 Anastasia Bragina
Anastasia Bragina Massimiliano Cardinale
Massimiliano Cardinale Christian Berg
Christian Berg Gabriele Berg
Gabriele Berg- 1Institute of Environmental Biotechnology, Graz University of Technology, Graz, Austria
- 2Institute of Plant Sciences, Karl-Franzens University of Graz, Graz, Austria
The betaproteobacterial genus Burkholderia is known for its versatile interactions with its hosts that can range from beneficial to pathogenic. A plant-beneficial-environmental (PBE) Burkholderia cluster was recently separated from the pathogen cluster, yet still little is known about burkholderial diversity, distribution, colonization, and transmission patterns on plants. In our study, we applied a combination of high-throughput molecular and microscopic methods to examine the aforementioned factors for Burkholderia communities associated with Sphagnum mosses – model plants for long-term associations – in Austrian and Russian bogs. Analysis of 16S rRNA gene amplicons libraries revealed that most of the Burkholderia are part of the PBE group, but a minor fraction was closely related to B. glathei and B. andropogonis from the pathogen cluster. Notably, Burkholderia showed highly similar composition patterns for each moss species independent of the geographic region, and Burkholderia-specific fluorescent in situ hybridization of Sphagnum gametophytes exhibited similar colonization patterns in different Sphagnum species at multi-geographic scales. To explain these patterns, we compared the compositions of the surrounding water, gametophyte-, and sporophyte-associated microbiome at genus level and discovered that Burkholderia were present in the Sphagnum sporophyte and gametophyte, but were absent in the flark water. Therefore, Burkholderia is a part of the core microbiome transmitted from the moss sporophyte to the gametophyte. This suggests a vertical transmission of Burkholderia strains, and thus underlines their importance for the plants themselves.
Introduction
The genus Burkholderia, which was described by Yabuuchi et al. (1992), encompasses a diverse group of Betaproteobacteria with currently more than 60 validly described species. Burkholderia species are known for their beneficial as well as pathogenic interaction with plants, animals, and humans (Coenye and Vandamme, 2003). In the past, most studies focused on the pathogenic species for their enormous clinical importance (Mahenthiralingam et al., 2005). Recently, a specific plant-beneficial-environmental (PBE) Burkholderia cluster that contains non-pathogenic species was divided from the cluster that comprises human, animal and plant pathogens (Caballero-Mellado et al., 2007; Suárez-Moreno et al., 2010, 2012). However, there is no clear border between both groups especially within the plant-associated species; for example B. glathei was suggested to be transfered from the pathogenic to the PBE group (Verstraete et al., 2013). Many PBE members belong to Burkholderia species symbiotic to tropical plants; each nodulating plant species is colonized by a single unique endophytic Burkholderia species (Van Oevelen et al., 2002; Lemaire et al., 2011). Several species from the PBE cluster share characteristics that are of use in association with plants, such as quorum sensing systems, the presence of nitrogen fixation and/or nodulation genes, and the ability to degrade aromatic compounds (Suárez-Moreno et al., 2012), and many of them are characterized by an endophytic lifestyle (Sessitsch et al., 2005; Gasser et al., 2009; Mitter et al., 2013). While single strains of the PBE cluster are already well-characterized, little is known about the ecology and colonization pattern of Burkholderia species on plants.
Plants have been recognized as meta-organisms due to their close symbiotic relationship with their microbiome that fulfills important host functions (Berg, 2009; Bulgarelli et al., 2012; Hirsch and Mauchline, 2012; Lundberg et al., 2012; Berg et al., 2013). These advances were driven by both “omic”-technologies guided by next-generation sequencing (NGS) and microscopic insights (Berendsen et al., 2012; Jansson et al., 2012). Mosses belong to the phylogenetically oldest group of land plants on Earth, and their long-term intense relationship with their associated microbes has contributed to the co-evolution of a highly specific microbiome (Opelt and Berg, 2004; Opelt et al., 2007c; Bragina et al., 2012). Therefore, mosses are important models in studying plant-microbe interactions and the ecology of plant-associated bacteria. The genus Sphagnum is among the most abundant and cosmopolitan of bog vegetation in the Northern hemisphere, and greatly contributes to both global carbon turnover and global climate (Raghoebarsing et al., 2005; Dise, 2009). The ecological significance of bogs is directly related to the physical, morphological, and chemical characteristics of Sphagnum peat mosses which set Sphagnum apart from other mosses in practically every stage of the life cycle (Daniels and Eddy, 1985). Burkholderia species play an important role for Sphagnum mosses and peatland ecosystem (Opelt et al., 2007a,b), and new Burkholderia species, which belong to the PBE cluster, have recently been isolated from these mosses (Vandamme et al., 2007). However, their composition and occurrence on Sphagnum at various geographical scales—ranging from the moss gametophyte and sporophyte up to continental level—is not yet understood. We hypothesize that Sphagnum species are colonized by specific Burkholderia from the PBE cluster independent from the geographic region.
To study this hypothesis and understand the ecological role, composition, colonization, as well as distribution pattern on plants, we studied Burkholderia communities on two Sphagnum species (S. magellanicum and S. fallax) associated with different a-biotic parameters from different bogs in Austria and Russia. We used an assortment of methods combining the analysis of Burkholderia-specific 16S rRNA gene pyrosequencing libraries with FISH-CLSM analysis. Furthermore, we compared the compositions of water, gametophyte-, and sporophyte-associated microbiomes to understand the transmission and distribution patterns of the Burkholderia communities.
Materials and Methods
Sampling Design
To analyze the diversity and distribution pattern of the Sphagnum-associated Burkholderia community, S. magellanicum BRID. (section Sphagnum) and S. fallax H. KLINGGR. (section Cuspidata) were selected. Both bryophytes are members of the typical and cosmopolitan vegetation in peat bogs (Daniels and Eddy, 1985). Adult gametophytes of mosses were sampled in three acidic peat bogs in Austria and in three acidic peat bogs in Russia in September 2009 and July 2010, respectively (Table S1). Four single replicates per Sphagnum species (15–20 plantlets) were collected in each of the investigated bogs at a minimum distance of about 40 m from each other. The plant samples were placed into sterile plastic bags and transported to the laboratory. In addition, two sporophyte samples of S. fallax consisting of enclosed spore capsules, and one water sample from a small wet depression (flark) were collected into sterile screw cap tubes and processed separately.
Total-Community DNA Isolation
The microbial fractions associated with moss gametophytes and sporophytes were extracted as previously described (Bragina et al., 2012). In short, 5 g of plant material were physically disrupted and resuspended in 10 ml of 0.85% NaCl. 2 ml of suspension were centrifuged at 13,000 r.p.m. for 20 min at 4°C and the supernatant was discarded. For extraction of the sporophyte-associated microbial community, 10 enclosed spore capsules per sample were surface-sterilized and ground with 1.5 ml of 0.85% NaCl. The ground suspension was centrifuged at 13,000 r.p.m. for 20 min at 4°C and the supernatant was discarded. The pellet from the flark water sample was obtained through several rounds of centrifugation at 10,000 r.p.m. for 15 min at 4°C until a constant pellet size was obtained. The resulting cell pellets were applied for isolation of the total-community DNA using the FastDNA® SPIN Kit for Soil (MP Biomedicals, Solon, OH, USA). Final aliquots of the total-community DNA were further used for a deep sequencing-approach.
