN reduced the fluoroquinolone MICs of the HUE1 strain from 4- to 16-fold (Table 3). In mutants derived from plasmid curing in groups A and B, the effects of PAβN were less than in the parental strain, HUE1. Remarkably, the fluoroquinolone MICs of three mutants that lost oqxAB in group B (HUE1B-Curoqxqnr, HUE1B-Curoqx, and HUE1B-Reqnr) were barely affected by PAβN.
HUE1 exhibited higher mRNA expression of efflux pump genes (acrA, acrB, acrE, acrD, mdtK, mdfA, and tolC) and their global activators (soxS, marA, and rob) than the control strains, AG100 and DH5α. In contrast, ompF expression was lower in HUE1 than in the control strains (Table 4). Moreover, in HUE1A and HUE1A-Curoqxqnr, the mRNA expression of efflux pumps and their regulatory genes were approximately half of those in HUE1, while those of HUE1B-Curoqxqnr were similar to those in HUE1. The mRNA expression of qnrS1 and oqxB was not significantly different among the HUE1, HUE1A, HUE1A-Reoqxqnr, and HUE1B-Reoqxqnr strains (data not shown).
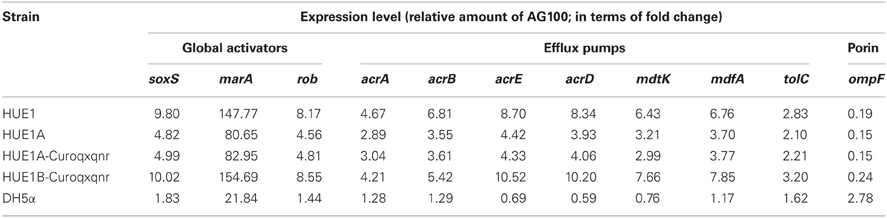
Table 4. mRNA expression levels of global activators, efflux pumps, and ompF genes in HUE1, HUE1A, HUE1A-Curoqxqnr, and HUE1B-Curoqqnr.
Next, we determined the full DNA sequences of efflux pump genes (acrA, acrB, acrE, acrF, and tolC) and their regulatory genes (marR, acrS, soxS, soxR, rob, and acrR). Although most genes in HUE1 and its plasmid-cured mutants were wild-type mutants, we found some mutations. acrR was disrupted by the insertion of a transposon, an IS3–IS629 element, in HUE1 and group A HUE1A-Curoqxqnr (Figure 2). HUE1B-Curoqxqnr (group B) had a deletion across acrR (corresponding to amino acids from Met-1 to Leu-73 of AcrR) and acrA (corresponding to amino acids from Met-1 to Asp-106 of AcrA), in addition to the insertion of the IS3–IS629 element. In addition, a nucleotide deletion of cytosine at position 223 of marR caused a frameshift in HUE1, HUE1A-Curoqxqnr, and HUE1B-Curoqxqnr.
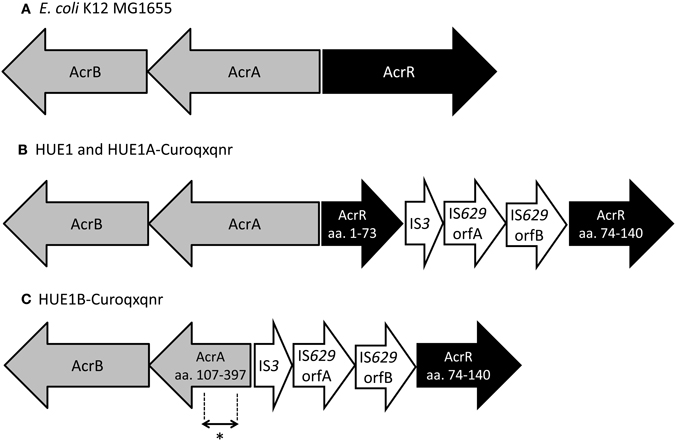
Figure 2. Schematic representation of the acrAB−acrR region in HUE1, group A mutants, and group B mutants. (A) Escherichia coli K12 MG1655 (accession no U00096). (B) HUE1 and HUE1A-Curoqxqnr (group A). (C) HUE1B-Curoqxqnr (group B). aa., Amino acids. *Position of primer pairs (acrART-F and acrART-R) used in RT-PCR (corresponding to amino acids 272–294 in AcrA).
