 Maria R. Petrara
Maria R. Petrara Riccardo Freguja
Riccardo Freguja Ketty Gianesin
Ketty Gianesin Marisa Zanchetta
Marisa Zanchetta Anita De Rossi
Anita De Rossi- 1Viral Oncology Unit and AIDS Reference Center, Section of Oncology and Immunology, Department of Surgery, Oncology and Gastroenterology, University of Padova, Padova, Italy
- 2Istituto Oncologico Veneto - Istituto di Ricovero e Cura a Carattere Scientifico, Padova, Italy
Epstein–Barr virus (EBV) is a ubiquitous human γ-herpes virus which establishes a life-long asymptomatic infection in immunocompetent hosts. In human immunodeficiency virus type 1 (HIV-1) infected patients, the impaired immunosurveillance against EBV may favor the development of EBV-related diseases, ranging from lymphoproliferative disorders to B cell non-Hodgkin’s lymphomas (NHL). Antiretroviral therapy (ART) has significantly modified the natural course of HIV-1 infection, resulting in decreased HIV-1 plasmaviremia, increased CD4 lymphocytes, and decreased opportunistic infections, indicating a restoration of immune functions. However, the impact of ART appears to be less favorable on EBV-related malignancies than on other AIDS-defining tumors, such as Kaposi’s sarcoma, and NHL remains the most common cancer during the ART era. EBV-driven tumors are associated with selective expression of latent oncogenic proteins, but uncontrolled lytic cycle with virus replication and/or reactivation may favor cell transformation, at least in the early phases. Several host’s factors may promote EBV reactivation and replication; besides immunodepression, inflammation/chronic immune stimulation may play an important role. Microbial pathogen-associated molecular patterns and endogenous damage-associated molecular patterns, through Toll-like receptors, activate the immune system and may promote EBV reactivation and/or polyclonal expansion of EBV-infected cells. A body of evidence suggests that chronic immune stimulation is a hallmark of HIV-1 pathogenesis and may persist even in ART-treated patients. This review focuses on lymphomagenesis driven by EBV both in the context of the natural history of HIV-1 infection and in ART-treated patients. Understanding the mechanisms involved in the expansion of EBV-infected cells is a premise for the identification of prognostic markers of EBV-associated malignancies.
Introduction
Epstein–Barr virus (EBV) is a ubiquitous human γ-herpes virus which establishes a life-long asymptomatic infection in immunocompetent hosts. Most individuals contract EBV infection by early adulthood; primary EBV infection is usually asymptomatic, but sometimes results in infectious mononucleosis, a self-limiting disease due to the EBV-specific immunological response. Regardless of its iatrogenic or infectious origin, immunodeficiency increases the risk of tumor development, particularly of tumors etiologically linked to viral agents. In post-transplant patients and in individuals infected with human immunodeficiency virus type 1 (HIV-1), impaired immunosurveillance against EBV may favor the development of EBV-associated diseases, ranging from lymphoproliferative disorders to B cell non-Hodgkin’s lymphomas (NHL; Ometto et al., 1997; Beral and Newton, 1998). Antiretroviral therapy (ART) partially restores immune functions and has significantly changed the course of HIV-1 infection (Palella et al., 1998). However, the impact of ART seems to be less favorable on EBV-related diseases than on other AIDS-defining events, including Kaposi’s sarcoma (KS; Simard et al., 2011). Notably, NHL has been considered as an AIDS-defining cancer (ADC) since 1985, and remains one of the main causes of death in HIV-1 infected patients even in the ART era [Collaboration of Observational HIV Epidemiological Research Europe (COHERE) Study Group, 2009; Simard et al., 2011].
Incidence of EBV-Related Malignancies in HIV-1 Infected Patients
There are several subtypes of AIDS-associated NHL, including Burkitt’s lymphoma (BL), immunoblastic lymphoma (IBL), diffuse large B cell lymphoma (DLBCL), and primary central nervous system lymphoma (PCNSL). The frequency of EBV detection in these tumors ranges from 60% in BL to 100% in PCNSL (Carbone et al., 2009). Before the introduction of ART, the incidence of NHL in HIV-1 infected subjects was 100 times higher than in the general population (Goedert et al., 1998; International Collaboration on HIV and Cancer, 2000), PCNSL and high grade IBL being the most common NHL subtypes, followed by BL and intermediate grade DLBCL (Kaplan, 2012).
High levels of HIV-1 plasmaviremia and low CD4 cell counts are both risk factors for the onset of NHL. The risk substantially increases in patients with >100,000 HIV-1 RNA copies/ml and/or with <50 CD4 cells/μl (Engels et al., 2010). However, systemic lymphomas may occur at virtually any level of CD4 cell count, BL occurring more frequently in patients with moderate/advanced immunodepression (200–500 CD4 cells/μl) and PCNSL occurring in patients with severe immunodepression (<200 CD4 cells/μl).
The introduction of ART into clinical practice has modified the natural course of HIV-1 infection, resulting in the decrease of HIV-1 plasmaviremia, increase in CD4 T lymphocytes, and decrease in HIV-1-associated opportunistic infections. Deaths from AIDS-related diseases have fallen by 75% (Tirelli and Bernardi, 2001) and people diagnosed with HIV-1 can expect to live for 30 or 40 years after contracting the infection, equivalent to a reduction in lifespan of about one-third (Grulich, 2009). Therefore, patients with HIV-1 infection are now living longer, with improved immune functions and reduced risk of developing AIDS; nevertheless, ADCs remain higher when compared with the rate of these cancers in the general population (Petoumenos et al., 2013).
After the introduction of ART, the overall incidence of NHL declined by nearly 50% compared with ART-untreated patients (International Collaboration on HIV and Cancer, 2000; Biggar et al., 2007; Simard et al., 2011); the adjusted incidence rate of NHL was estimated to decline from 6.2% pre-ART (1992–1999) to 3.2% in the ART era (1997–1999; International Collaboration on HIV and Cancer, 2000). Five-year cumulative incidence of NHL declined from 3.8% during the 1990–1995 period to 2.2% during 1996–2006 (Simard et al., 2011). This pattern is quite different from that observed with KS, associated with human herpes virus 8 (HHV8; Cattelan et al., 1999; Simard et al., 2011). The adjusted incidence rate of KS declined from 14.3% pre-ART to 1.8% in the ART era (Simard et al., 2011). After the introduction of ART, NHL is still the most common ADC. In addition, the decline is not equally distributed among the various NHL subtypes, but is higher for PCNSL and lower for the other subtypes.