454-Pyrosequencing and Bioinformatic Processing
The diversity of the Sphagnum-associated microbiome with a special focus on the genus Burkholderia was investigated using a barcoded pyrosequencing technology. For this purpose, 16S rDNA amplicons were generated using Taq-&Go™ Ready-to-use PCR Mix (MP Biomedicals, Solon, OH, USA). The total-community DNA of gametophyte samples was selectively amplified with Burkholderia-specific primers BKH143Fw/BKH1434Rw (Schönmann et al., 2009) followed by amplification with universal bacterial primers Unibac-II-515f/Unibac-II-927r (Lieber et al., 2003). In addition, the total-community DNA of S. fallax gametophyte samples from the bog Pürgschachen Moor (Table S1) and flark water sample was amplified with universal bacterial primers Unibac-II-515f/Unibac-II-927r. The total-community DNA of sporophyte samples was amplified with universal bacterial primers 799f/1492r (Lane, 1991; Chelius and Triplett, 2001) because application of the Unibac-II-515f/Unibac-II-927r achieved mostly plant-derived sequences (data of preliminary experiments). Primer sequences are listed in Table 1. Duplicate PCR products from all templates were purified with Wizard® SV Gel and PCR Clean-Up System (Promega, Madison, WI, USA). Amplicons derived from the same Sphagnum sp. and sampling site were pooled in equimolar ratios and subjected to pyrosequencing using the Roche 454 GS FLX and FLX+ Titanium platforms performed by LGC Genomics (Berlin, Germany) and Eurofins MWG (Ebersberg, Germany), respectively. In total, we produced 12 pyrosequencing libraries specific for Burkholderia and four general bacterial pyrosequencing libraries.
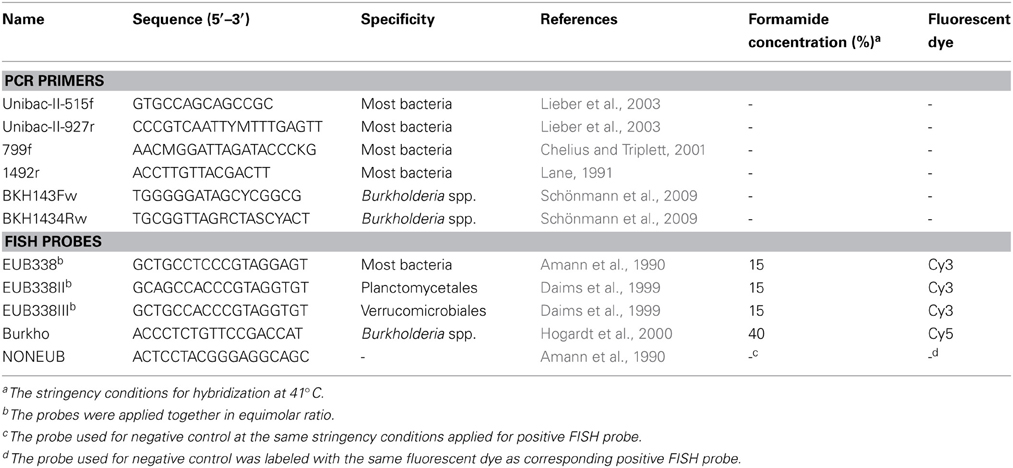
Table 1. Nucleotide probes used for the PCR and FISH.
The 16S rDNA pyrosequencing libraries were processed using the open source software package Quantitative Insights Into Microbial Ecology (QIIME) version 1.6.0 (Caporaso et al., 2010) with default parameters. The raw datasets were de-multiplexed, the primer sequences were truncated, and the datasets were filtered by removing sequences of low-quality (quality score, <25), short sequences (<200 bp), and sequences containing ambiguous characters and/or homopolymers (>6 bp). The quality-filtered datasets were de-noised and chimeras were removed if present. Sequences were clustered into operational taxonomic units (OTUs) using the UCLUST algorithm with a 97% similarity cut-off (Schloss and Handelsman, 2006; Edgar, 2010). The most abundant sequence within each OTU was taxonomically assigned using the Ribosomal Database Project (RDP) classifier with 80% confidence threshold (Wang et al., 2007). To refine the analysis, generated OTU-tables were filtered based on taxonomic metadata: OTUs classified to genera other than Burkholderia and OTUs containing chloroplast-derived sequences were removed from the burkholderial and general bacterial OTU-tables, correspondingly. Rarefaction analysis was performed for the complete datasets, while richness and diversity estimations were performed by calculating Chao1 and Shannon (H′) indices for the datasets normalized to the same number of sequences. For the general bacterial datasets, the occurrence of bacterial taxa was analyzed using the normalized datasets. Beta-diversity of the burkholderial datasets was analyzed using weighted UniFrac distance metric (Lozupone et al., 2010) and jackknife re-sampling (1,781 sequences per sample × 100 times). Statistical analysis was performed for the normalized datasets using the adonis test with 999 permutations (http://qiime.org/tutorials/category_comparison.html).
Representative sequences of the burkholderial OTUs were aligned with reference sequences from the non-redundant nucleotide database (nt) of the NCBI server using the BLASTN algorithm. A bootstrapped neighbor-joining phylogenetic tree of the representative sequences and the closest database matches was constructed using software packages ClustalX version 2.0.12 (Larkin et al., 2007), Phylip version 3.69 (Felsenstein, 1989), and MEGA version 4.0 (Tamura et al., 2007) as previously described (Bragina et al., 2012).
Sequence Accession Numbers
The raw pyrosequencing data was deposited in the European Nucleotide Archive (ENA) under the project number PRJEB4660 with the accession numbers ERR361316–ERR361331.
Fluorescent In Situ Hybridization and Confocal Laser Scanning Microscopy
Single plants of S. magellanicum and S. fallax were fixed with 4% paraformaldehyde/phosphate buffered salt (3:1, v/v) and stained by in-tube FISH (Grube et al., 2009). The samples were hybridized with rRNA-targeting probes (genXpress, Wiener Neudorf, Austria) specific for Burkholderia and with a set of universal bacterial probes. Hybridization was carried out at 41°C. The probes and corresponding stringency conditions are listed in Table 1. Confocal laser scanning microscopy (CLSM) was performed with a Leica TCS SPE confocal microscope (Leica Microsystems, Mannheim, Germany) as previously described (Bragina et al., 2012) followed by volume rendering of confocal stacks using the software Imaris 7.3 (Bitplane, Zurich, Switzerland).
