

- Department of Pharmaceutical Sciences, University of Kentucky, Lexington, KY, USA
Tuberculosis (TB) has become a worldwide threat, mainly due to the emergence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) strains of Mycobacterium tuberculosis (Mtb). This mini-review is focused on the various mechanisms of resistance to the currently available anti-TB drugs and provides perspective on novel strategies and lead scaffolds/compounds aimed at inhibiting/overcoming these resistance mechanisms.
Introduction
Approximately 1.4 million deaths were attributed to Mtb infections in 2012. When compared to other infectious agents, only HIV claims more lives (WHO, 2012). While recent efforts have resulted in a global decline in TB incidence and mortality (WHO, 2011), the number of individuals infected with drug-resistant isolates continues to increase, presenting a serious global health threat. Generally, drug-susceptible Mtb infections are treated with a combination of four compounds; rifampicin (RIF), ethambutol (EMB), pyrazinamide (PZA), and isoniazid (INH) for 2 months, followed by treatment with RIF and INH for 4 additional months (Lienhardt et al., 2012) (Figure 1A). By definition, MDR-TB is resistant to the most potent first-line drugs, RIF and INH. The WHO reports that ~60,000 cases of MDR-TB were diagnosed in 2011, likely an underestimate. To treat MDR-TB, second-line drugs including fluoroquinolones (FQs), amikacin (AMK), kanamycin (KAN), and capreomycin (CAP) are employed. These drugs are administered for ~20 months and can be toxic, poorly tolerated, and difficult to procure. Approximately 95% of MDR-TB is XDR-TB, having additional resistance to at least one FQ and one injectable drug (AMK, KAN, or CAP), and for which treatment options are limited. Therefore, new therapeutics are critically needed to overcome drug resistance and to eliminate TB as a public health threat.
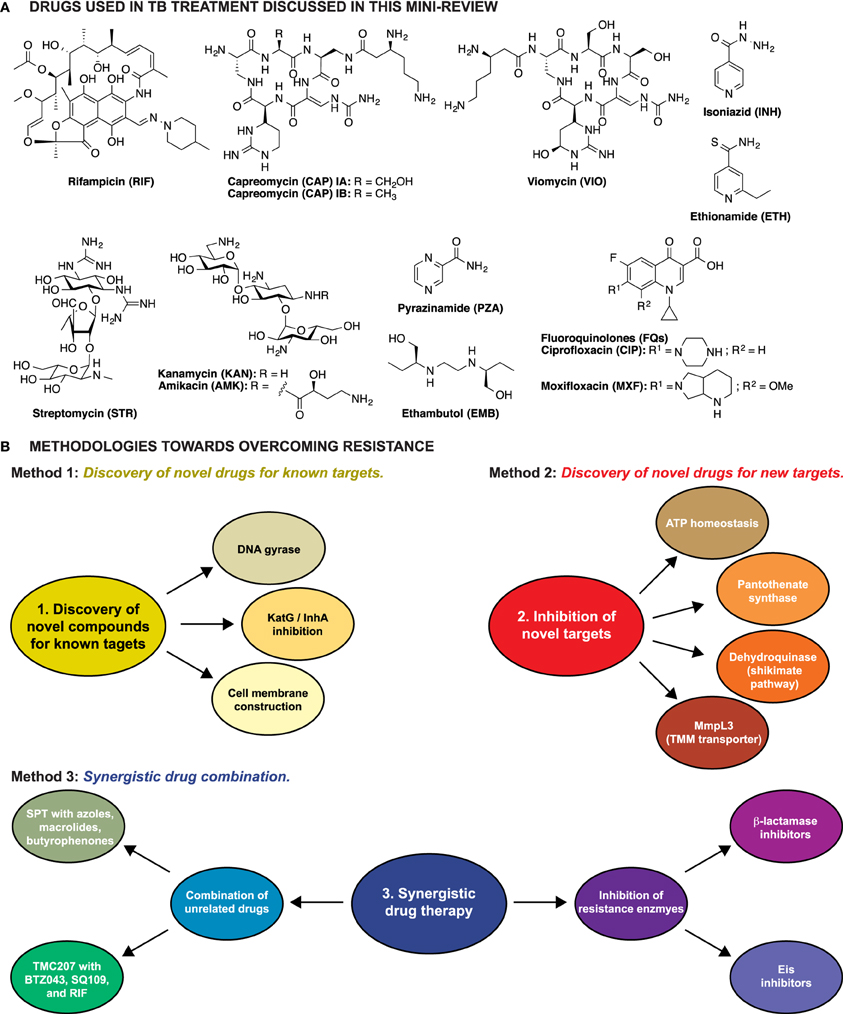
Figure 1. (A) Structures of the anti-TB drugs discussed in this mini-review. (B) Schematic representation of the three methodologies explored to overcome resistance in Mtb.
Herein we briefly discuss the drugs currently used to treat TB and the respective mechanisms of resistance followed by a review of approaches aimed toward overcoming TB drug resistance.
Current Drugs and Resistance
Mtb is intrinsically resistant to many antibiotics due to the low permeability of its mycolic acid-rich waxy cell envelope, the action of efflux pumps (Banerjee et al., 1996; Silva et al., 2001; Singh et al., 2011), and the presence of chromosomally encoded resistance genes. Drug resistance in Mtb has been emerging due to the accumulation of chromosomal mutations and not acquisition of mobile genetic elements. The major mechanisms of acquired drug resistance in Mtb can be broken down into several categories: (1) mutations or modifications (e.g., RNA methylation) of the drug targets (RIF, EMB, KAN, AMK, CAP, and the FQs), (2) the inability to activate a prodrug [INH, PZA, ethionamide (ETH)] due to mutations leading to a loss of function, and (3) enzymatic inactivation of the drug (KAN).
