

- Department of Immunology, National Institute of Infectious Diseases, Tokyo, Japan
Although dendritic cells (DCs) represent a small cell population in the body, they have been recognized as professional antigen presenting cells and key players of both innate and acquired immunity. The recent expansion of basic knowledge concerning differentiation and function of various DC subsets will greatly help to understand the nature of protective immunity required in designing acquired immunodeficiency syndrome (AIDS) vaccines. However, human immunodeficiency virus (HIV) not only targets CD4+ T cells but also myeloid cells, including macrophages and DC. When HIV infects DC, its replication is highly restricted in DC. Nevertheless, even a low level of HIV production is sufficient to enhance HIV replication in activated CD4+ T cells, through antigen presentation activity by HIV-infected DC. Considering how antiviral immunity is initiated and memory response is maintained, such efficient DC–T cell transmission of HIV should play an important role in the disturbed immune responses associated with HIV infection. Recently, accessory proteins encoded by HIV have been shown to interact with various proteins in DC, and thereby affect DC–T cell transmission. In this review, we summarize the current understanding about DC biology, antiviral immune responses and DC restriction factors, all of which will be important issues for the development of an effective AIDS vaccine in the future.
Introduction
Since the discovery of dendritic cells (DCs) in the 1970s (Steinman, 2007), DC have long been recognized as: (1) professional antigen presenting cells expressing high levels of major histocompatibility complex (MHC) class II and other costimulatory molecules, (2) localizing to various tissues/organs, and (3) migrating to lymphoid tissues after antigen acquisition, to either initiate immune responses or induce tolerance by interaction with T cells (see review by Banchereau and Steinman, 1998).
In the 1990s, we (Tsunetsugu-Yokota et al., 1995) and several others demonstrated that DC are easily susceptible to human immunodeficiency virus type 1 (HIV-1) infection and can therefore also efficiently transmit virus to antigen-specific CD4+ T cells, in spite of low levels of virus production. These earlier studies lead to further research into the role of DC in the pathogenesis of HIV infection, because DC–T cell interaction is considered to better reflect the physiological mode of cell-to-cell HIV-1 infection in vivo as compared to T-to-T or T-to-adherent cell interactions, which involve env-mediated membrane fusion (Sattentau, 2008). As our laboratory previously demonstrated, the close contact site between DC–T cells is consolidated by the presence of T cell receptor binding to MHC–antigen complex, followed by the interaction and mutual signaling of various costimulatory molecules (Tsunetsugu-Yokota et al., 1997).
A substantial number of reviews, focusing on HIV-1 transmission from DC to T cells (Rinaldo and Piazza, 2004; Wu and KewalRamani, 2006; Piguet and Steinman, 2007) have illustrated distinct features of cell-to-cell transmission. However, although the majority of studies focus on the close contact as a virological synapse (VS: a neuronal synapse like cell–cell contact structure for spreading virus infection), most of them do not consider the importance of antigen-dependent DC–T cell interaction, which is well-recognized as an Immunological synapse (IS: a synapse like cell–cell contact structure for activating immune response).
In this review, we will summarize the current understanding of various human DC subsets based on several outstanding recent findings in DC biology and of antiviral immune responses initiated by DCs, and discuss about newly identified DC restriction factors counteracting HIV-1 accessory proteins (e.g., Vif, Vpu, and Nef), which may have an impact on (1) the susceptibility of DC to HIV-1 infection and (2) the transmissibility of HIV from DC to T cells via VS or IS.
Biology of Dendritic Cells
DC originate from common myeloid precursor cells in the bone marrow, but are quite heterogeneous in terms of their localization, surface phenotype, and function. The major DC subsets are classical or conventional DC (cDC) and plasmacytoid DC (pDC). The development pathway and the lineage relationship of these DCs have been subjects of extensive investigation (see review by Steinman and Idoyaga, 2010). By analyzing bone marrow precursors in vivo, a current view of DC development and homeostasis has been established. In the bone marrow, monocytes and DC precursors (MDP) first develop into a common DC precursor (CDP) before continuing development into either a monocyte, or a cDC precursor (PreDC) and pDC, respectively (Fogg et al., 2006; Liu et al., 2009). PreDC, pDC, and monocytes subsequently migrate through blood to the spleen and lymph nodes, whereby preDC differentiate into cDC in the presence of Flt3L, and monocytes differentiate into either macrophages or DC, presumably depending on the inflammatory stimuli present (Cheong et al., 2010).
With regards to lineage, there is considerable difference between mouse and human DC subsets, as defined by surface markers. Five major DC subsets are known in mice: CD8+DC, CD4+DC, and pDC in lymphoid tissues, and CD103+ and CD11b+DC in non-lymphoid tissues (Steinman and Idoyaga, 2010). Alternately, the source of DC in humans is more limited, originating from either skin or blood. In human skin, three DC subsets have been identified: epidermal CD207+ Langerhans cells (LCs), CD14+ dermal DC, and CD14-CD207-CD1a+ DC. Amongst them, LC are the most potent inducer of Th2 cytokines and able to cross prime CD8+ effector cells (Klechevsky et al., 2008). With respect to the origin of LC localized in the skin epidermis, Sere et al. (2012) have recently reported that LC are replenished in two waves, one by monocyte-derived, short lived LC upon inflammation, and the other by non-blood monocyte origin with long life expectancy (Sere et al., 2012). In other tissues and blood, two subsets of human cDC are known, which express either CD1c (BDCA-1) or CD141 (BDCA-3). The other group, pDC, expresses human-specific pDC markers, BDCA-2 and BDCA-4, in addition to CD123 [interleukin-3 receptor (IL-3R)], and produces a large amount of type I interferon (IFN; Steinman and Idoyaga, 2010).
A unique and important function of DC is their cross-presentation ability by phagocytosing dead cells, tumor cells, or infected cells, which results in the activation of MHC class I-restricted effector CD8+ T cells (Bevan, 2006). A particular subset of DC is known to efficiently cross-present antigens. In mice, splenic CD8α DC are the most potent DC subset for cross-presentation, and the human counterpart of mouse CD8α has been recently identified (Bachem et al., 2010; Jongbloed et al., 2010; Poulin et al., 2010).
The minor DC population in human blood, CD141+ DC, shares several phenotypic and functional properties with mouse CD8α DC, such as: (1) the expression of DC natural killer (NK) lectin group receptor-1 (DNGR-1) or CLEC9A, a sensor of necrotic cells (Sancho et al., 2009), and (2) the selective expression of the chemokine receptor XCR1 (Bachem et al., 2010), transcriptional factor ATF (activating transcription factor)-like-3, and IFN regulatory factor 8 (IRF-8). CD141+ DC also express a high level of Toll-like receptor 3 (TLR3) and upon TLR3 stimulation, produce IL-12 and IFN-β. More recently, by using comparative genomics to align human and mouse cell types, Haniffa et al. (2012) demonstrated that cutaneous CD141 hi CLEC9A hiXCR1+ DC, which are closely related to blood CD141 hi CLEC9A hiXCR1+ DC, are much more potent at cross-presentation than LC. Taken together, in humans, CD141+DC, CD1c+DC, and pDC localize within lymphoid tissues, while the former two types of DC and CD14+ DC are present in non-lymphoid tissues. In addition, human tissue CD141+ DC are functional homologues of mouse CD103+(α chain of the αEβ7 integrin) non-lymphoid DC, with high cross-presentation activity (Haniffa et al., 2012).