Results
Sphagnum Mosses are Preferentially Colonized by Burkholderial Strains From the PBE Cluster and A Minor Community Fraction Belongs to the Plant-Pathogenic Cluster
High-throughput analysis of the Burkholderia community was achieved via an amplicon pyrosequencing approach targeting the V4–V5 region of the 16S rRNA gene. The pyrosequencing of 12 amplicon libraries of S. fallax and S. magellanicum samples from Austria and Russia retrieved 149,024 raw sequences (Table 2). After initial processing, 87,917 quality sequences (average length, 405 bp) specific for Burkholderia genus were subjected to a detailed investigation. Rarefaction analysis of the pyrosequencing libraries, which were clustered with 97% sequence similarity, resulted in similar saturation profiles for all Sphagnum samples (Figure S1). Richness estimation of the normalized datasets revealed that the current pyrosequencing survey attained 81.1-100% of the estimated richness (Table 2). Low values of the Shannon diversity index (0.21–0.90, Table 2) indicated that the retrieved burkholderial communities contained a low number of highly abundant phylotypes. Through the use of automatic classification of the representative sequences, these phylotypes were assigned to B. bryophila, B. andropogonis, and B. glathei with several of them remaining unclassified at species level (Figure 1). According to the division of the genus Burkholderia sensu Suárez-Moreno et al. (2012), the most abundant B. bryophila species belongs to the plant-beneficial cluster, while minor B. andropogonis and B. glathei species are within the plant-pathogenic cluster.
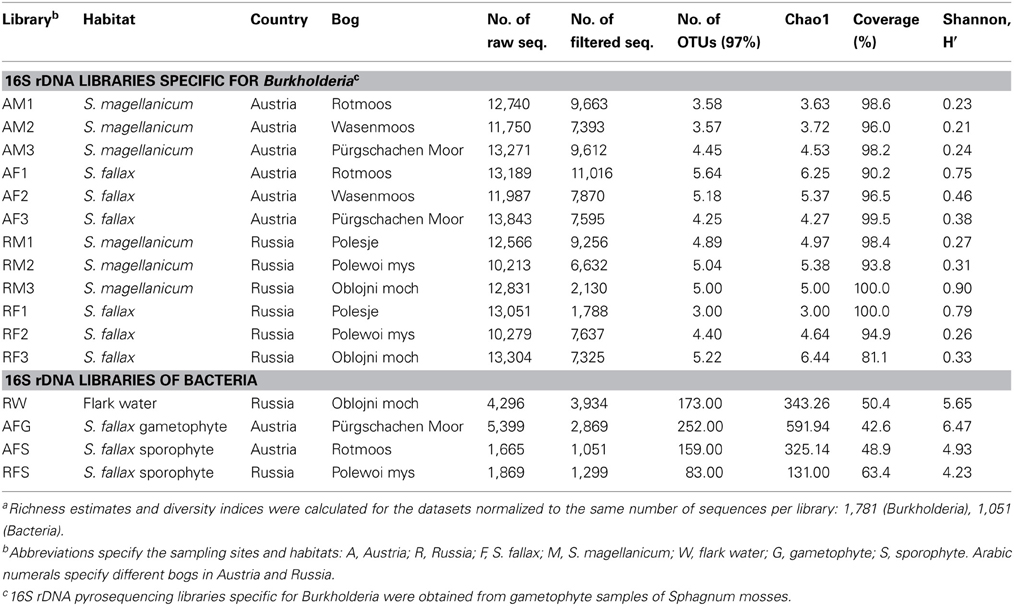
Table 2. Description and alpha-diversity estimation of the 16S rDNA pyrosequencing libraries of Sphagnum samplesa.
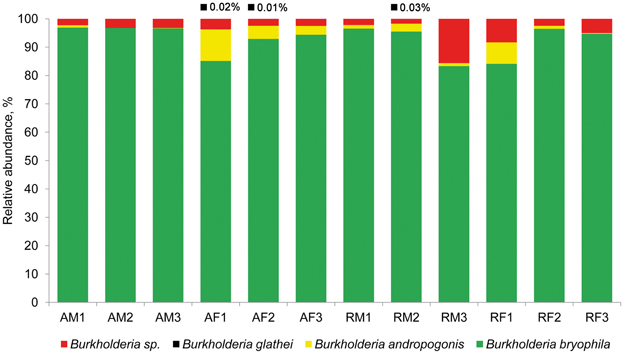
Figure 1. Taxonomic classification of burkholderial communities associated with Sphagnum mosses. Bar charts represent the composition of Burkholderia-specific 16S rDNA pyrosequencing libraries classified using RDP-classifier with a confidence threshold of 80%. The burkholderial sequences that remained unclassified at the species level are shown as Burkholderia sp. (red). Black squares and percentage values above the bar charts show occurrence and abundance of B. glathei. Abbreviations: A, Austria; R, Russia; F, S. fallax; M, S. magellanicum. Arabic numerals specify different bogs in Austria and Russia.
To achieve a deeper insight into burkholderial diversity, we performed a phylogenetic analysis of the partial 16S rRNA gene sequences from pyrosequencing libraries and closely related environmental strains (Figure 2). The closest database matches showed ≥96% of sequence identity to representative burkholderial sequences from pyrosequencing libraries. Clustering of the representative and reference sequences on the phylogenetic tree reflected several ecological traits of the examined burkholderial community. Specifically, cluster 2 was formed from representative sequences (this study) and the B. bryophila strain LMG 23648, a plant growth-promoting and antagonistic bacterium that was originally isolated from mosses in a nature reserve bog in Germany (Vandamme et al., 2007). This cluster also contained burkholderial strains PB1, F4W, F4, and SB1 which were isolated from acidic peat bogs in Russia (Belova et al., 2006). The phylogenetic clusters 1, 3, 4, 5, and 6 were represented by various endophytic and rhizospheric bacteria. These bacteria included the endophytic mycorrhizal B. phenazinium clone WT1 5, burkholderial clones sen290 and mat480 associated with lupin cluster roots (Weisskopf et al., 2011), and burkholderial endophytes M1U5b and M1U23 that were isolated from the arctic plants (Nissinen et al., 2012). Within the clusters 5 and 6, we detected B. andropogonis strain W20, a causative agent of the leaf spot in betel palm, and SFecto-B3clone1 clone of B. glathei species, a free-living or moss-associated bacterium that is not considered a member of the PBE cluster (Opelt et al., 2007a; Suárez-Moreno et al., 2012). Interestingly, the representative sequence in these clusters showed higher sequence similarity (98–100%) with the harmless burkholderial strains M1U5b and M1U23 than with B. andropogonis and B. glathei sequences. Furthermore, cluster 7 contained bacteria from acidic and alpine soils. Overall, the phylogenetic analysis revealed that Sphagnum-associated Burkholderia are phylogenetically closely related to plant-beneficial and non-pathogenic Burkholderia from various acidic habitats, especially peat bogs, but also potential plant pathogens.