Mutations or Modifications of the Drug Targets
Mutations
Antibiotics target cellular processes that are vital in bacteria by binding their targets at a specific site, often directly interacting with key functional residues of the target. The most common mechanism of resistance in Mtb is the alteration of the target's binding site through the accumulation of mutations. These mutations decrease the binding affinity of the drug to its target and typically occur in a very defined region of the gene termed the resistance-determining region. This mechanism is used by Mtb to confer resistance to RIF, EMB, and FQs by altering the binding site of their respective targets: the β-subunit of RNA polymerase (Campbell et al., 2001), a glycosyltransferase (Telenti et al., 1997), and DNA gyrase (Takiff et al., 1994). Similarly, ribosomal mutations (e.g., A1401G) in the 16S rRNA have been found to confer resistance to AMK, KAN, and CAP (Wachino et al., 2010).
Modifications
An alternative mode of resistance in Mtb is the inactivation of rRNA methyltransferase enzymes. Mutations in TlyA hindering the methylation activity of rRNA 2′-O-methyltransferase at nucleotides C1409 of 16S rRNA and C1920 of 23S rRNA have been linked to resistance to the ribosome-targeting drugs CAP and viomycin (VIO) (Johansen et al., 2006). Mutations in a putative 16S rRNA methyltransferase, GidB, were found to confer low-level resistance to the aminoglycoside (AG) streptomycin (STR) (Spies et al., 2011; Wong et al., 2011).
Inability to Activate a Prodrug
Three first-line drugs, INH, PZA, and ETH, are prodrugs that must be metabolized to be active against Mtb. INH is activated by the catalase-peroxidase, KatG, and produces isonicotinic-acyl radicals that react with NADH to form an INH-NADH adduct. The adduct binds its main target, enoyl-acyl carrier protein reductase (InhA), and inhibits mycolic acid biosynthesis. ETH is a structural analog of INH and is activated by the monooxygenase EthA, which oxidizes ETH to its active form, 2-ethyl-4-amidopyridine. This activated compound also targets InhA and blocks mycolic acid biosynthesis similarly to INH. PZA is activated by pyrazinamidase/nicotinamidase, PncA, to pyrazinoic acid (POA). The proposed targets of POA range from cell membrane energy to fatty acid synthesis, but the exact killing mechanism of POA remains unclear. Resistance to any of the prodrugs can arise due to reduced metabolism by their corresponding activator. Structural mutations that lower or abolish the enzymatic activity of KatG (Ramaswamy et al., 2003; Hazbon et al., 2006), EthA (Baulard et al., 2000; Debarber et al., 2000; Morlock et al., 2003), and PncA (Stoffels et al., 2012; Rajendran and Sethumadhavan, 2013) were found to lead to INH, ETH, and PZA resistance, respectively. Mutations in InhA causing overexpression of the target (Baulard et al., 2000; Debarber et al., 2000; Larsen et al., 2002) or preventing binding of the active form of INH and ETH to the target (Banerjee et al., 1994; Vilcheze et al., 2008) also confer cross-resistance to these drugs.
Inactivation of the Drug
Drug modification is perhaps the most prevalent and well-studied resistance mechanism employed by bacteria. Many bacteria either degrade or modify the offending compound thereby generating an inactive molecule. Resistance to penicillin is mediated by a class of enzymes known as the β-lactamases, which inactivate the antibiotic by destroying the β-lactam ring. Mtb naturally harbors a chromosomally encoded class A β-lactamase, BlaC, which is constitutively expressed providing intrinsic resistance to penicillin (Hugonnet and Blanchard, 2007). One mechanism of acquired resistance to AGs, such as KAN and AMK, is their modification and inactivation by a family of enzymes known as AG-modifying enzymes (AMEs) (Ramirez and Tolmasky, 2010; Labby and Garneau-Tsodikova, 2013). Mtb expresses two AMEs, the AG 2′-N-acetyltransferace AAC(2′)-Ic (Hegde et al., 2001) and the enhanced intracellular survival (Eis) protein (Zaunbrecher et al., 2009). Both enzymes acetylate AGs, reducing the ability of these drugs to inhibit the ribosome. In the case of Eis, increased expression due to mutations in the eis promoter or the 5′-untranslated region of the transcriptional activator WhiB7 leads to clinically relevant, low-level resistance to KAN (Zaunbrecher et al., 2009; Reeves et al., 2013). A unique feature of Eis resides in its ability to modify AGs at multiple sites in vitro, completely inactivating these compounds (Chen et al., 2011). A recent report has shown that Eis also acetylates CAP at its β-lysine side chain in vitro, but the clinical relevance of this finding remains unclear (Houghton et al., 2013).
Methodologies to Overcome Resistance
Three major strategies have been explored to overcome resistance in TB: (1) identification of novel drugs for well-established targets, (2) identification and characterization of novel compounds that target unexplored vital cell processes or enzymes, thus killing the cell by preventing regular metabolism, and (3) identification of compounds that behave synergistically with current anti-TB drugs (Figure 1B).
Discovery of Novel Drugs for Known Targets
DNA gyrase
Among the known Mtb drug targets, DNA gyrase remains attractive for development of inhibitors interacting with either GyrA or GyrB subunits. To overcome the FQ resistance of DNA gyrase mutants, a series of 30 ofloxacin derivatives with various substituents ortho to the fluoro moiety of the FQ scaffold were synthesized (Dinakaran et al., 2008). These compounds displayed MIC99 values <10 μM and retained anti-mycobacterial activity against MDR-Mtb strains. While ofloxacin displayed a 17-fold lower activity against MDR-Mtb relative to susceptible Mtb strains, its derivatives displayed at most a 4-fold lower activity and, in some cases, higher activity against MDR-Mtb. The toxicity of selected compounds in mammalian Vero cells displayed a range of IC50 values (60 to >150 μ M), 6-fold higher than the highest MIC99 value. When tested in culture, the compound with the best MIC99 value showed reduction in Mtb colonies similar to that observed with ofloxacin.