The increased risk of NHL cannot be explained by immunodepression alone. Patients treated with ART may survive longer, with continued B cell stimulation, leading to an increased incidence of lymphoma over time. Emerging data also indicate that HIV-1 infection, even among long-term ART-treated patients, is associated with a higher rate of non-AIDS-defining malignancies (NADC; Deeken et al., 2012); this evidence further supports the concept that immunodeficiency is not sufficient to explain the higher risk of cancer in HIV-1 infected subjects. The substantial reduction in HIV-1-associated mortality and the increase in an aging population with HIV-1 may contribute to the rise in NADC, including some tumors associated with co-infections [e.g., anal and oropharyngeal cancer associated with human papillomavirus, liver cancer associated with hepatitis B and C viruses, and Hodgkin’s lymphoma (HL) associated with EBV]. In a cohort of HIV-1 infected European patients, the incidence of HL was 4.5 times higher than in the general population pre-ART (1983–1985) and 32 times higher in the ART era (2002–2007; Powles et al., 2009). These results have been replicated in a Swiss HIV-1 cohort, in which the incidence of HL increased 9 times pre-ART, 21 times in the early ART era, and 28 times in the late ART era (Franceschi et al., 2010). Also, in a United States cohort, the 5-year cumulative incidence of HL increased from 0.09% pre-ART (1980–1989) to 0.19% in the ART era (1996–2006; Simard et al., 2011). HIV-1-associated HL differs from HL in HIV-1 uninfected patients, because it is almost always associated with EBV and more often presents systemic “B” symptoms, and extranodal involvement (Jacobson and Abramson, 2012).
EBV-Driven Tumorigenesis
Epstein–Barr virus is associated with both B cell and epithelial cell malignancies. Like all herpes viruses, EBV has both latent and lytic replication programs, allowing it to evade immune clearance and persist for the host’s lifetime. Latent infection is characterized by replication of the viral genome as an integral part of the host cell chromosomes and the absence of production of infectious virus. Since lytic EBV replication triggers the death of infected cells, tumors require the expression of latent programs. Latent proteins include nuclear antigens (EBNA-1, -2, -3A, -3B, and -3C), leader protein (LP), and latent membrane proteins (LMP-1, -2A, and -2B). LMP-1 is the main oncogenic protein of EBV and is essential for EBV-driven tumorigenesis. It is expressed in all NHL, except BL, and also in HL. Functionally, LMP-1 mimics CD40, a member of the tumor necrosis factor receptor (TNFR) superfamily, thus providing both growth and differentiation signals to B cells. LMP-1 does in fact activate several downstream signaling pathways which contribute to the induction of genes encoding anti-apoptotic proteins (e.g., BCL-2 and A20) and cytokines, such as interleukin (IL)-1, and CD40L (Young and Rickinson, 2004; Chen, 2011).
In vitro, EBV infection of B cells may generate immortalized lymphoblastoid B cell lines (LCL) which express all the latent viral genes, as most EBV-NHL do; this is why LCL are often employed as a model to study EBV-driven lymphomagenesis. Besides the expression of viral latent proteins, the transformation of primary B lymphocytes by EBV requires the activation of telomerase (Dolcetti and De Rossi, 2012). Telomerase is a ribonucleoprotein complex, containing an internal RNA, used as a template to elongate telomeres, and a catalytic protein with telomerase reverse transcriptase (TERT) activity. Telomerase is critically involved in maintaining telomere length and its activation is required for cells to overcome replicative senescence and acquire unlimited replicative potential, a prerequisite for neoplastic transformation (Blackburn et al., 2006). It has been demonstrated that LMP-1 activates TERT at transcriptional level via nuclear factor kappa B (NF-κB) and MAPK/ERK1/2 pathways (Terrin et al., 2007, 2008). In turn, TERT expression confers resistance to apoptotic stimuli and to the induction of the lytic cycle (Giunco et al., 2013).
Although latency programs predominate in EBV-driven tumors, recent evidence suggests that lytic EBV replication has some pathogenic importance, at least in the early phases of cell transformation. Several lytic proteins expressed during the lytic cycle of infection are involved in immune evasion. They include the BCRF-1 gene which encodes a viral IL-10 cytokine which, like human IL-10, inhibits the synthesis of interferon-γ (IFN-γ) and suppresses CD8 cytotoxic T cell (Draborg et al., 2012). Recent studies also suggest that the lytic cycle contributes to the growth of EBV-associated malignancies by enhancing angiogenesis. Indeed, B cells infected with a virus competent for the expression of the lytic protein BZLF-1 released more vascular endothelial growth factor and IL-6 than cells infected with BZLF-1-defective virus (Hong et al., 2005). A new humanized mouse model, in which both human fetal CD34 hematopoietic stem cells and thymus/liver tissue were transplanted, was employed to test EBV-driven lymphomagenesis; mice injected with lytic replication-defective BZLF1-deleted virus developed fewer lymphomas than animals infected with lytic BZLF1-competent virus, suggesting that lytically infected cells promote EBV-driven lymphomagenesis (Ma et al., 2011).
Direct and Indirect Interactions of HIV-1 with B Cells and EBV
Besides impaired immunosurveillance against EBV-infected cells due to the loss of T cell function, HIV-1 may contribute to the genesis of EBV-associated NHL through chronic B cell hyperactivation. Chronic systemic immune activation is a hallmark of HIV-1 pathogenesis; being one of the strongest predictors of disease progression (Deeks et al., 2004; Hunt et al., 2008) and it is also associated with impaired immune reconstitution during ART (Anselmi et al., 2007).
Immune activation involves both T cell and B cell compartments. In the former, immune activation may be assessed by the high frequency of T cells expressing markers of activation, such as CD38 and HLA-DR (Hazenberg et al., 2000; Anselmi et al., 2007), high levels of T cell proliferation (Hellerstein et al., 2003), and activation-induced apoptosis of HIV-1 uninfected T cells (Gougeon et al., 1996). In the B cell compartment, B cells express high levels of CD80 and CD86 co-stimulatory molecules. Several changes in the B cell subsets have also been described in association with HIV-1 plasmaviremia and/or CD4 cell decline. These changes include an increase in activated (CD27+CD21low) and exhausted (CD27-CD21low) B cells (Moir and Fauci, 2009), a decrease in circulating memory (CD27+) B cells (Titanji et al., 2006), and an increase in immature/transitional (CD27-CD10+) B cells (Malaspina et al., 2006). Immune activation may result in polyclonal B cell activation, hypergammaglobulinemia, increased cell turnover (Moir and Fauci, 2009), and ultimately increased frequency of B cell malignancies (Grulich et al., 2000).