However, the relationship between DC and monocytes/macrophages can sometimes be confusing when based only on cell surface markers and functional properties. Two types of human monocytes are present in the blood: “patrolling” CD14 dimCD16+ and inflammatory CD14+ CD16- or CD14+CD16+monocytes, which resemble mouse Gr- (Ly6C low) and Gr+ (Ly6C high) cells, respectively (Auffray et al., 2009; Cros et al., 2010). When tissue inflammation occurs, CD14+inflammatory monocytes migrate to the site of inflammation, and may differentiate into CD11b+CD14+ monocyte-derived DCs (MDDCs) as demonstrated in mice (Cheong et al., 2010).
Currently, four major DC subsets comprise and make up the category of DC: cDC, pDC, LCs, and MDDCs (Satpathy et al., 2012). A recent large scale collaborative study focusing on the transcriptional network of DC and monocyte/macrophage subsets, highlighted on some lineage-specific key transcriptional factors and the mutual relationship between DC and monocytes/macrophages (Miller et al., 2012), though the definition of DC by molecular signatures has raised further discussion (Hume et al., 2013).
Incorporating recent findings, the development and lineage relationship of human myeloid precursors is illustrated in Figure 1.
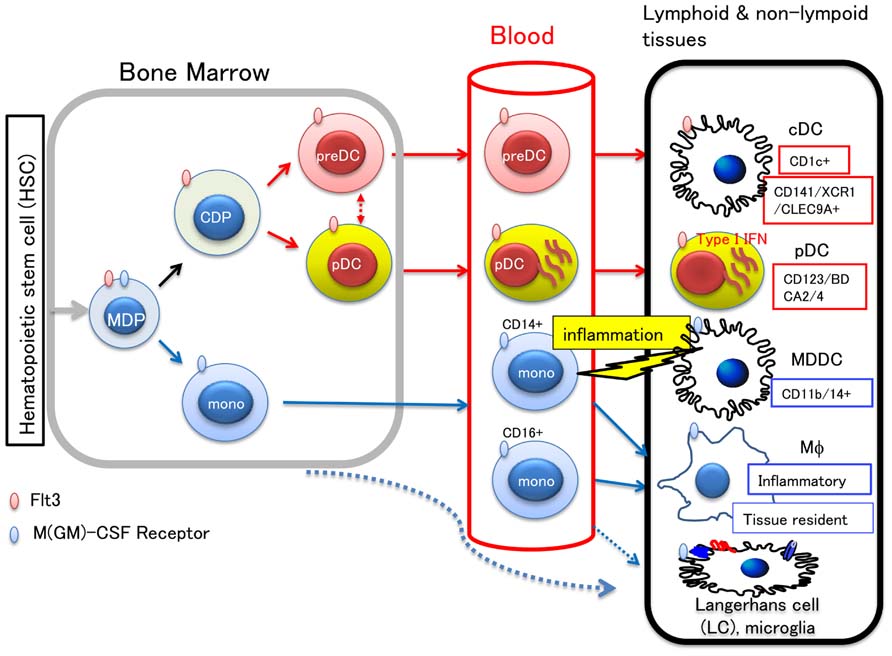
FIGURE 1. Development of myeloid cells and lineage relationship between DC subsets. Hematopoietic stem cells in the bone marrow differentiate into monocyte/macrophage and DC precursor (MDP), which commits to a distinct lineage cell, either becoming a monocyte or common DC precursor (CDP). CDP differentiates to preDC or pDC, and migrates to the peripheral tissues through blood. In the lymphoid or non-lymphoid tissues, preDC can become either of two types of cDC: CD1c+ DC and CD141+/XCR1+/CLEC9A+ DC, the latter of which has high cross-presenting activity. In the blood, CD14+ or CD16+ monocytes are also circulating. Blood monocytes are source of macrophages (Mϕ), LC, and microglias. The other LC precursor in the bone marrow, was also shown to migrate to the skin. Upon tissue inflammation, CD14+ monocytes migrate to the inflamed tissue and differentiate into either migratory MDDC or inflammatory Mϕ. It is known that the cells committed to become DC express Flt3, whereas those that commit to monocytes/macrophages express M-CSF receptor. Therefore, Flt3L and M-CSF are key molecules for differentiation and proliferation of DC and monocytes/macrophages, respectively.
Overview of Antiviral Immune Responses in Acute and Chronic HIV Infection
The natural infection of HIV-1 mostly occurs through vaginal or rectal routes. Because these submucosal spaces are rich in DC, they have been argued to be the primary targets of HIV-1 infection (see reviews by Wu and KewalRamani, 2006; Piguet and Steinman, 2007). However, detection of HIV-1-producing DC in these tissues is rare, which is in contrast to the rapid and massive HIV/simian immunodeficiency virus (SIV) infection detected in CD4+CCR5+ memory T cells (Brenchley et al., 2004; Mehandru et al., 2004; Li et al., 2005; Mattapallil et al., 2005). Nonetheless, the scenario that mucosally residing, non-producing HIV-infected DC or phagocytosed HIV-1-infected DC migrate to regional lymph nodes, and subsequently spread HIV-1 infection there to contacting T cells, in association with nominal antigen presentation, well reflects what maybe occurring in lymphoid organs (Wu and KewalRamani, 2006; Piguet and Steinman, 2007).
In a chronic stage of HIV-1 infection, a major reservoir of latent HIV-1 infection is considered to be the circulating resting memory T cells, carrying integrated HIV-1 DNA (review by Pierson et al., 2000). When resting memory cells enter a lymphoid organ, they interact with resident DC, which may induce T cell activation and concomitant transmission of reactivated HIV-1 to DC. There is a possibility that newly HIV-infected DC can transmit virus to intact naïve or memory T cells. Although these HIV-1-infected T cells and DC can be eliminated by a cytotoxic T cell (CTL) response, a continuous cycle of such aforementioned events may cause intermittent surges of plasma viral load under chronic infection. Henceforth, lymphoid organs are considered to be major sites of HIV-1 production.
During the induction of primary immune responses in lymphoid organs, an inflammatory response can occur, as an innate immune mechanism at the invasion site, attracting a variety of immune cells, including T cells and monocytes. As previously described, so-called inflammatory monocytes are recruited into peripheral tissues subsequently, differentiating into inflammatory MDDC which can regulate immune responses locally (Cheong et al., 2010).
It is well-known that effector CD8+ T cells play an essential role regarding protection against HIV/SIV infection (Appay et al., 2008). Once infection primes in lymphoid organs, memory CD8+ T cells can act as effector cells by circulating in the blood, in order to migrate to infected tissues upon re-exposure, and quickly eliminate infected cells. The importance of long-term resident memory CD8+T (TRM) cells has been highlighted with respect to efficacy of the local memory response (Gebhardt and Mackay, 2012). By utilizing an elegant mouse model of herpes simplex virus (HSV) infection, memory CD8+ T cells were shown to be initiated in extra-lymphoid tissues, independent of migratory memory CD8+ T cells (Wakim et al., 2008). In this model, HSV-infected dorsal root ganglia (DRG) was surgically transplanted to naïve mice, because DRG is known to be latently infected with HSV, after acute virus resolution, and to be reactivated by surgical extraction. Stimulation of these CD8+T RM cells was dependent on recruited DC in the DRG, and CD4+ T cell help was required. These CD8+ TRM cells represent a self-renewing and highly protective population of memory T cells distinct from circulating memory CD8+ T cells, and express CD103 which is known to be widely expressed in non-lymphoid tissues (Gebhardt et al., 2009). Interestingly, a study using intravital two-photon microscopy revealed that while memory CD4+ T cells are trafficked rapidly, CD8+ TRM cells are removing slowly in the original skin site during HSV infection (Gebhardt et al., 2011).