Characterization of Knockout Mutants Prepared by Red/ET Recombination
We obtained a series of gene knockout mutants by Red/RT recombination. The fluoroquinolone MICs of HUE1-ΔoqxAB, HUE1-ΔqnrS1, and HUE1-ΔoqxAB qnrS1 were reduced by 2- or 4-fold, 16- or 32-fold, and 32- or 64-fold, as compared to those of HUE1, respectively (Table 5). HUE1-ΔacrA, HUE1-ΔacrB, and HUE1-ΔacrAB demonstrated 4- or 8-fold lower MICs than HUE1. HUE1-ΔacrA oqxAB exhibited 32- or 64-fold lower fluoroquinolone MICs and HUE1-ΔacrA qnrS1 exhibited 64- or 128-fold lower fluoroquinolone MICs than HUE1. HUE1-ΔoqxAB did not exhibit marked changes in fluoroquinolone MICs compared with those of HUE1, while HUE1-ΔacrAB oqxAB demonstrated larger fold changes, ranging from 4- to 16-fold lower, than HUE1-ΔacrAB.
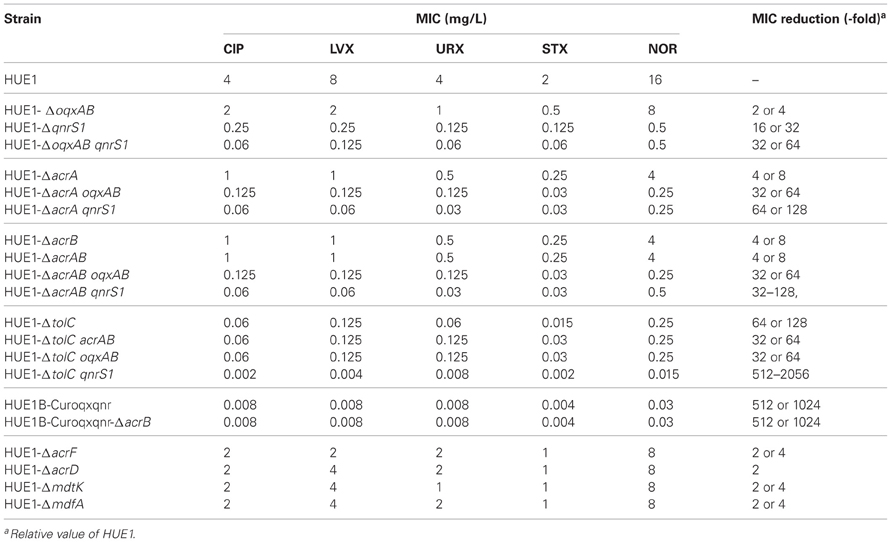
Table 5. Fluoroquinolones susceptibilities of Red/ET recombination mutants derived from HUE1.
Knockout of tolC markedly decreased fluoroquinolone MICs by 64- or 128-fold, and the MICs of the tolC knockout mutant (HUE1-ΔtolC) were not altered by additional knockout of acrAB or oqxAB (HUE1-ΔtolC acrAB or HUE1-ΔtolC oqxAB). HUE1-ΔtolC qnrS1 showed the lowest fluoroquinolone MICs, ranging from 512- to 2056-fold lower than those of HUE1. Mutants in which other efflux pump-associated genes, acrF, acrD, mdtK, and mdfA, were knocked out showed 2- to 4-fold lower fluoroquinolone MICs than did HUE1 (Table 5). The mutant derived from HUE1B-Curoqxqnr with knockout acrB (HUE1B-Curoqxqnr-ΔacrB) did not show altered fluoroquinolone MICs, when compared to HUE1B-Curoqxqnr.