The factors contributing to B cell hyperactivation and expansion of EBV-infected B cells remain largely unknown. The mechanisms responsible for polyclonal B cell activation seem to be multiple, and due to a combination of both direct and indirect interaction of B cells with HIV-1. HIV-1 itself can induce polyclonal B cell activation in vitro (Schnittman et al., 1986). It has been demonstrated that CD40L is incorporated into the HIV-1 envelope (Ambinder et al., 2010), and that B cells derived from the peripheral blood of HIV-1 infected viremic patients carry replication-competent virus on their membrane (Moir et al., 2000). There is evidence that HIV-1 binds to B cells through interactions between the complement receptor CD21, which is expressed on most mature B cells (Moir et al., 2000); the virus bound to B cells can efficiently infect CD4 T cells through cell–cell contact (Malaspina et al., 2002). A model of “self-limiting” HIV-1 infection of B cells has been described. After in vitro exposure to HIV-1, a steady-state fraction of LCL resulted positive for HIV-1 gp120 envelope protein. These cells underwent down-regulation of BCL-2 and death by apoptosis; the persistent fraction of gp120+ cells was maintained by HIV-1 transmission to B cells newly arising from the proliferation of gp120- cells (De Rossi et al., 1994). It was also demonstrated that a fraction of B cells may bind gp120 through mannose C-type lectin receptors (MCLRs). In the presence of gp120, MCLR-expressing B cells up-regulate the activation-induced cytidine deaminase, and undergo immunoglobulin class switching (He et al., 2006). By contrast, HIV-1 protein Nef suppresses class switch recombination on B cells by interfering with CD40L-mediated signaling (Qiao et al., 2006). Nef also promotes polyclonal B cell activation and increases CD4 T cell susceptibility to HIV-1 infection by inducing macrophages to secrete pro-inflammatory cytokines (Swingler et al., 2008). Additional in vitro evidence of direct interaction between HIV-1 and several receptors on B cells have been reported (Berberian et al., 1993; Rappocciolo et al., 2006), however, to date there is still little evidence that HIV-1 can productively replicate in B cells in vivo.
The transactivator Tat protein of HIV-1 is released by HIV-1 infected cells and is taken up by surrounding HIV-1-uninfected cells through interactions with cell membrane heparan sulfate proteoglycans (Frankel and Pabo, 1988; Tyagi et al., 2001). HIV-1 binds to B cells which are in close contact with HIV-1-infected macrophages and to dendritic cells in the extranodal sites where NHL usually occur (Sparano, 2001). This cell–cell contact may favor B cell uptake of the protein; Tat protein has in fact been consistently detected in the neoplastic cells of NHL occurring in HIV-1 infected subjects (Lazzi et al., 2002). Tat modulates the expression of several cellular genes, including cytokines and their receptors, and increases the expression of IL-6 and IL-10 (Scala et al., 1994; Blazevic et al., 1996), which in turn promote B cell activation. It has been demonstrated that Tat prevents cell cycle arrest and confers resistance to apoptosis in EBV-infected B cells cultured in vitro in serum starvation conditions (Colombrino et al., 2004).
Chronic B cell hyperactivation is driven by overproduction of B cell stimulatory cytokines, such as IL-6, IL-10, interferon-α (IFN-α), and tumor necrosis factor (TNF; Rieckmann et al., 1991; Takeshita et al., 1995; Mandl et al., 2008). The serum levels of these B cell stimulatory cytokines and other molecules, such as soluble sCD27 and sCD30 generated from the corresponding B cell receptors during the process of immune activation, were significantly high 1–5 years prior to diagnosis of systemic AIDS-NHL (Ambinder et al., 2010; Breen et al., 2011). Elevated serum levels of IL-6 have also been observed 1–3 years prior to the onset of AIDS-NHL, thus supporting the role of IL-6-driven B cell stimulation in the development of these lymphomas (Breen et al., 2011).
Lastly, an important source of immune activation in HIV-1 infected individuals is provided by microbial translocation due to damage to intestinal mucosa caused by massive HIV-1-induced T cell depletion in the gut (Brenchley et al., 2006). Translocation of intestinal bacteria and bacterial products into the bloodstream can activate the immune system by binding to receptors involved in the host inflammatory response, such as Toll-like receptors (TLRs). TLRs are pattern recognition receptors which recognize structural components belonging to bacteria, fungi, and viruses, known as “pathogen-associated molecular patterns” (PAMPs), and activate the innate immune response (Janeway and Medzhitov, 2002). PAMPs include bacterial lipopolysaccharide (LPS), 16S ribosomal DNA (16S rDNA), and CpG DNA. A recent study demonstrated that levels of PAMPs, generated by microbial translocation (sCD14 and LPS) are associated with the risk of NHL (Marks et al., 2013). TLR9 substantially suppresses BZLF-1-mRNA expression by histone modifications in acute EBV infection ex vivo and in latently BL cells in vitro, suggesting that immune activation can also promote EBV-driven lymphomagenesis by suppressing the viral lytic cycle (Ladell et al., 2007). Patients who developed PCNSL expressed more CD80 and CD86 in their B cells and responded to TLR9 agonist better than patients without tumors (Audigé et al., 2010).
The loss of mucosal surface integrity in the gut, due to the massive depletion of CD4 T cells, involves not only increased mucosal permeability and consequent microbial translocation, but also an increase in “damage-associated molecular patterns” (DAMPs), endogenous molecules released after cell death, such as mitochondrial DNA (mtDNA; Zhang et al., 2010), high mobility group box 1 (HMGB1) protein (Scaffidi et al., 2002) and defensins (Sørensen et al., 2005). The binding of PAMP and DAMP ligands to the extra- or intra-cellular domain of TLRs initiates a complex-signal transduction cascade which, via the NF-κB pathway, ultimately leads to increased transcription of pro-inflammatory cytokines, such as IL-6 and TNF. During chronic infection, PAMPs and DAMPs may act in synergy on the TLR response. Thus, chronic inflammation may favor an environment which triggers tumor development.
Impact of Antiretroviral Therapy on EBV Lymphomagenesis
The impact of ART is less favorable on EBV-related malignancies than on other ADC. In addition, the decline after the introduction of ART was more substantial in PCNSL than in other NHL subtypes. Because of the role played by chronic immune activation in the genesis of NHL, it may be argue that this mechanism (partially) persists even during ART. A study performed on a cohort of HIV-1-infected patients under ART disclosed a decline in both EBV plasmaviremia and citoviremia in patients with immunological (increase in CD4 cells) and virological (decline in HIV-1 plasmaviremia to undetectable levels) responses. Notably, a decrease in EBV load was also detected in patients with only virological response to ART. By contrast, the increase in CD4 cells without suppression of HIV-1 plasmaviremia was accompanied by an increase in EBV load, paralleled by an increase in immunoglobulin levels (Righetti et al., 2002). In agreement, EBV-DNA load was higher in patients with detectable HIV-1 plasmaviremia, despite good immunological status (>500 CD4 cells/μl) than in patients with undetectable HIV-1 plasmaviremia, regardless of immunological status (Petrara et al., 2012). It has been observed that ART only partially normalizes the serum cytokine levels; IL-6 remain at high levels even 2–3 years after ART initiation (Regidor et al., 2011). These findings strongly suggest that immune reconstitution without reduction in HIV-1 viremia may increase B cell stimulation and the number of EBV-infected cells; this may be important for new therapeutic strategies against HIV-1 to implement immune reconstitution and/or to alleviate adverse drug effects.