The functional properties of CD8+ TRM cells have just begun to be elucidated in influenza virus infection. The innate antiviral function of IFN-induced transmembrane protein 3 (IFTM3) was first discovered by using RNA interference screening for factors modifying influenza virus infection (Brass et al., 2009). The IFITM family is: (1) made up of IFN-stimulated genes (ISGs) with diverse biological functions, (2) comprises of multiply closed members of four genes in both humans and mice, and (3) restricts various virus infections at a site of viral fusion (Diamond and Farzan, 2012). Recently, IFITM3 was reported to be expressed in lung CD8+ TRM cells after influenza virus infection, endowing greater resistance to the secondary influenza virus infection (Wakim et al., 2013). Sustained expression of IFITM3 is intrinsically regulated in CD8+ TRM, but not CD4+ memory T cells, and enhances the survival of CD8+ TRM cells only at sites of viral infection. Although the involvement of recruited DCs on the activation of CD8+ TRM cells is demonstrated (Wakim et al., 2008), the mechanisms in which memory and effector CD8+ T cells develop and localize to target peripheral tissue such as intestinal mucosa and the involvement of intestinal DCs need to be addressed in future for the development of effective anti-HIV vaccines.
The overview of this section is illustrated as Figure 2.
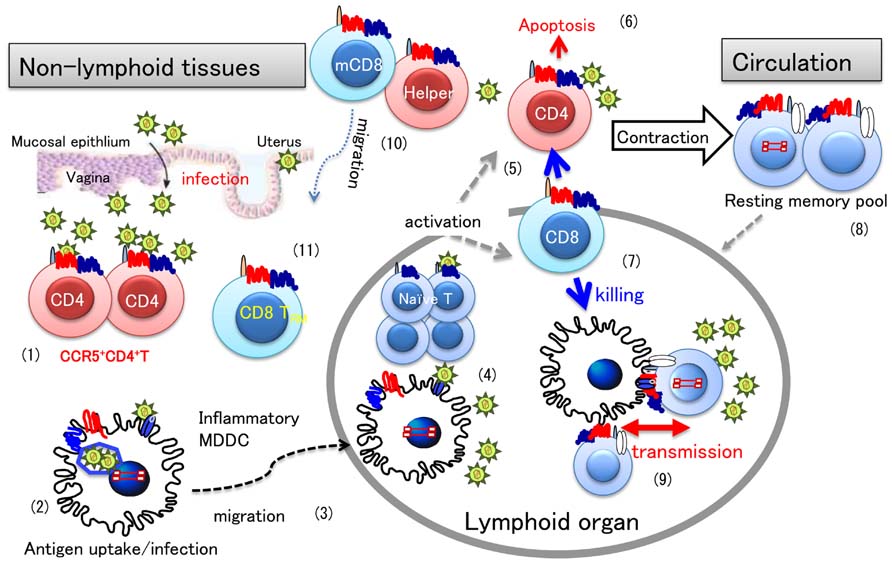
FIGURE 2. HIV infection and T cell differentiation during primary and memory antiviral immune responses. A general view of antiviral immune responses focusing on HIV-1 infection is illustrated. Once HIV invades the vaginal mucosa, infection mostly occurs in activated CD4+CCR5+ T cells (1). Resident DCs in the submucosa engulf virions or HIV-infected cells (2), and then migrate to the draining lymph nodes (3), where an antiviral immune response is initiated (4). In a primary immune response, antigen-specific T cells are differentiate into effector cells. During the effector phase, memory T cells are also produced, the mechanism of which is not fully understood. Once activated, CD4+ T cells are infected with HIV (5), however, most effector cells will die of apoptosis (6), or killed by effector CD8+ T cells (7). When virus-infected cells are eliminated in the infection site, the immune response enters into the contraction phase. After some time, memory T cells revert to quiescent or resting state (8) and circulate back to peripheral tissues for checking for next infection. Latent infection of HIV is known to occur in such quiescent memory CD4+ T cells. When these latently HIV-infected memory CD4+ T cells interact with uninfected DCs and T cells, for secondary antigen exposure, HIV-1 may spread to CD4+ T cells in the lymphoid organ through DC–T cell interaction (9). In the case of circulating memory CD8+ T cells (mCD8), they may quickly enter into tissues to eliminate the secondary infection (10). However, recent evidence indicate that tissue resident CD8+ T cells (TRM) (11) primed in situ play a more important role than circulating mCD8 in protective immunity, which may be a distinct feature from memory CD4+ T cells.
Distinct Susceptibility of DCs to HIV: CIS- and Trans-Infection Modes
When MDDC are activated by various stimuli, such as lipopolysaccharide (LPS), TLR ligands, cytokines like type I IFN, DC express higher levels of MHC class II and other costimulatory molecules, thereby engaging in antigen presentation rather than antigen uptake. This maturation status and difference in lineage can profoundly affect the susceptibility of DC to HIV-1 infection (see review by Wu and KewalRamani, 2006).
Dendritic cell express various C-type lectin receptors (CLRs) which bind to HIV-1, such as DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin), Langerin, and dectin (Turville et al., 2002), and recently discovered CLEC9A (Caminschi et al., 2008; Huysamen et al., 2008; Sancho et al., 2009). Each lectin receptor appears to have a unique and distinct function. HIV-1 transfer in trans, through the capture of virus by a CLR, such as DC-SIGN, occurs early in in vitro experiments (Cavrois et al., 2007; Dong et al., 2007; Izquierdo-Useros et al., 2007), and is designated trans-infection. However, DC lectin receptors are important molecules involved in the presentation of foreign antigens. In fact, the most of the HIV-1 virions captured by DCs were known to be rapidly degraded (Turville et al., 2004; Nobile et al., 2005). Moreover, the interaction of leukocyte-specific protein 1 (LSP-1), a protein directing internalized virus to the proteasome, with the cytoplasmic region of DC-SIGN may further facilitate the degradation of HIV-1 (Smith et al., 2007).
On the other hand, HIV-1 replication in DCs, but not DC-SIGN, is required for long-term transfer of HIV from DCs to CD4+ T cells (Lore et al., 2005; Nobile et al., 2005; Burleigh et al., 2006; Wang et al., 2007). Although DCs can support only minimal replication of HIV-1 (DC restriction, see Restrictions in DCs), it is considered that the antigen-dependent close DC–T cell contact, forming IS, would support the efficient virus transmission followed by massive virus replication in CD4+ T cells (Tsunetsugu-Yokota et al., 1995; Lore et al., 2005). We assume that such cis-infection is more likely to occur in vivo, as opposed to trans-infection.
Thus, the trans-infection in vitro needs to be discriminated from HIV-1 transmission in cis or cis-transmission through genuine infection via DC as shown in Figure 3.