Transformation of HUE1 with plasmids encoding wild-type acrR or wild-type marR (HUE1-wt-acrR and HUE1-wt-marR) resulted in fluoroquinolone MICs that were 2- or 4-fold lower than those for HUE1 (data not shown). HUE1-wt-acrR and HUE1-wt-marR resulted in reduced mRNA expression levels of acrA and acrB, and HUE1-wt-marR also exhibited a significant reduction in the expression of marA compared with that for HUE1 (Figure 3).
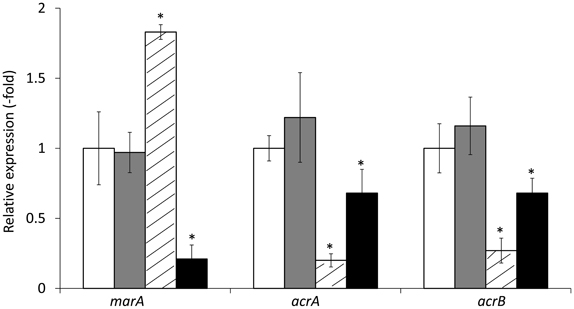
Figure 3. mRNA expression levels of marA, acrA, and acrB in HUE1 and its acrR or marR transformants. The values indicate the expression levels of indicated genes relative to those in HUE1 (fold-changes). White, HUE1; gray, HUE1-pUC19; diagonal line, HUE1-wt-acrR (HUE1 strain transformed wild-type of acrR); black, HUE1-wt-marR (HUE1 strain transformed wild-type marR). *p < 0.05 compared to HUE1. Data represent the mean ± standard deviation values calculated from at least three independent experiments.
Intracellular Fluoroquinolone Concentrations
The intracellular CIP concentration in HUE1 was approximately 2.3-fold lower than that in DH5α (p < 0.05; Figure 4). Intracellular CIP concentrations in HUE1A and HUE1A-Curoqxqnr were minimally, albeit significantly, higher than those of the parental strain, HUE1 (p < 0.05). In contrast, the intracellular CIP concentration in HUE1B-Curoqxqnr was markedly higher than those in HUE1, HUE1A, and HUE1A-Curoqxqnr (p < 0.05), and was similar to those of HUE1-ΔacrA oqxAB, and HUE1-ΔacrAB oqxAB (Figure 4). HUE1-ΔacrA and HUE1-ΔacrAB exhibited significantly increased intracellular CIP concentrations compared with that for HUE1 (p < 0.05). However, the intracellular CIP concentrations of acrD-, acrF-, mdtK-, and mdfA-knockout HUE1 mutants were slightly higher, but not significantly different from that for HUE1 (p > 0.05; data not shown). The intracellular CIP concentration in HUE1-ΔoqxAB was also not significantly different from that of HUE1; however, HUE1-ΔacrA oqxAB and HUE1-ΔacrAB oqxAB exhibited a clear increase in intracellular CIP levels compared to that in HUE1-ΔacrA or HUE1-ΔacrAB (p < 0.05). HUE1-ΔtolC showed the highest intracellular CIP concentrations (Figure 4), which were not altered by additional knockout of acrAB or oqxAB (HUE1-ΔtolC acrAB or HUE1-ΔtolC oqxAB; data not shown). Addition of CCCP resulted in increased intracellular CIP concentrations in all strains, with levels ranging from 21.8 ± 2.3 to 25.8 ± 5.2 ng/mg wet cells; there were no statistical differences (p > 0.05, data not shown).