HIV-1 infected patients, treated with ART in combination with IL-2 to improve immunological reconstitution, showed a decrease in EBV plasmaviremia and citoviremia when treated with low-intermittent IL-2 doses, but presented an increase in EBV in both cells and plasma when treated with high doses of IL-2 (Burighel et al., 2006). Thus, according to dose, IL-2 may significantly affect EBV dynamics. Low doses of IL-2, by binding to high-affinity receptors expressed on T lymphocytes, may promote the activity of cytotoxic T lymphocytes, thus restoring protective immunity against EBV. Notably, low doses of IL-2 prevented the development of EBV-associated disease in severe combined immunodeficiency (SCID) mice reconstituted with peripheral blood cells from EBV-seropositive subjects (Baiocchi and Caligiuri, 1994). Conversely, high doses of IL-2, by binding low-affinity receptors expressed on natural killer (NK) cells, may enhance several pro-inflammatory cytokines, and consequently B cell stimulation and activation (Jacobson et al., 1996; Malnati et al., 2002; Table 1).
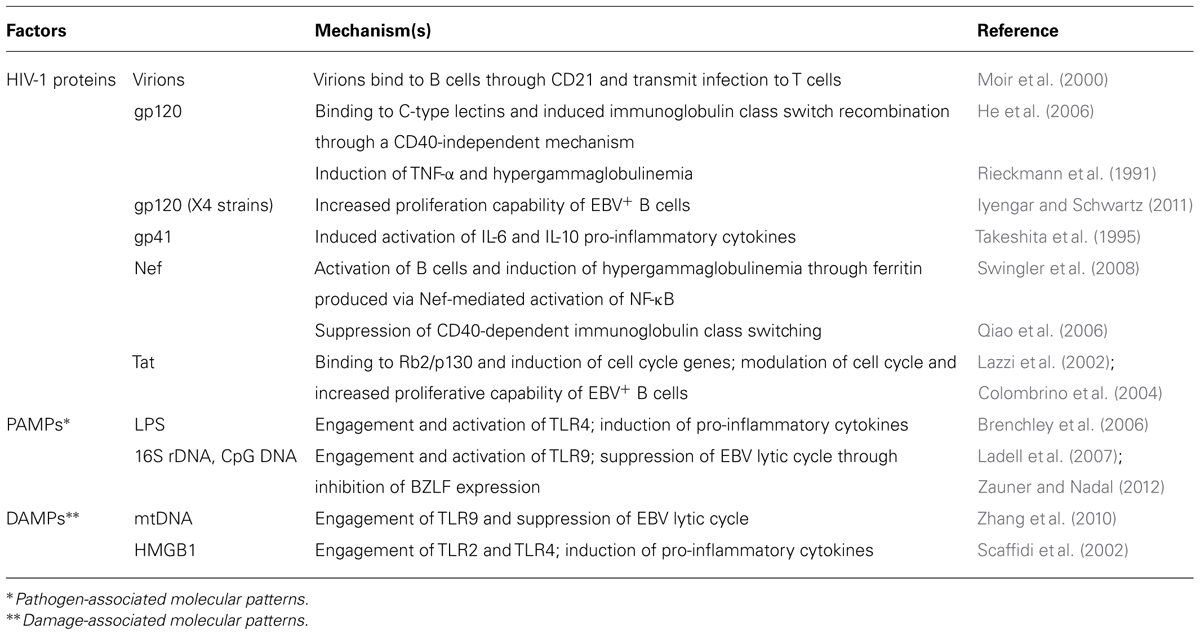
TABLE 1. Factors involved in HIV-1 and EBV interplay through B cell activation.
It has been demonstrated that patients with high levels of EBV have higher levels of pro-inflammatory cytokines (IL-6, IL-10, TNF-α) and PAMPs (LPS) than patients with low EBV level, and that EBV load strongly correlates with the percentage of activated B cells (Petrara et al., 2012). In addition, patients under CD4-guided interruption of ART to reduce adverse drug effects show similar levels of HIV-1 plasmaviremia to ART-untreated patients and high levels of EBV (Petrara et al., 2012). The association between markers of B cell activation and the risk of NHL persists even after adjustment for CD4 T cell count, HIV-1 load and ART, thus supporting the independent role of these markers with NHL risk (Guiguet et al., 2009; Engels et al., 2010; Marks et al., 2013). These findings additionally support the concept that B cell activation, regardless of CD4 immune reconstitution, favors expansion of EBV-infected cells and the onset of EBV-related malignancies.
Concluding Remarks
The risk of EBV-associated NHL in HIV-1 infected patients compared with the general population is mainly increased by impaired immunosurveillance against EBV and B cell chronic immune activation. The two mechanisms may contribute to the development of NHL subtypes in different ways. In HIV-1 infected subjects with low CD4 T cell count and severe immunedepression, a notable proportion of NHL is represented by PCNSL. These tumors are almost 100% EBV-positive, are less significantly linked to circulating levels of pro-inflammatory cytokines than systemic NHL (Breen et al., 2011), and represent the NHL subtype which most significantly declined after the introduction of ART. These observations are consistent with the concept that PCNSL primarily results from the loss of immunosurveillance against EBV.
Systemic HL may occur relatively early in HIV-1 disease and the impact of ART in reducing these tumors is lower than that observed for PCNSL and other ADC. Chronic immune activation may considerably affect the onset of systemic NHL; chronic B cell activation, generated by HIV-1 antigens and the products of microbial translocations, may result in expansion of B cells and accumulation of genetic lesions over a prolonged period of time. According to this model, EBV does not represent a prerequisite for systemic lymphomagenesis; unlike PCNSL, systemic lymphomas may also be EBV-negative. Nonetheless, the oncogenic properties of EBV play a critical role; EBV-infected B cells may overcome EBV-negative B cells due to the proliferating activity and activation of telomerase (Terrin et al., 2007, 2008; Dolcetti and De Rossi, 2012); most systemic tumors are in fact EBV-positive (Figure 1). Current strategies to treat HIV-1 are efficient in decreasing HIV-1 plasmaviremia and restoring immune functions but may be not efficient enough in reducing immune activation (Regidor et al., 2011). The new strategies to treat HIV-1 should take into account this important parameter. The prevention of chronic immune activation may prevent the onset of EBV-related diseases.
