 Tomoo Sawabe1*
Tomoo Sawabe1* Yoshitoshi Ogura2
Yoshitoshi Ogura2 Yuta Matsumura1Gao Feng1
Yuta Matsumura1Gao Feng1 AKM Rohul Amin1
AKM Rohul Amin1 Sayaka Mino1
Sayaka Mino1 Satoshi Nakagawa1Toko Sawabe3
Satoshi Nakagawa1Toko Sawabe3 Ramesh Kumar4 Yohei Fukui5Masataka Satomi5Ryoji Matsushima5Fabiano L. Thompson6Bruno Gomez-Gil7Richard Christen8,9
Ramesh Kumar4 Yohei Fukui5Masataka Satomi5Ryoji Matsushima5Fabiano L. Thompson6Bruno Gomez-Gil7Richard Christen8,9 Fumito Maruyama10
Fumito Maruyama10 Ken Kurokawa11Tetsuya Hayashi2
Ken Kurokawa11Tetsuya Hayashi2- 1Laboratory of Microbiology, Faculty of Fisheries Sciences, Hokkaido University, Hakodate, Japan
- 2Division of Genomics and Bioenvironmental Science, Frontier Science Research Center, University of Miyazaki, Miyazaki, Japan
- 3Department of Food and Nutrition, Hakodate Junior College, Hakodate, Japan
- 4National Institute for Interdisciplinary Science and Technology (CSIR), Kerala, India
- 5National Research Institute of Fisheries Science, Fisheries Research Agency, Yokohama, Japan
- 6Department of Genetics, Center of Health Sciences, Federal University of Rio de Janeiro (UFRS), Rio de Janeiro, Brazil
- 7A.C. Unidad Mazatlán, CIAD, Mazatlán, México
- 8CNRS UMR 7138, Systématique-Adaptation-Evolution, Nice, France
- 9Systématique-Adaptation-Evolution, Université de Nice-Sophia Antipolis, Nice, France
- 10Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, Tokyo, Japan
- 11Earth-Life Science Institute, Tokyo Institute of Technology, Tokyo, Japan
To date 142 species have been described in the Vibrionaceae family of bacteria, classified into seven genera; Aliivibrio, Echinimonas, Enterovibrio, Grimontia, Photobacterium, Salinivibrio and Vibrio. As vibrios are widespread in marine environments and show versatile metabolisms and ecologies, these bacteria are recognized as one of the most diverse and important marine heterotrophic bacterial groups for elucidating the correlation between genome evolution and ecological adaptation. However, on the basis of 16S rRNA gene phylogeny, we could not find any robust monophyletic lineages in any of the known genera. We needed further attempts to reconstruct their evolutionary history based on multilocus sequence analysis (MLSA) and/or genome wide taxonomy of all the recognized species groups. In our previous report in 2007, we conducted the first broad multilocus sequence analysis (MLSA) to infer the evolutionary history of vibrios using nine housekeeping genes (the 16S rRNA gene, gapA, gyrB, ftsZ, mreB, pyrH, recA, rpoA, and topA), and we proposed 14 distinct clades in 58 species of Vibrionaceae. Due to the difficulty of designing universal primers that can amplify the genes for MLSA in every Vibrionaceae species, some clades had yet to be defined. In this study, we present a better picture of an updated molecular phylogeny for 86 described vibrio species and 10 genome sequenced Vibrionaceae strains, using 8 housekeeping gene sequences. This new study places special emphasis on (1) eight newly identified clades (Damselae, Mediterranei, Pectenicida, Phosphoreum, Profundum, Porteresiae, Rosenbergii, and Rumoiensis); (2) clades amended since the 2007 proposal with recently described new species; (3) orphan clades of genomospecies F6 and F10; (4) phylogenetic positions defined in 3 genome-sequenced strains (N418, EX25, and EJY3); and (5) description of V. tritonius sp. nov., which is a member of the “Porteresiae” clade.
Introduction
Bacterial systematics has evolved alongside the development of innovative methodologies and techniques (Wayne et al., 1987; Stackebrandt et al., 2002; Gevers et al., 2005). The first definition of bacterial species in “phylogenetic terms” was developed in 1987 using the DNA-DNA reassociation and DNA sequencing. These approaches to bacterial systematics provided us with a uniform definition of prokaryotic species (Wayne et al., 1987). In 2002, an ad hoc committee listed additional innovative methods that could be used for bacterial systematics, such as 16S rRNA gene sequence analysis, DNA typing methods (AFLP, RAPD, Rep-PCR, PFGE), MLSA, WGS analysis, and proteomics (Stackebrandt et al., 2002). The primary purpose of the committee's statement was to promote dialogue among systematists, population and evolutionary geneticists, ecologists and microbiologists for the benefit of bacterial systematics in general, and to create a more transparent species concept in particular (Stackebrandt et al., 2002). Among those innovative methodologies, MLSA and the WGS analysis have become the most important and successful methodologies; their strong impact on bacterial systematics is due to data reproducibility and portability (see, e.g., Maiden et al., 1998; Aanensen and Spratt, 2005; Gevers et al., 2005; Konstantinidis and Tiedje, 2005; Staley, 2006; Goris et al., 2007; Richer and Rosselló-Móra, 2009; Auch et al., 2010).
MLST, the prototype for MLSA-based methodology, was used for the first highly portable typing of Neisseria meningitides from invasive disease and healthy carriers, and it yielded the first understanding of the epidemiology and population structure of that infectious agent (Maiden et al., 1998). Its high levels of discriminatory power between those strains, which required half the loci typically required for a classical allozyme electrophoresis, and its superior application to evolutionary, phylogenetic, or population genetic studies, allowed researchers to develop MLST schemes for a number of bacteria taxa (Aanensen and Spratt, 2005) (also refer to the MLST website; http://www.mlst.net/). It also opened the use of MLSA for bacterial systematics (e.g., Sawabe et al., 2007; Thompson et al., 2007; Bishop et al., 2009), and it guided the reconsideration and re-evaluation of prokaryotic species concepts (Gevers et al., 2005; Staley, 2006; Preheim et al., 2011). Even now, MLSA provides a better understanding of taxonomically controversial bacterial taxa; for example, the human origins of the Agrobacterium (Rhizobium) radiobacter clustered as a well-separated genovar (Aujoulat et al., 2011) and the highly versatile aeromonads consisting of 3 major clades (Roger et al., 2012).
In the genome era, genome sequencing has been used to characterize new bacterial species (Haley et al., 2010; Hoffmann et al., 2012), to reclassify bacterial taxons such as Neisseria (Bennett et al., 2012), Acinetobacter (Chan et al., 2012) and Vibrio (Lin et al., 2010), and to challenge defined prokaryotic species (Konstantinidis and Tiedje, 2005; Thompson et al., 2009; Chan et al., 2012). Using these WGSs, in-silico DDH calculations can also be emulated, mainly in two ways: high-scoring segment pairs (HSPs) (Konstantinidis and Tiedje, 2005; Goris et al., 2007) and the genome-to-genome distance calculation, called the Digital DDH measurement (Auch et al., 2010). The criterion of more than 95% ANI is currently a widely used similarity value for species delineation. The WGS analysis also supercedes the limitations of MLSA, which is only capable of including genes that are successfully amplified by designed primers (Gevers et al., 2005; Thompson et al., 2005; Sawabe et al., 2007).
Vibrionaceae are at the forefront of bacterial taxons being tested with new innovative methodologies and techniques for bacterial systematics (Thompson et al., 2001, 2005, 2009; Sawabe et al., 2007). The number of species described in Vibrionaceae has increased remarkably since the establishment of genome fingerprinting techniques (Thompson et al., 2001) and MLSA schemes (Thompson et al., 2005, 2007; Sawabe et al., 2007, 2009). Now, a total of 142 species are recognized in the family Vibrionaceae (Association of Vibrio Biologists website; http://www.vibriobiology.net/). Vibrionaceae are defined as a group of strains with the following characteristics: they are Gram-negative rods with a polar flagellum enclosed in a sheath, have facultative anaerobic metabolisms, are capable of fermenting D-glucose, and grow at 20°C. The bacteria are primarily aquatic, and most species are oxidase positive, can reduce nitrate to nitrite, require Na+ for growth, and ferment D-fructose, maltose, and glycerol (Gomez-Gil et al., 2014). In addition, most vibrio species ferment a variety of carbohydrates without gas production, and grow on TCBS medium (Farmer III et al., 2005; Thompson et al., 2009). As we experience a rapid expansion in the number of known species in the family Vibrionaceae, we face a number of unique vibrio isolates that lack one or more of the above common properties. Vibrionaceae species are metabolically versatile, and the number of species showing gas production, nitrogen fixation, phototrophy, and non-motility is increasing (Gomez-Gil et al., 2014). Considerably more attention should be paid to the biological and genetic plasticity of vibrios to help understand the dynamics of vibrio evolution (Sawabe et al., 2007; Grimes et al., 2009; Thompson et al., 2009).