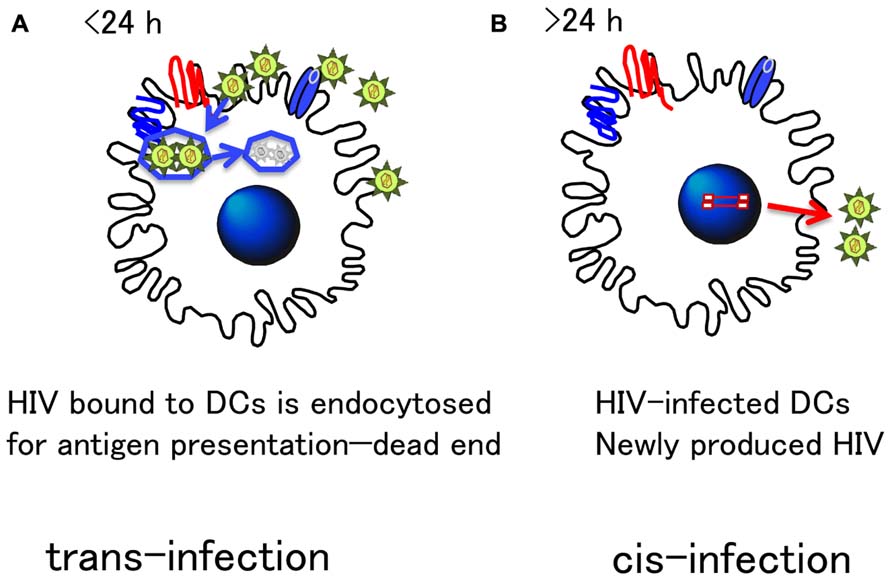
FIGURE 3. Distinct HIV transmission mode by DCs: Cis- and Trans-infection. There are two modes of transmission of HIV from DC to T cells. (A) Trans-infection: DCs express various receptors to bind foreign antigens, which is important to efficiently endocytose and process for antigen presentation. Such machinery is highlighted for HIV transfer in trans in in vitro experiments. However, because endocytosed antigens are quickly degraded within 24 h (Turville et al., 2004), infectious HIV particles may not retain their infectivity for a long time. Thus, the physiological relevancy of trans- infection in vivo remains unclear. (B) Cis-infection: On the other hand, because DC express CD4 and chemokine receptors essential for HIV infection, DC are naturally susceptible to HIV infection. Although HIV replication is limited in DC, their strong antigen-presenting activity allows HIV to replicate more in antigen-specific CD4+ T cells tightly interacting with DC. This DC–T cell interaction is a genuine immunological synapse (IS). The precise molecular mechanism of HIV transmission from DC to T cells has yet to be clarified, but the cis-infection mode, is what most likely occurs in vivo.
The Role of Tetherin in HIV Transmission: Immunological Synapse Versus Virological Synapse
Various intrinsic antiviral mechanisms or factors have evolved in eukaryotes to fight against virus invasion (see review by Yan and Chen, 2012). To countermeasure these intrinsic cellular factors (restriction factors), HIV-1 evolved to encode several accessory proteins, such as Vpu, Vif, Vpr, and Vpx. BST-2/CD317/tetherin was recently identified as one such cellular factor, antagonized by HIV-1 Vpu (Neil et al., 2008; Van Damme et al., 2008). Normally, BST-2/tetherin inhibits virion release by anchoring virus at the cell membrane, but Vpu can directly bind to BST-2/tetherin, internalizing and degrading it, probably in the endolysosomal compartment (Arias et al., 2012).
It has been known for some time that vpu mutant virus can be efficiently spread by T cell to T cell transfer (Gummuluru et al., 2000) in a rapid turnover HIV-1 replication culture. As such, this observation raises the important question of whether Vpu affects cell-to-cell spread of HIV-1 by inhibiting the membrane tethering function of BST-2/tetherin? This issue was addressed under the setting of a VS, using HIV-infected cells directly contacting with an uninfected target cell system. Jolly et al. (2010) found that Vpu-defective HIV-1 disseminates more efficiently through cell-to-cell contact, despite the premise that BST-2/tetherin inhibits cell-free virion release, which corroborates with a previous report by Gummuluru et al. (2000), in which the authors speculated that BST-2/tetherin either mediates the accumulation of virions or regulation of VS integrity. On the other hand, Casartelli et al. (2010) studied virus transfer to T cells (targets) from HeLa or 293T cells, expressing a high level of BST-2/tetherin either naturally or by transfection (donors). They observed that BST-2/tetherin does not prevent VS formation, but assists the accumulation of Gag at the contact zone in the absence of Vpu (Casartelli et al., 2010). Important to note, however, is since the authors used mostly vesicular stomatitis virus (VSV)-pseudotyped HIV-1 for the infection of donors, their system is a transient infection event without new infectious virus production and reflect a cell-to-cell trans-infection as discussed in a previous section. Regardless, in both studies (Casartelli et al., 2010; Jolly et al., 2010), the accumulation of HIV-1 virions, in the presence of a high level of tetherin expression was observed.
Alternately, Kuhl et al. (2011) reported that a Vpu-mediated effect in viral spread among CD4+ T cells is independent of the extent of Vpu-mediated BST-2/tetherin cell surface downmodulation. They postulated the presence of an additional Vpu-independent mechanism for BST-2/tetherin cell surface downmodulation following HIV-1 infection in T cell lines, which is consistent with a report by Miyagi et al. (2009).
Vpu interacts with BST-2/tetherin to promote virion release, whereas BST-2/tetherin normally causes accumulation of virus particle on the membrane surface. However, the efficiency of cell-to-cell HIV-1 transfer or transmission appears largely dependent on experimental conditions. As previously described, DC–T cell contact is not equal to the VS form of contact. Therefore, we should perhaps consider the physiological function of BST-2/tetherin, under the setting of an antigen-mediated IS instead.
In this context, Coleman et al. (2011) reported that immature MDDC does not express BST-2/tetherin, however, after HIV-1 infection, BST-2/tetherin expression was upregulated by HIV-1 Nef. Therefore, HIV-1 transmission from DC to T cells does not appear to be restricted by BST-2/tetherin (Coleman et al., 2011). However, because their target is the HUT/CCR5 T cell line and culture conditions favor trans infection (immediate co-culture after HIV-1 infection), the question of whether cell surface expression of BST-2/tetherin assists or inhibits virus transmission to CD4+T cells via IS needs to be clarified.
Restrictions in DCs
The poor replication of HIV in DC is partly explained by a block during virus fusion with MDDC (Cavrois et al., 2006). The restriction of HIV-1 infection in DC can also occur at a post-entry level. Several cell factors known to interfere with HIV-1 infection and/or replication step in DC are illustrated in Figure 4. APOBEC3 was originally discovered as a potent intrinsic antiviral factor interacting with HIV-1 Vif (Sheehy et al., 2002). APOBEC3G (A3G) is a member of the cytidine deaminase family, which edits C to U in a single stranded HIV DNA, causing G-to-A hyper mutation of the HIV-1 genome. HIV-1 Vif counteracts this deaminase function by inhibiting A3G incorporation into virions and promoting A3G degradation by ubiquitination (Sheehy et al., 2003).