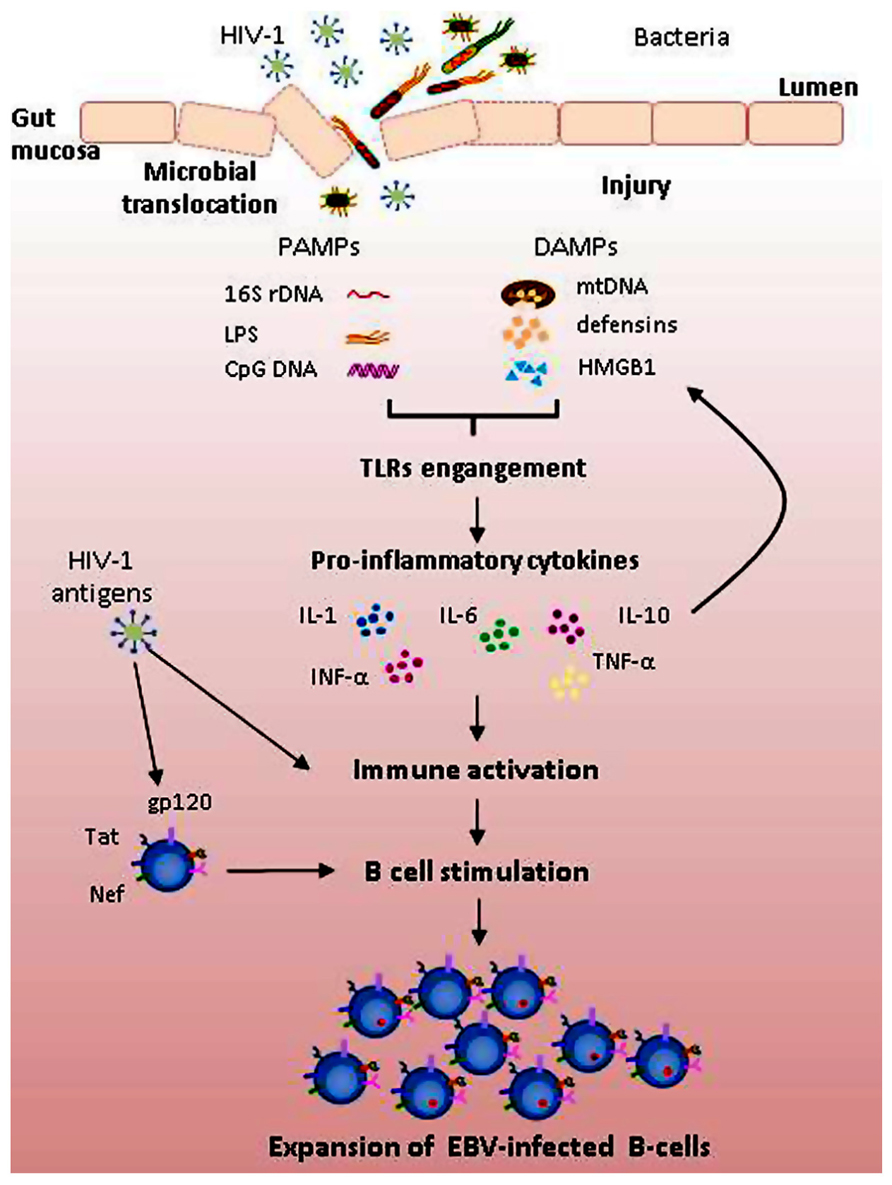
FIGURE 1. Mechanisms potentially contributing to polyclonal B cell activation and expansion of EBV-infected B cells in HIV-1 infected patients. HIV-1 acts as both a direct or indirect activator of B cells. Direct effects of HIV-1 include binding to B cells of HIV-1 viral proteins, such as gp120, Tat, and Nef, promoting polyclonal B cell activation. Indirect effects result from HIV-1-induced immune activation. Breakdown of gut mucosa induced by HIV-1 antigens increases intestinal permeability, resulting in translocation of microbial products. Pathogen-associated molecular patterns (PAMPs), such as LPS, 16S rDNA, and CpG DNA, and endogenous molecules created upon tissue injury (damage-associated molecular patterns, DAMPs), such as mitochondrial DNA (mtDNA), defensins and high mobility group box 1 (HMGB1) protein, by engaging Toll-like receptors (TLRs) activate signaling cascade leading to increased transcription of pro-inflammatory cytokines. In this context, both EBV-infected and uninfected B cells are activated and may undergo proliferation; EBV-infected cells may overcome uninfected ones due to higher replicative capacity and telomerase activation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the PENTA Foundation, and the Associazione Italiana per la Ricerca sul Cancro (AIRC).
References
Ambinder, R. F., Bhatia, K., Martinez-Maza, O., and Mitsuyasu, R. (2010). Cancer biomarkers in HIV patients. Curr. Opin. HIV AIDS 5, 531–536. doi: 10.1097/COH.0b013e32833f327e
Anselmi, A., Vendrame, D., Rampon, O., Giaquinto, C., Zanchetta, M., and De Rossi, A. (2007). Immune reconstitution in human immunodeficiency virus type 1-infected children with different virological responses to anti-retroviral therapy. Clin. Exp. Immunol. 150, 442–450. doi: 10.1111/j.1365-2249.2007.03526.x
Audigé, A., Schlaepfer, E., von Wyl, V., Miller, R. C., Vernazza, P., Nadal, D., et al. (2010). B cells from HIV-infected patients with primary central nervous system lymphoma display an activated phenotype and have a blunted TNF-α response to TLR9 triggering. AIDS Res. Hum. Retroviruses 26, 1063–1074. doi: 10.1089/aid.2009.0288
Baiocchi, R. A., and Caligiuri, M. A. (1994). Low-dose interleukin 2 prevents the development of Epstein–Barr virus (EBV)-associated lymphoproliferative disease in scid/scid mice reconstituted i.p. with EBV-seropositive human peripheral blood lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 91, 5577–5581. doi: 10.1073/pnas.91.12.5577
Beral, V., and Newton, R. (1998). Overview of the epidemiology of immunodeficiency-associated cancers. J. Natl. Cancer Inst. Monogr. 23, 1–6. doi: 10.1093/oxfordjournals.jncimonographs.a024164
Berberian, L., Goodglick, L., Kipps, T. J., and Braun, J. (1993). Immunoglobulin VH3 gene products: natural ligands for HIV gp120. Science 261, 1588–1591. doi: 10.1126/science.7690497
Biggar, R. J., Chaturvedi, A. K., Goedert, J. J., and Engels, E. A.; HIV/AIDS Cancer Match Study. (2007). AIDS-related cancer and severity of immunosuppression in persons with AIDS. J. Natl. Cancer Inst. 99, 962–972. doi: 10.1093/jnci/djm010
Blackburn, E. H., Greider, C. W., and Szostak, J. W. (2006). Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 12, 1133–1138. doi: 10.1038/nm1006-1133
Blazevic, V., Heino, M., Lagerstedt, A., Ranki, A., and Krohn, K. J. (1996). Interleukin-10 gene expression induced by HIV-1 Tat and Rev in the cells of HIV-1 infected individuals. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 13, 208–214. doi: 10.1097/00042560-199611010-00002
Breen, E. C., Hussain, S. K., Magpantay, L., Jacobson, L. P., Detels, R., Rabkin, C. S., et al. (2011). B-cell stimulatory cytokines and markers of immune activation are elevated several years prior to the diagnosis of systemic AIDS-associated non-Hodgkin B-cell lymphoma. Cancer Epidemiol. Biomarkers Prev. 20, 1303–13014. doi: 10.1158/1055-9965.EPI-11-0037
Brenchley, J. M., Price, D. A., Schacker, T. W., Asher, T. E., Silvestri, G., Rao, S., et al. (2006). Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 12, 1365–1371. doi: 10.1038/nm1511
Burighel, N., Ghezzi, S., Nozza, S., Del Bianco, P., Lazzarin, A., Tambussi, G., et al. (2006). Differential dynamics of Epstein–Barr virus in HIV-1 infected individuals receiving intermittent interleukin-2 and antiretroviral therapy. Haematologica 91, 244–247.