Due to the limitations of using 16S rRNA gene phylogeny (Gomez-Gil et al., 2014) to elucidate an “Integrated Vibrio Biology” that includes a biodiversity assessment, an inferred evolutionary history, the population biology, and genomics, Sawabe et al. (2007) developed an MLSA scheme for the family Vibrionaceae using nine gene sequences (ftsZ, gapA, gyrB, mreB, pyrH, recA, rpoA, topA and the 16S rRNA gene). The analysis involved the complete sequence sets of 9 genes from 58 vibrio taxa, and it revealed 14 monophyletic clades with a significant bootstrap support. The species within each clade shared >20% DDH, <5% G+C (mol%), >85% MLSA sequence similarity, and >89% AAI (Sawabe et al., 2007).
Recent extraordinary progress in biodiversity studies and WGS projects in vibrios has resulted a substantial leap in novel vibrio species and taxonomically unassigned strains. In fact, after the proposal of 14 robust clades in 2007, more than 60 new species have been described. The number of genome-sequenced strains has exceeded 1000. It is, therefore, obvious that the phylogenetic tree based on the multilocus gene sequences reported in 2007 is insufficient to show the most recent molecular phylogenetic structure of vibrios.
In our previous analysis of multilocus gene phylogeny, eight housekeeping protein coding genes and the 16S rRNA gene were included in the MLSA. However, the 16S rRNA gene has a rather low interspecies resolution (100, 99.5, and 95.3% of maximum, median, and minimum resolution, respectively) (see Figure 5S, in Sawabe et al., 2007). It was also difficult to include the 16S rRNA gene sequences for the calculation of the radiation time of each clade. We have been debating whether the inclusion of the 16S rRNA gene sequences is necessary to infer the evolutional history of vibrios, but there are no other proper and fast tools to check the tree topologies constructed using a rather large data set (58 species).
A variety of methods have been proposed to tackle the problem of gene tree reconciliation to reconstruct a species tree. When the taxa in all the trees are identical, the problem can be stated as a consensus tree problem (Guénoche, 2013). The comparison of gene trees and their assembly into a unique tree representing the species tree is a general problem in phylogeny. However, in this study, we faced a different problem: determining whether the inclusion or exclusion of a given gene in the analysis would substantially change the outcome. This was the method used in this analysis to investigate if the inclusion or exclusion of the 16S rRNA gene sequences in the MLSA analysis would, or would not, affect the final result.
The aims of this study were to re-evaluate how the 16S rRNA gene sequence affects the final phylogenetic tree, as based on multilocus gene sequencing analysis; to update our knowledge in vibrio biodiversity and evolution on the basis of an 8-gene MLSA; and to reconstruct a better vibrio phylogeny. The analysis provided a further opportunity to propose additional eight clades to the most up-to-date vibrio phylogeny.
Materials and Methods
Subtree Incongruence Test of Multilocus Gene Phylogeny
The usual approach to compare trees is to count how many subtrees they share; a given subtree often has a different topology according to the method used (NJ, ML, or MP) but the differences are often subtle and generally not well-supported. Accordingly, it is best to compare trees according to bipartitions (a tree is considered as a set of bipartitions, each one corresponding to an internal edge of the tree, the external ones connecting the leaves to the tree) (Guénoche, 2013). This was the method used in this analysis to investigate whether the inclusion or exclusion of the 16S rRNA gene sequences in the MLSA analysis would, or would not, affect the final result. TreeDyn (Chevenet et al., 2006) was used to compare trees to subtrees sharing the same topology. A dedicated python script (using libraries from Huerta-Cepas et al., 2010) was used to compare trees with shared sub-trees, independently of their topologies.
Our previous MLSA revealed that the 16S rRNA gene, in contrast to the other gene sequences used, has a rather low interspecies resolution (Sawabe et al., 2007). To increase the sensitivity and reduce the time taken by MLSA to update the vibrio phylogeny, we compared the subtree topologies obtained from a nine-gene data set (that included the 16S rRNA gene) and an eight-gene data set (that excluded the 16S rRNA gene). We used the gene sequence data set from the 58 species, for which we had the sequence of every gene, and which is identical to the data set used in Sawabe et al. (2007). The method selected, (1) shared subtrees only if they had the same topology, and (2) subtrees that shared the same species, independently of their topology. The results were visualized using TreeDyn (Chevenet et al., 2006), and congruent subtrees with the same topologies were indicated using the same color.
Sequencing of Housekeeping Protein-Coding Genes
An additional eight housekeeping genes of type strains of the genera Vibrio and Photobacterium were sequenced manually according to Sawabe et al. (2007) with newly designed primers (Vgap150f:ACTCAYGGYCGTTTCAACGGYAC, Vgap957r:RCCGATTTCGTTRTCGTACCAAG, VftsZf55f:GTKGGTGGCGGCGGCGGTAA, VftsZ782r:ACACCACGWGCACCAGCAA GATCG, VftsZ-9f:ACCGATGATGGAAATGTCTGACGATGC, VmreB225f:RATGAAA GACGGCGTWATYGC, VmreB1025r:TCGCCRCCGTGCATRTCGATCA) (Table S1). All strains were maintained in ZoBell 2216E agar and stored with 20% glycerol at −80°C. Whole genome sequencing was performed in nine type strains (V. aerogenes LMG 19650T, V. gazogenes ATCC 29988T, V. halioticoli IAM 14596T, V. neonatus HDD3-1T, V. porteresiae MSSRF 30T, V. rhizosphaerae MSSRF 3T, V. ruber LMG 23124T, V. tritonius sp. nov. AM2T, and V. superstes G3-29T) using the Roche 454 FLX titanium genome sequencer alone or in combination with the Illumina MiSeq sequencer. The sequence reads were assembled using the Newbler software version 2.3 or later. Illumina reads were used only for sequence error correction. After auto-annotation by Microbial Genome Annotation Pipeline (MiGAP, http://www.migap.org/; Sugawara et al., 2009), relevant housekeeping gene sequences were retrieved and used for the MLSA. The house keeping genes necessary for updating the vibrio phylogeny were also retrieved from the latest version of the NCBI microbial genome and GenBank database (Release 197.0, 15 August 2013), and used in the analysis. All sequence data used in this study are listed in Table S1.
Sequence Analysis
MLSA was performed the same way as in Sawabe et al. (2007). The sequences were aligned using the ClustalX program (Larkin et al., 2007). The domains used to construct the phylogenetic trees shown in Figures 2, 3 were regions of the ftsZ, gapA, gyrB, mreB, pyrH, recA, rpoA, and topA genes of Vibrionaceae: positions 195–630, 225–861, 441–1026, 390–897, 171–543, 429–915, 87–873, and 570–990 (V. cholerae O1 Eltor N16961 (AE003852) numbering), respectively. The regions were within those used in the previous study (Sawabe et al., 2007). Sequence similarity and the number of nucleotide and amino acid mutation were determined using MEGA version 5 (Tamura et al., 2011).
Split Decomposition Analysis (SDA) was also performed as described in Sawabe et al. (2007) using SplitsTree version 4.12.8, with a neighbor net drawing and Jukes-Cantor correction (Bandelt and Dress, 1992; Huson and Bryant, 2005). The concatenated sequences of the eight housekeeping genes were also generated using the program and used for a phylogenetic analysis combined with NJ, MP, and ML analyses (Sawabe et al., 1998).
Phylogenetic, Genetic, Phenotypic, and Chemotaxonomic Characterization of Vibrio tritonius sp. nov
Four isolates of V. tritonius sp. nov., JCM 16456T = LMG 25401T = AM2T, JCM 16457 = LMG 25402 = MA12, JCM 16458 = LMG 25403 = MA17, and JCM 16459 = LMG 25404 = MA35, isolated from gut of a sea hare, Aplysia kurodai, were used in this study. The strains were cultured on ZoBell 2216E agar (Oppenheimer and ZoBell, 1952) and stored at −80°C in 10% glycerol-supplemented broth.
