

- School of Biological Sciences, The University of Hong Kong, Hong Kong, China
The gadE-mdtEF operon encodes a central acid resistance regulator GadE and two multidrug efflux proteins MdtEF. Although transcriptional regulation of gadE in the context of acid resistance under the aerobic growth environment of Escherichia coli has been extensively studied, regulation of the operon under the physiologically relevant environment of anaerobic growth and its effect on the expression of the multidrug efflux proteins MdtEF in the operon has not been disclosed. Our previous study revealed that anaerobic induction of the operon was dependent on ArcA, the response regulator of the ArcBA two-component system, in the M9 glucose minimal medium. However, the detailed regulatory mechanism remains unknown. In this study, we showed that anaerobic activation of mdtEF was driven by the 798 bp unusually long gadE promoter. Deletion of evgA, ydeO, rpoS, and gadX which has been shown to activate the gadE expression during acid stresses under aerobic condition did not have a significant effect on the anaerobic activation of the operon. Rather, anaerobic activation of the operon was largely dependent on the global regulator ArcA and a GTPase MnmE. Under aerobic condition, transcription of gadE was repressed by the global DNA silencer H-NS in M9 minimal medium. Interestingly, under anaerobic condition, while ΔarcA almost completely abolished transcription of gadE-mdtEF, further deletion of hns in ΔarcA mutant restored the transcription of the full-length PgadE-lacZ, and P1- and P3-lacZ fusions, suggesting an antagonistic effect of ArcA on the H-NS mediated repression. Taken together, we conclude that the anaerobic activation of the gadE-mdtEF was primarily mediated by the two-component system ArcBA through antagonizing the H-NS mediated repression.
Introduction
Drug efflux constitutes an important mechanism in bacterial drug resistance. Efflux activity is mediated by a class of membrane protein transporters called multidrug efflux pumps, which actively extrude a variety of cytotoxic substances including antibiotics out of bacterial cells (Li and Nikaido, 2009; Allen et al., 2010). Based on phylogenetic analysis and functional properties, drug efflux pumps of bacteria are classified into five families: the ATP-binding cassette (ABC), the major facilitator (MFS), the multidrug and toxic compound extrusion (MATE), the small multidrug resistance (SMR), and the resistance nodulation division (RND) family (Piddock, 2006). Among them, the tripartite RND pumps are particularly noteworthy in gram-negative bacteria owing to their capability of exporting a broad range of structurally diverse drugs directly to the outside of bacterial cells (Poole, 2008). The three components of RND efflux pumps include cytoplasmic membrane transporter component of RND family, outer membrane factor (OMF) component, and periplasmic component belonging to the membrane fusion protein (Murakami et al., 2006; Piddock, 2006). The constitutive activity of these efflux systems, such as the AcrAB-TolC pump in Escherichia coli and the MexAB-OprM and MexXY-OprM systems in Pseudomonas aeruginosa, renders the bacteria a low level, intrinsic resistance to a wide range of toxic substances (Poole, 2005).
Previous genome wide studies revealed that there are 20 efflux systems encoded in the E. coli K-12 genome (Nishino and Yamaguchi, 2001). However, except for the housekeeping AcrAB-TolC pump, the expression of other 19 pumps is largely inactive under ordinary laboratory growth condition, i.e., at 37°C in rich medium with aeration (Nishino and Yamaguchi, 2001). Increasing evidence suggests that bacterial stress response, i.e., bacterial growth in the adverse environment of their natural ecological niches and human host, induces the expression of specific efflux pumps (Piddock, 2006; Li and Nikaido, 2009; Poole, 2012a). Indeed, our previous studies revealed that MdtEF (previously known as YhiUV), an RND family pump, is activated during the anaerobic growth of E. coli, a signature environment of human gut where the bacterium primarily colonizes (Zhang et al., 2011). The pump was shown to expel the cytotoxic indole nitrosative compounds from E. coli cells accumulated during the anaerobic respiration of nitrate and thus protect the bacterium from nitrosative stress under this physiological condition (Zhang et al., 2011). Activation of MdtEF under this condition was shown to be dependent on ArcA, the response regulator of the ArcBA two-component system, which is a global regulatory system dedicated to the anaerobic adaptation of E. coli (Green and Paget, 2004), but the detailed regulatory mechanism remains unknown.
MdtEF forms an RND type multidrug efflux system with the common outer membrane channel TolC in which MdtF forms the cytoplasmic membrane transporter and is connected to TolC by the periplasmic protein MdtE (Nishino and Yamaguchi, 2002). Over-expression of the pump from a multicopy plasmid has been shown to confer resistance to a broad range of antimicrobial agents such as some β-lactams (e.g., oxacillin, cloxacillin, and nafcillin), macrolide antibiotic erythromycin, as well as doxorubicin, crystal violet, ethidium bromide, rhodamine 6G, tetraphenylphosphonium bromide (TPP), benzalkonium, SDS, deoxycholate, suggesting that MdtEF is a multidrug resistance determinant (Nishino and Yamaguchi, 2001; Nishino et al., 2003; Lennen et al., 2013). Recently, MdtEF was also found to be involved in the efflux of physiological substance fatty acids (Lennen et al., 2013), and their expression can be induced by various of environmental and physiological signals, such as the entry of the stationary growth phase, N-acetyl-glucosomine, indole, as well as the combined environmental challenges of oxygen limitation and acid stress (Hirakawa et al., 2005, 2006; Hayes et al., 2006; Kobayashi et al., 2006), highlighting the physiological relevance of the MdtEF-TolC efflux pump.
MdtEF genes are located in the gadE-mdtEF operon where mdtE is 339 bp downstream of the gadE gene (Keseler et al., 2011). Interestingly, the gadE gene in this operon encodes a key regulator of the major acid resistance system in E. coli which is composed of the glutamate decarboxylase isoenzymes GadA and GadB, and a dedicated glutamic acid/γ-aminobutyrate (GABA) antiporter GadC (Hommais et al., 2004). During bacterial response to acid stresses, GadA and GadB catalyze the decarboxylation of glutamic acid, yielding GABA, which is subsequently exported by GadC in exchange for another molecule of glutamic acid (Foster, 2004). Since the reaction consumes a proton and releases CO2, it effectively limits the intracellular acidification during acid stresses, thus plays an important role in the adaptation and survival of E. coli in certain host niches, such as the extremely low pH environment (pH = 2) of gastric acid (Foster, 2004). The expression of this system is primarily subject to the control of the acid resistance regulator GadE in the gadE-mdtEF operon (Masuda and Church, 2003).
Owing to its significant roles in acid resistance, transcriptional regulation from the gadE promoter has been extensively studied. It was demonstrated that the gadE promoter encompasses an unusually large 798 bp intergenic region between the hdeD and gadE gene (Ma et al., 2004), representing one of the eight similarly large intergenic regions in the entire E. coli genome (Tjaden et al., 2002). Transcription from at least four starting sites which are located at -21 (T), -124 (T1), -324/-317 (T2), and -566 (T3) relative to the gadE start codon have been identified (Ma et al., 2004; Itou et al., 2009; Sayed and Foster, 2009). More than ten transcription regulators including EvgA, YdeO, GadE, TorR, H-NS, PhoP, RpoS, CRP, MnmE, GadX, and GadW have been found to participate in the regulation of gadE during various circumstances of acid stresses through at least five different regulatory circuits (Ma et al., 2004; Sayed and Foster, 2009). Involvement of EvgA, YdeO, H-NS, CRP, GadX, as well as a small RNA DsrA in the regulation of the mdtEF gene in the same operon has also been reported (Nishino and Yamaguchi, 2002, 2004; Oshima et al., 2006; Nishino et al., 2008a,b, 2009, 2011). However, little is known about the regulation of the gadE-mtEF expression in the anaerobic physiological condition of E. coli which is the predominant environmental challenge encountered by the Entericbacteria family of bacteria when they are being transmitted from the status of free living to human intestinal tract, except for the involvement of the global transcription regulator ArcA as shown in our previous study (Zhang et al., 2011). Moreover, it remains unclear whether the various of regulatory circuits that activate the expression of gadE in the context of acid resistance also lead to the expression of the multidrug efflux genes mdtEF present in the same operon.
In the current study we set out to investigate the detailed regulatory circuits governing the anaerobic transcription from the gadE promoter in E. coli and examine its effect on the expression of the multidrug efflux proteins MdtEF present in the operon. We found that unlike the regulatory patterns occurred during aerobic acid stresses, anaerobic activation of the gadE-mdtEF operon is primarily mediated by ArcA through its antagonization of the H-NS mediated repression.
Materials and Methods
Bacterial Strains and Plasmids
The bacteria strains and plasmids used in this study are listed in Table 1. Gene deletion mutants were constructed either by P1 phage transduction from the Keio collection (Baba et al., 2006) or using the method described by Datsenko and Wanner (2000). All of the constructed strains were verified by colony PCR and DNA sequencing (BGI, Hong Kong). E. coli was cultured in Luria Bertani (LB) broth (USB) or M9 minimum medium supplemented with 0.2% glucose (USB), 0.2% casamino acids (CAA) (USB), 4 μg/ml thiamin (Sigma), 0.1 mM CaCl2, 1 mM MgCl2, 10 μg/ml ferric ammonium citrate (Sigma), and 0.5 μM (NH4)6Mo7O24 (Sigma). Without specific indication, exponential phase cells were harvested when A600 of cultures reached around 0.3. Antibiotic concentrations used for bacterial culture or colony screening were 100 μg/ml ampicillin (USB), 20 μg/ml kanamycin (USB), or 25 μg/ml chloramphenicol (USB). All other chemicals without specification were purchased from USB.