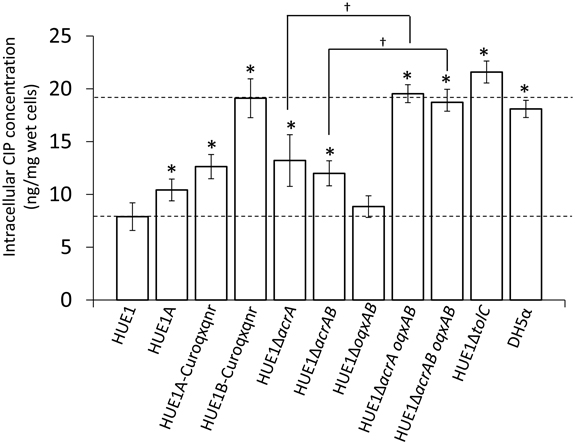
Figure 4. Intracellular concentrations of CIP in HUE1 and its mutants obtained from plasmid curing and Red/ET recombination. Data represent the mean ± standard deviation values of the results from three independent experiments. *p < 0.05 compared to HUE1. †p < 0.05 between HUE1-ΔacrA and HUE1-ΔacrA oqxAB and between HUE1-ΔacrAB and HUE1-ΔacrAB oqxAB, respectively.
Discussion
It is known that QRDR mutations are required for the fluoroquinolone MICs to exceed the breakpoints (Jacoby, 2005; Hansen et al., 2007; Cesaro et al., 2008; Zhao et al., 2010). To our knowledge, a fluoroquinolone-resistant strain without QRDR mutations has been reported only in a laboratory-derived resistant mutant strain of Acinetobacter baumannii that had been selected by in vitro fluoroquinolone exposure experiments; however, the resistance mechanism of this strain remains unclear (Chopra and Galande, 2011). In the current study, we demonstrated the fluoroquinolone-resistance mechanisms in an E. coli clinical isolate, HUE1, which lacks mutations in QRDRs (Sato et al., 2011).
HUE1 possessed two PMQRs on different plasmids, pHFQ1 (harboring oqxAB) and pHFQ2 (harboring qnrS1). Transformation experiments using DH5α as a host clearly showed that oqxAB and qnrS1 did in fact contribute to fluoroquinolone resistance. However, transformants containing both plasmids still did not exceed the MIC breakpoints.
It has been suggested that some efflux pumps are associated with fluoroquinolone resistance in HUE1. AcrAB-TolC is known to be the most important chromosomally encoded efflux pump related to fluoroquinolone resistance in Escherichia coli (Poole, 2005). During plasmid curing, a series of HUE1 group B mutants showed markedly lower fluoroquinolone MICs and higher intracellular CIP concentrations than did HUE1 and its group A mutants, whereas these mutants had mRNA expression levels of efflux pumps similar to those of HUE1. We found that group B mutants possessed deletions across parts of acrA. AcrA is an outer membrane protein that binds to AcrB (which plays a role in fluoroquinolone excretion) and TolC (an outer membrane factor) (Kobayashi et al., 2001; Elkins and Nikaido, 2003; Ge et al., 2009). The deleted region of AcrA in group B mutants involves an α−β barrel (53–61 amino acid residues), which forms the AcrB-binding domain, and a part of an α-helical hairpin (99–172 amino acid residues), which forms the TolC-binding domain (Ge et al., 2009). Fluoroquinolone MICs of the ΔacrA-knockout HUE1 mutant (HUE1-ΔacrA) were similar to those of HUE1B-Reoqxqnr (a group B mutant that possesses oqxAB and qnrS1), HUE1-ΔacrB, and HUE1-ΔacrAB. Moreover, the intracellular CIP concentration in HUE1B-Curoqxqnr (a group B mutant that had been cured of oqxAB and qnrS1) was not different from that in HUE1B-Curoqxqnr-ΔacrB. These results suggested that reduction of fluoroquinolone MICs in group B strains was caused by functional disruption of AcrAB–TolC, which did not allow cooperation between AcrA and AcrB and TolC, indicating that fluoroquinolone resistance in HUE1 is partially due to AcrAB–TolC.