A total of 1400 bp 16S rRNA gene sequences of the four strains were determined according to Sawabe et al. (1998) using four sequence primers (24F, 1100F, 920R, and 1509R). The 16S rRNA gene sequences were blasted to the latest release ver. 197 of GenBank and related sequences were retrieved. Finally, 16S rRNA gene sequences of V. aerogenes X74705, V. brasiliensis AJ316172, V. cholerae X76337, V. fluvialis X74703, V. furnissii X76336, V. gazogenes X74705, V. hepatarius AJ345063, V. nereis X74716, V. porteresiae EF488079, V. rhizosphaerae DQ847123, V. ruber AF462458, V. tubiashii X74725, and V. xuii AJ316181 were included in the phylogenetic analysis (Figure 4). Phylogenetic trees were constructed using three different methods (NJ, ML, and MP). For NJ analysis, distance matrices were calculated using the Kimura two parameters correction and using MEGA version 5.0 (Tamura et al., 2011). ML and MP analysis was conducted using PHYLIP (Phylogeny Inference Package, version 3.573c, distributed by J. Felsenstein, Department of Genetics, UW, Seattle, WA, USA). Sequences corresponding to positions 86–1420 of the E. coli gene (NC_000913) were used in this analysis. Figure 4 represents a subset of the final tree obtained using the NJ method with 500 bootstrap replications. Nodes supported by ML and MP analyses are indicated by the bootstrap values in Figure 4.
DNAs of bacterial strains were prepared following the procedures of Marmur (1961), with minor modifications. The mol% G+C content of DNAs was determined by HPLC (Tamaoka and Komagata, 1984). DNA-DNA hybridization experiments were performed in microdilution wells using a fluorometric direct binding method described by Ezaki et al. (1988); Ezaki et al. (1989). DNA-DNA similarity data were shown as the average value of triplicate experiments. V. brasiliensis, V. furnissii, V. fluvialis, V. tubiashii, V. hepatarius, V. mytili, V. nereis, V. porteresiae, and V. xuii were selected as the reference species of these DNA-DNA hybridization experiments, based on the results from the MLSA molecular phylogenetic assessment and the 16S rRNA gene phylogeny of V. tritonius sp. nov.
A total of 62 phenotypic characteristics were determined using the standard manual characterization method established in our laboratory (Sawabe et al., 1998). The carbon assimilation test was conducted using a basal seawater medium, as previously described (Sawabe et al., 1998). These phenotypic characterizations were performed at 25°C. O/129 sensitivity was determined using the sensitivity basal agar medium (Nissui Pharmaceutical, Tokyo, Japan) at 30°C.
Results
The Subtree Incongruence Test between the 9-Gene and 8-Gene Phylogenies
Subtrees obtained in the 8-gene phylogeny were compared to those in the 9-gene phylogeny and the 16S rRNA gene phylogeny (Figure 1). Among the 13 subtrees reconstructed in the 8-gene phylogeny, 12 were retained in the 9-gene phylogeny; the only difference observed was the inclusion of V. proteolyticus in the V. cholera subtree (Figures 1A,B). Most of the 13 subtrees in the 8-gene phylogeny corresponded to the clades that we previously proposed based on the 9-gene phylogeny (Figure 1B). The results of the subtree incongruence test using the 58 vibrio taxa data showed that the inclusion of the 16S rRNA gene sequence is not a critical factor in optimizing the vibrio phylogeny on the basis of MLSA.
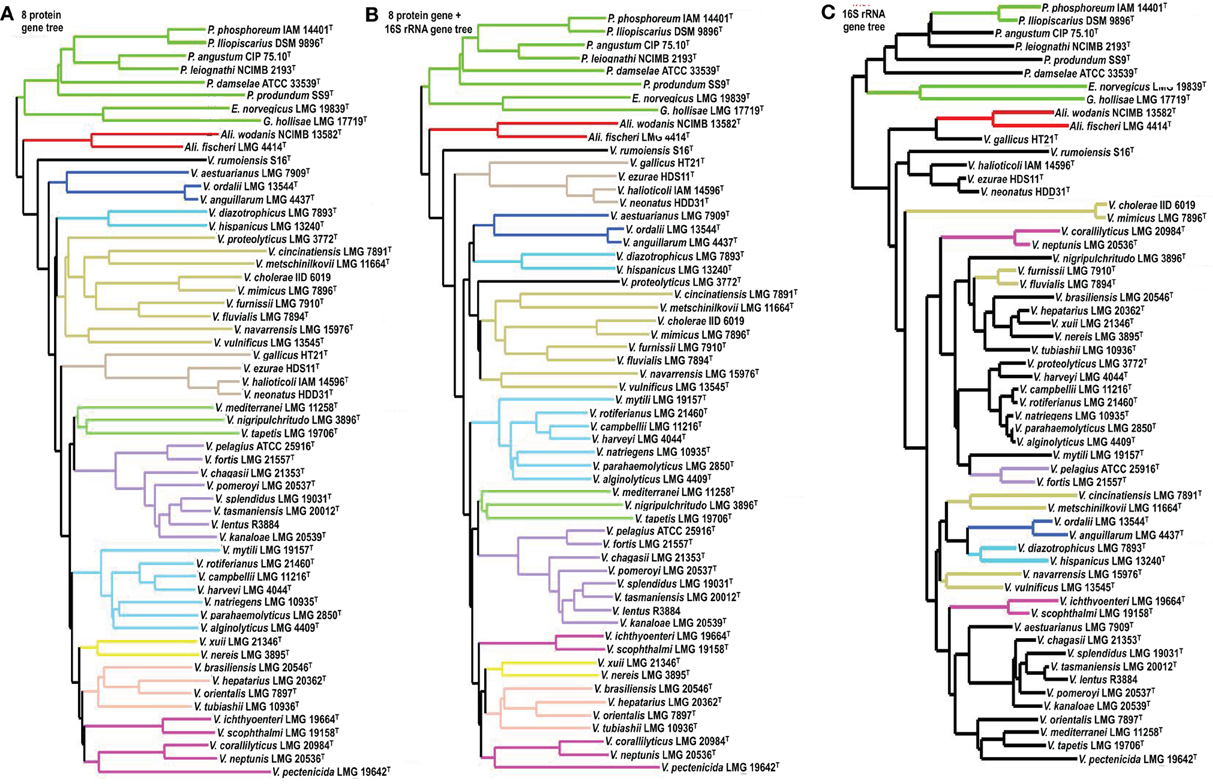
Figure 1. Subtree incongruence test. Using the 58 vibrio taxon data set reported in Sawabe et al. (2007), subtree topology was compared between 8 gene (−16S rRNA) (A) and 9 gene (+16S rRNA) phylogeny (B). The topology was also compared between 8 gene and only 16S rRNA gene phylogeny (C).
The Latest Vibrio Phylogeny Based on Multilocus Housekeeping Protein-Coding Gene Sequences
WGSs of key vibrios species that were resistant to the gene amplification, e.g., V. gazogenes, S. costicola, V. porteresiae, V. caribbenthicus, are now available. This result indicated that we could use the complete set of 8 housekeeping protein-coding gene sequences currently available from 86 described vibrio species and 10 genome-sequenced Vibrionaceae strains for the MLSA updating of the 8-gene phylogeny, on the basis of Splits Decomposition Analysis (SDA) (Bandelt and Dress, 1992; Huson and Bryant, 2005) (Figure 2) and a supertree reconstruction (Figure 3). On the basis of SDA, we could retain the 14 distinct monophyletic clades that were previously defined, and we were able to further define 8 new clades: Damselae, Mediterranei, Pectenicida, Phosphoreum, Profundum, Porteresiae, Rosenbergii, and Rumoiensis (Figures 2, 3, Table 1). The robustness of these clades was high enough to propose their monophyly in the supertree reconstruction using three different molecular phylogenetic analyses (Figure 3). Using an 8-housekeeping protein-coding gene analysis, most of clades shared >80.5% ANI and >92% AAI, and the highest ANI (98.3%) was observed in the sequence comparison between V. anguillarum and V. ordalii (Table 1).