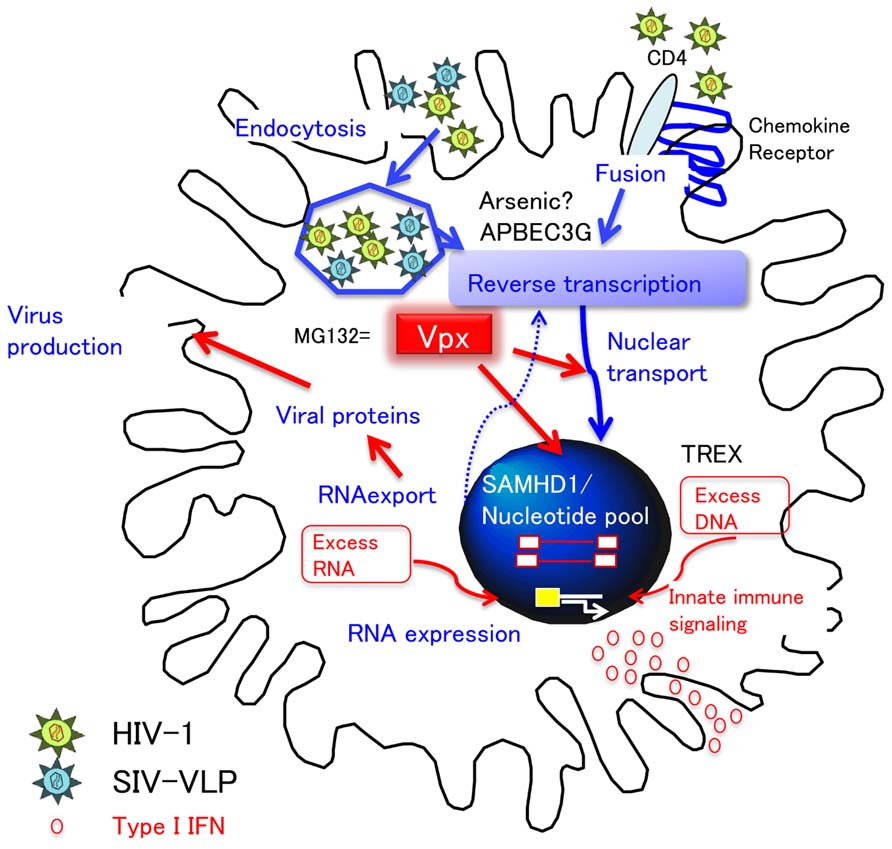
FIGURE 4. DC restriction and type I IFN response. HIV-1 utilizes CD4 and chemokine receptors for entry. The restriction of cDC to HIV-1 infection occurs at the entry (fusion) and post-entry level. Of note, in order to investigate the step at the post-entry level, VSV-pseudotyped virus is frequently utilized, which enters cells by endocytosis. After entry, APOBEC3G and arsenic sensitive factor inhibit reverse transcription. However, Vpx can counteract this restriction in DC by either degrading (1) SAMHD1-nucleotide pool-reverse transcription pathway or (2) nuclear transport. In addition, because the proteasome inhibitor MG132 works in a similar fashion to Vpx, it is possible that Vpx interferes with the proteasomal degradation step of endocytosed virions by degrading DC factors other than SAMHD1. Whatever the case, Vpx causes the accumulation of numerous virus products in the cytosol (red arrows). Importantly, the accumulation of viral DNA and RNA may trigger the innate immune signaling, resulting in type I IFN production, followed by the maturation of DC.
Myeloid cells differentiate from monocytes to either macrophages or immature MDDC, and susceptibility to HIV-1 infection amongst these cell types has been inversely correlated to A3G expression, with highest A3G expression and lowest HIV-1 susceptibility in monocytes (Peng et al., 2007). In MDDC, A3G and APOBEC3F were shown to restrict HIV-1 infection at a post-entry step by Pion et al. (2006) and can be mainly ascribed to the deaminase-independent A3G function that inhibits reverse transcription (Bishop et al., 2008). However, Pion et al. (2006) also observed that arsenic trioxide enhanced the reverse transcription of HIV-1 in MDDC and CD1c+ blood cDC in an A3G-independent manner (Pion et al., 2007). Although the mechanism of arsenic in DC is currently unknown, these results suggest that cellular factors, other than APOBEC3, are definitely playing important roles in the restriction of HIV-1 replication, most likely at a reverse transcription step, in DC.
Coincidentally, a novel DC restriction factor was discovered from a study on Vpx. SIV virus like particles (VLPs) containing Vpx are known to enhance the expression of VSV-G-pseudotyped lentivirus in DC (Santini et al., 2000), and this finding lead to the identification of SAMHD1, a myeloid cell restriction factor (Caminschi et al., 2008; Hrecka et al., 2011). SAMHD1 is a potent triphosphohydrolase that converts deoxynucleoside triphosphates (dNTPs) to deoxynucleoside and triphosphate (Goldstone et al., 2011). Therefore, the current working hypothesis for the relationship between Vpx and SAMHD1 is that virus containing Vpx can replicate well in macrophages by degrading SAMHD1, which would otherwise reduce the dNTP pool and inhibit reverse transcription (Lahouassa et al., 2012). In SIV SM or HIV-2 infection, Vpx was shown to be essential for viral replication and critical for reverse transcription of the viral RNA genome in macrophages (Fujita et al., 2008). However, a lack of Vpx in HIV-1 has negligible consequence with HIV-1, which can substantially replicate in macrophages. Therefore, Fujita et al. (2012) hypothesized that because of the lower activity of reverse transcriptase in HIV-2 versus HIV-1, HIV-2 had evolved to carry Vpx for compensation (Fujita et al., 2012).
However, it should be mentioned that immature MDDC do not support HIV-1 replication, in a similar manner, as macrophages do in R5-type HIV-1 infection (Tsunetsugu-Yokota et al., 1995) and that the susceptibility of HIV-2 containing native Vpx is even lower in MDDC and blood cDC (Smith et al., 2007). Furthermore, although remarkable Vpx-induced enhancement is observed in the case of VSV-pseudotyped lentivirus infection, it is not so obvious when native HIV was co-infected with SIV-VLP (Manel et al., 2010). Because it is known that the way virus enters via HIV envelope and/or VSV glycoprotein can lead to distinct outcomes in CD4+ T cells (Yu et al., 2009), the HIV envelope and its signaling in DC may contribute to different effects of Vpx.
There is no convincing explanation as to why the DC restriction, common to natural HIV-1 and HIV-2 infections, would necessarily be mediated by Vpx causing SAMHD1 degradation per se. Considering that the proteasome inhibitor, MG132, exerts similar effects with Vpx (Groot et al., 2006), we speculate that Vpx delivered into endosomes may inhibit endosomal degradation of endocytosed VSV-based lentivirus vector, which results in the release of numerous viral particles into the cytosol followed by production of a high copy number of RT products, thereby enhancing all subsequent steps of HIV-1 infection (integration, RNA synthesis, nuclear export, etc.). This scenario still needs to be proven and validated. Determination of whether Vpx induces the degradation of DC factors, other than SAMHD1, will prove to be an interesting future endeavor.
Vpx and Type I IFN
Another interesting feature of Vpx is that SIV-VLP containing Vpx induces a high level of Type I IFN in DC (Manel et al., 2010). Genetic diseases lacking SAMHD1 and TREX, a recently identified 3′-exonuclease which can suppress excess DNA accumulation in the cell (Yan et al., 2010), are known to develop similar autoimmune diseases due to a high level of type I IFN production (Lim and Emerman, 2011), indicating that these two distinct cellular proteins are important to regulate type I IFN responses. Thus, it is safe to assume that SAMHD1 degradation by Vpx can contribute to the induction of a high level of type I IFN production in DC. However, because Vpx enhances intracellular virus replication events as described in the previous section, excess accumulation of proviral DNA, as well as viral RNA, will occur, which might act as triggers for innate immune signaling in DC as depicted in Figure 4.
In contrast to other RNA viruses such as measles and influenza infection (Zilliox et al., 2006), HIV-1 infection, and other retroviruses also, do not induce type I IFN responses in DC (Luban, 2012). As regards to the mechanism, IRF-3, a pathogen-sensing pathway component, was shown to be depleted in HIV-infected T cells (Doehle et al., 2009) but not in DC, whereas Vpr-dependent inhibition of IRF-3 nuclear translocation was reported to occur in DC (Harman et al., 2011). The stimulation of DNA- and/or RNA-sensing pathway by Vpx may overcome this Vpr effect in DCs.