While many efforts have been directed toward the development of FQ analogs to target the GyrA subunit of DNA gyrase, development of inhibitors targeting GyrB has also been pursued. A series of aminopyrazinamides were found to target the ATPase site of GyrB and inhibit the growth of Mtb in the low micromolar/high nanomolar range (Shirude et al., 2013). A variety of naphthoquinones were also discovered to inhibit the ATPase domain of the GyrB subunit of Mtb DNA gyrase with IC50 values between 15 and >200 μ M (Karkare et al., 2013). The most promising naphthoquinone, diospyrin, was tested against DNA gyrase from other bacterial species and found to be a broad spectrum DNA gyrase inhibitor. The aminobenzimidazole AB-1 was also investigated for its inhibitory properties of GyrB and compared to the well-characterized GyrB inhibitor novobiocin (Chopra et al., 2012). AB-1 displayed lower MIC values than novobiocin with Mtb strains resistant to STR, INH, CIP, and p-aminosalicylic acid. AB-1 also displayed improved activity in aerobic and anaerobic conditions while not interfering with RIF or INH mechanisms.
KatG/inhA inhibition
To overcome mutations of KatG and other mechanisms of INH resistance, analogs of INH have been synthesized. Early generations of INH analogs incorporated aryloxyacetonitriles onto the hydrazide of the INH scaffold (Bukowski et al., 1999). These compounds were then evolved into 1,2,3-triazole derivatives with MIC values slightly higher than those of INH (Boechat et al., 2011).
As an alternative strategy to overcoming KatG mutations or deletion, InhA inhibitors, not needing KatG activation, have been investigated. Triclosan (Parikh et al., 2000) served as a lead compound for the design of a series of 5-alkyl-diphenyl ethers (Sullivan et al., 2006). These ethers displayed IC50 values for InhA ranging from 5 nM to 2 μ M, displayed higher bactericidal activity than the parent triclosan, and were further optimized to slow the release time from the InhA active site (Luckner et al., 2010). More recently, from screening 300 molecules, CD117 was identified as an InhA inhibitor that likely targets other enzymes (Vilcheze et al., 2011). CD117 was active against hypoxic Mtb and yielded 80% Vero cell viability at the highest concentration tested. This year, virtual screening efforts led to the identification of 7 potential InhA inhibitors (Kinjo et al., 2013). In model Mycobacterium species the top hit compounds displayed similar growth inhibition to that of INH and directly inhibited Mtb InhA.
Cell membrane construction
The inhibition of cell membrane construction also remains a target of choice for the development of new drugs. Nitroimidazoles such as PA-824 and OPC-67683 have been found to inhibit the synthesis of membrane proteins and lipids (Mukherjee and Boshoff, 2011). Much like INH and PZA, PA-824 is a prodrug that requires activation by reduction of its nitro group (Stover et al., 2000). PA-824 and OPC-67683 have demonstrated activity against both susceptible and resistant-Mtb strains (Stover et al., 2000; Rivers and Mancera, 2008a,b). PA-824 showed activity in the high ng/mL range for both susceptible and resistant-Mtb (Ginsberg et al., 2009) and OPC-67683 displayed better MIC values in the low ng/mL range (Matsumoto et al., 2006).
Benzothiazinones (BTZs) (Makarov et al., 2006), such as BTZ043, have also been shown to interfere with cell membrane construction. Like EMB, these compounds disrupt arabinan's biosynthesis by targeting decaprenylphosphoryl-β-D-ribose 2′-epimerase (DprE) (Makarov et al., 2009). In infected macrophages, BTZ043 showed a faster decrease in percentage of infected cells and bacterial load compared to INH or RIF. BTZ043 has also been tested in 240 clinical isolates of Mtb from European hospitals and was active against all strains (Pasca et al., 2010).
Discovery of Novel Drugs for New Targets
ATP homeostasis
One of the challenges in treating TB is the organism's extended period(s) of dormancy. One key difference in the states of growing and hypoxic non-growing Mtb is a reduced, but significant, ATP pool in the non-growing organism (Rao et al., 2008). A plausible way to attack the non-growing bacilli is to affect their ATP storage by inactivating ATP synthase to deplete the ATP and eventually kill the bacilli (Andries et al., 2005). This strategy led to the identification of the ATP synthase inhibitor TMC207 (bedaquiline or sirturo), which was approved by the FDA in December 2012 for TB treatment (Diacon et al., 2009; Avorn, 2013). TMC207 is the first FDA-approved anti-TB drug in 40 years (Mahajan, 2013). TMC207 targets the C-subunit of ATP synthase proton pump and mutations are known that are resistant to this novel compound (Segala et al., 2012). There are also some mycobacterial strains that are naturally resistant to TMC207. Because of the potential risks associated with this drug, it is recommended that it be utilized only in patients for which other treatment options have failed. A high-throughput screen (HTS) to identify additional compounds that block ATP synthesis and kill dormant Mtb resulted in 0.5% hit rate from a library of 600,000 compounds (Mak et al., 2012).
Pantothenate synthetase
Pantothenate synthetase has also been explored as an antibiotic target of Mtb. Actinomycin D was identified via HTS as an inhibitor of this de novo synthesis enzyme (Yang et al., 2011). Futher in silico HTS led to the discovery of two compounds that inhibit the panthothenate synthesis 10× more efficiently than actinomycin D does. These compounds displayed identical MIC values against susceptible and MDR-Mtb strains. Analogs of the pantothenate synthetase reaction intermediate were shown to bind the enzyme more tightly than the nucleotide substrate (Ciulli et al., 2008). Another essential enzyme, pantothenate kinase (CoaA) has also been considered as a target. Three classes of compounds were found to target CoaA: triazoles, quinoline carboxamides, and biaryl acetic acids (Venkatraman et al., 2012). All compounds inhibited CoaA with IC50 values ranging from 70 nM to 8.4 μ M with various modes of inhibition.