AcrAB-TolC expression is controlled by several activators (i.e., soxS, marA, and rob) and repressors (i.e., acrR, marR, and soxR) (White et al., 1997; Poole, 2005; Keeney et al., 2008). HUE1 exhibited higher expression of acrA, acrB, tolC and these activators. In addition, HUE1 had concomitant disruption of the acrR and marR repressors. AcrR is a local repressor of AcrAB (Wang et al., 2001), and MarR is a repressor of MarA, which is one of the global activators of AcrAB (Keeney et al., 2008). Introduction of wild-type acrR or wild-type marR to HUE1 actually reduced acrA and acrB mRNA expression and reduced fluoroquinolone MICs. These data suggested that overexpression of AcrAB–TolC in HUE1 was mediated by the concomitant disruptions of AcrR and MarR. However, other mechanisms are also responsible for the overexpression of AcrAB–TolC and other chromosomally encoded efflux pumps, since the group A series of HUE1 mutants was found to have only approximately half of the mRNA expression levels of chromosomal efflux pump genes, including those encoding AcrAB, despite having the same disruptions in acrR and marR as HUE1. This will require further investigation.
Other chromosomally encoded efflux pumps, such as AcrEF–TolC and AcrD, belonging to the RND family, MdtK (also known as YdhE or NorE), belonging to the multidrug and toxic compound extrusion family, and MdfA, belonging to the major facilitator superfamily, also excrete fluoroquinolones (Nishino and Yamaguchi, 2001; Poole, 2005; Nishino et al., 2006). The mRNA expression of the related genes were also higher in HUE1, and HUE1 knockout mutants of these genes exhibited reduced fluoroquinolone MICs compared to HUE1, although the influence of these knockouts was weaker than that of AcrAB. These results indicated that the fluoroquinolone-resistance mechanism in HUE1 was most likely associated with the overexpression of AcrAB–TolC, with minor contributions of other chromosomally encoded efflux pumps, in addition to 2 PMQRs, viz., oqxAB and qnrS1.
OqxAB belongs to the RND-type efflux pump family, as does AcrAB (Hansen et al., 2004). Although OqxAB did not cause marked changes in fluoroquinolone MICs and intracellular CIP concentrations, we observed more distinct changes following acrAB deletion. OqxAB requires TolC to excrete AMP, CHL, and OLA, as does AcrAB (Hansen et al., 2004). In this study, knockout of tolC markedly decreased fluoroquinolone MICs and intracellular CIP concentrations and negated the effects of both acrAB and oqxAB. This finding indicated that OqxAB, in conjunction with TolC, was also involved in mediating decreased fluoroquinolone susceptibility. TolC is therefore implicated in the functions of both AcrAB and OqxAB. Although OqxAB minimally contributed to fluoroquinolone resistance in HUE1, it may largely contribute to supplementation of AcrAB functions. TolC is also required for the function of several efflux pumps (other than AcrAB), such as AcrEF and AcrD, in S. enterica serovar Typhimurium (Horiyama et al., 2010). Therefore, overexpression of TolC should also be an important element in the fluoroquinolone-resistance mechanism in HUE1 cells, as verified by our finding of the greatest reduction in fluoroquinolone MICs by knockout of tolC.
In conclusion, this study is the first to reveal the existence of a fluoroquinolone-resistance mechanism that is mediated without QRDR mutations in the E. coli clinical isolate, HUE1. This mechanism involved the concomitant presence of oqxAB and qnrS1 and was associated with the overexpression of AcrAB, other chromosomally encoded efflux pumps, and TolC. HUE1 was identified as phylogenic group A−O125:H37−ST48. This clonal group has also been isolated from humans and animals worldwide (Jorgensen et al., 2010; Bortolaia et al., 2011; Croxall et al., 2011). However, the susceptibilities of ST48 strains to fluoroquinolone compounds vary and have not yet been defined in all cases. Therefore, further epidemiological and molecular biology analyses of the ST48 lineage are required.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Hirotsugu Akizawa (Hokkaido University Hospital) for providing E. coli clinical isolates. This work was supported in part by a Grant-in-Aid from the Japanese Ministry of Health, Labour, and Welfare (H24-Shokuhin-Ippan-008) and a grant from the Program for Developing the Supporting System for Upgrading Education and Research from the Japan Ministry of Education, Culture, Sports, Science, and Technology.