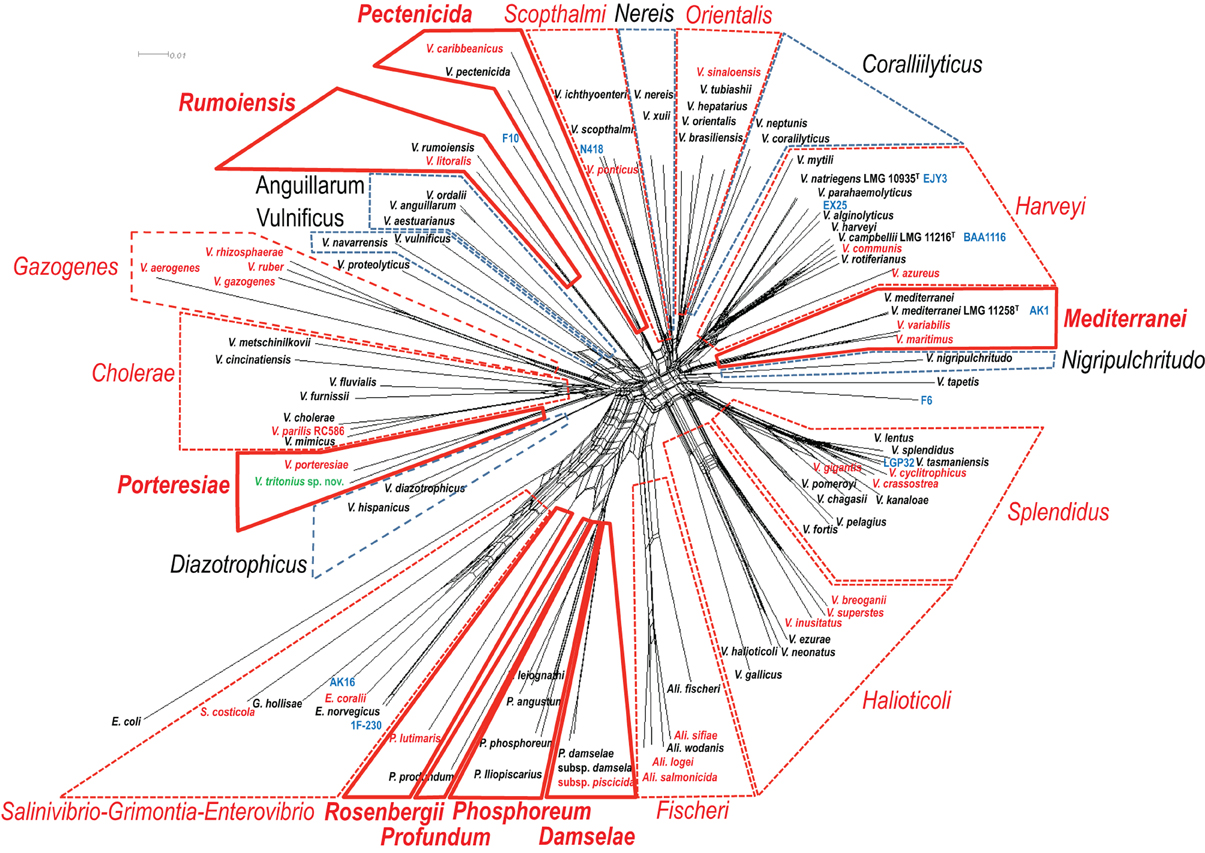
Figure 2. Concatenated split network tree based on eight gene loci. The ftsZ, gapA, gyrB, mreB, pyrH, recA, and topA gene sequences from 96 taxa were concatenated, and a tree was reconstructed using the SplitsTree4 program. Clades indicated by a solid red line were the “new” clades proposed in this study. Clades indicated by a dotted red line or by a dotted black line are the clades “emended” and “un-changed,” respectively.
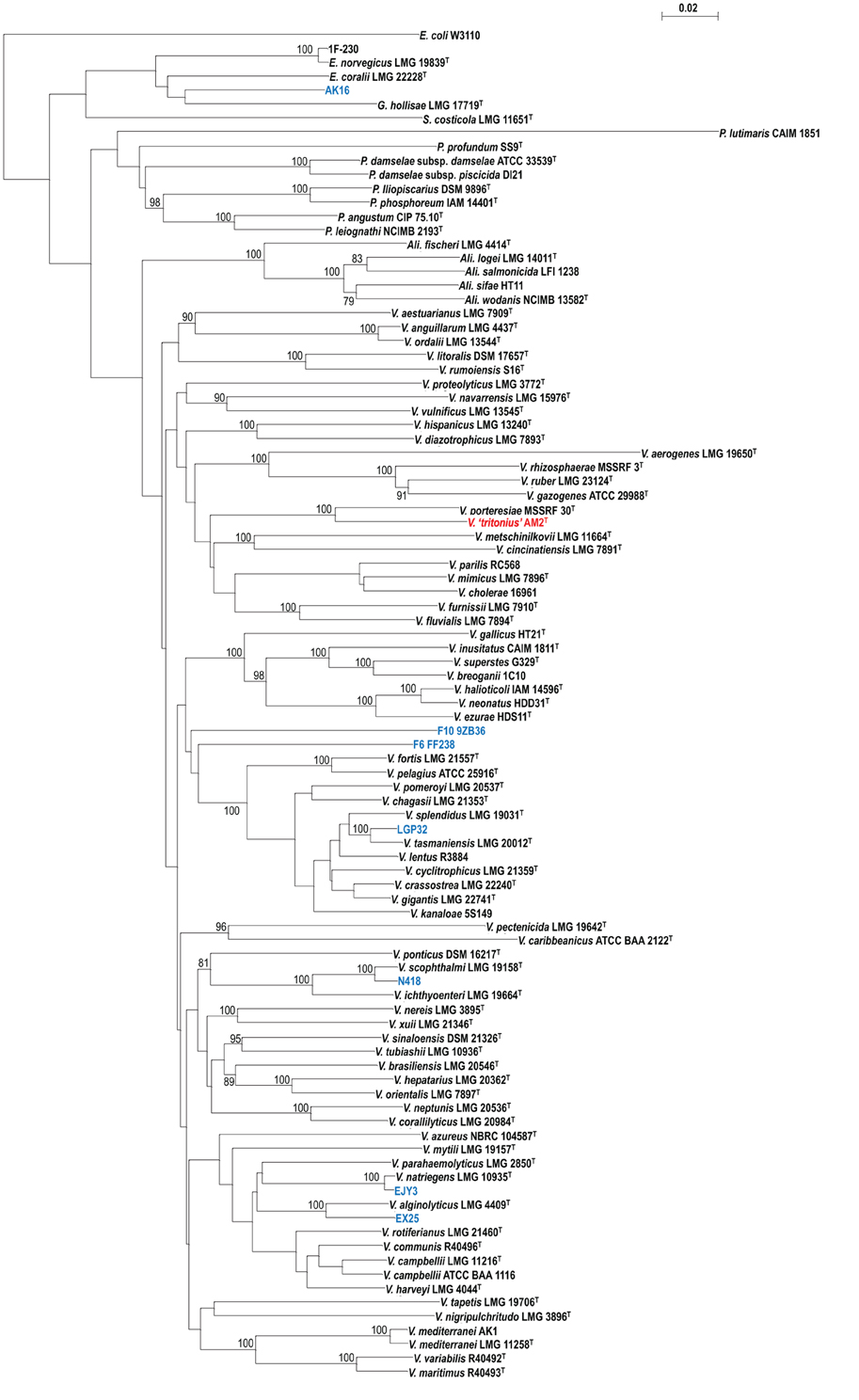
Figure 3. Supertree reconstructed on the basis of the same data set drawn for Figure 2.