Finally, it is well-known that type I IFN is one of a multitude of cytokines which can induce the maturation of DC (Santini et al., 2000), and the maturation of MDDC has been shown to block HIV-1 infection at a post-entry level (Dong et al., 2007). However, in pDC, type I IFN production is induced by HIV-1 infection, as with other virus infections, resulting in inhibition on HIV-1 replication (Groot et al., 2006). Therefore, the lack of innate immune responses in cDC will be compensated in vivo in the early phase of HIV infection. Surprisingly, Schlafen 11 (SLFN11), a molecule induced by type I IFN, was shown to inhibit the translation of HIV-1 based on its virus-specific codon usage (Li et al., 2012). This finding indicates that there are still many undiscovered factors related to type I IFN signaling in DC which may be exploited to fight against retrovirus infections. Alternately, from the HIV side of things, by counteracting these restriction factors, HIV is able to establish a low level infection in DC. However, once type I IFN is induced in DC, it will help to generate more potent antigen-presenting DC, as demonstrated by Manel et al. (2010). Vaccine strategies utilizing Vpx for the enhancement of antigen presenting cell (APC) function in DC have already began and look promising (Durand et al., 2013).
Conclusion
We now recognize that the majority of recently identified cellular factors interacting with HIV accessory proteins are related to the type I IFN or innate immune response. The remarkable antiviral activity of type I IFN has been well-known for a long time, but we are just beginning to understand its precise mechanism, which is not necessarily common to all cell types, especially in DC. It is has been postulated that HIV has evolved to escape from potent antigen presenting activity of DC, and took advantage of subverting within DC with a minimum level of replication, for easy transmission of virus to T cells, during the T cell activation process. As described in this review, our knowledge concerning the biology of various subsets of DC has advanced enormously. However, we need to further apply this basic knowledge and understanding to manipulate DC by placing them in crucial sites (tissues), within proper time frames, for the development ofa protective acquired immunodeficiency syndrome (AIDS) vaccine.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all of our colleagues in the First laboratory, Department of Immunology, NIID Japan and other collaborators for their help and discussions. We are also grateful to Dr. Peter Lee for editing the manuscript. This paper was supported by a grant for AIDS Research from the Ministry of Health, Labor and Welfare of Japan.
References
Appay, V., Douek, D. C., and Price, D. A. (2008). CD8+ T cell efficacy in vaccination and disease. Nat. Med. 14, 623–628. doi:10.1038/nm.f.1774
Arias, J. F., Iwabu, Y., and Tokunaga, K. (2012). Sites of action of HIV-1 Vpu in BST-2/tetherin downregulation. Curr. HIV Res. 10, 283–291. doi:10.2174/157016212800792423
Auffray, C., Sieweke, M. H., and Geissmann, F. (2009). Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu. Rev. Immunol. 27, 669–692. doi:10.1146/annurev.immunol.021908.132557
Bachem, A., Guttler, S., Hartung, E., Ebstein, F., Schaefer, M., Tannert, A., et al. (2010). Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J. Exp. Med. 207, 1273–1281. doi:10.1084/jem.20100348
Banchereau, J., and Steinman, R. M. (1998). Dendritic cells and the control of immunity. Nature 392, 245–252. doi:10.1038/32588
Bishop, K. N., Verma, M., Kim, E. Y., Wolinsky, S. M., and Malim, M. H. (2008). APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 4:e1000231. doi:10.1371/journal.ppat.1000231
Brass, A. L., Huang, I. C., Benita, Y., John, S. P., Krishnan, M. N., Feeley, E. M., et al. (2009). The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139, 1243–1254. doi:10.1016/j.cell.2009.12.017
Brenchley, J. M., Schacker, T. W., Ruff, L. E., Price, D. A., Taylor, J. H., Beilman, G. J., et al. (2004). CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J. Exp. Med. 200, 749–759. doi:10.1084/jem.20040874
Burleigh, L., Lozach, P. Y., Schiffer, C., Staropoli, I., Pezo, V., Porrot, F., et al. (2006). Infection of dendritic cells (DCs), not DC-SIGN-mediated internalization of human immunodeficiency virus, is required for long-term transfer of virus to T cells. J. Virol. 80, 2949–2957. doi:10.1128/JVI.80.6.2949-2957.2006
Caminschi, I., Proietto, A. I., Ahmet, F., Kitsoulis, S., Shin Teh, J., Lo, J. C., et al. (2008). The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood 112, 3264–3273. doi:10.1182/blood-2008-05-155176
Casartelli, N., Sourisseau, M., Feldmann, J., Guivel-Benhassine, F., Mallet, A., Marcelin, A. G., et al. (2010). Tetherin restricts productive HIV-1 cell-to-cell transmission. PLoS Pathog. 6:e1000955. doi:10.1371/journal.ppat.1000955
Cavrois, M., Neidleman, J., Kreisberg, J. F., Fenard, D., Callebaut, C., and Greene, W. C. (2006). Human immunodeficiency virus fusion to dendritic cells declines as cells mature. J. Virol. 80, 1992–1999. doi:10.1128/JVI.80.4.1992-1999.2006
Cavrois, M., Neidleman, J., Kreisberg, J. F., and Greene, W. C. (2007). In vitro derived dendritic cells trans-infect CD4 T cells primarily with surface-bound HIV-1 virions. PLoS Pathog. 3:e4. doi:10.1371/journal.ppat.0030004
Cheong, C., Matos, I., Choi, J. H., Dandamudi, D. B., Shrestha, E., Longhi, M. P., et al. (2010). Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell 143, 416–429. doi:10.1016/j.cell.2010.09.039
Coleman, C. M., Spearman, P., and Wu, L. (2011). Tetherin does not significantly restrict dendritic cell-mediated HIV-1 transmission and its expression is upregulated by newly synthesized HIV-1 Nef. Retrovirology 8, 26. doi:10.1186/1742-4690-8-26
Cros, J., Cagnard, N., Woollard, K., Patey, N., Zhang, S. Y., Senechal, B., et al. (2010). Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity 33, 375–386. doi:10.1016/j.immuni.2010.08.012
Diamond, M. S., and Farzan, M. (2012). The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 13, 46–57. doi:10.1038/nri3344
Doehle, B. P., Hladik, F., Mcnevin, J. P., Mcelrath, M. J., and Gale, M. Jr. (2009). Human immunodeficiency virus type 1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J. Virol. 83, 10395–10405. doi:10.1128/JVI.00849-09
Dong, C., Janas, A. M., Wang, J. H., Olson, W. J., and Wu, L. (2007). Characterization of human immunodeficiency virus type 1 replication in immature and mature dendritic cells reveals dissociable cis- and trans-infection. J. Virol. 81, 11352–11362. doi:10.1128/JVI.01081-07
Durand, S., Nguyen, X. N., Turpin, J., Cordeil, S., Nazaret, N., Croze, S., et al. (2013). Tailored HIV-1 vectors for genetic modification of primary human dendritic cells and monocytes. J. Virol. 87, 234–242. doi:10.1128/JVI.01459-12