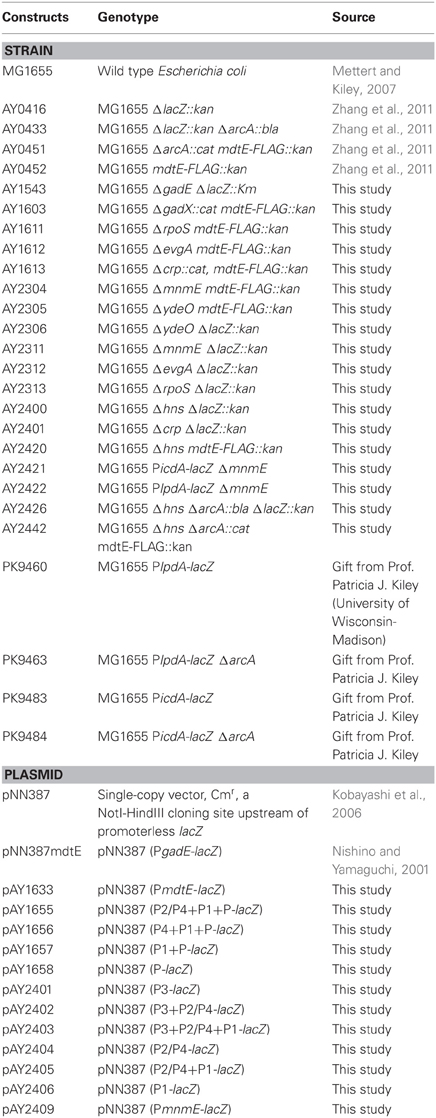
Table 1. Bacterial strains and plasmids used in this study.
Construction of Promoter-lacZ Transcription Reporter in pNN387
Promoter-lacZ fusions were constructed based on the methods described previously (Zhang et al., 2011). Firstly, DNA fragments corresponding to the desired promoter regions were amplified using iProof High Fidelity DNA polymerase (Bio-Rad). The PCR condition and program is as follows: initial denaturation at 98°C for 30 s followed by 35 cycles of denaturation, annealing, and extension which are achieved by the program of: 98°C for 10 s, 60°C for 30 s, 72°C for 45 s, and the final extension at 72°C for 5 min. The pair of primers used in PCR includes protective bases (lowercase), restriction enzyme site (NotI or HidIII), followed by ~20 bp homologous to the 5′- and 3′-end of the desirable promoter regions respectively (e.g., PgadE-F: 5′-aaggaaaaaaGCGGCCGCTTACCCCGGTTGTCACCCGGAT-3′; PgadE-R: 5′-cccAAGCTTAACTTGCTCCTTAGCCGTTATC-3′). Primers for construction of other promoter-lacZ fusions were designed similarly and the detailed sequences are available upon request. The PCR fragments were purified using illustra GFX PCR DNA and Gel Band Purification Kit (GE) followed by NotI/HindIII (NEB) digestion, and then were ligated into NotI/HindIII (NEB) digested single copy plasmid pNN387 using Quick Ligase (NEB). Colonies were screened by chloramphenicol resistance and then verified by colony PCR and DNA sequencing (BGI, Hong Kong).
Construction of Chromosomal mdtE-Flag by P1 Vir Transduction
Construction of chromosomal mdtE-FLAG in various of deletion mutants was achieved by P1 phage transduction using the lysate from the strain AY0451 (Zhang et al., 2011). To prepare the lysate of the donor strain, AY0451 was grown in 2 ml LB medium till A600 achieved 0.1–0.2, and then 40 μl P1vir and 10 mM CaCl2 was added to the culture. The mixture was shaken at 37°C for ~3 h or till cells lysed. 2 drops of chloroform per ml of culture was then added to the lysate to diminish any chance of cell viability. After a vortex and centrifugation at 16,000 g for 2 min, the supernatant was transferred to a fresh tube and stored at 4°C in the presence of couple of drops of chloroform. Culture of the recipient strain was prepared as follows: the recipient strain was grown in LB medium till A600 exceeded 0.7. 10 mM CaCl2 was then added followed by continuing growth for 15 min. 200 μl of the culture was then mixed with 100 μl lysate of the donor strain and incubated at 37°C for 20 min. Following the incubation, 100 μl of 1 M citrate was added to terminate the infection. Following the addition of 600 μl LB medium, the transductant was incubated at 37°C for 1 h before being spread onto LB agar plates containing kanamycin and 4 mM citrate. Desirable colonies were screened by colony PCR and were verified by DNA sequencing (BGI, Hong Kong).
β-Galactosidase Activity Assay
β-galactosidase activity assay was performed based on the method by Miller (1972) and detailed experimental procedure has been described previously (Zhang et al., 2011). Briefly, cells were grown anaerobically by inoculating a small number of cell cultures (initial cell density of 103 cells/ml) into screw-capped culture tubes filled with M9 medium, and subsequently cultured in 37°C water bath without aeration. When A600 of the cultures reached ~0.3, tetracycline (10 μg/ml) was added to terminate protein synthesis and cell growth, and the cultures were placed on ice until assayed. The assays were performed in triplicate, and results were presented as the mean in either Miller unit or the percentages relative to the activity of full length promoter-lacZ fusion in WT (wild type) strain. Error bars indicate the standard deviation.
Total RNA Extraction
8 ml anaerobically grown (the same condition as used in β-galactosidase activity assay) culture of E. coli MG1655 was mixed with 1.25 ml ice-cold ethnol/phenol stop solution (5% water-saturated phenol pH 4.5 in ethanol) and placed on ice for 10 min before being harvested by centrifugation at 4000 g for 9 min at 4°C. After removing supernatant, the cell pellet was frozen in liquid nitrogen and stored at −80°C to aid lysis. Cells were lysed by resuspending in 800 μl TE buffer (pH 8.0) containing 1.4 μl 36 kU μl−1 lysozyme (epicenter), and then placed in 64°C water bath for 2 min. After incubation, 88 μl 3M NaOAc (pH 5.2) was added to adjust the pH and ion strength of the lysate solution. Subsequently, acid-phenol/chloroform extraction followed by ethanol precipitation was performed to obtain the total RNA following the manufacturer's instruction. To remove trace amount of genomic DNA contamination, the extracted RNA was subject to DNase I treatment using the turbo DNA Free Kit (Amibion). Absence of genomic DNA contamination was confirmed by PCR suing the prepared RNA as template. The quantity of RNA was determined using NanoDrop 2000 (Thermo Scientific).
RNA Ligase-Mediated Rapid Amplification of 5′ cDNA Ends (5′ RLM-RACE)
1 μg of RNA which was treated with the turbo DNA Free Kit (Amibion) as above and shown as tight, no smearing band on the agrose gel (following the instructions of the FirstChoice RLM-RACE Kit (Ambion)) was treated with the Tobacco Acid Pyrophosphatase (TAP), ligated to the 5′ RACE adapter, and reverse transcripted with reagents supplied in the FirstChoice RLM-RACE Kit (Ambion) following the manufacturer's instruction. The obtained cDNA was subsequently utilized to perform outer (primer: PgadEout: 5′-TCCAGAAATTTAATCGCTTCTTCATC-3′) and inner (primer: PgadEin: 5′-GTACTCGAGGTGATTATCTTTCAACTGCCAAAAGC-3′) PCR using Fastart Taq (Roche) DNA polymerase. The PCR products were gel band purified using PCR DNA and Gel Band Purification Kit (GE) and cloned into BamHI and XhoI sites of pPK7035 (Kang et al., 2005) followed by DNA sequencing. The first nucleotide being sequenced following the 5′ RACE adapter sequence was determined as the transcription start site.
Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Western Blot
E. coli strains containing chromosomal FLAG tagged MdtE were grown anaerobically (the same condition as used in β-galactosidase activity assay) to log phase (A600 = 0.3) in M9 minimal medium supplemented with 0.2% glucose, 0.2% CAA, 4 μg/ml thiamin, 0.1 mM CaCl2, 1 mM MgCl2, 10 μg/ml ferric ammonium citrate, and 0.5 μM (NH4)6Mo7O24. Cells were harvested by centrifugation at 3800 g at 4°C for 10 min. Cell pellet was resuspended in lysis mix (BugBuster reagent (Merck) supplemented with 2.5 mg/mL lysozyme and 10 U/mL DNase I (Invitrogen)). Following the cell lysis, a small fraction of the lysis was subject to the Dc Protein Assay kit (BioRad) to measure the concentration of total proteins in the lysate. The volumes of the cell lysis loaded onto the SDS-PAGE were then determined based on the concentrations of each of the samples such that each of the lanes contains the same amount of total protein. The lysis was then heated at 55°C for 25 min prior to being subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Separated proteins were electrically (Wet/Tank Blotting Systems, Bio-Rad; 110V) transferred to a nitrocellulose membrane (Bio-Rad) for 1.5 h. After blocking with 5% non-fat milk in 1% TBST (Tris Buffered Saline with Tween 20, 0.1 M Tris, 0.15 M NaCl, 0.1% Tween 20, and pH was adjusted with HCl to 7.6), the membrane was incubated with monoclonal anti-FLAG antibody (Sigma), and then secondary antibody (goat anti-mouse IgG HRP conjuate, Bio-Rad). After treatment of the membrane by the ECL Plus Western Blotting Detection Reagents (GE health), the protein bands were visualized by X-ray film development. Signals corresponding to MdtE-FLAG proteins were quantified using ImageJ (National Institutes of Health) and are presented as percentages relative to the level of MdtE-FLAG in the wild type strain.