References
Bin Kim, H., Wang, M. H., Park, C. H., Kim, E. C., Jacoby, G. A., and Hooper, D. C. (2009). oqxAB encoding a multidrug efflux pump in human clinical isolates of enterobacteriaceae. Antimicrobial Agents Chemother. 53, 3582–3584.
Bortolaia, V., Larsen, J., Damborg, P., and Guardabassi, L. (2011). Potential pathogenicity and host range of extended-spectrum beta-lactamase-producing Escherichia coli isolates from healthy poultry. Appl. Environ. Microbiol. 77, 5830–5833.
Breines, D. M., Ouabdesselam, S., Ng, E. Y., Tankovic, J., Shah, S., Soussy, C. J., et al. (1997). Quinolone resistance locus nfxD of Escherichia coli is a mutant allele of the parE gene encoding a subunit of topoisomerase IV. Antimicrob. Agents Chemother. 41, 175–179.
Cattoir, V., Poirel, L., Rotimi, V., Soussy, C. J., and Nordmann, P. (2007). Multiplex PCR for detection of plasmid-mediated quinolone resistance qnr genes in ESBL-producing enterobacterial isolates. J. Antimicrob. Chemother. 60, 394–397.
Cesaro, A., Bettoni, R. R., Lascols, C., Mérens, A., Soussy, C. J., and Cambau, E. (2008). Low selection of topoisomerase mutants from strains of Escherichia coli harbouring plasmid-borne qnr genes. J. Antimicrob. Chemother. 61, 1007–1015.
Chopra, S., and Galande, A. (2011). A fluoroquinolone-resistant Acinetobacter baumannii without the quinolone resistance-determining region mutations. J. Antimicrob. Chemother. 66, 2668–2670.
Cizman, M., Orazem, A., Krizan-Hergouth, V., and Kolman, J. (2001). Correlation between increased consumption of fluoroquinolones in outpatients and resistance of Escherichia coli from urinary tract infections. J. Antimicrob. Chemother. 47, 502.
Clinical and Laboratory Standards Institute. (2011). Performance Standards for Antimicrobial Susceptibility Testing. CLSI M100–S21. Wayne, PA: Clinical and Laboratory Standards Institute.
Conrad, S., Oethinger, M., Kaifel, K., Klotz, G., Marre, R., and Kern, W. V. (1996). GyrA mutations in high-level fluoroquinolone-resistant clinical isolates of Escherichia coli. J. Antimicrob. Chemother. 38, 443–455.
Croxall, G., Hale, J., Weston, V., Manning, G., Cheetham, P., Achtman, M., et al. (2011). Molecular epidemiology of extraintestinal pathogenic Escherichia coli isolates from a regional cohort of elderly patients highlights the prevalence of ST131 strains with increased antimicrobial resistance in both community and hospital care settings. J. Antimicrob. Chemother. 66, 2501–2508.
Deane, S. M., and Rawlings, D. E. (2004). Plasmid evolution and interaction between the plasmid addiction stability systems of two related broad-host-range IncQ-like plasmids. J. Bacteriol. 186, 2123–2133.
Edgar, R., Friedman, N., Molshanski-Mor, S., and Qimron, U. (2012). Reversing bacterial resistance to antibiotics by phage-mediated delivery of dominant sensitive genes. Appl. Environ. Microbiol. 78, 744–751.