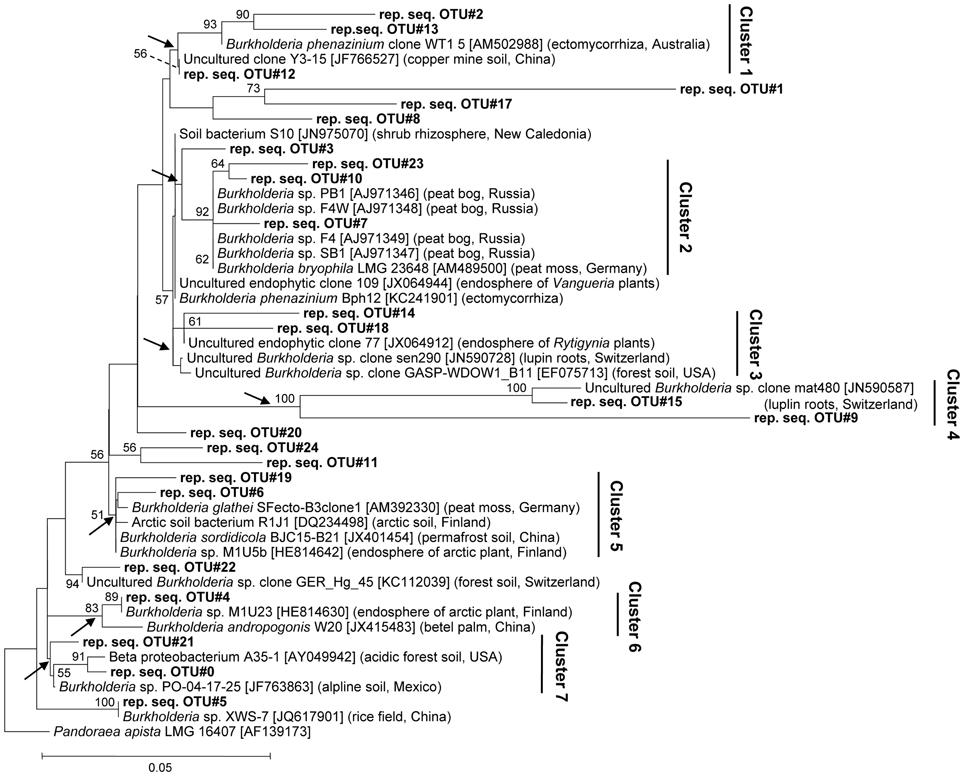
Figure 2. Neighbor-joining tree of Sphagnum-associated burkholderial communities. Phylogenetic relationships are shown for 16S rDNA sequence representing burkholderial OTUs (97%) (bold) and the nearest ecologically related reference sequences. The isolation sources and accession numbers of reference sequences are shown in brackets. Partial 16S rDNA sequence of Pandoraea apista strain LMG 16407 (AF139173) was used as an out-group. Numbers at nodes indicate bootstrap values out of 100 data re-samplings exceeding 50%. Arrows show monophyletic branches for cluster designation. Distance bar: 0.05 substitutions per site.
Burkholderia Communities of Sphagna Exhibit Similar Distribution and Colonization Patterns Independent of the Geographic Region
Biogeographical distribution of the Burkholderia communities was examined for the peat mosses S. fallax and S. magellanicum collected from Austrian and Russian bogs (Table S1). Burkholderia showed highly similar distribution patterns for the analyzed moss species independent of the geographic region (Figure 3). An average weighted UniFrac distance was 0.47% with a maximum value of 1.08% for S. magellanicum-associated communities in Russian bogs (Table S2). The statistical analysis using an adonis test confirmed that neither Sphagnum species (P = 0.261) nor geographic position (P = 0.363) had a significant influence on the beta-diversity of burkholderial communities.
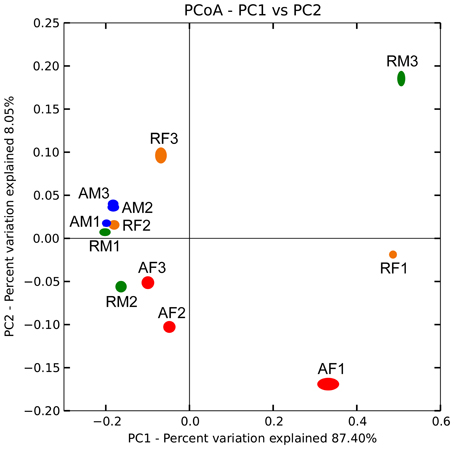
Figure 3. Comparison of burkholderial communities on Sphagnum mosses in Austrian and Russian bogs by principal coordinate analysis (PCoA). PCoA biplot is based on the weighted UniFrac distance matrix of the 16S rDNA pyrosequencing libraries specific for Burkholderia and supported by 100 jackknife data re-samplings using 1781 sequences per library. The single libraries are shown by colored ellipses: blue, S. magellanicum (Austria); red, S. fallax (Austria); green, S. magellanicum (Russia); orange, S. fallax (Russia). Letters and arabic numerals specify library names. Variation explained by each principal coordinate (PC) is defined on the biplot, respectively.
The general distribution of Burkholderia was confirmed through fluorescent in situ hybridization (FISH) of Sphagnum gametophytes with genus-specific and universal bacterial probes (Figure 4). Sphagnum mosses are characterized by a unique morphology (Daniels and Eddy, 1985) which makes them easily accessible for microbial colonization (Bragina et al., 2012). CLSM observation of hybridized plants showed that the Burkholderia community inhabited the leaves, but not the stem tissues of mosses. Straight and slightly curved rods of Burkholderia were detected in hyalocyte cells of leaves being likely attached to their cell walls. Inside the hyalocytes, Burkholderia remained as individual cells or formed microcolonies composed of few cells. Burkholderial cells were also found in association with other bacteria of unidentified taxonomy as shown in Figure 4A. Analysis of FISH-CLSM data showed that Burkholderia communities established similar colonization patterns in different Sphagnum species across the examined geographic scales.
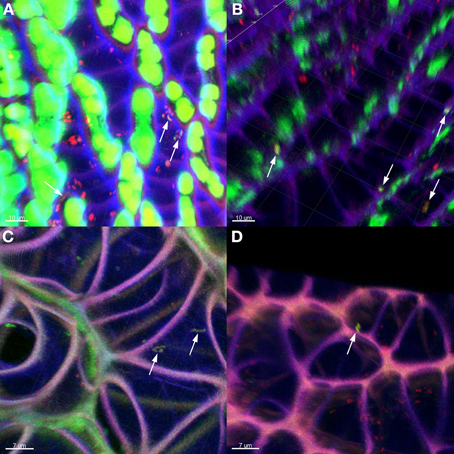
Figure 4. Detection of Burkholderia in Sphagnum plants by FISH-CLSM visualization. Branch leaves of S. fallax (A,B) and S. magellanicum (C,D) from Austrian (A,C) and Russian (B,D) bogs hybridized with Burkholderia-specific and universal bacterial probes. Yellow: Burkholderia spp. indicated by arrows; red: other bacteria; green: algae, Sphagnum chlorocytes; violet: moss cell walls. Scale bar = 10 μm (A,B); 7 μm (C,D).