Carbone, A., Cesarman, E., Spina, M., Gloghini, A., and Schulz, T. F. (2009). HIV-associated lymphomas and gamma-herpesviruses. Blood 113, 1213–1224. doi: 10.1182/blood-2008-09-180315
Cattelan, A. M., Calabrò, M. L., Aversa, M. L., Zanchetta, M., Meneghetti, F., De Rossi, A., et al. (1999). Regression of AIDS-related Kaposi’s sarcoma following antiretroviral therapy with protease inhibitors: biological correlates of clinical outcome. Eur. J. Cancer 35, 1809–1815. doi: 10.1016/ S0959-8049(99)00161-6
Chen, M. R. (2011). Epstein–Barr virus, the immune system, and associated diseases. Front. Microbiol. 2:5. doi: 10.3389/fmicb.2011.00005
Collaboration of Observational HIV Epidemiological Research Europe (COHERE) Study Group. (2009). Incidence and risk factors of HIV-related non-Hodgkin’s lymphoma in the era of combination antiretroviral therapy: a European multicohort study. Antivir. Ther. 14, 1065–1074. doi: 10.3851/IMP1462
Colombrino, E., Rossi, E., Ballon, G., Terrin, L., Indraccolo, S., Chieco-Bianchi, L., et al. (2004). Human immunodeficiency virus type 1 Tat protein modulates cell cycle and apoptosis in Epstein–Barr virus-immortalized B cells. Exp. Cell Res. 295, 539–548. doi: 10.1016/j.yexcr.2004.01.018
Deeken, J. F., Tjen-A-Looi, A., Rudek, M. A., Okuliar, C., Young, M., Little, R. F., et al. (2012). The rising challenge of non-AIDS-defining cancers in HIV-infected patients. Clin. Infect. Dis. 55, 1228–1235. doi: 10.1093/cid/cis613
Deeks, S. G., Kitchen, C. M., Liu, L., Guo, H., Gascon, R., Narváez, A. B., et al. (2004). Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood 104, 942–947. doi: 10.1182/blood-2003-09-3333
De Rossi, A., Ometto, L., Roncella, S., D’Andrea, E., Menin, C., Calderazzo, F., et al. (1994). HIV-1 induces down-regulation of bcl-2 expression and death by apoptosis of EBV-immortalized B cells: a model for a persistent “self-limiting” infection. Virology 198, 234–244. doi: 10.1006/viro.1994.1026
Dolcetti, R., and De Rossi, A. (2012). Telomere/telomerase interplay in virus-driven and virus-independent lymphomagenesis:pathogenetic and clinical implications. Med. Res. Rev. 32, 233–253. doi: 10.1002/med.20211
Draborg, A. H., Duus, K., and Houen, G. (2012). Epstein–Barr virus and systemic lupus erythematosus. Clin. Dev. Immunol. 2012, 370516. doi: 10.1155/2012/370516
Engels, E. A., Pfeiffer, R. M., Landgren, O., and Moore, R. D. (2010). Immunologic and virologic predictors of AIDS-related non-Hodgkin lymphoma in the highly active antiretroviral therapy era. J. Acquir. Immune Defic. Syndr. 54, 78–84. doi: 10.1097/01.qai.0000371677.48743.8d
Franceschi, S., Lise, M., Clifford, G. M., Rickenbach, M., Levi, F., Maspoli, M., et al. (2010). Changing patterns of cancer incidence in the early- and late-HAART periods: the Swiss HIV Cohort Study. Br. J. Cancer 103, 416–422. doi: 10.1038/sj.bjc.6605756
Frankel, A. D., and Pabo, C. O. (1988). Cellular uptake of the tat protein from human immunodeficiency virus. Cell 55, 1189–1193. doi: 10.1016/0092-8674(88)90263-2
Giunco, S., Dolcetti, R., Keppel, S., Celeghin, A., Indraccolo, S., Dal Col, J., et al. (2013). hTERT inhibition triggers Epstein–Barr virus lytic cycle and apoptosis in immortalized and transformed B cells: a basis for new therapies. Clin. Cancer Res. 19, 2036–2047. doi: 10.1158/1078-0432.CCR-12-2537
Goedert, J. J., Coté, T. R., Virgo, P., Scoppa, S. M., Kingma, D. W., Gail, M. H., et al. (1998). Spectrum of AIDS-associated malignant disorders. Lancet 351, 1833–1839. doi: 10.1016/S0140-6736(97)09028-4
Gougeon, M. L., Lecoeur, H., Dulioust, A., Enouf, M. G., Crouvoiser, M., Goujard, C., et al. (1996). Programmed cell death in peripheral lymphocytes from HIV-infected persons: increased susceptibility to apoptosis of CD4 and CD8 T cells correlates with lymphocyte activation and with disease progression. J. Immunol. 156, 3509–3520.
Grulich, A. E. (2009). Living longer with HIV: what does it mean for cancer risk? Curr. Opin. HIV AIDS 4, 1–2. doi: 10.1097/COH.0b013e32831c508a
Grulich, A. E., Wan, X., Law, M. G., Milliken, S. T., Lewis, C. R., Garsia, R. J., et al. (2000). B-cell stimulation and prolonged immune deficiency are risk factors for non-Hodgkin’s lymphoma in people with AIDS. AIDS 14, 133–140. doi: 10.1097/00002030-200001280-00008
Guiguet, M., Boué, F., Cadranel, J., Lang, J. M., Rosenthal, E., Costagliola, D., et al. (2009). Effect of immunodeficiency, HIV viral load, and antiretroviral therapy on the risk of individual malignancies (FHDH-ANRS CO4): a prospective cohort study. Lancet Oncol. 10, 1152–1159. doi: 10.1016/S1470-2045(09)70282-7
Hazenberg, M. D., Stuart, J. W., Otto, S. A., Borleffs, J. C., Boucher, C. A., de Boer, R. J., et al. (2000). T-cell division in human immunodeficiency virus (HIV)-1 infection is mainly due to immune activation: a longitudinal analysis in patients before and during highly active antiretroviral therapy (HAART). Blood 95, 249–255.
He, B., Qiao, X., Klasse, P. J., Chiu, A., Chadburn, A., Knowles, D. M., et al. (2006). HIV-1 envelope triggers polyclonal Ig class switch recombination through a CD40-independent mechanism involving BAFF and C-type lectin receptors. J. Immunol. 176, 3931–3941.