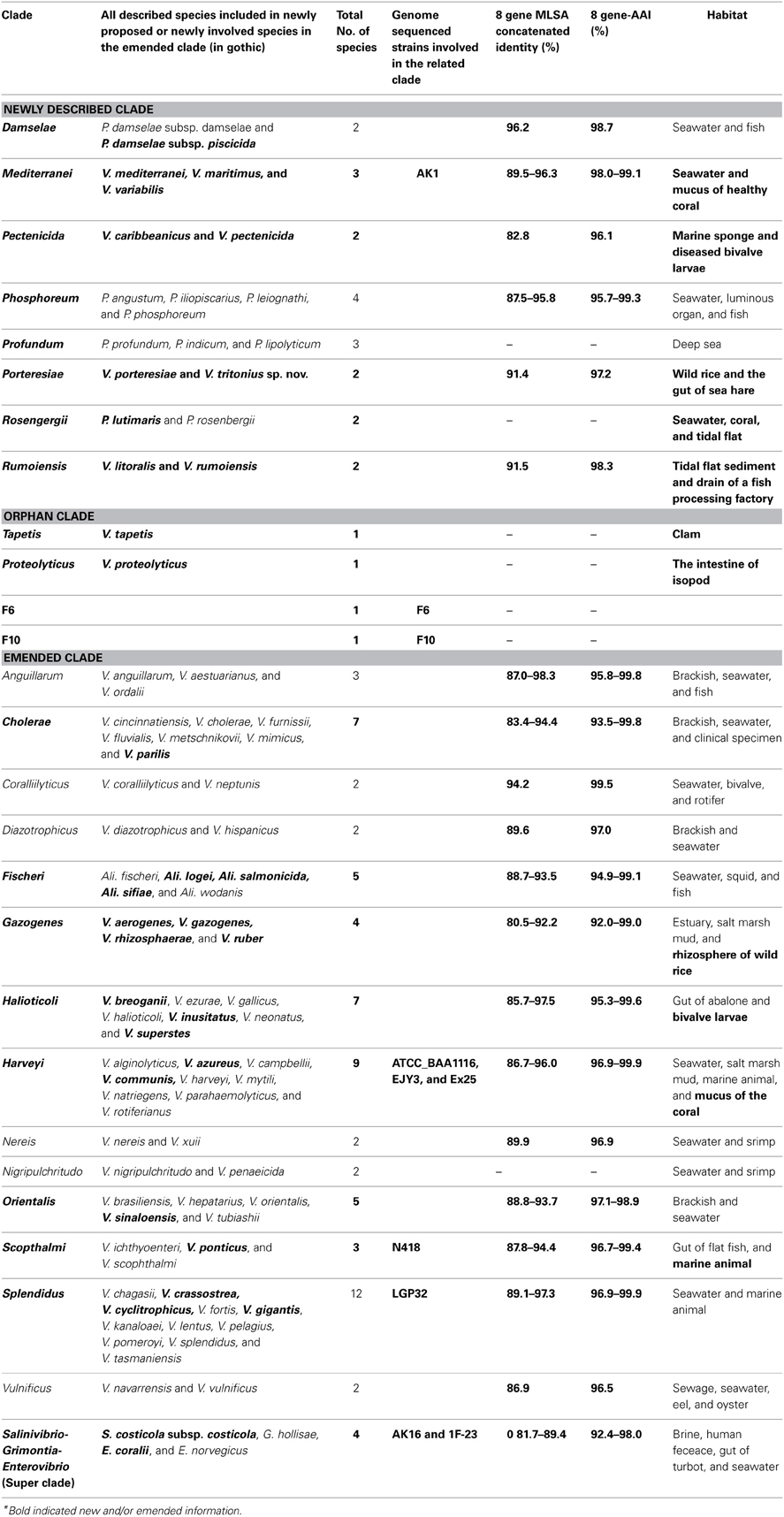
Table 1. Newly proposed and emended clades by means of 8 gene MLSA for vibrios.
New Clades
Mediterranei consisted of three species: V. mediterranei, V. maritimus, and V. variabilis. The 8-gene ANI and AAI were 89.5–96.3% and 98.0–99.1%, respectively. The mol% G+C range of the clade members was 42–46.3 mol%. The genome-sequenced strain V. mediterranei AK1 showed 98.7% ANI and 99.9% AAI against the V. mediterranei type strain. Their known habitats are warm seawater and coral mucus.
Porteresiae consisted of V. porteresiae and the newly described V. tritonius sp. nov. Detailed information for this new species is described in the section “The bacterial taxonomical remarks of V. tritonius sp. nov.” below. These two species shared 91.4% ANI and 97.2% AAI. Two of the unique phenotypes in these species were an efficient H2 production and nitrogen fixation. While the genome sequences of these two species are highly conserved (unpublished data), they have distinct habitats (Table 1). The mol% G+C ranged from 44.2 to 45.5, and the DDH value of V. tritonius type strain against V. porteresiae type strain was 59% (Table 2).
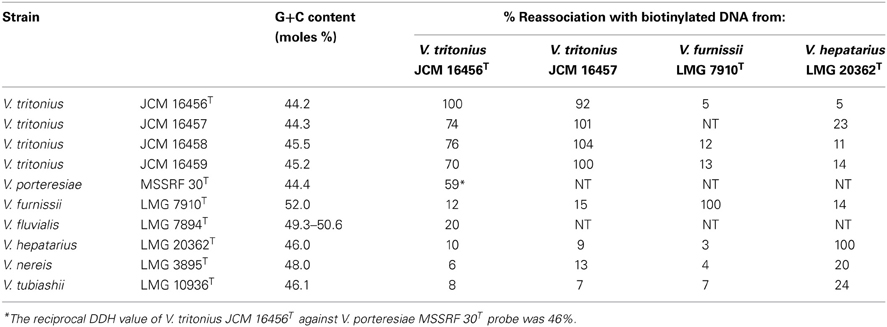
Table 2. DNA relatedness among Vibrio tritonius and the related vibiro species.
Pectenicida consisted of two species, V. caribbeanicus and V. pectenicida showing 82.8% ANI and 96.1% AAI. The reported habitats were tidal flats and diseased larvae, respectively (Table 1).
Rumoiensis consisted of two species, V. litoralis and V. rumoiensis showing 91.5% ANI and 98.3% AAI. These species were isolated from a tidal flat and sewage from a fishery product factory, respectively (Table 1). The reported DDH value between V. litoralis and V. rumoiensis was below 7.4%.
Damselae, Phosphoreum, Profundum, and Rosenbergii were the newly proposed clades that are included in Photobacterium spp. These four new clades are based on ANI (87.5–96.2% in range), AAI (95.7–99.3% in range), and branch separation according to the supertree analysis in comparison with those ranges and branch separations of other Vibrio clades. The Damselae clade consisted of two subspecies of P. damselae. The Rosenbergii clade consisted of P. lutimaris and P. rosenbergii.
Defining Orphan Clades
These are the clades that are formed by only one species. V. tapetis and V. proteolyticus were not grouped with any other species (Figure 2 and Table 1). The recently proposed genomospecies F6 and F10 also did not belong to any of the clades proposed in this analysis. Previously reported singletons, V. agarivorans, and V. pacinii were not included in this analysis due to the lack of some gene sequences.
Emended Clades
We can find emendations in most of the clades previously defined (Figures 2, 3, and Table 1): (1) Cholerae (inclusion of V. parilis); (2) Fischeri (incl. Ali. sifae); (3) Gazogenes (incl. V. rhizosphaerae); (4) Halioticoli (incl. V. breoganii, and V. inusitatus); (5) Harveyi (incl. V. azureus, and V. communis); (6) Orientalis (incl. V. sinaloensis); (7) Scophthalmi (incl. V. ponticus); and (8) Splendidus (incl. V. cyclitrophicus, V. gigantis, and V. crassostrea).
We first included the complete set of the 8-housekeeping protein-coding gene sequences of Salinivibrio costicola subsp. costicola in the MLSA for vibrio phylogeny, because its WGS data (ASAI01000001) are available. However, for the current analysis of the Salinivibrio/Grimontia/Enterovibrio grouping, we could use only the data set including a single species of Salinivibrio, 2 species of Enterovibrio, and a single species of Grimontia for the 8-gene phylogeny. As both SDA and supertree analysis showed less robustness in these genera/species grouping, we decided to tentatively define them as the Salinivibrio-Grimontia-Enterovibrio (SGE) super-clade.
Defining the Clade of Genome Sequenced Strains
Vibrios are one of the most advanced groups in WGS analysis; currently more than 900 genomes are available in the public database (http://www.vibriobiology.net/). The MLSA of the 10 genome sequenced strains revealed: (1) LGP32 and EX25 formed a robust cluster with V. tasmaniensis and V. alginolyticus, respectively; (2) N418 and EJY3 (Roh et al., 2012) were related to V. scopthalmi and V. natriegens, respectively; and (3) The orphan positions of genomospecies F6 and F10, and AK16 (Figures 2, 3, and Table 1).
The Bacterial Taxonomical Remarks of Vibrio tritonius sp. nov.
On the basis of the 8-gene MLSA, the sea hare (Aplysia kurodai) isolates were highly likely to represent a new species within the family Vibrionaceae, more precisely within a new clade “Porteresiae.” Four strains of V. tritonius formed a robust cluster within the Porteresiae clade on the basis of 4-gene sequence SDA (data not shown). To confirm the taxonomic status of the sea hare strains, a standard polyphasic taxonomy was conducted.
The results of our phylogenic analyses based on the 16S rRNA gene sequence clearly showed that these strains belong to class Gammaproteobacteria, and more precisely to the family Vibrionaceae. The closest phylogenic neighbor of the four sea hare isolates was the V. furnissii-V. fluvialis cluster (Figure 4). V. porteresiae was not closely related, as shown by the 16S rRNA gene sequence phylogeny. Intra-species sequence similarities of the 16S rRNA gene among V. tritonius sp. nov. were above 99.5%. Four strains of V. tritonius sp. nov. showed 98.0–98.1% similarity, and 98.1% similarity toward V. furnissii (X76336) and V. fluvialis (X74703), respectively. Sequence similarities of V. tritonius sp. nov. to the other phylogenetic neighbors and to gas-producing vibrios were below 98%. The 16S rRNA gene sequence similarity between V. tritonius sp. nov. and “Allomonas enterica” AJ550855 was 98.3%. The 16S rRNA gene sequences of V. fluvialis X74703 and “A. enterica“ AJ550855 were identical.