EB Stained Electrophoresis Gel Mobility Shift Assay (EMSA)
Purification and subsequent in vitro phosphorylation of His6-ArcA protein was following the method described previously (Bekker et al., 2010). Briefly, E. coli strain BL21 transformed with pET-His6ArcA were grown in LB to A600 of 0.4 before adding 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) to induce the overexpress of His6-ArcA. After grown for additional 2.5 h, cells were harvested by centrifugation. Cell pellet was then resuspended in 4 ml of buffer A (0.5 MNaCl, 20 mMTris-HCl, pH 7.9) containing 1.3 mg/ml lysozyme, 30 g/ml DNase and RNase. Cells were then incubated at room temperature for 30 min followed by sonication to lyse the cells. Cell lysate was obtained by centrifugation at 15,000 g for 30 min and then was subjected to a 1.5-ml Ni-nitrilotriacetic acid–agarose column (Qiagen) equilibrated with buffer A. Following washing the column with 10 ml buffer A containing 10 mM imidazole, His6-ArcA was eluted with buffer A containing 50 mM imidazole. Purified His6-ArcA protein was phosphorylated by incubating the protein in TEGD buffer (50 mM Tris-HCl [pH 7.5], 0.5 mM EDTA, 10% glycerol) supplemented with 5 mM MgCl2 and 50 mM (final concentration) carbamoyl phosphate. The mixture was incubated for 90 min at 25°C, and then the phosphorylated ArcA was used immediately for DNA-binding reaction. To initiate the DNA binding reaction, various concentration of DNA probes and phosphorylated proteins were mixed and subsequently incubated at 37°C for 20 min in EMSA binding buffer (pH 7.2, 20 mM Tris, 50 mM NaCl, 10 mM EDTA, 4 mM DTT, 5% glycerol, 0.5 mg/mL BSA). The reaction mixture was then subjected to 6% non-denaturing polyacrylamide gel electrophoresis in 0.5 × TBE buffer. The polyacrylamide gel was visualized under UV light (254–366 nm) following the staining in 0.5 × TBE buffer containing 0.5 μg/ml ethidium bromide (EB) for 10 min.
Results
Anaerobic Transcription of mdtEF is Initiated from the gadE Promoter
MdtEF are located downstream of the gadE gene, which encodes a central regulator of the glutamic acid dependent acid resistance system in E. coli (Nishino and Yamaguchi, 2002). Previous RT-PCR analysis revealed that mdtEF were co-transcribed with gadE, thus were annotated in the operon of gadE-mdtEF (Hirakawa et al., 2006). However, an EMSA study by Nishino et al. showed that EvgA, one of the regulators that can activate the expression of mdtEF, could bind to the intergenic region between gadE and mdtEF, indicating that this region might serve as a potential promoter of mdtEF under certain conditions (Nishino and Yamaguchi, 2002). Thus, we first examined whether this intergenic region (namely PmdtE) is involved in the transcription of mdtEF under anaerobic condition. To address this, we constructed lacZ fusion of the intergenic region (denoted as PmdtE-lacZ) in the single copy plasmid pNN387 and performed β-galactosidase activity assay. It was shown that no transcription from PmdtE-lacZ was detected under either aerobic or anaerobic condition, while transcription from PgadE-lacZ constructed in the same vector was significantly induced under anaerobic condition (data not shown). These results suggested that anaerobic transcription of mdtEF was primarily initiated from the gadE promoter. Therefore we focused on the 798 bp PgadE region in the following investigation to examine its contribution to the expression of mdtEF under anaerobic condition.
Transcription from the Same Four Starting Sites of gadE were Identified in the Anaerobically Grown E. coli
In previous studies, three transcription starting sites, located at -124 (T1), -324/-317 (T2), and -566 (T3) relative to the gadE start codon respectively, have been identified in E. coli grown in LB glucose to early stationary phase (A600 = 1.75) (Sayed and Foster, 2009) and one transcription starting site located at the -21 position was detected from E. coli grown in the EG minimal buffer at pH 5.5 (Ma et al., 2004). To examine whether transcription from all these starting sites, and whether transcription from additional starting site, is active under anaerobic growth condition, we performed 5′ RLM-RACE using RNA isolated from E. coli grown anaerobically in M9 glucose medium which has been shown to significantly induce the expression of MdtEF in our previous study (Zhang et al., 2011). 5′-RACE revealed that transcription from the three starting sites identified previously was detected from several clones (data not shown). Transcription starting site of -21 was identified from one clone, but a weak -35 and -10 element was present upstream of this starting site. Two putative new transcription starting sites were also detected, however, since no recognizable -35 and -10 elements were present upstream of the sites, they are probably the digestive products from the established four transcripts. This result suggested that transcription from all four starting sites of gadE-mdtEF operon, -21 (T), -124 (T2/T4), -566 (T3), is active under anaerobic conditions. We next investigated the transcription regulatory mechanism of gadE-mdtEF from all these sites under anaerobic condition.
The P3 Region is Important in the Anaerobic Activation of the gadE-mdtEF Operon
Following the designation of the PgadE region in previous studies (Sayed and Foster, 2009), the four transcription starting sites from 5′- to 3′-end of the 798 bp intergenic region between hdeD and gadE are designated as T3, T2, T4, T1, and T, respectively (Figure 1A) (Ma et al., 2004; Itou et al., 2009; Sayed and Foster, 2009), and the promoter regions immediately upstream of these transcription start sites are designated as P3, P2, P4, P1, and P accordingly. To examine which promoter regions are important for the anaerobic transcription of the gadE-mdtEF operon, we first constructed three PgadE truncations which lacked P3, P2/P4, P1 from 5′-end of the region successively, resulting in the promoter-lacZ fusion containing the intact P2/P4+P1+P, P1+P, and P regions, respectively (the upper four constructs in Figure 1A). Transcription activity assay showed that promoter-lacZ fusions lacking the P3 region, i.e., lacZ fusions of P2/P4+P1+P, and P1+P, displayed only ~1/3 activity of that of the full length PgadE-lacZ fusion (Figure 1B), and that of P region showed almost no activity. These results suggested that the P3 region is important in anaerobic activation of the gadE-mdtEF operon and that transcription from the P promoter region is not active under this condition. This result also indicated that the -21 transcription starting site (driven by the P promoter) detected from the RACE analysis might be a digestive product from the transcripts initiated from the other three sites.
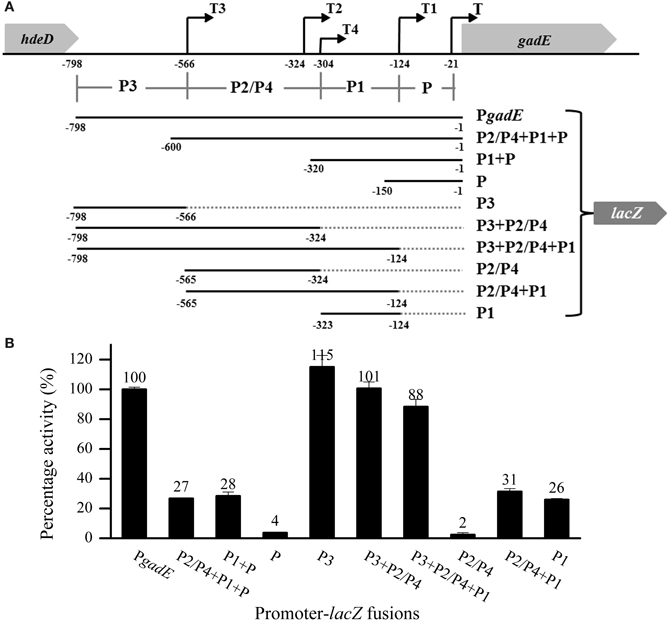
Figure 1. The P3 region is important in the anaerobic up-regulation of gadE-mdtEF. (A) Schematic diagram of the constructed promoter-lacZ fusions. PgadE is defined as the 798 bp intergenic region between the hdeD and gadE genes. Labeled numbers indicate the nucleotides positions relative to the ATG start codon of gadE. Transcription starting sites are indicated as black arrows, and the regions upstream of each of the transcription starting sites are designated as its individual promoters, labeled as P3, P2/P4, P1, and P, respectively. (B) Promoter activity of all PgadE truncations determined by β-galactosidase activity of corresponding lacZ fusions. Cells were grown anaerobically to exponential phase (A600 = 0.3) in minimal M9 glucose medium. Results are presented as percentages relative to that of full length gadE promoter activity (100%), and error bars represent the standard errors of triplicate experiments (n = 3).