Fogg, D. K., Sibon, C., Miled, C., Jung, S., Aucouturier, P., Littman, D. R., et al. (2006). A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 311, 83–87. doi:10.1126/science.1117729
Fujita, M., Nomaguchi, M., Adachi, A., and Otsuka, M. (2012). SAMHD1-Dependent and -Independent Functions of HIV-2/SIV Vpx Protein. Front. Microbiol. 3:297. doi:10.3389/fmicb.2012.00297
Fujita, M., Otsuka, M., Miyoshi, M., Khamsri, B., Nomaguchi, M., and Adachi, A. (2008). Vpx is critical for reverse transcription of the human immunodeficiency virus type 2 genome in macrophages. J. Virol. 82, 7752–7756. doi:10.1128/JVI.01003-07
Gebhardt, T., and Mackay, L. K. (2012). Local immunity by tissue-resident CD8(+) memory T cells. Front. Immunol. 3:340. doi:10.3389/fimmu.2012.00340
Gebhardt, T., Wakim, L. M., Eidsmo, L., Reading, P. C., Heath, W. R., and Carbone, F. R. (2009). Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 10, 524–530. doi:10.1038/ni.1718
Gebhardt, T., Whitney, P. G., Zaid, A., Mackay, L. K., Brooks, A. G., Heath, W. R., et al. (2011). Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature 477, 216–219. doi:10.1038/nature10339
Goldstone, D. C., Ennis-Adeniran, V., Hedden, J. J., Groom, H. C., Rice, G. I., Christodoulou, E., et al. (2011). HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 480, 379–382. doi:10.1038/nature10623
Groot, F., Van Capel, T. M., Kapsenberg, M. L., Berkhout, B., and De Jong, E. C. (2006). Opposing roles of blood myeloid and plasmacytoid dendritic cells in HIV-1 infection of T cells: transmission facilitation versus replication inhibition. Blood 108, 1957–1964. doi:10.1182/blood-2006-03-010918
Gummuluru, S., Kinsey, C. M., and Emerman, M. (2000). An in vitro rapid-turnover assay for human immunodeficiency virus type 1 replication selects for cell-to-cell spread of virus. J. Virol. 74, 10882–10891. doi:10.1128/JVI.74.23.10882-10891.2000
Haniffa, M., Shin, A., Bigley, V., Mcgovern, N., Teo, P., See, P., et al. (2012). Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity 37, 60–73. doi:10.1016/j.immuni.2012.04.012
Harman, A. N., Lai, J., Turville, S., Samarajiwa, S., Gray, L., Marsden, V., et al. (2011). HIV infection of dendritic cells subverts the IFN induction pathway via IRF-1 and inhibits type 1 IFN production. Blood 118, 298–308. doi:10.1182/blood-2010-07-297721
Hrecka, K., Hao, C., Gierszewska, M., Swanson, S. K., Kesik-Brodacka, M., Srivastava, S., et al. (2011). Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474, 658–661. doi:10.1038/nature10195
Hume, D. A., Mabbott, N., Raza, S., and Freeman, T. C. (2013). Can DCs be distinguished from macrophages by molecular signatures? Nat. Immunol. 14, 187–189. doi:10.1038/ni.2516
Huysamen, C., Willment, J. A., Dennehy, K. M., and Brown, G. D. (2008). CLEC9A is a novel activation C-type lectin-like receptor expressed on BDCA3+ dendritic cells and a subset of monocytes. J. Biol. Chem. 283, 16693–16701. doi:10.1074/jbc.M709923200
Izquierdo-Useros, N., Blanco, J., Erkizia, I., Fernandez-Figueras, M. T., Borras, F. E., Naranjo-Gomez, M., et al. (2007). Maturation of blood-derived dendritic cells enhances human immunodeficiency virus type 1 capture and transmission. J. Virol. 81, 7559–7570. doi:10.1128/JVI.02572-06
Jolly, C., Booth, N. J., and Neil, S. J. (2010). Cell–cell spread of human immunodeficiency virus type 1 overcomes tetherin/BST-2-mediated restriction in T cells. J. Virol. 84, 12185–12199. doi:10.1128/JVI.01447-10
Jongbloed, S. L., Kassianos, A. J., Mcdonald, K. J., Clark, G. J., Ju, X., Angel, C. E., et al. (2010). Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 207, 1247–1260. doi:10.1084/jem.20092140
Klechevsky, E., Morita, R., Liu, M., Cao, Y., Coquery, S., Thompson-Snipes, L., et al. (2008). Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity 29, 497–510. doi:10.1016/j.immuni.2008.07.013
Kuhl, B. D., Sloan, R. D., Donahue, D. A., Liang, C., and Wainberg, M. A. (2011). Vpu-mediated tetherin antagonism of ongoing HIV-1 infection in CD4+ T-cells is not directly related to the extent of tetherin cell surface downmodulation. Virology 417, 353–361. doi:10.1016/j.virol.2011.06.012
Lahouassa, H., Daddacha, W., Hofmann, H., Ayinde, D., Logue, E. C., Dragin, L., et al. (2012). SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 13, 223–228. doi:10.1038/ni.2236
Li, Q., Duan, L., Estes, J. D., Ma, Z. M., Rourke, T., Wang, Y., et al. (2005). Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature 434, 1148–1152. doi:10.1038/nature03513
Li, M., Kao, E., Gao, X., Sandig, H., Limmer, K., Pavon-Eternod, M., et al. (2012). Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 491, 125–128. doi: 10.1038/nature11433
Lim, E. S., and Emerman, M. (2011). HIV: going for the watchman. Nature 474, 587–588. doi:10.1038/474587a
Liu, K., Victora, G. D., Schwickert, T. A., Guermonprez, P., Meredith, M. M., Yao, K., et al. (2009). In vivo analysis of dendritic cell development and homeostasis. Science 324, 392–397. doi:10.1126/science.1170540
Lore, K., Smed-Sorensen, A., Vasudevan, J., Mascola, J. R., and Koup, R. A. (2005). Myeloid and plasmacytoid dendritic cells transfer HIV-1 preferentially to antigen-specific CD4+ T cells. J. Exp. Med. 201, 2023–2033. doi:10.1084/jem.20042413
Luban, J. (2012). Innate immune sensing of HIV-1 by dendritic cells. Cell Host Microbe 12, 408–418. doi:10.1016/j.chom.2012.10.002
Manel, N., Hogstad, B., Wang, Y., Levy, D. E., Unutmaz, D., and Littman, D. R. (2010). A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature 467, 214–217. doi:10.1038/nature09337
Mattapallil, J. J., Douek, D. C., Hill, B., Nishimura, Y., Martin, M., and Roederer, M. (2005). Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 434, 1093–1097. doi:10.1038/nature03501
Mehandru, S., Poles, M. A., Tenner-Racz, K., Horowitz, A., Hurley, A., Hogan, C., et al. (2004). Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J. Exp. Med. 200, 761–770. doi:10.1084/jem.20041196
Miller, J. C., Brown, B. D., Shay, T., Gautier, E. L., Jojic, V., Cohain, A., et al. (2012). Deciphering the transcriptional network of the dendritic cell lineage. Nat. Immunol. 13, 888–899. doi:10.1038/ni.2370
Miyagi, E., Andrew, A. J., Kao, S., and Strebel, K. (2009). Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc. Natl. Acad. Sci. U.S.A. 106, 2868–2873. doi:10.1073/pnas.0813223106
Neil, S. J., Zang, T., and Bieniasz, P. D. (2008). Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451, 425–430. doi:10.1038/nature06553
Nobile, C., Petit, C., Moris, A., Skrabal, K., Abastado, J. P., Mammano, F., et al. (2005). Covert human immunodeficiency virus replication in dendritic cells and in DC-SIGN-expressing cells promotes long-term transmission to lymphocytes. J. Virol. 79, 5386–5399. doi:10.1128/JVI.79.9.5386-5399.2005