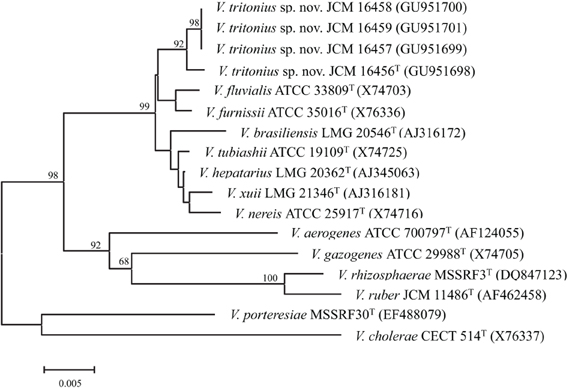
Figure 4. Unrooted phylogenetic tree on the basis of 16S rRNA gene sequences. Scale bar: 0.005 accumulated change per nucleotide. This figure combines the results of three analyses i.e., neighbor-joining, maximum parsimony, and maximum likelihood. The topology shown was obtained using neighbor-joining and 500 bootstrap replications. Percentages indicate the branches that were also obtained both in the maximum likelihood analysis (P < 0.01) and in the most parsimonious tree.
Mutual DDH experiments showed that the four strains of V. tritonius sp. nov., JCM 16456T, JCM 16457, JCM 16458, and JCM 16459, were conspecific and clearly separated from their phylogenetic neighbors, e.g., V. porteresiae, V. fluvialis V. furnissii, V. tubiashii, V. hepatarius, and V. nereis (Table 2). The mol% G+C content was 44.8 ± 0.6, which was within the range of the genus Vibrio.
Description of Vibrio tritonius sp. nov
Etymology of the newly describing Vibrio species was provided here: Vibrio tritonius (tri.to'ni.us. L. masc. adj. tritonius, named after Triton (a sea-god, son of Neptune and the nymph Salacia, referring to the habitat of the bacteria).
Major phenotypic features of V. tritonius sp. nov. are shown in Table 3. The four sea hare strains have the major phenotypic features of the genus Vibrio (except for no growth on TCBS and gas production). These strains required salt for their growth, and they were motile, fermentative and oxidase positive. Apparent catalase activity was not observed. The four strains of V. tritonius sp. nov. were phenotypically most similar to V. porteresiae, but they differed from V. porteresiae in four traits (catalase production, and the assimilation of D-mannose, γ-aminobutyrate and pyruvate), out of 62 tested traits (Table 3). The four V. tritonius strains were sensitive to the vibrio-static agent O/129 (150 μ g). Positive assimilation of glucose, mannitol, gluconate, glucuronate, and xylose indicated the presence of three major carbohydrate metabolic pathways, the Embden-Meyerhof, Entner-Doudoroff, and pentose-phosphate pathways, in V. tritonius sp. nov. Presence of the gene set for those three central metabolic pathways of carbohydrates was supported by our preliminary WGS analysis of V. tritonius JCM 16456T (data not shown). Phenotypic traits differentiating V. tritonius sp. nov. from V. aerogenes, which shows a gas production phenotype, included nitrate reduction, amylase production, and arginine dihydrolase activity. Inability to grow on TCBS was a common trait of V. tritonius sp. nov. and V. porteresiae (Table 3).
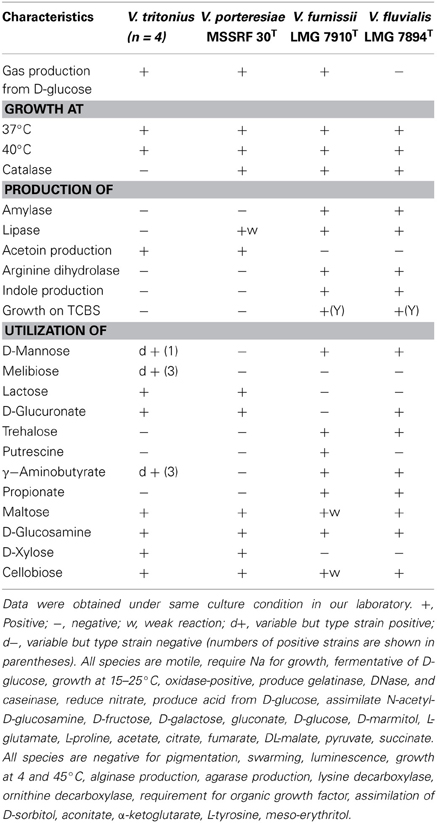
Table 3. Phenotypic characteristics for distinguishing Vibrio tritonius from the related Vibrio species.
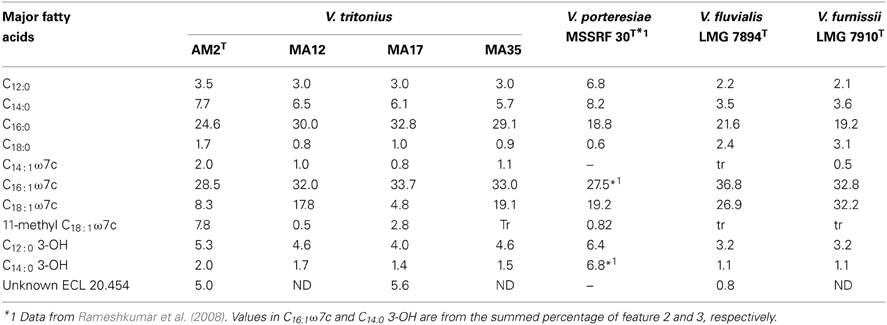
Table 4. FAME dominance (%) of Vibrio tritonius and the related species.
The other phenotypic traits were also described below. No swarming cells were observed. Gas production from glucose and mannitol occurred. Cells are curved rods, with rounded ends, are 0.7–0.9 μm in diameter and 2.6–2.7 μm in length when the organism is grown on ZoBell 2216E medium; the cells occur singly on the agar. No endospores or capsules are formed. Colonies on ZoBell 2216E agar medium are beige, circular, and smooth and convex with an entire edge. Sodium ions are essential for growth. The bacterium can grow in presence of 0.5% to 6% NaCl. The bacterium is a mesophilic chemoorganotroph which grows at temperatures between 15 and 40°C. Optimal growth is observed from 25 to 30°C. Growth occurs from pH 4.5 to pH 9, and optimal growth is at pH 7.5–8.0. No growth occurs at 45°C. The bacterium is positive for acid production from glucose and mannitol; for nitrate reduction, acetoin production, and hydrolysis of gelatin, DNA and casein. The bacterium also can assimilate N-acetyl-D-glucosamine, cellobiose, D-fructose, maltose, D-mannitol, D-galactose, lactose, L-glutamate, L-proline, acetate, citrate, fumarate, DL-malate, pyruvate, and succinate. The bacterium is negative for catalase; indole production; arginine dihydrolase, lysine decarboxylase, ornithine decarboxylase, luminescence, and pigmentation; the requirement of organic growth factors; hydrolysis of agar, alginate, starch, and Tween 80; and assimilation of D-glucosamine, D-sorbitol, aconitate, α-ketoglutarate, L-tyrosine, meso-erythritol, trehalose, putrescine, propionate, and D-glucosamine. The G+C content of DNA is 44.2–45.5 mol%. The type strain is JCM 16459T = LMG 25401T = AM2T.