Hellerstein, M. K., Hoh, R. A., Hanley, M. B., Cesar, D., Lee, D., Neese, R. A., et al. (2003). Subpopulations of long-lived and short-lived T cells in advanced HIV-1 infection. J. Clin. Invest. 112, 956–966. doi: 10.1172/JCI17533
Hong, G. K., Kumar, P., Wang, L., Damania, B., Gulley, M. L., Delecluse, H. J., et al. (2005). Epstein–Barr virus lytic infection is required for efficient production of the angiogenesis factor vascular endothelial growth factor in lymphoblastoid cell lines. J. Virol. 79, 13984–13992. doi: 10.1128/JVI.79.22.13984-13992.2005
Hunt, P. W., Brenchley, J., Sinclair, E., McCune, J. M., Roland, M., Page-Shafer, K., et al. (2008). Relationship between T cell activation and CD4+ T cell count in HIV-seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. J. Infect. Dis. 197, 126–133. doi: 10.1086/524143
International Collaboration on HIV and Cancer. (2000). Highly active antiretroviral therapy and incidence of cancer in human immunodeficiency virus-infected adults. J. Natl. Cancer Inst. 92, 1823–1830. doi: 10.1093/jnci/92.22.1823
Iyengar, S., and Schwartz, D. H. (2011). Potentiation of EBV-induced B cell transformation by CXBR4-tropic, but not CBR5-tropic, HIV gp120: implications for HIV-associated lymphomagenesis. AIDS Res. Hum. Retroviruses 27, 519–523. doi: 10.1089/aid.2010.0131
Jacobson, C. A., and Abramson, J. S. (2012). HIV-associated Hodgkin’s lymphoma: prognosis and therapy in the era of cART. Adv. Hematol. 2012, 507257. doi: 10.1155/2012/507257
Jacobson, L. E., Pilaro, F., and Smith, K. A. (1996). Rational interleukin 2 therapy for HIV positive individuals: daily low doses enhance immune function without toxicity. Proc. Natl. Acad. Sci. U.S.A. 93, 10405–10410. doi: 10.1073/pnas.93.19.10405
Janeway, C. A. Jr., and Medzhitov, R. (2002). Innate immune recognition. Annu. Rev. Immunol. 20, 197–216. doi: 10.1146/annurev.immunol.20.083001.084359
Kaplan, L. D. (2012). HIV-associated lymphoma. Best Pract. Res. Clin. Haematol. 25, 101–117. doi: 10.1016/j.beha.2012.01.001
Ladell, K., Dorner, M., Zauner, L., Berger, C., Zucol, F., Bernasconi, M., et al. (2007). Immune activation suppresses initiation of lytic Epstein–Barr virus infection. Cell. Microbiol. 9, 2055–2069. doi: 10.1111/j.1462-5822.2007.00937.x
Lazzi, S., Bellan, C., De Falco, G., Cinti, C., Ferrari, F., Nyongo, A., et al. (2002). Expression of RB2/p130 tumor-suppressor gene in AIDS-related non-Hodgkin’s lymphomas: implications for disease pathogenesis. Hum. Pathol. 33, 723–731. doi: 10.1053/hupa.2002.125372
Ma, S. D., Hegde, S., Young, K. H., Sullivan, R., Rajesh, D., Zhou, Y., et al. (2011). A new model of Epstein–Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 85, 165–177. doi: 10.1128/JVI.01512-10
Malaspina, A., Moir, S., Ho, J., Wang, W., Howell, M. L., O’Shea, M. A., et al. (2006). Appearance of immature/transitional B cells in HIV-infected individuals with advanced disease: correlation with increased IL-7. Proc. Natl. Acad. Sci. U.S.A. 103, 2262–2267. doi: 10.1073/pnas.0511094103
Malaspina, A., Moir, S., Nickle, D. C., Donoghue, E. T., Ogwaro, K. M., Ehler, L. A., et al. (2002). Human immunodeficiency virus type 1 bound to B cells: relationship to virus replicating in CD4+ T cells and circulating in plasma. J. Virol. 76, 8855–8863. doi: 10.1128/JVI.76.17.8855-8863.2002
Malnati, M., Broccolo, F., Nozza, S., Sarmati, L., Ghezzi, S., Locatelli, G., et al. (2002). Retrospective analysis of HHV-8 viremia and cellular viral load in HIV-seropositive patients receiving interleukin 2 in combination with antiretroviral therapy. Blood 100, 1575–1578.
Mandl, J. N., Barry, A. P., Vanderford, T. H., Kozyr, N., Chavan, R., Klucking, S., et al. (2008). Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat. Med. 14, 1077–1087. doi: 10.1038/nm.1871
Marks, M. A., Rabkin, C. S., Engels, E. A., Busch, E., Kopp, W., Rager, H., et al. (2013). Markers of microbial translocation and risk of AIDS-related lymphoma. AIDS 27, 469–474. doi: 10.1097/QAD.0b013e32835c1333
Moir, S., and Fauci, A. S. (2009). B cells in HIV infection and disease. Nat. Rev. Immunol. 9, 235–245. doi: 10.1038/nri2524
Moir, S., Malaspina, A., Li, Y., Chun, T. W., Lowe, T., Adelsberger, J., et al. (2000). B cells of HIV-1-infected patients bind virions through CD21 complement interactions and transmit infectious virus to activated T cells. J. Exp. Med. 192, 637–646. doi: 10.1084/jem.192.5.637
Ometto, L., Menin, C., Masiero, S., Bonaldi, L., Del Mistro, A., Cattelan, A. M., et al. (1997). Molecular profile of Epstein–Barr virus (EBV) in human immunodeficiency virus type 1 (HIV-1)-related lymphadenopathies and lymphomas. Blood 90, 313–322.