To dissect the contribution of each of these promoter regions, we performed another series of truncation on PgadE, resulting in lacZ fusions containing individual or combined promoters (the lower six constructs in Figure 1A). Transcription activities of these truncations showed that promoter-lacZ fusions containing the P3 region, i.e., P3, P3+P2/P4 and P3+P2/P4+P1, displayed similar or even higher activity than that of the full length, whereas promoter-lacZ fusions lacking the P3 region, i.e., P2/P4, P2/P4+P1, and P1, displayed very low activities (Figure 1B), confirming the important role of the P3 region in the up-regulation of gadE-mdtEF operon under anaerobic condition. This result indicated that a strong activator binding site may exist at the P3 region. Furthermore, transcription of lacZ fusion containing the P2/P4 promoter region was almost undetectable, suggesting that similar as the transcription starting site T which is driven by the individual P promoter region, transcription from the individual P2/P4 promoter is also inactive under anaerobic conditions. These results combined explained the observed very low activities of the promoter-lacZ fusions of P2/P4+P1, P1, P2/P4+P1+P and P1+P (Figure 1B).
Involvement of Different Regulators in the Anaerobic Expression of mdtEF from that of Aerobic Condition
Previous studies focusing on the expression of gadE-mdtEF operon during acid resistance have revealed that several regulators, such as EvgA, YdeO, MnmE, GadE, GadX, CRP, and H-NS, are involved in the regulation of gadE-mdtEF operon (Gong et al., 2004; Ma et al., 2004; Hirakawa et al., 2006; Tramonti et al., 2008). However, all those studies were carried out under aerobic condition and primarily focused on the transcription of the gadE gene. A previous study from our group focusing on the expression of the multidrug efflux genes mdtEF present in the same operon showed that under anaerobic condition the global regulator ArcA activates the expression of mdtEF, since ΔarcA caused significant decrease of both transcription and protein levels of mdtE and mdtF (Zhang et al., 2011). However, whether other regulators also participate in the regulation of mdtEF under this condition is unknown. Thus, in the present study, we first examined whether previously identified regulators, such as EvgA, YdeO, MnmE, GadE, GadX, and H-NS, also participate in the anaerobic activation of mdtEF. Since our previous finding on the anaerobic activation of mdtEF occurred in M9 glucose minimal medium in which the effect of CRP on gene expression is minimal, we focused on the effect of other regulators in the current study.
We constructed strains containing deletion of each of these genes and measured β-galactosidase activities of PgadE-lacZ in these strains. As shown in Figure 2A, deletion of arcA caused dramatic decrease of the β-galactosidase activity of PgadE-lacZ, confirming the role of ArcA in activating the gadE-mdtEF expression under anaerobic condition. Interestingly, except for mnmE, deletion of the regulators previously shown to participate in the regulation of mdtEF under aerobic condition, such as EvgA, YdeO, GadE, and GadX, caused no or only a moderate decrease (17–40%) of the transcription of PgadE-lacZ, and deletion of H-NS caused a moderate increase of PgadE-lacZ activity, suggesting that these regulators are not responsible for the activation of mdtEF under anaerobic condition. Interestingly, deletion of the GTPase MnmE caused a significant decrease of the transcription of gadE-mdtEF, suggesting that MnmE may also contribute to the activation of gadE-mdtEF under this condition. Since the current study focuses on the regulation of the multidrug efflux genes mdtEF present in the same operon, we next examined whether deletion of these genes caused alteration of MdtEF protein levels. We constructed chromosomal mdtE-flag in these strains (except for ΔgadE) and performed Western blot to examine the level of MdtE-FLAG. As shown in Figure 2B, production of MdtE-FLAG in the corresponding deletion mutants showed a similar pattern as that of β-galactosidase activity assay except ΔmnmE, which caused a more significant reduction of MdtE expression at translational level. Together these results confirmed that ArcA and MnmE are primarily responsible for the anaerobic activation of mdtEF.
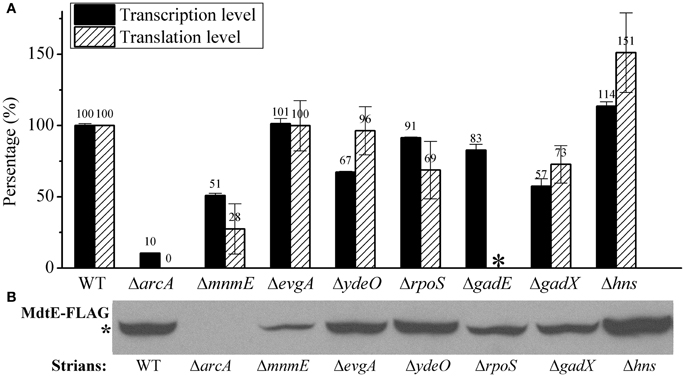
Figure 2. MdtEF expression in various mutants at both transcription and translation level under anaerobic condition. (A) Transcription of gadE-mdtEF was determined by β-galactosidase activity of PgadE-lacZ, and translation of the gene is determined by Western blot to detect the production of MdtE-FLAG. (B) Production of chromosomal MdtE-FLAG in various mutants. Cells were grown anaerobically to exponential phase (A600 = 0.3) in minimal M9 glucose medium. All results were presented as percentages relative to the transcription of PgadE-lacZ or production of MdtE-FLAG in wild type strain. Error bars represent the standard errors of triplicate experiments (n = 3). *mdtE-flag in ΔgadE mutant was not constructed because of the close proximity of gadE and mdtEF which is not applicable by the P1 phage transduction.
As a control, we also measured the effect of deletion of these regulators on the aerobic expression of gadE-mdtEF in E. coli grown in M9 glucose medium at log phase. As shown in Figure 3, PgadE-lacZ activities in wild type strain, and in ΔarcA, ΔmnmE, ΔevgA, ΔydeO, ΔgadE, ΔgadX mutants are very low (around 20 Miller units), consistent with the fact that ArcA is not active under this condition. However, in Δhns mutants, the PgadE-lacZ activities increased more than 10-fold to about 250 Miller units, suggesting H-NS strongly represses the expression of gadE-mdtEF under aerobic condition in the M9 glucose minimal medium. This result is consistent with previous findings that H-NS represses the transcription from PgadE through its interaction with various sites in the gadE promoter region (Sayed and Foster, 2009). It is noted that deletion of evgA, ydeO, gadE, and gadX did not show obvious effect on the transcription of PgadE-lacZ in this assay. This is not unexpected as previous studies on the regulation of gadE-mdtEF by these regulators was found to be effective in the acidified minimal medium (pH 5.5) containing glucose or during stationary phase in rich medium (Ma et al., 2004), whereas the current study is carried out in the neutral M9 minimal medium with glucose, the identical culture medium as in our previous assay which showed increased expression of MdtEF under anaerobic condition. These results together suggested that the regulatory network of mdtEF expression under anaerobic condition is different from that under aerobic condition, and anaerobic activation of mdtEF is largely dependent on ArcA and MnmE.
Figure 3. β-galactosidase activity of PgadE-lacZ fusion in WT (MG1655 ΔlacZ), ΔarcA, ΔmnmE, ΔevgA, ΔydeO, ΔgadE, ΔgadX, and Δhns mutant strains under aerobic condition.
Deletion of EvgA, YdeO, H-NS Does not Have Obvious Effect on the Transcription from Individual Promoters Under Anaerobic Condition
Our investigation above showed that ArcA and MnmE are the major activators responsible for the expression of gadE-mdtEF under anaerobic condition, whereas other regulators tested did not show obvious regulatory effects. However, since four promoter regions (P, P1, P2/P4, and P3) are involved in the transcription initiation of gadE-mdtEF, we cannot rule out the possibility that they might participate in the regulation of individual promoters contained in the complete 798 bp promoter of gadE. For example, it is possible that a certain regulator activates the transcription from one promoter region while represses transcription from another promoter, consequently cancels its effect on the transcription from the full promoter in our assay. To verify this, we examined the anaerobic transcription activity of those PgadE truncations in the deletion mutants constructed in this study except for GadX and GadW since they were indicated to be active in low pH, complex medium or stationary phase (Ma et al., 2003). It was shown that deletion of evgA, ydeO, and hns did not cause significant changes on the transcription activity from each of the individual promoters (data not shown), indicating that these regulators had no effect on the transcript from the individual promoters contained in the 798 bp full promoter of gadE either under anaerobic condition.
GadE Autoregulates Transcription from P1 Region Under Anaerobic Condition
Interestingly, deletion of gadE caused different effects on the transcription from different promoters contained in the full 798 bp PgadE region, which is dissimilar from that observed in the case of ΔevgA, ΔydeO, and Δhns. As shown in Figure 4 (stripe bars), while gadE deletion had no significant effect on the transcription from the lacZ fusions of the full length, P3, P3+P2/P4, as well as P3+P2/P4+P1, deletion of gadE caused almost complete abolishment of transcription from the P2/P4+P1 and the P1 promoters. This observation combined with the results above which showed that individual P2/P4 was not active under anaerobic conditions suggested that GadE autoactivates the transcription of gadE-mdtEF from its P1 promoter. This result is consistent with a previous truncation study performed under aerobic condition, where deletion of gadE caused decrease of transcription from P2/P4+P1 and P1 promoters (Sayed and Foster, 2009), and the presence of a conserved GAD box to which GadE binds in the P1 region (Ma et al., 2004). Moreover, the fact that deletion of acrA and mnmE caused decrease of transcription from P1 at variable degrees whereas deletion of gadE almost completely abolished transcription from this promoter suggested that activation of P1 is dependent on the production of GadE which is transcribed from the ArcA and MnmE activated other promoters, such as P3, in the case of the full length, native promoter in the cell.