Discussion
Considerable biodiversity can be found within the family Vibrionaceae (Gomez-Gil et al., 2014), even after the first proposal of vibrio phylogeny and evolution was inferred on the basis of MLSA in 2007 (Sawabe et al., 2007). More than 60 species of Vibrionaceae, with a surprising level of biodiversity, have been described since 2007. These include a marine invertebrate isolates such as coral associated vibrios (Chimetto et al., 2011; Gomez-Gil et al., 2014), introduction of nitrogen-fixing vibrios within an endophyte-like ecological niche (Rameshkumar et al., 2008), and an isolation of new vibrio species from the surface of cheese (Bleicher et al., 2010) have been reported. In addition to the increasing number of newly described vibrio species, many strains showing interesting ecophysiological features have been genome sequenced. However, taxonomic information appears to be insufficient to push the elucidation of vibrio biodiversity and evolution forward. Such a rapid progress in the study of vibrio biodiversity, genomics and evolution prompted us to update the vibrio phylogeny on the basis of MLSA. We have retrieved the complete sets of 8 house-keeping protein-coding gene sequences for 30 additional Vibrionaceae species including a newly described vibrio species, V. tritonius sp. nov., as well as for 10 as yet unnamed Vibrio/Enterovibrio spp. The MLSA led us to propose eight new clades (Damselae, Mediterranei, Pectenicida, Phosphoreum, Profundum, Porteresiae, Rosenbergii, and Rumoiensis) in the family Vibrionaceae, in addition to those previously proposed in the report of Sawabe et al. (2007). In 2007, V. mediterranei, V. petenicida, V. rumoiensis, and P. rosenbergii were affiliated as singlet species, but they have now been grouped. Four orphan clades (Tapetis, Proteolyticus, F6 and F10) were newly defined. More efforts are required to isolate the closest neighbors of these orphan species. Strains EJY3, EX25, N418, and LGP32 clustered robustly with V. natriegens, V. alginolyticus, V. scopthalmi, and V. tasmaniensis, but a further systematic survey is required to analyze the phylogenetic position of AK16 and 1F-230 (Figure 3).
We are still facing a lack of Photobacterium spp. sequences to infer their precise evolutionary history. Among the 23 described Photobacterium spp., we could include only half of them in this study. This situation has arisen mainly due to the “primer problems” in MLSA. Unfortunately, there are also limited numbers of WGS of Photobacterium spp. available in public databases. However, in this analysis, considering the results of SDA, supertree analysis, and the ANI and AAI similarity ranges in comparion to the other Vibrio spp. clades, we proposed four new clades for the Photobacterium spp.; (1) Damselae, (2) Phosphoreum, (3) Profundum, and (4) Rosenbergii. A Salinivibrio/Grimontia/Enterovibrio super-clade is also proposed.
In molecular phylogenetics, the use of minimum gene set is crucial to reduce time and cost, as well as to improve the accuracy, of analyses. This is of particular importance when identifying species and elucidating population structure and evolution in a super bacterial taxon such as the family Vibrionaceae, which has more than 140 species. The previous MLSA of 58 vibrio taxa (Sawabe et al., 2007) showed that 16S rRNA gene sequences have an extremely low species/strain discriminating power compared to the other genes tested. Therefore, before conducting the current vibrio MLSA, we evaluated whether the 16S rRNA gene data set could be eliminated from the MLSA. For this analysis, we developed a subtree incongruence test algorithm. The algorithm is a fast and reliable method for selecting subtrees that share the same topology or those that have different topologies but share the same species. The results of this analysis indicate that inclusion of 16S rRNA gene sequences is not necessary for reconstructing the vibrio phylogeny on the basis of MLSA.
We have experienced the first case in which the molecular phylogenies resulting from 16S rRNA gene sequences and from housekeeping gene sequences were largely incongruent in the species descriptions of V. porteresiae and V. tritonius (Figures 2, 4). For the affiliation of the clade of V. porteresiae, we used only four genes (the pyrH, recA, rpoA, and 16S rRNA genes), and we confirmed that V. porteresiae was affiliated with the Cholerae clade (Rameshkumar et al., 2008). The 16S rRNA gene sequence phylogeny revealed that V. tritonius sp. nov. was the most closely related to V. furnissi and V. fulvialis with ca. 98% sequence similarity, and V. tritonius sp. nov. and V. porteresiae were distantly related in their phylogenetic relationship (Figure 4). Less phylogenetic relatedness in the 16S rRNA gene sequence tree between both the species and the lack of housekeeping gene sequences of V. porteresiae prevented no direct comparison of V. porteresiae and V. tritonius sp. nov. Fortunately, the whole genome nucleotide sequences of V. porteresieae and V. tritonius sp. nov. were determined in this study, and the first direct comparison of both species by MLSA and whole genome comparison was achieved. Surprisingly, the MLSA with 8-housekeeping genes phylogeny led to the conclusion that both Vibrio species, V. porteresiae and V. tritonius sp. nov., share a common ancestry and that they can be proposed as a new vibrio clade, “Porteresiae.” Our preliminary genome comparison of both species also supported monophyly because a strong synteny was observed between both genomes (unpublished data). Incongruences between the 16S rRNA gene sequence tree and the MLSA tree were also observed in Mediterranei and Pectenicida clades (Lambert et al., 1998; Chimetto et al., 2011; Hoffmann et al., 2012) in this study. Therefore, we conclude that at present the “8-housekeeping-gene phylogeny” is the most powerful method for delineating vibrio species description/biodiversity/population study/evolution, until alternative genome-based approaches are proposed. This analysis reduces the misidentification of ancestry clades of vibrios.
The polyphasic taxonomic approach reveals that the Aplysia gut isolates, V. tritonius sp. nov., to be a novel Vibrio species showing gas producing ability (Figures 2–4, Tables 2, 3) and forming a robust clade, Porteresiae, with V. porteresiae. Production of gas during the fermentation of carbohydrates is not a prevalent property in the Vibrio genus (Shieh et al., 2000, 2003; Farmer III et al., 2005; Kumar and Nair, 2007; Rameshkumar et al., 2008). V. aerogenes, V. furnissii, V. gazogenes, V. porteresiae, V. ruber, and V. rhizosphaerae are the species in which the gas production phenotype is retained in a stable way among 98 Vibrio species. For this reason, gas production is an atypical property in the genus Vibrio. The current phylogenetic network analysis using the 8-gene MLSA confirmed that V. aerogenes, V. gazogenes, V. rhizosphaerae, and V. ruber form a robust clade, Gazogenes (Figure 2) (Kumar and Nair, 2007; Sawabe et al., 2007). V. furnissii belongs to the Cholerae clade. The newly described V. tritonius sp. nov. belongs to a new clade, Porteresiae. The gas compositions of Porteresiae clade species but also of the Gazogenes and Cholerae clades, were identical for H2 and CO2, but the H2 production efficiencies differ between these clades. The H2 production efficiency of V. tritonius sp. nov. and V. porteresiae was high and comparable to that of enterobacterial species, such as Escherichia coli, Salmonella, Enterobacter, and Klebsiella (Nakashimada et al., 2002). Our preliminary genome comparisons suggests that V. tritonius sp. nov. and V. porteresiae contain very similar gene clusters responsible for H2 production and nitrogen fixation machinery (unpublished data). It would be intriguing to understand how those vibrios acquire and/or lose gas producing abilities from the standpoint of both evolutionary dynamics and metabolic diversity.
In conclusion, 8-gene MLSA is a reliable tool for delineating a species and a monophyletic group or “clade.” Using the current data set reported in this study, <98% of 8-gene-concatenated nucleotide sequence identity may allow us to define a species boundary. We also showed that WGSs overcome the limitations of gene-by-gene multilocus sequencing tasks found in the MLSA for Vibrionaceae (primer problem, Chromosome 2 gene inclusion). In fact, we could not include V. aerogenes, V. gazogenes, V. rhizosphaerae, and V. superstes for the previous 9-gene MLSA by the primer problems (Sawabe et al., 2007), but the successful WGS for these species and the inclusion of 8 housekeeping protein gene sequences retrieved from the genome sequences can provide the better picture of current vibrio molecular phylogeny. More efforts to sequence the individual housekeeping genes and WGS of all remaining species in the family Vibrionaceae not involved in this study allow the possibility of elucidating the ultimate clade structure of these Vibrios. The ultimate phylogenetic trees allow us to provide the ideal phylogenetic backbone to elucidate the evolutional history, genome dynamics, and plasticity in the family Vibrionaceae.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Ministry of Agriculture, Forestry, and Fisheries, Japan, and KAKENHI (Grants-in-Aid for Scientific Research from Ministry of Education, Culture, Sports, Science, and Technology of Japan) (No. 21380129, 2365817201, 2529212203, and 2529212283). This work was also supported by the National Council for Science and Technology (CONACYT) of Mexico (Grant No. 132328 awarded to Bruno Gomez Gil), by Grant in Aid for Scientific Research on Innovative Area “Genome Science” from Ministry of Education, Culture, Sports, Science, and Technology of Japan (No. 221S0002), and by JST-CNPq Strategic Japanese-Brazilian Cooperative Program, Biomass and Biotechnology. We gratefully thank Professor Jean Euzéby at the École Nationale Vétérinaire, Toulouse for his advice and suggestions on naming Vibrio tritonius, and Professor Elena P. Ivanova, Swinburne University of Technology, for a critical reading of the manuscript. We also thank Fumito Testukawa, Takashi Wakabayashi, and Yoshiko Kawahara for technical contributions.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2013.00414/abstract
The GenBank accession numbers for the ftsZ, gapA, gyrB, mreB, recA, rpoA, pyrH, and topA gene sequences used in this analysis, as well as those used in Sawabe et al. (2007), are listed in Table S1.