Dehydroquinase of the shikimate pathway
The essential shikimate pathway, responsible for the biosynthesis of aromatic amino acids, folate, and vitamins E/K, is also an antibacterial target (Gonzalez-Bello and Castedo, 2007; Payne et al., 2007a,b; Tran et al., 2011). Recently, inhibitors of the dehydroquinase enzyme of the shikimate pathway have been optimized against Mtb and a series of 3,4-dihydroxyacetophenone and acetonide cores with MIC values in the low-micromolar range were identified (Tran et al., 2012). Similarly, a series of anhydroquinones were synthesized and found to be low-micromolar to high-nanomolar Mtb dehydroquinase inhibitors (Payne et al., 2007a; Prazeres et al., 2007). Other studies have focused on generating dehydroquinic acid derivatives (Lence et al., 2013) or 3-alkyl enol mimics (Blanco et al., 2012) to inhibit dehydroquinase and disrupt the shikimate pathway. Dehydroquinic acid analogs showed IC50 values in the mid-nanomolar range (26–100 nM), while the 3-alkyl enol mimics had a broader nanomolar range (28–780 nM).
MmpL3 (TMM transporter)
SQ109 is an EMB analog with an adamantyl moiety that targets cell well-biosynthesis. SQ109 was discovered in a screen of ~63,000 compounds and showed high ng/mL MIC values (Sacksteder et al., 2012). Recent studies revealed that SQ109 acts on MmpL3, the trehalose monomycolate (TMM) transporter of Mtb (Tahlan et al., 2012). Adamantyl ureas, discovered by high-throughput screening, also displayed anti-tubercular activity (Brown et al., 2011) by targeting MmpL3 (Grzegorzewicz et al., 2012). Since their discovery, the structure of organic ureas has undergone several optimizations for solubility (Scherman et al., 2012) and pharmacokinetic properties (North et al., 2013). Additional scaffolds based on the structure of the pyrrole derivative BM212 have also recently emerged as MmpL3 inhibitors (Poce et al., 2013).
Synergistic Drug Combination
Combination of unrelated drugs
In an effort to reduce emerging resistance, combinations of small molecules have been explored. In a search for compounds that would work synergistically with spectinomycin (SPT), a high-throughput synergy screen (HTSS) was performed with a library of molecules with known pharmacological properties. Three structural cores were found to enhance the antibiotic activity of SPT: the macrolides, azoles, and butyrophenones (Ramon-Garcia et al., 2011). Bromperidol displayed bactericidal activity, was effective against Mtb in a macrophage model, and displayed similar enhancement of the activities of RIF, STR, clofazimine, and clarithromycin. In addition to its anti-TB activity, TMC207 has also been shown to speed up treatment with the first-line anti-TB drugs (Diacon et al., 2009). Lechartier et al. combined BTZ043 with known and experimental TB treatments (Lechartier et al., 2012). In most cases additive interactions of BTZ043 with the second TB treatment were observed. However, BTZ043 was shown to act synergistically with TMC207. Both drugs used at one-quarter of the MIC concentration showed more significant growth retardation than TMC207 at its MIC value alone. The authors suggest that BTZ043 inhibits DprE1 leading to weaker cell walls allowing transmission of TMC207 across the cell membrane. In a recent study, SQ109 acted synergistically with RIF and TMC207 (Reddy et al., 2010).
Inhibition of resistance enzymes
The use of inhibitors of drug-modifying enzymes along with currently approved anti-TB drugs complements the drug combination strategy described above. Perhaps the best-known combination of this type is a β-lactam and a β-lactamase inhibitor. A combination of clavulanate and meropenem proved to be active against both susceptible and XDR-Mtb strains (Hugonnet et al., 2009) A novel β-lactamase inhibitor, NXL104, was recently reported and found to bind BlaC in a 1:1 molar ratio (Xu et al., 2012). Recently, inhibitors of the AG-resistance enzyme Eis were discovered (Green et al., 2012). These compounds encompass a wide variety of chemical structures and offer a plethora of scaffolds to be studied for development of novel anti-TB drug adjuvants. While these compounds have not yet been tested in AG-resistant Mtb strains, combinations of inhibitors with AGs are predicted to restore the antibacterial activity of AGs in cells.
Conclusion
Even though current TB treatments are still broadly employed, the rapid spread of MDR-TB and XDR-TB is a rising threat. Based on our current understanding of the resistance mechanisms, contemporary strategies are focused on generating compounds that will avoid or overcome the defenses of Mtb. Research is being carried out to find novel compounds that will disrupt DNA, cell membrane biosynthesis, and general cellular metabolism. Other lines of inquiry are focusing on pathways currently not used to treat TB: cellular energetics, cellular transport, and other aspects of cellular metabolism. A third facet relies on current TB treatment and combines drugs to achieve synergistic effects, either blocking two separate pathways or inhibiting a drug-modifying enzyme. While many of the compounds discussed herein are far from being approved as drugs, the recent approval of TMC207 and the multitude of efforts in searching for novel antibiotics give hope for the discovery of novel TB treatments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work on TB in our group is supported by NIH Grant AI090048 (Sylvie Garneau-Tsodikova). We thank James Posey and Melisa Willby (Centers for Disease Control and Prevention) and Oleg Tsodikov (University of Kentucky) for critical proof-reading of this mini-review.
References
Andries, K., Verhasselt, P., Guillemont, J., Gohlmann, H. W., Neefs, J. M., Winkler, H., et al. (2005). A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307, 223–227. doi: 10.1126/science.1106753
Avorn, J. (2013). Approval of a tuberculosis drug based on a paradoxical surrogate measure. JAMA 309, 1349–1350. doi: 10.1001/jama.2013.623
Banerjee, A., Dubnau, E., Quemard, A., Balasubramanian, V., Um, K. S., Wilson, T., et al. (1994). InhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 263, 227–230. doi: 10.1126/science.8284673
Banerjee, S. K., Bhatt, K., Rana, S., Misra, P., and Chakraborti, P. K. (1996). Involvement of an efflux system in mediating high level of fluoroquinolone resistance in Mycobacterium smegmatis. Biochem. Biophys. Res. Commun. 226, 362–368. doi: 10.1006/bbrc.1996.1362
Baulard, A. R., Betts, J. C., Engohang-Ndong, J., Quan, S., McAdam, R. A., Brennan, P. J., et al. (2000). Activation of the pro-drug ethionamide is regulated in mycobacteria. J. Biol. Chem. 275, 28326–28331.