Burkholderia are Vertically Transmitted Within the Core Microbiome Over Entire Life Cycle of the Host Plants
The 16S rDNA pyrosequencing libraries from the moss sporophyte, gametophyte, and flark water samples were compared to reveal potential transmission mechanisms of Sphagnum-associated bacteria with a special focus on the genus Burkholderia. The libraries were rarefied as shown in Figure S1. The pyrosequencing survey achieved 42.6–63.4% of total richness as estimated by the Chao1 index (Table 2). Classification of the normalized datasets revealed the occurrence of certain bacterial taxa in S. fallax and water microhabitats (Figure 5). Thus, Proteobacteria, Bacteroidetes, and Acidobacteria were among the most abundant phyla in all examined microhabitats. At class level, Alphaproteobacteria (within the phylum Proteobacteria) comprised the dominant portion of the plant-associated microbiome, while the water microbiome was dominated by Sphingobacteria (Bacteroidetes). Furthermore, a comparison of microbiome structure at family level revealed several different occurrence patterns. For instance, Acidobactriaceae (within the class Acidobacteria) were ubiquitously distributed unlike the family Xanthomonadaceae (Gammaproteobacteria) that specifically colonized the moss-associated microhabitats, gametophyte, and sporophyte. Moreover, several bacterial taxa, namely Methylocystaceae (Alphaproteobacteria), inhabited moss gametophytes and flark water.
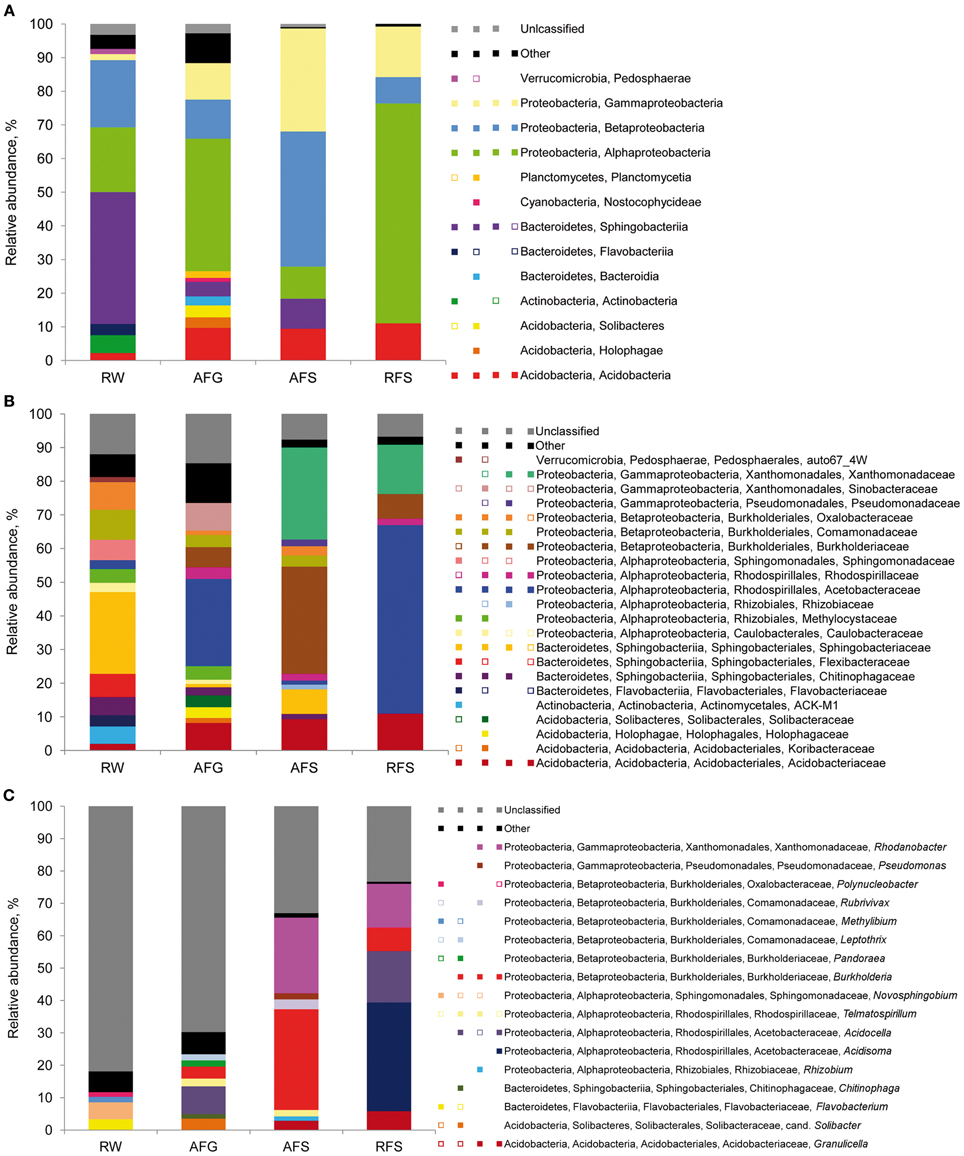
Figure 5. Taxonomic classification of bacterial communities of Sphagnum mosses and flark water. Bar charts represent the composition of 16S rDNA pyrosequencing libraries of Bacteria classified at class (A), family (B) and genus (C) level using RDP-classifier with a confidence threshold of 80%. Multi-colored charts in the legend represent occurrence of each taxon in each library correspondingly. Taxons below 1% of relative abundance are included in ‘Other’ and depicted as empty squares on the multi-colored charts. Abbreviations: A, Austria; R, Russia; W, flark water; F, S. fallax; G, gametophyte, S, sporophyte.
To study the occurrence of Burkholderia in various microhabitats, we compared the compositions of water, gametophyte-, and sporophyte-associated microbiomes at genus level. Consequently, Burkholderia were detected in the Sphagnum sporophyte and gametophyte, but were absent in the flark water. To ensure that normalization did not influence the Burkholderia occurrence pattern, the non-normalized pyrosequencing libraries were checked for the presence of this genus. The occurrence pattern of Burkholderia coincided between the normalized and non-normalized datasets (data not shown). However, pyrosequencing of the flark water microbiome achieved partial coverage of the estimated diversity (Table 2) and therefore additional experiments would be required to confirm this finding. Altogether, the obtained results indicated that the moss microbiome exhibits potential for both water-mediated and host-mediated transmission and that Sphagnum-associated Burkholderia are potentially transmitted over the entire life cycle of the host plants.
Discussion
The genus Burkholderia is very important for plant and human health (Coenye and Vandamme, 2003; Suárez-Moreno et al., 2012; Mitter et al., 2013). We found that the microbiome of our model Sphagnum plant is preferentially enriched by the plant-beneficial and non-pathogenic Burkholderia from the PBE cluster, but also contains minor fraction of potential plant pathogens. We have provided new ecological insights into these important plant inhabitants including their composition, distribution, colonization, and transmission pattern.