Palella, F. J. Jr., Delaney, K. M., Moorman, A. C., Loveless, M. O., Fuhrer, J., Satten, G. A., et al. (1998). Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N. Engl. J. Med. 338, 853–860. doi: 10.1056/NEJM199803263381301
Petoumenos, K., van Leuwen, M. T., Vajdic, C. M., Woolley, I., Chuah, J., Templeton, D. J., et al. (2013). Cancer, immunodeficiency and antiretroviral treatment: results from the Australian HIV Observational Database (AHOD). HIV Med. 14, 77–84. doi: 10.1111/j.1468-1293.2012.01038.x
Petrara, M. R., Cattelan, A. M., Zanchetta, M., Sasset, L., Freguja, R., Gianesin, K., et al. (2012). Epstein–Barr virus load and immune activation in human immunodeficiency virus type 1-infected patients. J. Clin. Virol. 53, 195–200. doi: 10.1016/j.jcv.2011.12.013
Powles, T., Robinson, D., Stebbing, J., Shamash, J., Nelson, M., Gazzard, B., et al. (2009). Highly active antiretroviral therapy and the incidence of non-AIDS-defining cancers in people with HIV infection. J. Clin. Oncol. 27, 884–890. doi: 10.1200/JCO.2008.19.6626
Qiao, X., He, B., Chiu, A., Knowles, D. M., Chadburn, A., and Cerutti, A. (2006). Human immunodeficiency virus 1 Nef suppresses CD40-dependent immunoglobulin class switching in bystander B cells. Nat. Immunol. 7, 302–310. doi: 10.1038/ni1302
Rappocciolo, G., Piazza, P., Fuller, C. L., Reinhart, T. A., Watkins, S. C., Rowe, D. T., et al. (2006). DC-SIGN on B lymphocytes is required for transmission of HIV-1 to T lymphocytes. PLoS Pathog. 2:e70. doi: 10.1371/journal.ppat.0020070
Regidor, D. L., Detels, R., Breen, E. C., Widney, D. P., Jacobson, L. P., Palella, F., et al. (2011). Effect of highly active antiretroviral therapy on biomarkers of B-lymphocyte activation and inflammation. AIDS 25, 303–314. doi: 10.1097/QAD.0b013e32834273ad
Rieckmann, P., Poli, G., Fox, C. H., Kehrl, J. H., and Fauci, A. S. (1991). Recombinant gp120 specifically enhances tumor necrosis factor-alpha production and Ig secretion in B lymphocytes from HIV-infected individuals but not from seronegative donors. J. Immunol. 147, 2922–2927.
Righetti, E., Ballon, G., Ometto, L., Cattelan, A. M., Menin, C., Zanchetta, M., et al. (2002). Dynamics of Epstein–Barr virus in human immunodeficiency virus type 1-infected subjects on highly active antiretroviral therapy. AIDS 16, 63–73. doi: 10.1097/00002030-200201040-00009
Scaffidi, P., Misteli, T., and Bianchi, M. E. (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195. doi: 10.1038/nature00858
Scala, G., Ruocco, M. R., Ambrosino, C., Mallardo, M., Giordano, V., Baldassarre, F., et al. (1994). The expression of the interleukin 6 gene is induced by the human immunodeficiency virus 1 TAT protein. J. Exp. Med. 179, 961–971. doi: 10.1084/jem.179.3.961
Schnittman, S. M., Lane, H. C., Higgins, S. E., Folks, T., and Fauci, A. S. (1986). Direct polyclonal activation of human B lymphocytes by the acquired immune deficiency syndrome virus. Science 233, 1084–1086. doi: 10.1126/science.3016902
Simard, E. P., Pfeiffer, R. M., and Engels, E. A. (2011). Cumulative incidence of cancer among individuals with acquired immunodeficiency syndrome in the United States. Cancer 117, 1089–1096. doi: 10.1002/cncr.25547
Sørensen, O. E., Thapa, D. R., Rosenthal, A., Liu, L., Roberts, A. A., and Ganz, T. (2005). Differential regulation of beta-defensin expression in human skin by microbial stimuli. J. Immunol. 174, 4870–4879.
Sparano, J. A. (2001). Clinical aspects and management of AIDS-related lymphomas. Eur. J. Cancer 37, 1296–1305. doi: 10.1016/S0959-8049(01)00111-3
Swingler, S., Zhou, J., Swingler, C., Dauphin, A., Greenough, T., Jolicoeur, P., et al. (2008). Evidence for a pathogenic determinant in HIV-1 Nef involved in B cell dysfunction in HIV/AIDS. Cell Host Microbe 4, 63–76. doi: 10.1016/j.chom.2008.05.015
Takeshita, S., Breen, E. C., Ivashchenko, M., Nishanian, P. G., Kishimoto, T., Vredevoe, D. L., et al. (1995). Induction of IL-6 and IL-10 production by recombinant HIV-1 envelope glycoprotein 41 (gp41) in the THP-1 human monocytic cell line. Cell. Immunol. 165, 234–242. doi: 10.1006/cimm.1995.1210
Terrin, L., Dal Col, J., Rampazzo, E., Zancai, P., Pedrotti, M., Ammirabile, G., et al. (2008). Latent membrane protein 1 of Epstein–Barr virus activates the hTERT promoter and enhances telomerase activity in B lymphocytes. J. Virol. 82, 10175–10187. doi: 10.1128/JVI.00321-08
Terrin, L., Dolcetti, R., Corradini, I., Indraccolo, S., Dal Col, J., Bertorelle, R., et al. (2007). hTERT inhibits the Epstein–Barr virus (EBV) lytic cycle and promotes the proliferation of primary B lymphocytes: implications for EBV-driven lymphomagenesis. Int. J. Cancer 121, 576–587. doi: 10.1002/ijc.22661
Tirelli, U., and Bernardi, D. (2001). Impact of HAART on the clinical management of AIDS-related cancers. Eur. J. Cancer 37, 1320–1324. doi: 10.1016/S0959-8049(01)00106-X
Titanji, K., De Milito, A., Cagigi, A., Thorstensson, R., Grützmeier, S., Atlas, A., et al. (2006). Loss of memory B cells impairs maintenance of long-term serologic memory during HIV-1 infection. Blood 108, 1580–1587. doi: 10.1182/blood-2005-11-013383
Tyagi, M., Rusnati, M., Presta, M., and Giacca, M. (2001). Internalization of HIV-1 Tat requires cell surface heparan sulphate proteoglycans. J. Biol. Chem. 276, 3254–3256. doi: 10.1074/jbc.M006701200
Young, L. S., and Rickinson, A. B. (2004). Epstein–Barr virus: 40 years on. Nat. Rev. Cancer 4, 757–768. doi: 10.1038/nrc1452
Zauner, L., and Nadal, D. (2012). Understanding TLR9 action in Epstein–Barr virus infection. Front. Biosci. (Landmark Ed.) 17: 1219–1231. doi: 10.2741/3982
Keywords: EBV, HIV-1, B cell activation, chronic immune activation, EBV-related malignancies, antiretroviral therapy, EBV lytic reactivation
Citation: Petrara MR, Freguja R, Gianesin K, Zanchetta M and De Rossi A (2013) Epstein–Barr virus-driven lymphomagenesis in the context of human immunodeficiency virus type 1 infection. Front. Microbiol. 4:311. doi: 10.3389/fmicb.2013.00311
Received: 09 May 2013; Accepted: 28 September 2013;
Published online: 18 October 2013.
Edited by:
Mei-Ru Chen, National Taiwan University, TaiwanReviewed by:
Mei-Ru Chen, National Taiwan University, TaiwanChi-Ju Chen, National Yang-Ming University, Taiwan
Copyright © 2013 Petrara, Freguja, Gianesin, Zanchetta and De Rossi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anita De Rossi, Viral Oncology Unit and AIDS Reference Center, Section of Oncology and Immunology, Department of Surgery, Oncology and Gastroenterology, University of Padova, Via Gattamelata 64, 35128 Padova, Italy e-mailYW5pdGEuZGVyb3NzaUB1bmlwZC5pdA==