Peng, G., Greenwell-Wild, T., Nares, S., Jin, W., Lei, K. J., Rangel, Z. G., et al. (2007). Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood 110, 393–400. doi:10.1182/blood-2006-10-051763
Pierson, T., Mcarthur, J., and Siliciano, R. F. (2000). Reservoirs for HIV-1: mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu. Rev. Immunol. 18, 665–708. doi:10.1146/annurev.immunol.18.1.665
Piguet, V., and Steinman, R. M. (2007). The interaction of HIV with dendritic cells: outcomes and pathways. Trends Immunol. 28, 503–510. doi:10.1016/j.it.2007.07.010
Pion, M., Granelli-Piperno, A., Mangeat, B., Stalder, R., Correa, R., Steinman, R. M., et al. (2006). APOBEC3G/3F mediates intrinsic resistance of monocyte-derived dendritic cells to HIV-1 infection. J. Exp. Med. 203, 2887–2893. doi:10.1084/jem.20061519
Pion, M., Stalder, R., Correa, R., Mangeat, B., Towers, G. J., and Piguet, V. (2007). Identification of an arsenic-sensitive block to primate lentiviral infection of human dendritic cells. J. Virol. 81, 12086–12090. doi:10.1128/JVI.00800-07
Poulin, L. F., Salio, M., Griessinger, E., Anjos-Afonso, F., Craciun, L., Chen, J. L., et al. (2010). Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J. Exp. Med. 207, 1261–1271. doi:10.1084/jem.20092618
Rinaldo, C. R. Jr., and Piazza, P. (2004). Virus infection of dendritic cells: portal for host invasion and host defense. Trends Microbiol. 12, 337–345. doi:10.1016/j.tim.2004.05.003
Sancho, D., Joffre, O. P., Keller, A. M., Rogers, N. C., Martinez, D., Hernanz-Falcon, P., et al. (2009). Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 458, 899–903. doi:10.1038/nature07750
Santini, S. M., Lapenta, C., Logozzi, M., Parlato, S., Spada, M., Di Pucchio, T., et al. (2000). Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J. Exp. Med. 191, 1777–1788. doi:10.1084/jem.191.10.1777
Satpathy, A. T., Wu, X., Albring, J. C., and Murphy, K. M. (2012). Re(de)fining the dendritic cell lineage. Nat. Immunol. 13, 1145–1154. doi:10.1038/ni.2467
Sattentau, Q. (2008). Avoiding the void: cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 6, 815–826. doi:10.1038/nrmicro1972
Sere, K., Baek, J. H., Ober-Blobaum, J., Muller-Newen, G., Tacke, F., Yokota, Y., et al. (2012). Two distinct types of Langerhans cells populate the skin during steady state and inflammation. Immunity 37, 905–916. doi:10.1016/j.immuni.2012.07.019
Sheehy, A. M., Gaddis, N. C., Choi, J. D., and Malim, M. H. (2002). Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418, 646–650. doi:10.1038/nature00939
Sheehy, A. M., Gaddis, N. C., and Malim, M. H. (2003). The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9, 1404–1407. doi:10.1038/nm945
Smith, A. L., Ganesh, L., Leung, K., Jongstra-Bilen, J., Jongstra, J., and Nabel, G. J. (2007). Leukocyte-specific protein 1 interacts with DC-SIGN and mediates transport of HIV to the proteasome in dendritic cells. J. Exp. Med. 204, 421–430. doi:10.1084/jem.20061604
Steinman, R. M. (2007). Lasker Basic Medical Research Award. Dendritic cells: versatile controllers of the immune system. Nat. Med. 13, 1155–1159. doi:10.1038/nm1643
Steinman, R. M., and Idoyaga, J. (2010). Features of the dendritic cell lineage. Immunol. Rev. 234, 5–17. doi:10.1111/j.0105-2896.2009.00888.x
Tsunetsugu-Yokota, Y., Akagawa, K., Kimoto, H., Suzuki, K., Iwasaki, M., Yasuda, S., et al. (1995). Monocyte-derived cultured dendritic cells are susceptible to human immunodeficiency virus infection and transmit virus to resting T cells in the process of nominal antigen presentation. J. Virol. 69, 4544–4547.
Tsunetsugu-Yokota, Y., Yasuda, S., Sugimoto, A., Yagi, T., Azuma, M., Yagita, H., et al. (1997). Efficient virus transmission from dendritic cells to CD4+ T cells in response to antigen depends on close contact through adhesion molecules. Virology 239, 259–268. doi:10.1006/viro.1997.8895
Turville, S. G., Cameron, P. U., Handley, A., Lin, G., Pohlmann, S., Doms, R. W., et al. (2002). Diversity of receptors binding HIV on dendritic cell subsets. Nat. Immunol. 3, 975–983. doi:10.1038/ni841
Turville, S. G., Santos, J. J., Frank, I., Cameron, P. U., Wilkinson, J., Miranda-Saksena, M., et al. (2004). Immunodeficiency virus uptake, turnover, and 2-phase transfer in human dendritic cells. Blood 103, 2170–2179. doi:10.1182/blood-2003-09-3129
Van Damme, N., Goff, D., Katsura, C., Jorgenson, R. L., Mitchell, R., Johnson, M. C., et al. (2008). The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3, 245–252. doi:10.1016/j.chom.2008.03.001
Wakim, L. M., Gupta, N., Mintern, J. D., and Villadangos, J. A. (2013). Enhanced survival of lung tissue-resident memory CD8(+) T cells during infection with influenza virus due to selective expression of IFITM3. Nat. Immunol. 14, 238–245. doi:10.1038/ni.2525
Wakim, L. M., Waithman, J., Van Rooijen, N., Heath, W. R., and Carbone, F. R. (2008). Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science 319, 198–202. doi:10.1126/science.1151869
Wang, J. H., Janas, A. M., Olson, W. J., and Wu, L. (2007). Functionally distinct transmission of human immunodeficiency virus type 1 mediated by immature and mature dendritic cells. J. Virol. 81, 8933–8943. doi:10.1128/JVI.00878-07
Wu, L., and KewalRamani, V. N. (2006). Dendritic-cell interactions with HIV: infection and viral dissemination. Nat. Rev. Immunol. 6, 859–868. doi:10.1038/nri1960
Yan, N., and Chen, Z. J. (2012). Intrinsic antiviral immunity. Nat. Immunol. 13, 214–222. doi:10.1038/ni.2229
Yan, N., Regalado-Magdos, A. D., Stiggelbout, B., Lee-Kirsch, M. A., and Lieberman, J. (2010). The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 11, 1005–1013. doi:10.1038/ni.1941
Yu, D., Wang, W., Yoder, A., Spear, M., and Wu, Y. (2009). The HIV envelope but not VSV glycoprotein is capable of mediating HIV latent infection of resting CD4 T cells. PLoS Pathog. 5:e1000633. doi:10.1371/journal.ppat.1000633
Keywords: DC–T transmission, HIV, DC subsets, accessory proteins
Citation: Tsunetsugu-Yokota Y and Muhsen M (2013) Development of human dendritic cells and their role in HIV infection: antiviral immunity versus HIV transmission. Front. Microbiol. 4:178. doi: 10.3389/fmicb.2013.00178
Received: 23 April 2013; Paper pending published: 24 May 2013;
Accepted: 13 June 2013; Published online: 09 July 2013.
Edited by:
Mei-Ru Chen, National Taiwan University, TaiwanReviewed by:
Ai Kawana-Tachikawa, University of Tokyo, JapanRyuta Sakuma, Tokyo Medical and Dental University, Japan
Copyright: © 2013 Tsunetsugu-Yokota and Muhsen. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Yasuko Tsunetsugu-Yokota, Department of Immunology, National Institute of Infectious Diseases, 1-23-1 Toyama, Shinjuku, Tokyo 162-8640, Japan e-mail:eXlva290YUBuaWguZ28uanA=