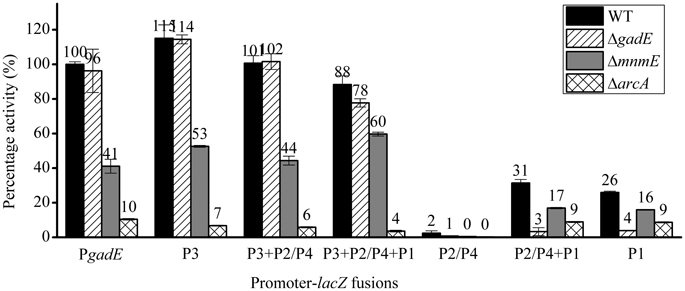
Figure 4. β-galactosidase activity of PgadE truncations in WT (black bars), ΔgadE (stripe bars), ΔmnmE (gray bars) and ΔarcA (grid bars) strains under anaerobic growth condition.
ArcA Directly Binds to Multiple Sites at the gadE Promoter to Activate the Expression of the gadE-mdtEF
Since ArcA and MnmE were identified as transcription activators of the gadE-mdtEF operon, we next asked how these two regulators activate gadE expression, i.e., through which promoters these two regulators affect the transcription of the gadE-mdtEF operon under anaerobic conditions. To address this, we measured the transcription of promoter-lacZ fusions containing various lengths of the PgadE region in ΔarcA and ΔmnmE strains, respectively (Figure 4). Surprisingly, deletion of arcA (grid bars) or mnmE (gray bars) caused decrease of transcription from the full length and all the truncation mutants with variable promoter lengths, indicating ArcA and MnmE might affect transcription from multiple promoters, especially the P3 and P1 regions, since P2/P4 and P individual promoters were not active under this condition as shown above. Considering that the activation of the P1 promoter is dependent on GadE, it is possible that ArcA and MnmE primarily activate the P3 promoter.
To examine whether the ArcA dependent activation of PgadE is through its direct binding on the gadE promoter region, we performed EMSA to measure the direct binding of ArcA-P (phosphorylated His6-ArcA) to the P1 and P3 promoters. As shown in Figure 5, retarded bands corresponding to the DNA-protein complexes of ArcA-P with both P3 and P1 were observed, suggesting AcrA-P binds to multiple sites at the gadE promoter encompassed in the individual P1 and P3 promoters and activates the expression of gadE under anaerobic condition. Genome wide CHIP-chip (Chromatin immunoprecipitation with microarray) and CHIP-seq study which determines all the ArcA binding sites across the E. coli K-12 MG1655 genome under anaerobic conditions in glucose minimum media also identified two broad ArcA binding peaks in the gadE promoter. Furthermore, the presence of the ArcA peaks correlated with the presence of a strong σ70 peak comparing with the very low σ70 peak on this region under aerobic condition (Park and Kiley, et al., unpublished data, pers. communication), further confirming the ArcA dependent activation of the gadE-mdtEF operon through its direct binding of multiple sites on the gadE promoter.
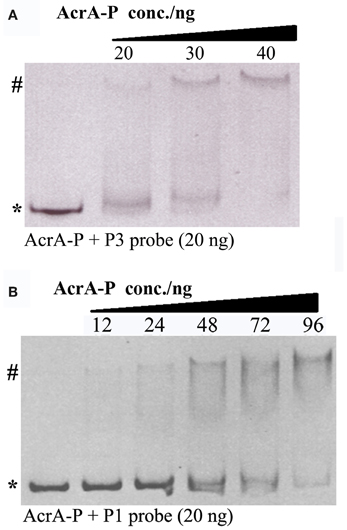
Figure 5. Binding of ArcA-P to P1 and P3 promoters using EB stained EMSA assay. Various concentrations of AcrA-P were incubated with P3 (A) or P1 (B). AcrA-P refers to phosphorylated His6-ArcA protein. Asterisk indicates free DNA probe and pound indicates protein–DNA complex.
ArcA Antagonizes the H-NS Mediated Repression of gadE-mdtEF During the Transition from Aerobic to Anaerobic Growth
Our results above showed that the expression of gadE-mdtEF is largely repressed by the DNA silencer H-NS under aerobic conditions in M9 glucose medium, whereas under anaerobic condition in the same medium its expression is activated primarily in an ArcA dependent manner. This led us to ask how the H-NS mediated repression is replaced by the ArcA dependent activation during the transition of the facultative bacterium E. coli from its aerobic to anaerobic growth. To answer this question, we first examined which individual promoter region is responsible for the H-NS mediated repression under aerobic condition. As shown in Figure 6A, in addition to the full length promoter PgadE, deletion of hns caused de-repression of transcription from the P1 and P3 promoters, but no effect on the transcription from the P2/P4 and P promoters, indicating that H-NS represses the transcription of PgadE from both the P1 and P3 promoters under aerobic condition in the M9 glucose medium. This pattern is similar to that of how ArcA achieves its activation of PgadE under anaerobic condition which is also through its interaction with the P3 and P1 promoters.
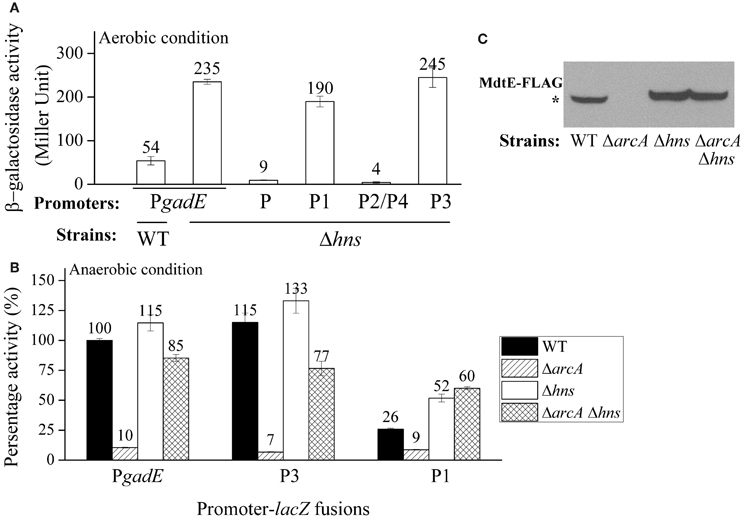
Figure 6. ArcA antagonizes the H-NS mediated repression of gadE under anaerobic growth. (A) Promoter activity of PgadE and its truncations under aerobic condition determined by β-galactosidase activity of corresponding lacZ fusions in WT and Δhns strains; (B) Promoter activity of PgadE and its truncations under anaerobic condition determined by β-galactosidase activity of corresponding lacZ fusions in WT, ΔarcA, Δhns, and ΔarcA Δhns strains; (C) Protein level of MdtE-FLAG in WT, ΔarcA, Δhns and ΔarcA Δhns strains determined by Western blot. Cells used for all assays were grown anaerobically in M9 minimal glucose medium to exponential phase (A600 = 0.3). Results of β-galactosidase activity assay are presented in Miller unit or percentage activity to that of wild type strain, error bars represent the standard errors of triplicate experiments (n = 3). * indicates the band of MdtE-FLAG.
Two possibilities may explain the transition from H-NS mediated repression to ArcA mediated activation of gadE-mdtEF during the transition from aerobic to anaerobic growth. One possibility is that H-NS is inactive under anaerobic conditions and it disassembles from the gadE promoter, allowing ArcA which is active under this condition to bind the promoter of gadE and initiate gene transcription. Alternatively, under anaerobic conditions H-NS can still bind to the gadE promoter, however, its binding is antagonized by the activated ArcA, resulting in the recruitment of RNAP and transcript initiation of the operon under this condition. To differentiate these two possibilities, we constructed ΔarcA Δhns double deletion and examined transcription of PgadE-lacZ in this strain. If H-NS is inactive under anaerobic condition and is disassembled from the gadE promoter, then transcription of PgadE-lacZ in ΔarcA Δhns should be similar to that of ΔarcA alone. However, if H-NS was not inactivated, rather, it can still bind to the promoter but its binding was antagonized by ArcA, then its binding to the gadE promoter should be resumed when ArcA is absent and an increase of PgadE-lacZ activity should be observed in Δhns ΔarcA than ΔarcA single deletion strain. As shown in Figure 6B, transcription of PgadE-lacZ in ΔarcA Δhns double deletion did display significantly higher activity than that in ΔarcA strain, suggesting that the anaerobically up-regulated gadE-mdtEF expression was through the antagonization of H-NS mediated repression by ArcA. Western blot analysis of chromosomal MdtE-FLAG showed the same pattern as that of transcription assay (Figure 6C). More interestingly, the increased transcription of lacZ fusions in ΔarcA Δhns double deletion strain than that in ΔarcA single deletion strain was also observed in the case of individual P3 and P1 promoters, suggesting that ArcA antagonization of H-NS takes place at multiple sites in the PgadE promoter, consistent with the notion that both H-NS and ArcA can oligomerize and bind to a broad region in its regulated gene promoters (Green and Paget, 2004; Dorman, 2006). CHIP-chip data of H-NS and ArcA are also consistent with this proposal, as characterized by a broad peak of H-NS binding to PgadE under aerobic conditions and the replacement of this broad peak by that of ArcA under anaerobic conditions (Park and Kiley et al., unpublished data, pers. communication).