Blanco, B., Sedes, A., Peon, A., Lamb, H., Hawkins, A. R., Castedo, L., et al. (2012). Synthesis of 3-alkyl enol mimics inhibitors of type II dehydroquinase: factors influencing their inhibition potency. Org. Biomol. Chem. 10, 3662–3676. doi: 10.1039/c2ob07081b
Boechat, N., Ferreira, V. F., Ferreira, S. B., De Lourdes, G. F. M., De, C. D. S. F., Bastos, M. M., et al. (2011). Novel 1,2,3-triazole derivatives for use against Mycobacterium tuberculosis H37Rv (ATCC 27294) strain. J. Med. Chem. 54, 5988–5999. doi: 10.1021/jm2003624
Brown, J. R., North, E. J., Hurdle, J. G., Morisseau, C., Scarborough, J. S., Sun, D., et al. (2011). The structure-activity relationship of urea derivatives as anti-tuberculosis agents. Bioorg. Med. Chem. 19, 5585–5595. doi: 10.1016/j.bmc.2011.07.034
Bukowski, L., Janowiec, M., Zwolska-Kwiek, Z., and Andrzejczyk, Z. (1999). Synthesis and some reactions of 2-acetylimidazo[4,5-b]pyridine. Antituberculotic activity of the obtained compounds. Pharmazie 54, 651–654.
Campbell, E. A., Korzheva, N., Mustaev, A., Murakami, K., Nair, S., Goldfarb, A., et al. (2001). Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 104, 901–912. doi: 10.1016/S0092-8674(01)00286-0
Chen, W., Biswas, T., Porter, V. R., Tsodikov, O. V., and Garneau-Tsodikova, S. (2011). Unusual regioversatility of acetyltransferase Eis, a cause of drug resistance in XDR-TB. Proc. Natl. Acad. Sci. U.S.A. 108, 9804–9808. doi: 10.1073/pnas.1105379108
Chopra, S., Matsuyama, K., Tran, T., Malerich, J. P., Wan, B., Franzblau, S. G., et al. (2012). Evaluation of gyrase B as a drug target in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 67, 415–421. doi: 10.1093/jac/dkr449
Ciulli, A., Scott, D. E., Ando, M., Reyes, F., Saldanha, S. A., Tuck, K. L., et al. (2008). Inhibition of Mycobacterium tuberculosis pantothenate synthetase by analogues of the reaction intermediate. Chembiochem 9, 2606–2611. doi: 10.1002/cbic.200800437
Debarber, A. E., Mdluli, K., Bosman, M., Bekker, L. G., and Barry, C. E. 3rd. (2000). Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 97, 9677–9682. doi: 10.1073/pnas.97.17.9677
Diacon, A. H., Pym, A., Grobusch, M., Patientia, R., Rustomjee, R., Page-Shipp, L., et al. (2009). The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N. Engl. J. Med. 360, 2397–2405. doi: 10.1056/NEJMoa0808427
Dinakaran, M., Senthilkumar, P., Yogeeswari, P., China, A., Nagaraja, V., and Sriram, D. (2008). Novel ofloxacin derivatives: synthesis, antimycobacterial and toxicological evaluation. Bioorg. Med. Chem. Lett. 18, 1229–1236. doi: 10.1016/j.bmcl.2007.11.110
Ginsberg, A. M., Laurenzi, M. W., Rouse, D. J., Whitney, K. D., and Spigelman, M. K. (2009). Safety, tolerability, and pharmacokinetics of PA-824 in healthy subjects. Antimicrob. Agents Chemother. 53, 3720–3725. doi: 10.1128/AAC.00106-09
Gonzalez-Bello, C., and Castedo, L. (2007). Progress in type II dehydroquinase inhibitors: from concept to practice. Med. Res. Rev. 27, 177–208. doi: 10.1002/med.20076
Green, K. D., Chen, W., and Garneau-Tsodikova, S. (2012). Identification and characterization of inhibitors of the aminoglycoside resistance acetyltransferase Eis from Mycobacterium tuberculosis. ChemMedChem 7, 73–77. doi: 10.1002/cmdc.201100332
Grzegorzewicz, A. E., Pham, H., Gundi, V. A., Scherman, M. S., North, E. J., Hess, T., et al. (2012). Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 8, 334–341. doi: 10.1038/nchembio.794
Hazbon, M. H., Brimacombe, M., Bobadilla Del Valle, M., Cavatore, M., Guerrero, M. I., Varma-Basil, M., et al. (2006). Population genetics study of isoniazid resistance mutations and evolution of multidrug-resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 50, 2640–2649. doi: 10.1128/AAC.00112-06
Hegde, S. S., Javid-Majd, F., and Blanchard, J. S. (2001). Overexpression and mechanistic analysis of chromosomally encoded aminoglycoside 2′-N-acetyltransferase (AAC(2′)-Ic) from Mycobacterium tuberculosis. J. Biol. Chem. 276, 45876–45881. doi: 10.1074/jbc.M108810200.