Our hypothesis that Sphagnum species are colonized by specific Burkholderia from the PBE cluster has to be slightly revised. Although the most abundant B. bryophila species belongs to the plant-beneficial cluster, a minor fraction of B. andropogonis and B. glathei species are within the pathogen cluster (Suárez-Moreno et al., 2012). However, the phylogenetic analysis was crucial for elucidating their intra-specific diversity and ecological background (Figure 2). Amazingly, the resolved phylogenetic clusters contained plant-beneficial and non-pathogenic burkholderial strain as well as environmental clone sequences. This fact led us to the conclusion that Burkholderia members from the PBE cluster are of a great importance for the health and growth of Sphagnum plants. Our conclusion was supported by the isolation of B. bryophila and B. phenazinium beneficial strains from Sphagnum mosses at the same sampling sites by Shcherbakov et al. (2013). The minor fraction of the burkholderial community was formed by Burkholderia spp. from the plant-pathogenic cluster sensu Suárez-Moreno et al. (2012). However, the collected Sphagnum plants did not exhibit any disease symptoms. Therefore, we support the transfer of B. glathei the PBE cluster as recently suggested by Verstraete et al. (2013), who identified the species as common endosymbiont in plants of the Rubiaceae family. In contrast, Burkholderia andropogonis is the causal agent of numerous plant diseases affecting a wide range of monocot and dicot plants, e.g., sweet and field corn, blueberry, sorghum, carnation, coffee, statice, rye, and clover. Bacterial leaf stripe is one of the three major bacterial diseases of sorghum, and strict quarantine regulations against importation of B. andropogonis-infested sorghum feed grains and seeds are imposed by numerous countries (Ramundo and Claflin, 2005). Sphagnum mosses seem to be a natural reservoir for this plant pathogen. This is important because dry Sphagnum is often used for orchid and ornamental cultivation and transferred world-wide. On the other side, there are also hints that saprophytic B. andropogonis exists (Estrada-de los Santos et al., 2013), and many disease outbreaks depend on the abundance of pathogens and the diversity of the indigenous microbiome. At last, it is impossible to predict any pathogenic or beneficial effect from 16S rDNA analysis, and additional studies would be required to prove or contradict the pathogenicity of Sphagnum-associated B. andropogonis.
In this study, we discovered similar distribution patterns of Sphagnum-associated burkholderial communities independent of the geographic region, which well-confirmed our hypothesis. To elucidate this distribution pattern, we aimed to answer the question—what factors shape this community? In terrestrial habitats, pH serves as both a primary driver of microbiome structure as well as a specific determinant of the genus Burkholderia as it is known to exhibit pH tolerance as a general phenotypic trait (Lauber et al., 2009; Stopnisek et al., 2013). In our previous study, the same sampling sites in Austria were characterized as extremely to moderately acidic by means of Ellenberg's indicator values for pH (expressed as soil reaction) (Bragina et al., 2012). For sampling sites in Russia, the Ellenberg's values for pH varied at the same range (data not shown) and therefore all examined sites possessed favorable a-biotic conditions for the burkholderial colonization. Apart from the a-biotic factors, we previously demonstrated that various Sphagnum species determine the microbiome composition to different extents (Bragina et al., 2011, 2012). Through statistical analysis, we showed that neither geographic location nor Sphagnum species had a significant influence on the distribution of Burkholderia. Moreover, the similar colonization patterns of the moss-associated Burkholderia were verified using FISH-CLSM in a semi-quantitative way.
For a better understanding of the distribution and colonization pattern revealed for Sphagnum-associated Burkholderia, we addressed the issue of bacterial transmission in the peat bog ecosystems. Recently, Putkinen et al. (2012) described a water dispersal of methane-oxidizing bacteria in the peat bogs. Moreover, our previous study revealed that nitrogen-fixing bacteria were transferred within the moss sporophyte (Bragina et al., 2013) As a result, we hypothesized that either host-mediated or water-mediated transmission is possible for Sphagnum-associated Burkholderia. Through the comparison of microbiome composition in various bog microhabitats, we found that burkholderial communities are potentially transmitted by the host plants. The violent spore discharge and wind dispersal of the Sphagnum spores would enable associated bacteria to migrate over the long distances and support spore germination at a new site (Szövényi et al., 2008; Sundberg, 2010). Altogether, the detected host-mediated transmission underlines the importance of Burkholderia for Sphagnum mosses themselves and defines their distribution pattern.
In recent decades, burkholderial community was considered a typical and well-adapted component of acidic peat bogs (Belova et al., 2006). In this study, we demonstrated that Burkholderia associated with the main vegetation of peat bogs, Sphagnum mosses, contain both plant-beneficial but also potentially pathogenic Burkholderia that are transmitted by the host plants over their life cycle. However, global warming and human disturbance may significantly shift the environmental conditions in the peat bog ecosystems and lead to the elimination or substitution of the beneficial microbes (Dise, 2009). The obtained data supports our knowledge on native plant microbiomes and can help for the maintenance of climate-relevant bog ecosystems.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Vladimir Chebotar and Andrey Shcherbakov (St. Petersburg) for their cooperation in the joint project and interesting discussions. We would also like to thank Ekaterina Kuzmina (St. Petersburg) for her bryological expertise during the sampling in Russia. We are grateful to Meg Starcher (Graz/Washington) for English revision of the manuscript. This study was supported by FWF Austrian Science Fund (grant no. E-1653100183) to Gabriele Berg.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/Journal/10.3389/fmicb.2013.00394/abstract
Table S1 | Sampling sites.
Table S2 | Weighted UniFrac distance matrix of 16S rDNA pyrosequencing libraries specific for Burkholderia.
Figure S1 | Rarefaction curves for 16S rDNA amplicon libraries of Sphagnum samples.
References
Amann, R. I., Binder, B. J., Olson, R. J., Chisholm, S. W., Devereux, R., and Stahl, D. A. (1990). Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 56, 1919–1925.