The Effect of MnmE on the Anaerobic Activation of gadE-mdtEF is not Through ArcA
As shown in Figure 4, ΔmnmE caused a very similar pattern on the transcription activities of the various PgadE truncations as that caused by ΔarcA, only to a less extent. This, combined with the fact that MnmE itself does not have DNA binding property whereas ArcA was shown to directly bind to PgadE and activate its expression, led us to speculate that the effect of ΔmnmE on gadE-mdtEF expression may be indirect, i.e., MnmE may affect the translation and/or activity of ArcA (such as through affecting its phosphorylation). To address this, we tested the effect of ΔmnmE on the transcription of two ArcA dependent promoters, PlpdA and the mutated PicdA. Transcription of lpdA gene has been shown to be mediated by the repression of ArcA under anaerobic conditions. In the case of icdA, it contains two known promoters, PI and PII, in which PI is the primary promoter and is repressed only by ArcA. In the mutated PicdA-lacZ, transcription from PII was eliminated by removal of its −10 element such that transcription repression of this PicdA-lacZ is exclusively ArcA dependent (Park and Kiley et al., unpublished work, pers. communication). If MnmE affects either the production or phosphorylation of ArcA (which affects the regulatory activity of ArcA), ΔmnmE should cause full or partial de-repression of these two promoters under anaerobic condition. As shown in Figure 7, as expected, transcription of PicdA-lacZ and PlpdA-lacZ fusions were significantly repressed by ArcA under anaerobic condition, since ΔarcA caused de-repression of their transcription under anaerobic condition in M9 glucose medium, the same condition used in this study. However, ΔmnmE had little or no effect on the transcription of these two promoters, suggesting that MnmE does not affect the production or phosphorylation of ArcA in vivo. Therefore, the observed MnmE mediated anaerobic activation of gadE-mdtEF was not due to its potential effects on ArcA.
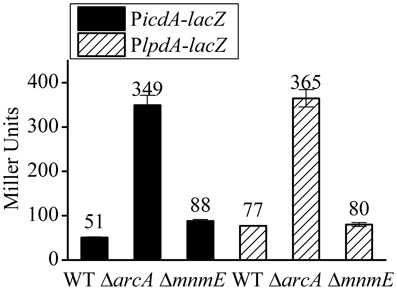
Figure 7. MnmE has no effect on the transcription of PicdA-lacZ and PlpdA-lacZ, two ArcA dependent promoters. β-galactosidase activities of chromosomal PicdA-lacZ (black bars) and PlpdA-lacZ (stripe bars) fusions were measured from cells grown anaerobically to exponential phase (A600 = 0.3) in minimal M9 glucose medium. Results are presented in Miller unit and error bars represent the standard errors of triplicate experiments (n = 3).
Discussion
Different Regulatory Circuits on the Expression of the gadE-mdtEF Operon Under Different Physiological Conditions
Owing to the important roles of GadE and MdtEF in acid resistance and drug efflux respectively in E. coli, transcriptional regulation of the gadE-mdtEF operon under various of growth phases and conditions has been extensively investigated. While previous studies largely focused on its relevance in bacterial acid resistance under aerobic growth environment, the current study aimed to dissect the up-regulation of the operon under the physiologically relevant environment of anaerobic growth and its effect on the production of the multidrug efflux protein MdtEF.
Several well-characterized regulatory circuits that activate gadE expression in the context of acid resistance have been disclosed. These include the EvgA-YdeO-GadE feed forward regulatory loop which is effective in minimal glucose medium under aerobic condition (Ma et al., 2004), the conditional activation of the operon in a MnmE dependent manner in LB containing glucose (LBG) (Gong et al., 2004), as well as the GadX or GadW dependent activation which functions primarily in rich media in stationary growth phase (Sayed et al., 2007). Here we show that the anaerobic activation of the operon, particularly the expression of the multidrug efflux protein MdtEF, is also dependent on the 798 bp gadE promoter and the activation is primarily dependent on the anaerobic global regulator ArcA, adding yet another regulatory circuit to the existing complex regulatory loops of the operon. These results reinforced the important role of the promoter region of gadE in integrating various of environmental and physiological signals to facilitate the adaptation of E. coli.
We also measured the transcription of PgadE-lacZ under anaerobic condition in stationary phase and examined whether the previously identified regulators, such as RpoS and GadE, participate in the regulation of the gadE-mdtEF expression under this growth phase anaerobically. Surprisingly, while a slightly higher transcription of PgadE-lacZ in stationary phase than in log phase (~700 Miller units vs. ~480 Miller units in log phase) was observed, deletion of rpoS or gadE did not have any effect on the transcription of PgadE-lacZ in this growth phase (data not shown), whereas ΔarcA still caused significant decrease of the gadE-mdtEF expression in the same growth phase. These results suggested that anaerobic expression of the gadE-mdtEF operon is primarily regulated by ArcA in both log phase and stationary phase under anaerobic condition. It is noted that previously a putative ArcA binding site located at the P2 region was proposed (Zhang et al., 2011), whereas in the current study, the P3 and P1 region of the gadE promoter was shown to be bound by ArcA and they play an important role in the anaerobic activation of the operon. This inconsistency is likely due to the inaccuracy of the bioinformatic search which utilized a sequence motif of “(5′ [A/T]GTTAATTA[A/T] 3′)” as the template and the motif was proposed as the ArcA binding site by Lynch and Lin (1996) following the DNA foot-printing analysis of a limited number of ArcA regulated promoters. Indeed, a genome-wide search of all the ArcA regulated promoters and subsequent characterization of them suggest a motif (Kiley et al., unpublished work, pers. communication) that deviates from the previously proposed ArcA binding site.
Interestingly, we demonstrated that the ArcA dependent activation of the operon is through its antagonistic effect on the H-NS mediated repression of the operon. This regulatory model explains the dynamic process taking place during the transition of E. coli from its aerobic to anaerobic growth, which mimics the transition of the bacterium from free living status to the anaerobic environment of the human host. H-NS mediated repression of the operon has also been implicated in previous studies (Ma et al., 2004; Sayed and Foster, 2009). However, the inter-relationship of this repression and gene activation mediated by other regulators has not been revealed. Our findings demonstrating that ArcA activates the expression of the operon through antagonizing the H-NS mediated repression probably provide a common model that underlies the dynamic and coordinated regulation of gadE-mdtEF in response to different environmental signals. Perhaps under unstressed conditions when the expression of the operon is unnecessary, the promoter of gadE is primarily occupied by H-NS and transcription from the various promoters in this region is repressed. Upon the activation of different transcription activators in response to different environmental or physiological conditions that presage acid stresses or efflux of certain metabolic by-products, the activators then displace H-NS binding on the promoter, resulting in the activation of the operon. Several lines of evidence support this proposal: first, bioinformatics have suggested the presence of an efficient DNA helix locking mechanism by H-NS in the promoter of gadE (Hommais et al., 2004); Second, H-NS mediated repression has been found to be common among acquired gene clusters which is consistent with the functions of these gene clusters in conferring the survival and adaptation of bacteria in various adverse growth environments (Oshima et al., 2006). The acid resistant island including the gadE-mdtEF operon indeed is one of the acquired gene clusters in E. coli with different GC content from the rest of E. coli genome; Third, the interplay between the H-NS mediated repression and ArcA mediated antagonizing of the repression is also observed in the case of another gene cydAB which is also involved in adapting E. coli to the microaerobic and anaerobic growth (Govantes et al., 2000). Thus, anaerobic activation of gadE-mdtEF through antagonizing the H-NS mediated repression by ArcA provides a regulatory model that is relevant to the dynamic lifestyle of the facultative bacterium E. coli. A model to explain this dynamic regulation is presented in Figure 8.
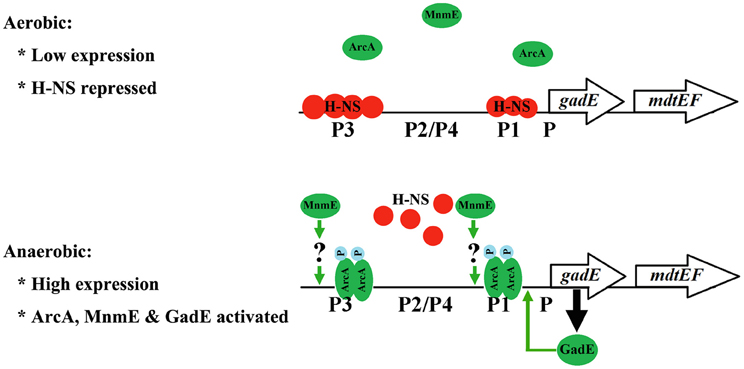
Figure 8. A regulatory model for the transcriptional regulation of gadE-mdtEF operon during the transition from aerobic to anaerobic growth in minimal M9 glucose medium. During aerobic growth, the gadE-mdtEF is expressed at a very low level due to the strong repression by H-NS. Upon transition to anaerobiosis, the response regulator ArcA is phosphorylated and activated by its sensor kinase ArcB. Phosphorylated ArcA (ArcA-P) antagonizes H-NS binding to gadE promoter, resulting in the relief of repression. ArcA-P also directly binds to the gadE promoter and activates the expression of gadE-mdtEF. ArcA-P activated gadE expression produces GadE, which activates the transcription from the P1 promoter and further up-regulate the gadE-mdtEF expression. MnmE also participates in the anaerobic activation of gadE-mdtEF, but in a yet uncharacterized manner.