Elkins, C. A., and Nikaido, H. (2003). Chimeric analysis of AcrA function reveals the importance of its C-terminal domain in its interaction with the AcrB multidrug efflux pump. J. Bacteriol. 185, 5349–5356.
Ge, Q., Yamada, Y., and Zgurskaya, H. (2009). The C-terminal domain of AcrA is essential for the assembly and function of the multidrug efflux pump AcrAB-TolC. J. Bacteriol. 191, 4365–4371.
Hansen, L. H., Jensen, L. B., Sorensen, H. I., and Sorensen, S. J. (2007). Substrate specificity of the OqxAB multidrug resistance pump in Escherichia coli and selected enteric bacteria. J. Antimicrob. Chemother. 60, 145–147.
Hansen, L. H., Johannesen, E., Burmolle, M., Sorensen, A. H., and Sorensen, S. J. (2004). Plasmid-encoded multidrug efflux pump conferring resistance to olaquindox in Escherichia coli. Antimicrob. Agents Chemother. 48, 3332–3337.
Hata, M., Suzuki, M., Matsumoto, M., Takahashi, M., Sato, K., Ibe, S., et al. (2005). Cloning of a novel gene for quinolone resistance from a transferable plasmid in Shigella flexneri 2b. Antimicrob. Agents Chemother. 49, 801–803.
Heisig, P. (1996). Genetic evidence for a role of parC mutations in development of high-level fluoroquinolone resistance in Escherichia coli. Antimicrob. Agents Chemother. 40, 879–885.
Horiyama, T., Yamaguchi, A., and Nishino, K. (2010). TolC dependency of multidrug efflux systems in Salmonella enterica serovar Typhimurium. J. Antimicrob. Chemother. 65, 1372–1376.
Jorgensen, R. L., Nielsen, J. B., Friis-Moller, A., Fjeldsoe-Nielsen, H., and Schonning, K. (2010). Prevalence and molecular characterization of clinical isolates of Escherichia coli expressing an AmpC phenotype. J. Antimicrob. Chemother. 65, 460–464.
Kado, C. I., and Liu, S. T. (1981). Rapid procedure for detection and isolation of Large and small Plasmids. J. Bacteriol. 145, 1365–1373.
Keeney, D., Ruzin, A., McAleese, F., Murphy, E., and Bradford, P. A. (2008). MarA-mediated overexpression of the AcrAB efflux pump results in decreased susceptibility to tigecycline in Escherichia coli. J. Antimicrob. Chemother. 61, 46–53.
Kobayashi, K., Tsukagoshi, N., and Aono, R. (2001). Suppression of hypersensitivity of Escherichia coli acrB mutant to organic solvents by integrational activation of the acrEF operon with the IS1 or IS2 element. J. Bacteriol. 183, 2646–2653.
Lindgren, P. K., Karlsson, A., and Hughes, D. (2003). Mutation rate and evolution of fluoroquinolone resistance in Escherichia coli isolates from patients with urinary tract infections. Antimicrob. Agents Chemother. 47, 3222–3232.
Nakaminami, H., Noguchi, N., and Sasatsu, M. (2010). Fluoroquinolone efflux by the plasmid-mediated multidrug efflux pump QacB variant QacBIII in Staphylococcus aureus. Antimicrob. Agents Chemother. 54, 4107–4111.
Nishino, K., Latifi, T., and Groisman, E. A. (2006). Virulence and drug resistance roles of multidrug efflux systems of Salmonella enterica serovar Typhimurium. Mol. Microbiol. 59, 126–141.
Nishino, K., and Yamaguchi, A. (2001). Analysis of a complete library of putative drug transporter genes in Escherichia coli. J. Bacteriol. 183, 5803–5812.
Okusu, H., Ma, D., and Nikaido, H. (1996). AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (MAR) mutants. J. Bacteriol. 178, 306–308.