Houghton, J. L., Green, K. D., Pricer, R. E., Mayhoub, A. S., and Garneau-Tsodikova, S. (2013). Unexpected N-acetylation of capreomycin by mycobacterial Eis enzymes. J. Antimicrob. Chemother. 68, 800–805. doi: 10.1093/jac/dks497
Hugonnet, J. E., and Blanchard, J. S. (2007). Irreversible inhibition of the Mycobacterium tuberculosis beta-lactamase by clavulanate. Biochemistry 46, 11998–12004. doi: 10.1021/bi701506h
Hugonnet, J. E., Tremblay, L. W., Boshoff, H. I., Barry, C. E. 3rd, and Blanchard, J. S. (2009). Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science 323, 1215–1218. doi: 10.1126/science.1167498
Johansen, S. K., Maus, C. E., Plikaytis, B. B., and Douthwaite, S. (2006). Capreomycin binds across the ribosomal subunit interface using tlyA-encoded 2′-O-methylations in 16S and 23S rRNAs. Mol. Cell 23, 173–182. doi: 10.1016/j.molcel.2006.05.044
Karkare, S., Chung, T. T., Collin, F., Mitchenall, L. A., McKay, A. R., Greive, S. J., et al. (2013). The naphthoquinone diospyrin is an inhibitor of DNA gyrase with a novel mechanism of action. J. Biol. Chem. 288, 5149–5156. doi: 10.1074/jbc.M112.419069
Kinjo, T., Koseki, Y., Kobayashi, M., Yamada, A., Morita, K., Yamaguchi, K., et al. (2013). Identification of compounds with potential antibacterial activity against Mycobacterium through structure-based drug screening. J. Chem. Inf. Model. 53, 1200–1212. doi: 10.1021/ci300571n
Labby, K. J., and Garneau-Tsodikova, S. (2013). Methods to identify compounds to overcome the action of aminoglycoside-modifying enzymes for treating resistant bacteria. Future. Med. Chem. [Epub ahead of print].
Larsen, M. H., Vilcheze, C., Kremer, L., Besra, G. S., Parsons, L., Salfinger, M., Jr. et al. (2002). Overexpression of inhA, but not kasA, confers resistance to isoniazid and ethionamide in Mycobacterium smegmatis, M. bovis BCG and M. tuberculosis. Mol. Microbiol. 46, 453–466. doi: 10.1046/j.1365-2958.2002.03162.x
Lechartier, B., Hartkoorn, R. C., and Cole, S. T. (2012). In vitro combination studies of benzothiazinone lead compound BTZ043 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 56, 5790–5793. doi: 10.1128/AAC.01476-12
Lence, E., Tizon, L., Otero, J. M., Peon, A., Prazeres, V. F., Llamas-Saiz, A. L., et al. (2013). Mechanistic basis of the inhibition of type II dehydroquinase by (2S)- and (2R)-2-benzyl-3-dehydroquinic acids. ACS Chem. Biol. 8, 568–577. doi: 10.1021/cb300493s
Lienhardt, C., Raviglione, M., Spigelman, M., Hafner, R., Jaramillo, E., Hoelscher, M., et al. (2012). New drugs for the treatment of tuberculosis: needs, challenges, promise, and prospects for the future. J. Infect. Dis. 205, S241–S249. doi: 10.1093/infdis/jis034
Luckner, S. R., Liu, N., Am Ende, C. W., Tonge, P. J., and Kisker, C. (2010). A slow, tight binding inhibitor of InhA, the enoyl-acyl carrier protein reductase from Mycobacterium tuberculosis. J. Biol. Chem. 285, 14330–14337. doi: 10.1074/jbc.M109.090373
Mahajan, R. (2013). Bedaquiline: First FDA-approved tuberculosis drug in 40 years. Int. J. Appl. Basic Med. Res. 3, 1–2. doi: 10.4103/2229-516X.112228
Mak, P. A., Rao, S. P., Ping Tan, M., Lin, X., Chyba, J., Tay, J., et al. (2012). A high-throughput screen to identify inhibitors of ATP homeostasis in non-replicating Mycobacterium tuberculosis. ACS Chem. Biol. 7, 1190–1197. doi: 10.1021/cb2004884
Makarov, V., Manina, G., Mikusova, K., Mollmann, U., Ryabova, O., Saint-Joanis, B., et al. (2009). Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 324, 801–804. doi: 10.1126/science.1171583
Makarov, V., Riabova, O. B., Yuschenko, A., Urlyapova, N., Daudova, A., Zipfel, P. F., et al. (2006). Synthesis and antileprosy activity of some dialkyldithiocarbamates. J. Antimicrob. Chemother. 57, 1134–1138. doi: 10.1093/jac/dkl095
Matsumoto, M., Hashizume, H., Tomishige, T., Kawasaki, M., Tsubouchi, H., Sasaki, H., et al. (2006). OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 3:e466. doi: 10.1371/journal.pmed.0030466
Morlock, G. P., Metchock, B., Sikes, D., Crawford, J. T., and Cooksey, R. C. (2003). ethA, inhA, and katG loci of ethionamide-resistant clinical Mycobacterium tuberculosis isolates. Antimicrob. Agents Chemother. 47, 3799–3805. doi: 10.1128/AAC.47.12.3799-3805.2003
Mukherjee, T., and Boshoff, H. (2011). Nitroimidazoles for the treatment of TB: past, present and future. Future. Med. Chem. 3, 1427–1454. doi: 10.4155/fmc.11.90
North, E. J., Scherman, M. S., Bruhn, D. F., Scarborough, J. S., Maddox, M. M., Jones, V., et al. (2013). Design, synthesis and anti-tuberculosis activity of 1-adamantyl-3-heteroaryl ureas with improved in vitro pharmacokinetic properties. Bioorg. Med. Chem. 21, 2587–2599. doi: 10.1016/j.bmc.2013.02.028
Parikh, S. L., Xiao, G., and Tonge, P. J. (2000). Inhibition of InhA, the enoyl reductase from Mycobacterium tuberculosis, by triclosan and isoniazid. Biochemistry 39, 7645–7650. doi: 10.1021/bi0008940
Pasca, M. R., Degiacomi, G., Ribeiro, A. L., Zara, F., De Mori, P., Heym, B., et al. (2010). Clinical isolates of Mycobacterium tuberculosis in four European hospitals are uniformly susceptible to benzothiazinones. Antimicrob. Agents Chemother. 54, 1616–1618. doi: 10.1128/AAC.01676-09
Payne, R. J., Peyrot, F., Kerbarh, O., Abell, A. D., and Abell, C. (2007a). Rational design, synthesis, and evaluation of nanomolar type II dehydroquinase inhibitors. ChemMedChem 2, 1015–1029. doi: 10.1002/cmdc.200700032
Payne, R. J., Riboldi-Tunnicliffe, A., Kerbarh, O., Abell, A. D., Lapthorn, A. J., and Abell, C. (2007b). Design, synthesis, and structural studies on potent biaryl inhibitors of type II dehydroquinases. ChemMedChem 2, 1010–1013. doi: 10.1002/cmdc.200700062.