Roles of the gadE-mdtEF Operon in Both Acid Resistance and Drug Efflux
It is interesting that the drug efflux genes mdtEF exist in the same operon with the central acid resistance regulator gadE, and their expression is driven by the same unusually long 798 bp promoter region upstream of the gadE gene. While functions of GadE in controlling the expression of the major acid resistant system GadA and GadBC is well established, the relevance of MdtEF to acid resistance has not been fully recognized. Our previous studies showed that MdtEF functions to expel indole nitrosative by-products accumulated during nitrate respiration under anaerobic condition (Zhang et al., 2011). It is known that anaerobic metabolism often causes pH reduction in growth medium, especially during glucose fermentation by E. coli which leads to the production of various organic acids. Although anaerobic induction of gadE-mdtEF occurs at a fairly neutral pH (6.9 at the log phase culture when the cells were harvested for transcription and Western blot analyses), significant accumulation of the indole nitrosative by-product occurred when pH dropped to 5.8 or below (Weiss, 2006). Thus, it seems that MdtEF does contribute to the detoxification of acid induced metabolic by-products. Yet, its activation does not necessarily depend on the occurrence of acid stresses. This is consistent with the findings from previous studies which propose that the gadE-mdtEF operon is rigged to be induced under many different circumstances that could presage an encounter with acid stresses (Hommais et al., 2004).
This raises the interesting question of whether the housekeeping pump AcrAB is also involved in the nitrosative and/or acid resistance by expelling metabolic by-products under anaerobic conditions. Although the broad substrate specificity of AcrAB-TolC was established in terms of its capability of exporting antibiotics, we speculate that the same degree of broad substrate specificity may not apply in terms of its capability of exporting physiological substances, since it has been well recognized that expression of certain efflux pump genes other than the housekeeping AcrAB pump is activated by specific environmental or physiological conditions (Piddock, 2006; Li and Nikaido, 2009; Poole, 2012b). We recently performed metabolic profiling of the WT vs. ΔmdtEF under the growth condition of anaerobic nitrate respiration and it was shown that ΔmdtEF cells accumulated more indole nitrosative by-products than the WT (data not shown) in the presence of the acrAB genes, suggesting that the MdtEF pump exhibits at least certain degree of substrate specificity even in the presence of the housekeeping pump AcrAB. It is highly likely that MdtEF also expels other metabolic by-products accumulated during acid stresses. If that is the case, there are probably more extensive relevance between MdtEF mediated drug efflux and the physiology of E. coli during acid resistance. This relevance probably can be disclosed through metabolic profiling by comparing the total metabolites in ΔmdtEF and WT strain grown during acid stresses.
The Unresolved Role of MnmE in the Up-Regulation of mdtEF Expression
MnmE is a GTP binding protein that is involved in tRNA modification (Cabedo et al., 1999). This protein was found to be in cytoplasm but can partially associate with the inner membrane (Cabedo et al., 1999). Its involvement in the conditional activation of gadE at exponential growth in LB medium with glucose (LBG) or stationary growth of E. coli in unbuffered LBG under aerobic condition has been reported (Gong et al., 2004). However, the detailed regulatory mechanism remains elusive. Since MnmE itself does not have DNA binding property, it was speculated that MnmE affects the gadE expression by controlling an unknown regulator which can bind to gadE promoter and mediate its transcription. Since ΔmnmE resulted in a similar pattern of decreased transcription from both full length and various truncation promoter fusions as in ΔarcA strain except to a less extent, and ArcA was shown to directly bind to PgadE, we had speculated that MnmE regulation of the gadE expression was through its effect on ArcA, i.e., through its mediation of the translation and/or phosphorylation of ArcA. However, our β-galactosidase activity results showing that ΔmnmE does not have any effect on two different ArcA dependent promoters (PicdA and PlpdA) suggested that MnmE does not affect the production and/or activity of ArcA under anaerobic conditions, and thus the observed reduction of gadE-mdtEF expression in ΔmnmE strain was not due to its potential effect on ArcA.
Studies also indicated that MnmE dependent conditional expression of gadE mainly depends on the P2 promoter region (Sayed and Foster, 2009). However, our study showed that the P2 region is not active under anaerobic condition at log growth phase; instead, the MnmE dependent gadE activation was likely through its effect on the P3 region, which is indispensable in the anaerobic activation of gadE. Furthermore, we found that MnmE dependent activation of the gadE-mdtEF expression also occurred in non-glucose media, such as LB and glycerol supplemented with nitrate (data not shown). This observation is not in agreement with a previous finding which suggested that MnmE dependent gadE expression was induced only in the presence of glucose. Thus, although several studies support the notion that the GTPase MnmE participates in the transcriptional activation of the gadE-mdtEF operon under certain growth conditions and phases, how exactly MnmE is involved in the process is still ambiguous. This is largely due to the fact that the exact biological function of MnmE itself is still elusive. Obviously, further studies of MnmE protein is needed before we can reveal its role in the up-regulation of gadE-mdtEF under the physiologically relevant conditions.
It is noteworthy that the 798 bp sensory integration region of the gadE-mdtEF promoter is highly conserved among the pathogenic E. coli strains, including pathogenic strains O157:H7, 2369, RS128 and CFT073 and Shigella (96–99% identity), and GadE has been demonstrated to be indispensable for the survival of E. coli O157::H7 in a simulated gastric environment (Kailasan Vanaja et al., 2009), underlying the importance of the gadE-mdtEF operon to the physiology of E. coli. Since both low pH, such as that of gastric acid, and oxygen limitation (anaerobic environment) activates the expression of the gadE-mdtEF operon, it is conceivable that synergetic activation of the system by both regulatory circuits may occur during the transition of E. coli to human host, particularly the passage of the gastrointestinal tract, thus greatly facilitate the adaptation and survival of this neutrophilic bacterium in its host niches.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful for Dr. Patricia J. Kiley (Department of Biomolecular Chemistry, University of Wisconsin-Madison) and Mr. Dan Park in the Kiley lab to share the unpublished data and the strains of PicdA-lacZ and PlpdA-lacZ. We appreciate Dr. Kunihiko Nishino (Institute of Scientific and Industrial Research, Osaka University) for the pNN387 vector and the PgadE-lacZ strain. This work was supported by the Hong Kong Research Fund for the Control of Infectious Diseases 11100982 (to Aixin Yan).
References
Allen, H. K., Donato, J., Wang, H. H., Cloud-Hansen, K. A., Davies, J., and Handelsman, J. (2010). Call of the wild: antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 8, 251–259. doi: 10.1038/nrmicro2312
Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., et al. (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2. doi: 10.1038/msb4100050
Bekker, M., Alexeeva, S., Laan, W., Sawers, G., Teixeira De Mattos, J., and Hellingwerf, K. (2010). The ArcBA two-component system of Escherichia coli is regulated by the redox state of both the ubiquinone and the menaquinone pool. J. Bacteriol. 192, 746–754. doi: 10.1128/JB.01156-09
Cabedo, H., Macián, F., Villarroya, M., Escudero, J. C., Martínez-Vicente, M., Knecht, E., et al. (1999). The Escherichia coli trmE (mnmE) gene, involved in tRNA modification, codes for an evolutionarily conserved GTPase with unusual biochemical properties. EMBO J. 18, 7063–7076. doi: 10.1093/emboj/18.24.7063
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Dorman, C. J. (2006). H-NS, the genome sentinel. Nat. Rev. Microbiol. 5, 157–161. doi: 10.1038/nrmicro1598
Foster, J. W. (2004). Escherichia coli acid resistance: tales of an amateur acidophile. Nat. Rev. Microbiol. 2, 898–907. doi: 10.1038/nrmicro1021
Gong, S., Ma, Z., and Foster, J. W. (2004). The Era-like GTPase TrmE conditionally activates gadE and glutamate-dependent acid resistance in Escherichia coli. Mol. Microbiol. 54, 948–961. doi: 10.1111/j.1365-2958.2004.04312.x
Govantes, F., Orjalo, A. V., and Gunsalus, R. P. (2000). Interplay between three global regulatory proteins mediates oxygen regulation of the Escherichia coli cytochrome d oxidase (cydAB) operon. Mol. Microbiol. 38, 1061–1073. doi: 10.1046/j.1365-2958.2000.02215.x
Green, J., and Paget, M. S. (2004). Bacterial redox sensors. Nat. Rev. Microbiol. 2, 954–966. doi: 10.1038/nrmicro1022
Hayes, E. T., Wilks, J. C., Sanfilippo, P., Yohannes, E., Tate, D. P., Jones, B. D., et al. (2006). Oxygen limitation modulates pH regulation of catabolism and hydrogenases, multidrug transporters, and envelope composition in Escherichia coli K-12. BMC Microbiol. 6:89. doi: 10.1186/1471-2180-6-89
Hirakawa, H., Inazumi, Y., Masaki, T., Hirata, T., and Yamaguchi, A. (2005). Indole induces the expression of multidrug exporter genes in Escherichia coli. Mol. Microbiol. 55, 1113–1126. doi: 10.1111/j.1365-2958.2004.04449.x
Hirakawa, H., Inazumi, Y., Senda, Y., Kobayashi, A., Hirata, T., Nishino, K., et al. (2006). N-acetyl-d-glucosamine induces the expression of multidrug exporter genes, mdtEF, via catabolite activation in Escherichia coli. J. Bacteriol. 188, 5851–5858. doi: 10.1128/JB.00301-06
Hommais, F., Krin, E., Coppée, J.-Y., Lacroix, C., Yeramian, E., Danchin, A., et al. (2004). GadE (YhiE): a novel activator involved in the response to acid environment in Escherichia coli. Microbiology 150, 61–72. doi: 10.1099/mic.0.26659-0
Itou, J., Eguchi, Y., and Utsumi, R. (2009). Molecular mechanism of transcriptional cascade initiated by the EvgS/EvgA system in Escherichia coli K-12. Biosci. Biotechnol. Biochem. 73, 870–878. doi: 10.1271/bbb.80795
Kailasan Vanaja, S., Bergholz, T. M., and Whittam, T. S. (2009). Characterization of the Escherichia coli O157:H7 Sakai GadE regulon. J. Bacteriol. 191, 1868–1877. doi: 10.1128/JB.01481-08
Kang, Y., Weber, K. D., Qiu, Y., Kiley, P. J., and Blattner, F. R. (2005). Genome-wide expression analysis indicates that FNR of Escherichia coli K-12 regulates a large number of genes of unknown function. J. Bacteriol. 187, 1135–1160. doi: 10.1128/JB.187.3.1135-1160.2005
Keseler, I. M., Collado-Vides, J., Santos-Zavaleta, A., Peralta-Gil, M., Gama-Castro, S., Muñiz-Rascado, L., et al. (2011). EcoCyc: a comprehensive database of Escherichia coli biology. Nucleic Acids Res. 39, D583–D590.