Peña, C., Albareda, J. M., Pallares, R., Pujol, M., Tubau, F., and Ariza, J. (1995). Relationship between quinolone use and appearance of ciprofloxacin-resistant Escherichia coli in bloodstream infections. Antimicrob. Agents Chemother. 39, 520–524.
Sanchez, G. V., Master, R. N., Karlowsky, J. A., and Bordon, J. M. (2012). In vitro antimicrobial resistance of urinary Escherichia coli isolates among U.S. outpatients from 2000 to 2010. Antimicrob. Agents Chemother. 56, 2181–2183.
Sato, T., Yokota, S., Uchida, I., Okubo, T., Ishihara, K., Fujii, N., et al. (2011). A fluoroquinolone-resistant Escherichia coli clinical isolate without quinolone resistance-determining region mutations found in Japan. Antimicrob. Agents Chemother. 55, 3964–3965.
Sorensen, A. H., Hansen, L. H., Johannesen, E., and Sorensen, S. J. (2003). Conjugative plasmid conferring resistance to olaquindox. Antimicrob. Agents Chemother. 47, 798–799.
Tamamura, Y., Uchida, I., Tanaka, K., Okazaki, H., Tezuka, S., Hanyu, H., et al. (2011). Molecular epidemiology of salmonella enterica serovar typhimurium isolates from cattle in Hokkaido, Japan: evidence of clonal replacement and characterization of the disseminated clone. Appl. Environ. Microbiol. 77, 1739–1750.
Usui, M., Uchiyama, M., Iwanaka, M., Nagai, H., Yamamoto, Y., and Asai, T. (2009). Intracellular concentrations of enrofloxacin in quinolone-resistant Salmonella enterica subspecies enterica serovar Choleraesuis. Int. J. Antimicrob. Agents 34, 592–595.
Wang, H., Dzink-Fox, J. L., Chen, M., and Levy, S. B. (2001). Genetic characterization of highly fluoroquinolone-resistant clinical Escherichia coli strains from China: role of acrR mutations. Antimicrob. Agents Chemother. 45, 1515.39–1521.39.
White, D. G., Goldman, J. D., Demple, B., and Levy, S. B. (1997). Role of the acrAB locus in organic solvent tolerance mediated by expression of marA, soxS, or robA in Escherichia coli. J. Bacteriol. 179, 6122–6126.
Yoshida, H., Bogaki, M., Nakamura, M., Yamanaka, L. M., and Nakamura, S. (1991). Quinolone resistance-determining region in the DNA gyrase gyrB gene of Escherichia coli. Antimicrob. Agents Chemother. 35, 1647–1650.
Keywords: AcrAB, efflux pump, Escherichia coli, fluoroquinolone resistance, oqxAB, qnrS
Citation: Sato T, Yokota S-i, Uchida I, Okubo T, Usui M, Kusumoto M, Akiba M, Fujii N and Tamura Y (2013) Fluoroquinolone resistance mechanisms in an Escherichia coli isolate, HUE1, without quinolone resistance-determining region mutations. Front. Microbiol. 4:125. doi: 10.3389/fmicb.2013.00125
Received: 23 March 2013; Accepted: 02 May 2013;
Published online: 24 May 2013.
Edited by:
Kunihiko Nishino, Osaka University, JapanReviewed by:
Axel Cloeckaert, Institut National de la Recherche Agronomique, FranceJunichi Yamagishi, Nihon Pharmaceutical University, Japan
Copyright © 2013 Sato, Yokota, Uchida, Okubo, Usui, Kusumoto, Akiba, Fujii and Tamura. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Yutaka Tamura, Laboratory of Food Microbiology and Food Safety, Department of Health and Environmental Sciences, School of Veterinary Medicine, Rakuno Gakuen University, 582 Midorimachi-Bunkyoudai, Ebetsu 069-8501, Japan. e-mail:dGFtdXJheUByYWt1bm8uYWMuanA=