Poce, G., Bates, R. H., Alfonso, S., Cocozza, M., Porretta, G. C., Ballell, L., et al. (2013). Improved BM212 MmpL3 inhibitor analogue shows efficacy in acute murine model of tuberculosis infection. PLoS ONE 8:e56980. doi: 10.1371/journal.pone.0056980
Prazeres, V. F., Sanchez-Sixto, C., Castedo, L., Lamb, H., Hawkins, A. R., Riboldi-Tunnicliffe, A., et al. (2007). Nanomolar competitive inhibitors of Mycobacterium tuberculosis and Streptomyces coelicolor type II dehydroquinase. ChemMedChem 2, 194–207. doi: 10.1002/cmdc.200600208
Rajendran, V., and Sethumadhavan, R. (2013). Drug resistance mechanism of PncA in Mycobacterium tuberculosis. J. Biomol. Struct. Dyn. doi: 10.1080/07391102.2012.759885. [Epub ahead of print].
Ramaswamy, S. V., Reich, R., Dou, S. J., Jasperse, L., Pan, X., Wanger, A., et al. (2003). Single nucleotide polymorphisms in genes associated with isoniazid resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 47, 1241–1250. doi: 10.1128/AAC.47.4.1241-1250.2003
Ramirez, M. S., and Tolmasky, M. E. (2010). Aminoglycoside modifying enzymes. Drug. Resist. Updat. 13, 151–171. doi: 10.1016/j.drup.2010.08.003
Ramon-Garcia, S., Ng, C., Anderson, H., Chao, J. D., Zheng, X., Pfeifer, T., et al. (2011). Synergistic drug combinations for tuberculosis therapy identified by a novel high-throughput screen. Antimicrob. Agents Chemother. 55, 3861–3869. doi: 10.1128/AAC.00474-11
Rao, S. P., Alonso, S., Rand, L., Dick, T., and Pethe, K. (2008). The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 105, 11945–11950. doi: 10.1073/pnas.0711697105
Reddy, V. M., Einck, L., Andries, K., and Nacy, C. A. (2010). In vitro interactions between new antitubercular drug candidates SQ109 and TMC207. Antimicrob. Agents Chemother. 54, 2840–2846. doi: 10.1128/AAC.01601-09
Reeves, A. Z., Campbell, P. J., Sultana, R., Malik, S., Murray, M., Plykaytis, B. B., et al. (2013). Aminoglycoside cross-resistance in Mycobacterium tuberculosis due to mutations in the 5'-untranslated region of whiB7. Antimicrob. Agents Chemother. 57, 1857–1865. doi: 10.1128/AAC.02191-12
Rivers, E. C., and Mancera, R. L. (2008a). New anti-tuberculosis drugs in clinical trials with novel mechanisms of action. Drug. Discov. Today 13, 1090–1098. doi: 10.1016/j.drudis.2008.09.004
Rivers, E. C., and Mancera, R. L. (2008b). New anti-tuberculosis drugs with novel mechanisms of action. Curr. Med. Chem. 15, 1956–1967. doi: 10.2174/092986708785132906
Sacksteder, K. A., Protopopova, M., Barry, C. E., 3rd, Andries, K., and Nacy, C. A. (2012). Discovery and development of SQ109a new antitubercular drug with a novel mechanism of action. Future Microbiol. 7, 823–837. doi: 10.2217/fmb.12.56
Scherman, M. S., North, E. J., Jones, V., Hess, T. N., Grzegorzewicz, A. E., Kasagami, T., et al. (2012). Screening a library of 1600 adamantyl ureas for anti-Mycobacterium tuberculosis activity in vitro and for better physical chemical properties for bioavailability. Bioorg. Med. Chem. 20, 3255–3262. doi: 10.1016/j.bmc.2012.03.058
Segala, E., Sougakof, W., Nevejans-Chauffour, A., Jarlier, V., and Petrella, S. (2012). New mutations in the mycobacterial ATP synthase: new insights into the binding of the diarylquinoline TMC207 to the ATP synthase C-ring structure. Antimicrobial. Agents Chemother. 56, 2326–2334. doi: 10.1128/AAC.06154-11.
Shirude, P. S., Madhavapeddi, P., Tucker, J. A., Murugan, K., Patil, V., Basavarajappa, H., et al. (2013). Aminopyrazinamides: novel and specific GyrB inhibitors that kill replicating and nonreplicating Mycobacterium tuberculosis. ACS Chem. Biol. 8, 519–523. doi: 10.1021/cb300510w
Silva, P. E., Bigi, F., Santangelo, M. P., Romano, M. I., Martin, C., Cataldi, A., et al. (2001). Characterization of P55, a multidrug efflux pump in Mycobacterium bovis and Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 45, 800–804. doi: 10.1128/AAC.45.3.800-804.2001
Singh, M., Jadaun, G. P. S., Srivastava, R. K., Chauhan, V., Mishra, R., Gupta, K., et al. (2011). Effect of efflux pump inhibitors on drug susceptibility of ofloxacin ressitant Mycobacterium tuberculosis isolates. Indian J. Med. Res. 133, 535–540.