Kobayashi, A., Hirakawa, H., Hirata, T., Nishino, K., and Yamaguchi, A. (2006). Growth phase-dependent expression of drug exporters in Escherichia coli and its contribution to drug tolerance. J. Bacteriol. 188, 5693–5703. doi: 10.1128/JB.00217-06
Lennen, R. M., Politz, M. G., Kruziki, M. A., and Pfleger, B. F. (2013). Identification of transport proteins involved in free fatty acid efflux in Escherichia coli. J. Bacteriol. 195, 135–144. doi: 10.1128/JB.01477-12
Li, X. Z., and Nikaido, H. (2009). Efflux-mediated drug resistance in bacteria: an update. Drugs 69, 1555–1623.
Lynch, A. S., and Lin, E. C. (1996). Transcriptional control mediated by the ArcA two-component response regulator protein of Escherichia coli: characterization of DNA binding at target promoters. J. Bacteriol. 178, 6238–6249.
Ma, Z., Masuda, N., and Foster, J. W. (2004). Characterization of EvgAS-YdeO-GadE branched regulatory circuit governing glutamate-dependent acid resistance in Escherichia coli. J. Bacteriol. 186, 7378–7389. doi: 10.1128/JB.186.21.7378-7389.2004
Ma, Z., Richard, H., and Foster, J. W. (2003). pH-Dependent modulation of cyclic AMP levels and GadW-dependent repression of RpoS affect synthesis of the GadX regulator and Escherichia coli acid resistance. J. Bacteriol. 185, 6852–6859. doi: 10.1128/JB.185.23.6852-6859.2003
Masuda, N., and Church, G. M. (2003). Regulatory network of acid resistance genes in Escherichia coli. Mol. Microbiol. 48, 699–712. doi: 10.1046/j.1365-2958.2003.03477.x
Mettert, E. L., and Kiley, P. J. (2007). Contributions of [4Fe-4S]-FNR and integration host factor to fnr transcriptional regulation. J. Bacteriol. 189, 3036–3043. doi: 10.1128/JB.00052-07
Miller, J. H. (1972). Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Murakami, S., Nakashima, R., Yamashita, E., Matsumoto, T., and Yamaguchi, A. (2006). Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 443, 173–179. doi: 10.1038/nature05076
Nishino, K., Senda, Y., Hayashi-Nishino, M., and Yamaguchi, A. (2009). Role of the AraC-XylS family regulator YdeO in multi-drug resistance of Escherichia coli. J. Antibiot. (Tokyo) 62, 251–257. doi: 10.1038/ja.2009.23
Nishino, K., Senda, Y., and Yamaguchi, A. (2008a). The AraC-family regulator GadX enhances multidrug resistance in Escherichia coli by activating expression of mdtEF multidrug efflux genes. J. Infect. Chemother. 14, 23–29. doi: 10.1007/s10156-007-0575-y
Nishino, K., Senda, Y., and Yamaguchi, A. (2008b). CRP regulator modulates multidrug resistance of Escherichia coli by repressing the mdtEF multidrug efflux genes. J. Antibiot. (Tokyo) 61, 120–127. doi: 10.1038/ja.2008.120
Nishino, K., Yamada, J., Hirakawa, H., Hirata, T., and Yamaguchi, A. (2003). Roles of TolC-dependent multidrug transporters of Escherichia coli in resistance to beta-lactams. Antimicrob. Agents Chemother. 47, 3030–3033. doi: 10.1128/AAC.47.9.3030-3033.2003
Nishino, K., and Yamaguchi, A. (2001). Analysis of a complete library of putative drug transporter genes in Escherichia coli. J. Bacteriol. 183, 5803–5812. doi: 10.1128/JB.183.20.5803-5812.2001
Nishino, K., and Yamaguchi, A. (2002). EvgA of the two-component signal transduction system modulates production of the YhiUV multidrug transporter in Escherichia coli. J. Bacteriol. 184, 2319–2323. doi: 10.1128/JB.184.8.2319-2323.2002
Nishino, K., and Yamaguchi, A. (2004). Role of histone-like protein H-NS in multidrug resistance of Escherichia coli. J. Bacteriol. 186, 1423–1429. doi: 10.1128/JB.186.5.1423-1429.2004
Nishino, K., Yamasaki, S., Hayashi-Nishino, M., and Yamaguchi, A. (2011). Effect of overexpression of small non-coding DsrA RNA on multidrug efflux in Escherichia coli. J. Antimicrob. Chemother. 66, 291–296. doi: 10.1093/jac/dkq420
Oshima, T., Ishikawa, S., Kurokawa, K., Aiba, H., and Ogasawara, N. (2006). Escherichia coli histone-like protein H-NS preferentially binds to horizontally acquired DNA in association with RNA polymerase. DNA Res. 13, 141–153. doi: 10.1093/dnares/dsl009
Piddock, L. J. V. (2006). Multidrug-resistance efflux pumps - not just for resistance. Nat. Rev. Microbiol. 4, 629–636. doi: 10.1038/nrmicro1464
Poole, K. (2005). Efflux-mediated antimicrobial resistance. J. Antimicrob. Chemother. 56, 20–51. doi: 10.1093/jac/dki171
Poole, K. (2012a). Bacterial stress responses as determinants of antimicrobial resistance. J. Antimicrob. Chemother. 67, 2069–2089. doi: 10.1093/jac/dks196
Poole, K. (2012b). Stress responses as determinants of antimicrobial resistance in Gram-negative bacteria. Trends Microbiol. 20, 227–234. doi: 10.1016/j.tim.2012.02.004
Sayed, A. K., and Foster, J. W. (2009). A 750 bp sensory integration region directs global control of the Escherichia coli GadE acid resistance regulator. Mol. Microbiol. 71, 1435–1450. doi: 10.1111/j.1365-2958.2009.06614.x
Sayed, A. K., Odom, C., and Foster, J. W. (2007). The Escherichia coli AraC-family regulators GadX and GadW activate gadE, the central activator of glutamate-dependent acid resistance. Microbiology 153, 2584–2592. doi: 10.1099/mic.0.2007/007005-0
Tjaden, B., Saxena, R. M., Stolyar, S., Haynor, D. R., Kolker, E., and Rosenow, C. (2002). Transcriptome analysis of Escherichia coli using high-density oligonucleotide probe arrays. Nucleic Acids Res. 30, 3732–3738. doi: 10.1093/nar/gkf505
Tramonti, A., De Canio, M., and De Biase, D. (2008). GadX/GadW-dependent regulation of the Escherichia coli acid fitness island: transcriptional control at the gadY–gadW divergent promoters and identification of four novel 42 bp GadX/GadW-specific binding sites. Mol. Microbiol. 70, 965–982.
Weiss, B. (2006). Evidence for mutagenesis by nitric oxide during nitrate metabolism in Escherichia coli. J. Bacteriol. 188, 829–833. doi: 10.1128/JB.188.3.829-833.2006
Keywords: multidrug efflux pump, acid resistance, anaerobic adaptation, H-NS, ArcBA two-component system, antagonization
Citation: Deng Z, Shan Y, Pan Q, Gao X and Yan A (2013) Anaerobic expression of the gadE-mdtEF multidrug efflux operon is primarily regulated by the two-component system ArcBA through antagonizing the H-NS mediated repression. Front. Microbiol. 4:194. doi: 10.3389/fmicb.2013.00194
Received: 07 May 2013; Accepted: 24 June 2013;
Published online: 11 July 2013.
Edited by:
Kunihiko Nishino, Osaka University, JapanCopyright © 2013 Deng, Shan, Pan, Gao and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Aixin Yan, School of Biological Sciences, The University of Hong Kong, Pokfulam Road, Hong Kong Special Administrative Region, Hong Kong Island, Hong Kong, China e-mail:YXlhbjhAaGt1Lmhr