Belova, S. E., Pankratov, T. A., and Dedysh, S. N. (2006). Bacteria of the genus Burkholderia as a typical component of the microbial community of Sphagnum peat bogs. Microbiology 75, 90–96. doi: 10.1134/S0026261706010164
Berendsen, R. L., Pieterse, C. M., and Bakker, P. A. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Berg, G. (2009). Plant-microbe interactions promoting plant growth and health: perspectives for conrolled use of microorganisms in agriculture. Appl. Microbiol. Biotechnol. 84, 11–18. doi: 10.1007/s00253-009-2092-7
Berg, G., Zachow, C., Müller, H., Philipps, J., and Tilcher, R. (2013). Next-generation bio-products sowing the seeds of success for sustainable agriculture. Agronomy 3, 648–656. doi: 10.3390/agronomy3040648
Bragina, A., Berg, C., Cardinale, M., Shcherbakov, A., Chebotar, V., and Berg, G. (2012). Sphagnum mosses harbour highly specific bacterial diversity during their whole lifecycle. ISME J. 6, 802–813. doi: 10.1038/ismej.2011.151
Bragina, A., Berg, C., Müller, H., Moser, D., and Berg, G. (2013). Insights into functional bacterial diversity and its effects on alpine bog ecosystem functioning. Sci. Rep. 3, 1955. doi: 10.1038/srep01955
Bragina, A., Maier, S., Berg, C., Müller, H., Chobot, V., Hadacek, F., et al. (2011). Similar diversity of Alphaproteobacteria and nitrogenase gene amplicons on two related Sphagnum mosses. Front. Microbiol. 2:275. doi: 10.3389/fmicb.2011.00275
Bulgarelli, D., Rott, M., Schlaeppi, K., Ver Loren van Themaat, E., Ahmadinejad, N., Assenza, F., et al. (2012). Revealing structure and assembly cues of Arabidopsis root-inhabiting bacterial microbiota. Nature 488, 91–95. doi: 10.1038/nature11336
Caballero-Mellado, J., Onofre-Lemus, J., Estrada-de Los Santos, P., and Martinez-Aguilar, P. (2007). The tomato rhizosphere, an environment rich in nitrogen-fixing Burkholderia species with capabilities of interest for agriculture and bioremediation. Appl. Environ. Microbiol. 73, 5308–5319. doi: 10.1128/AEM.00324-07
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chelius, M. K., and Triplett, E. W. (2001). The diversity of archaea and bacteria in association with the roots of Zea mays L. Microb. Ecol. 41, 252–263. doi: 10.1007/s002480000087
Coenye, T., and Vandamme, P. (2003). Diversity and significance of Burkholderia species occupying diverse ecological niches. Environ. Microbiol. 5, 719–729. doi: 10.1046/j.1462-2920.2003.00471.x
Daims, H., Brühl, A., Amann, R., Schleifer, K. H., and Wagner, M. (1999). The domain-specific probe EUB338 is insufficient for the detection of all bacteria: development and evaluation of amore comprehensive probe set. Syst. Appl. Microbiol. 22, 434–444. doi: 10.1016/S0723-2020(99)80053-8
Dise, N. B. (2009). Environmental science. Peatland response to global change. Science 326, 810–811. doi: 10.1126/science.1174268
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Estrada-de los Santos, P., Vinuesa, P., Martínez-Aguilar, L., Hirsch, A. M., and Caballero-Mellado, J. (2013). Phylogenetic analysis of burkholderia species by multilocus sequence analysis. Curr. Microbiol. 67, 51–60. doi: 10.1007/s00284-013-0330-9
Gasser, I., Müller, H., and Berg, G. (2009). Ecology and characterization of polyhydroxyalkanoate-producing microorganisms on and in plants. FEMS Microbiol. Ecol. 70, 142–150. doi: 10.1111/j.1574-6941.2009.00734.x
Grube, M., Cardinale, M., de Castro, J. V. Jr., Müller, H., and Berg, G. (2009). Species-specific structural and functional diversity of bacterial communities in lichen symbioses. ISME J. 3, 1105–1115. doi: 10.1038/ismej.2009.63
Hirsch, P. R., and Mauchline, T. H. (2012). Who's who in the plant root microbiome. Nat. Biotechnol. 30, 961–962. doi: 10.1038/nbt.2387
Hogardt, M., Trebesius, K., Geiger, A. M., Hornef, M., Rosenecker, J., and Heesemann, J. (2000). Specific and rapid detection by fluorescent in situ hybridization of bacteria in clinical samples obtained from cystic fibrosis patients. J. Clin. Microbiol. 38, 818–825.
Jansson, J. K., Neufeld, J. D., Moran, M. A., and Gilbert, J. A. (2012). Omics for understanding microbial functional dynamic. Environ. Microbiol. 14, 1–3. doi: 10.1111/j.1462-2920.2011.02518.x
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematic, eds E. Stackebrandt and M. Goodfellow (New York, NY: Wiley), 115–175.
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., Mcgettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Lauber, C. L., Hamady, M., Knight, R., and Fierer, N. (2009). Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75, 5111–5120. doi: 10.1128/AEM.00335-09
Lemaire, B., Vandamme, P., Merckx, V., Smets, E., and Dessein, S. (2011). Bacterial leaf symbiosis in angiosperms: host specificity without co-speciation. PLoS ONE 6:e24430. doi: 10.1371/journal.pone.0024430
Lieber, A., Kiesel, B., and Babel, W. (2003). “Microbial diversity of soil by SSCP fingerprinting technique using TGGE Maxi System,” in Ökophysiologie des Wurzelraumes, eds W. Merbach, B. W. Hütsch, and J. Augustin (Stuttgart: Teubner Verlag), 61–65.
Lozupone, C., Lladser, M. E., Knights, D., Stombaugh, J., and Knight, R. (2010). UniFrac: an effective distance metric for microbial community comparison. ISME J. 5, 169–172. doi: 10.1038/ismej.2010.133
Lundberg, D. S., Lebeis, S. L., Paredes, S. H., Yourstone, S., Gehring, J., Malfatti, S., et al. (2012). Defining the core Arabidopsis thaliana root microbiome. Nature 488, 86–90. doi: 10.1038/nature11237
Mahenthiralingam, E., Urban, T. A., and Goldberg, J. B. (2005). The multifarious, multireplicon Burkholderia cepacia complex. Nat. Rev. Microbiol. 3, 144–156. doi: 10.1038/nrmicro1085
Mitter, B., Petric, A., Shin, M. W., Chain, P. S. G., Hauberg-Lotte, L., Reinhold-Hurek, B., et al. (2013). Comparative genome analysis of Burkholderia phytofirmans PsJN reveals a wide spectrum of endophytic lifestyles based on interaction strategies with host plants. Front. Plant Sci. 4:120. doi: 10.3389/fpls.2013.00120
Nissinen, R. M., Männistö, M. K., and van Elsas, J. D. (2012). Endophytic bacterial communities in three arctic plants from low arctic fell tundra are cold-adapted and host-plant specific. FEMS Microbiol. Ecol. 82, 510–522. doi: 10.1111/j.1574-6941.2012.01464.x
Opelt, K., and Berg, G. (2004). Diversity and antagonistic potential of bacteria associated with bryophytes from nutrient-poor habitats of the Baltic Sea Coast. Appl. Environ. Microbiol. 70, 6569–6579. doi: 10.1128/AEM.70.11.6569-6579.2004
Opelt, K., Berg, C., and Berg, G. (2007b). The bryophyte genus Sphagnum is a reservoir for powerful and extraordinary antagonists and potentially facultative human pathogens. FEMS Microb. Ecol. 61, 38–53. doi: 10.1111/j.1574-6941.2007.00323.x
Opelt, K., Berg, C., Schönmann, S., Eberl, L., and Berg, G. (2007c). High specificity but contrasting biodiversity of Sphagnum-associated bacterial and plant communities in bog ecosystems independent of the geographical region. ISME J. 1, 502–516. doi: 10.1038/ismej.2007.58
Opelt, K., Chobot, V., Hadacek, F., Schönmann, S., Eberl, L., and Berg, G. (2007a). Investigations of the structure and function of bacterial communities associated with Sphagnum mosses. Environ. Microbiol. 9, 2795–2809. doi: 10.1111/j.1462-2920.2007.01391.x
Putkinen, A., Larmola, T., Tuomivirta, T. T., Siljanen, H. M. P., Bodrossy, L., Tuittila, E.-S., et al. (2012). Water dispersal of methanotrophic bacteria maintains functional methane oxidation in Sphagnum mosses. Front. Microbiol. 3:15. doi: 10.3389/fmicb.2012.00015.