Spies, F. S., Ribeiro, A. W., Ramos, D. F., Ribeiro, M. O., Martin, A., Palomino, J. C., et al. (2011). Streptomycin resistance and lineage-specific polymorphisms in Mycobacterium tuberculosis gidB gene. J. Clin. Microbiol. 49, 2625–2630. doi: 10.1128/JCM.00168-11
Stoffels, K., Mathys, V., Fauville-Dufaux, M., Wintjens, R., and Bifani, P. (2012). Systematic analysis of pyrazinamide-resistant spontaneous mutants and clinical isolates of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 56, 5186–5193. doi: 10.1128/AAC.05385-11
Stover, C. K., Warrener, P., Vandevanter, D. R., Sherman, D. R., Arain, T. M., Langhorne, M. H., et al. (2000). A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405, 962–966. doi: 10.1038/35016103
Sullivan, T. J., Truglio, J. J., Boyne, M. E., Novichenok, P., Zhang, X., Stratton, C. F., et al. (2006). High affinity InhA inhibitors with activity against drug-resistant strains of Mycobacterium tuberculosis. ACS Chem. Biol. 1, 43–53. doi: 10.1021/cb0500042
Tahlan, K., Wilson, R., Kastrinsky, D. B., Arora, K., Nair, V., Fischer, E., et al. (2012). SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 56, 1797–1809. doi: 10.1128/AAC.05708-11
Takiff, H. E., Salazar, L., Guerrero, C., Philipp, W., Huang, W. M., Kreiswirth, B., et al. (1994). Cloning and nucleotide sequence of Mycobacterium tuberculosis gyrA and gyrB genes and detection of quinolone resistance mutations. Antimicrob. Agents Chemother. 38, 773–780. doi: 10.1128/AAC.38.4.773
Telenti, A., Philipp, W. J., Sreevatsan, S., Bernasconi, C., Stockbauer, K. E., Wieles, B., et al. (1997). The emb operon, a gene cluster of Mycobacterium tuberculosis involved in resistance to ethambutol. Nat. Med. 3, 567–570. doi: 10.1038/nm0597-567
Tran, A. T., Cergol, K. M., West, N. P., Randall, E. J., Britton, W. J., Bokhari, S. A., et al. (2011). Synthesis and evaluation of potent ene-yne inhibitors of type II dehydroquinases as tuberculosis drug leads. ChemMedChem 6, 262–265. doi: 10.1002/cmdc.201000399
Tran, A. T., West, N. P., Britton, W. J., and Payne, R. J. (2012). Elucidation of Mycobacterium tuberculosis type II dehydroquinase inhibitors using a fragment elaboration strategy. ChemMedChem 7, 1031–1043. doi: 10.1002/cmdc.201100606.
Venkatraman, J., Bhat, J., Solapure, S. M., Sandesh, J., Sarkar, D., Aishwarya, S., et al. (2012). Screening, identification, and characterization of mechanistically diverse inhibitors of the Mycobacterium tuberculosis enzyme, pantothenate kinase (CoaA). J. Biomol. Screen 17, 293–302. doi: 10.1177/1087057111423069
Vilcheze, C., Av-Gay, Y., Attarian, R., Liu, Z., Hazbon, M. H., Colangeli, R., Jr. et al. (2008). Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol. Microbiol. 69, 1316–1329. doi: 10.1111/j.1365-2958.2008.06365.x
Vilcheze, C., Baughn, A. D., Tufariello, J., Leung, L. W., Kuo, M., Basler, C. F., Jr. et al. (2011). Novel inhibitors of InhA efficiently kill Mycobacterium tuberculosis under aerobic and anaerobic conditions. Antimicrob. Agents Chemother. 55, 3889–3898. doi: 10.1128/AAC.00266-11
Wachino, J., Shibayama, K., Kimura, K., Yamane, K., Suzuki, S., and Arakawa, Y. (2010). RmtC introduces G1405 methylation in 16S rRNA and confers high-level aminoglycoside resistance on Gram-positive microorganisms. FEMS Microbiol. Lett. 311, 56–60. doi: 10.1111/j.1574-6968.2010.02068.x
Wong, S. Y., Lee, J. S., Kwak, H. K., Via, L. E., Boshoff, H. I., and Barry, C. E. 3rd. (2011). Mutations in gidB confer low-level streptomycin resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 55, 2515–2522. doi: 10.1128/AAC.01814-10
Xu, H., Hazra, S., and Blanchard, J. S. (2012). NXL104 irreversibly inhibits the beta-lactamase from Mycobacterium tuberculosis. Biochemistry 51, 4551–4557. doi: 10.1021/bi300508r
Yang, Y., Gao, P., Liu, Y., Ji, X., Gan, M., Guan, Y., et al. (2011). A discovery of novel Mycobacterium tuberculosis pantothenate synthetase inhibitors based on the molecular mechanism of actinomycin D inhibition. Bioorg. Med. Chem. Lett. 21, 3943–3946. doi: 10.1016/j.bmcl.2011.05.021
Keywords: antibiotics, inhibitors, mechanisms of resistance, new drug targets, synergistic drug therapy
Citation: Green KD and Garneau-Tsodikova S (2013) Resistance in tuberculosis: what do we know and where can we go? Front. Microbiol. 4:208. doi: 10.3389/fmicb.2013.00208
Received: 14 May 2013; Paper pending published: 24 June 2013;
Accepted: 05 July 2013; Published online: 23 July 2013.
Edited by:
Jun Lin, The University of Tennessee, USACopyright © 2013 Green and Garneau-Tsodikova. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Sylvie Garneau-Tsodikova, Department of Pharmaceutical Sciences, University of Kentucky, BioPharm Complex, Room 423, 789 South Limestone Street, Lexington, KY 40536-0596, USA e-mail:c3lsdmllZ3Rzb2Rpa292YUB1a3kuZWR1