Raghoebarsing, A. A., Smolders, A. J., Schmid, M. C., Rijpstra, W. I., Wolters-Arts, M., Derksen, J., et al. (2005). Methanotrophic symbionts provide carbon for photosynthesis in peat bogs. Nature 436, 1153–1156. doi: 10.1038/nature03802
Ramundo, B. A., and Claflin, L. E. (2005). Identification of Burkholderia andropogonis with a repetitive sequence BOX element and PCR. Curr. Microbiol. 50, 52–56. doi: 10.1007/s00284-004-4354-z
Schloss, P. D., and Handelsman, J. (2006). Toward a census of bacteria in soil. PLoS Comput. Biol. 2:e92. doi: 10.1371/journal.pcbi.0020092
Schönmann, S., Loy, A., Wimmersberger, C., Sobek, J., Aquino, C., Vandamme, P., et al. (2009). 16S rRNA gene-based phylogenetic microarray for simultaneous identification of members of the genus Burkholderia. Environ. Microbiol. 11, 779–800. doi: 10.1111/j.1462-2920.2008.01800.x
Sessitsch, A., Coenye, T., Sturz, A. V., Vandamme, P., Barka, E. A., Salles, J. F., et al. (2005). Burkholderia phytofirmans sp. nov., a novel plant-associated bacteium with plant-beneficial properties. Int. J. Syst. Evol. Microbiol. 55, 1187–1192. doi: 10.1099/ijs.0.63149-0
Shcherbakov, A. V., Bragina, A. V., Kuzmina, E. Yu., Berg, C., Muntyan, A. N., et al. (2013). Endophytic bacteria of Sphagnum mosses as promising objects of agricultural microbiology. Microbiology 82, 306–315. doi: 10.1134/S0026261713030107
Stopnisek, N., Bodenhausen, N., Frey, B., Fierer, N., Eberl, L., and Weisskopf, L. (2013). Genus-wide acid tolerance accounts for the biogeographical distribution of soil Burkholderia populations. Environ. Microbiol. doi: 10.1111/1462–2920.12211
Suárez-Moreno, Z. R., Caballero-Mellado, J., Coutinho, B. G., Mendonça-Previato, L., James, E. K., and Venturi, V. (2012). Common features of environmental and potentially beneficial plant-associated Burkholderia. Microb. Ecol. 63, 249–266. doi: 10.1007/s00248-011-9929-1
Suárez-Moreno, Z. R., Devescovi, G., Myers, M., Hallack, L., Mendonça-Previato, L., Caballero-Mellado, J., et al. (2010). Commonalities and differences in regulation of N-acyl homoserine lactone quorum sensing in the beneficial plant-associated Burkholderia species clusters. Appl. Environ. Microbiol. 76, 4302–4317. doi: 10.1128/AEM.03086-09
Sundberg, S. (2010). Size matters for violent discharge height and settling speed of Sphagnum spores: important attributes for dispersal potential. Ann. Bot. 105, 291–300. doi: 10.1093/aob/mcp288
Szövényi, P., Terracciano, S., Ricca, M., Giordano, S., and Shaw, A. J. (2008). Recent divergence, intercontinental dispersal and shared polymorphism are shaping the genetic structure of amphi-Atlantic peatmoss populations. Mol. Ecol. 17, 5364–5377. doi: 10.1111/j.1365-294X.2008.04003.x
Tamura, K., Dudley, J., Nei, M., and Kumar, S. (2007). MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599. doi: 10.1093/molbev/msm092
Van Oevelen, S., De Wachter, R., Vandamme, P., Robbrecht, E., and Prinsen, E. (2002). Identification of the bacterial endosymbionts in leaf galls of Physcotria (Rubiaceae, angiosperms) and proposal of ‘Candidatus Burkholderia kirkii’ sp. nov. Int. J. Syst. Evol. Microbiol. 52, 2023–2027. doi: 10.1099/ijs.0.02103-0
Vandamme, P., Opelt, K., Knöchel, N., Berg, C., Schönmann, S., De Brandt, E., et al. (2007). Burkholderia bryophila sp. nov. and Burkholderia megapolitana sp. nov., moss-associated species with antifungal and plant-growth-promoting properties. Int. J. Syst. Evol. Microbiol. 57, 2228–2235. doi: 10.1099/ijs.0.65142-0
Verstraete, B., Janssens, S., Smets, E., and Dessein, S. (2013). Symbiotic ß-Proteobacteria beyond legumes: Burkholderia in Rubiaceae. PLoS ONE 8:e55260. doi: 10.1371/journal.pone.0055260
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Weisskopf, L., Heller, S., and Eberl, L. (2011). Burkholderia species are major inhabitants of white lupin cluster roots. Appl. Environ. Microbiol. 77, 7715–7720. doi: 10.1128/AEM.05845-11
Yabuuchi, E., Kosako, Y., Oyaizu, H., Yano, I., Hotta, H., Hashimoto, Y., et al. (1992). Proposal of Burkholderia gen. nov. and transfer of seven species of the genus Pseudomonas homology group II to the new genus, with the type species Burkholderia cepacia (Palleroni and Holmes 1981) comb. nov. Microbiol. Immunol. 36, 1251–1275. doi: 10.1111/j.1348-0421.1992.tb02129.x
Keywords: Sphagnum fallax, Sphagnum magellanicum, Burkholderia communities, amplicon pyrosequencing, FISH-CLSM
Citation: Bragina A, Cardinale M, Berg C and Berg G (2013) Vertical transmission explains the specific Burkholderia pattern in Sphagnum mosses at multi-geographic scale. Front. Microbiol. 4:394. doi: 10.3389/fmicb.2013.00394
Received: 01 November 2013; Accepted: 03 December 2013;
Published online: 18 December 2013.
Edited by:
Michael Schloter, Helmholtz Zentrum München, GermanyReviewed by:
David J. Studholme, University of Exeter, UKMichael Schmid, Helmholtz Zentrum München, Germany
Copyright © 2013 Bragina, Cardinale, Berg and Berg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriele Berg, Institute of Environmental Biotechnology, Graz University of Technology, Petersgasse 10-12/I, Graz, 8010, Austria e-mail:Z2FicmllbGUuYmVyZ0B0dWdyYXouYXQ=