Table S1 | List of strains and sequences accession number used for the MLSA.
Abbreviations
AFLP, amplified fragment length polymorphism; ANI, average nucleotide sequence identity; AAI, average amino acid identity; DDH, DNA-DNA hybridization; MLSA, multilocus sequence analysis; MLST, multilocus sequence typing; MP, maximum parsimony; NJ, neighbor joining; ML, maximum likelihood; PFGE, pulse field gel electrophoresis; RAPD, random amplified polymorphic DNA.
References
Aanensen, D. M., and Spratt, B. G. (2005). The multilocus sequence typing network: mlst.net. Nucleic Acids Res. 33, W728–W733. doi: 10.1093/nar/gki415
Auch, A. F., von Jan, M., Klenk, H.-P., and Göker, M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Aujoulat, F., Jumas-Bilak, E., Masnou, A., Sallé, F., Faure, D., Segonds, C., et al. (2011). Multilocus sequence-based analysis delineates a clonal population of Agrobacterium (Rhizobium) radiobacter (Agrobacterium tumefaciens) of human origin. J. Bacteriol. 193, 2608–2618. doi: 10.1128/JB.00107-11
Bandelt, H.-J., and Dress, A. W. M. (1992). Split decomposition: a new and useful approach to phylogenetic analysis of distance data. Mol. Phylogenet. Evol. 1, 242–252. doi: 10.1016/1055-7903(92)90021-8
Bennett, J. S., Jolley, K. A., Earle, S. G., Corton, C., Bentley, S. D., Parkhill, J., et al. (2012). A genomic approach to bacterial taxonomy: an examination and proposed reclassification of species within the genus Neisseria. Microbiology 158, 1570–1580. doi: 10.1099/mic.0.056077-0
Bishop, C. J., Aanensen, D. M., Jordan, G. E., Kilian, M., Hanage, W. P., and Spratt, B. G. (2009). Assigning strains to bacterial species via the internet. BMC Biol. 7:3. doi: 10.1186/1741-7007-7-3
Bleicher, A., Neuhaus, K., and Scherer, S. (2010). Vibrio casei sp. nov., isolated from the surfaces of two French red smear soft cheeses. Int. J. Syst. Evol. Microbiol. 60, 1745–1749. doi: 10.1099/ijs.0.016493-0
Chan, J. Z.-M., Halachev, M. R., Loman, N. J., Constantinidou, C., and Pallen, M. J. (2012). Defining bacterial species in the genomic era: insights from the genus Acinetobacter. BMC Microbiol. 12:302. doi: 10.1186/1471-2180-12-302
Chimetto, L. A., Cleenwerck, I., Moreira, A. P. B., Brocchi, M., Willems, A., De Vos, P., et al. (2011). Vibrio variabilis sp. nov. and Vibrio maritimus sp. nov., isolated from Palythoa caribaeorum. Int. J. Syst. Evol. Microbiol. 61, 3009–3015. doi: 10.1099/ijs.0.026997-0
Chevenet, F., Brun, C., Bañuls, A.-L., Jacq, B., and Christen, R. (2006). TreeDyn: towards dynamic graphics and annotations for analyses of trees. BMC Bioinfomatics 7:439. doi: 10.1186/1471-2105-7-439
Ezaki, T., Hashimoto, Y., Takeuchi, N., Yamamoto, H., Liu, S.-L., Miura, H., et al. (1988). Simple genetic method to identify viridans group streptococci by colorimetric dot hybridization and fluorometric hybridization in microdilution wells. J. Clin. Microbiol. 26, 1708–1713.
Ezaki, T., Hashimoto, Y., and Yabuuchi, E. (1989). Fluorometric deoxyribonucleic acid-deoxyribonucleic acid hybridization in microdilution wells as an alternative to membrane filter hybridization in which radioisotopes are used to determine genetic relatedness among bacterial strains. Int. J. Syst. Bacteriol. 39, 224–229. doi: 10.1099/00207713-39-3-224
Farmer, III, J. J., Janda, J. M., Brenner, F. W., Cameron, D. N., and Birkhead, K. M. (2005). “Genus I. Vibrio Pacini 1854, 411AL,” in Bergey's Manual of Systematic Bacteriology, 2nd Edn., Vol. 2, eds D. J. Brenner, N. R. Krieg, and J. T. Staley (New York, NY: Springer), 494–546.
Gevers, D., Cohan, F. M., Lawrence, J. G., Spratt, B. G., Coenye, T., Feil, E. J., et al. (2005). Re-evaluating prokaryotic species. Nat. Microbiol. 3, 733–739. doi: 10.1038/nrmicro1236
Gomez-Gil, B., Thompson, C. C., Matsumura, Y., Sawabe, T., Iida, T., Christen, R., et al. (2014). “Family Vibrionaceae (Chapter 225),” in The Prokaryotes, 4th Edn., eds E. Rosenberg, E. F. DeLong, F. L. Thonpson, S. Lory, and E. Stackebrandt (New York, NY: Springer), 88.
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Grimes, D. J., Johnson, C. N., Dillon, K. S., Flowers, A. R., Noriea III, N. F., and Berutti, T. (2009). What genomic sequence information has revealed about vibrio ecology in the ocean-a review. Microb. Ecol. 58, 447–460. doi: 10.1007/s00248-009-9578-9
Guénoche, A. (2013). Multiple consensus trees: a method to separate divergent genes. BMC Bioinformatics 14:46. doi: 10.1186/1471-2105-14-46
Haley, B. J., Grim, C. J., Hasan, N. A., Choi, S.-Y., Chun, J., Brettin, T. S., et al. (2010). Comparative genomic analysis reveals evidence of two novel Vibrio species closely related to V. cholera. BMC Microbiol. 10:154. doi: 10.1186/1471-2180-10-154
Hoffmann, M., Monday, S. R., Allard, M. W., Strain, E. A., Whittaker, P., Naum, M., et al. (2012). Vibrio caribbeanicus sp. nov., isolated from the marine sponge Scleritoderma cyanea. Int. J. Syst. Evol. Microbiol. 62, 1736–1743. doi: 10.1099/ijs.0.032375-0
Huerta-Cepas, J., Dopazo, J., and Gabaldón, T. (2010). ETE: a python environment for tree exploration. BMC Bioinformatics 11:24. doi: 10.1186/1471-2105-11-24
Huson, D. H., and Bryant, D. (2005). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Kumar, R. N., and Nair, S. (2007). Vibrio rhizosphaerae sp. nov., a novel red-pigmented species that antagonizes phytopathogenic bacteria. Int. J. Syst. Evol. Microbiol. 57, 2241–2246. doi: 10.1099/ijs.0.65017-0
Lambert, C., Nicolas, J. L., Cilia, V., and Corre, S. (1998). Vibrio pectenicida sp. nov., a pathogen of scallop (Pecten maximus) larvae. Int. J. Syst. Bacteriol. 48, 481–487. doi: 10.1099/00207713-48-2-481
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Lin, B., Wang, Z., Malanoski, A. P., O'Grady, E. A., Wimpee, C. F., Vuddhakul, V., et al. (2010). Comparative genomic analyses identify the Vibrio harveyi genome sequenced strains BAA-1116 and HY01 as Vibrio campbellii. Environ. Microbiol. Rep. 2, 81–89. doi: 10.1111/j.1758-2229.2009.00100.x
Maiden, M. C. J., Bygraves, J. A., Feil, E., Morelli, G., Russell, J. E., Urwin, R., et al. (1998). Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U.S.A. 95, 3140–3145. doi: 10.1073/pnas.95.6.3140
Marmur, J. (1961). A procedure for the isolation of deoxyribonucleic acid from microorganisms. J. Mol. Biol. 3, 208–218. doi: 10.1016/S0022-2836(61)80047-8
Nakashimada, Y., Rachman, M. A., Kakizono, T., and Nishio, N. (2002). Hydrogen production of Enterobacter aerogenes altered by extracellular and intracellular redox states. Int. J. Hydrogen Energy 27, 1399–1405. doi: 10.1016/S0360-3199(02)00128-3
Oppenheimer, C. H., and ZoBell, C. E. (1952). The growth and viability of sixty-three species of marine bacteria as influenced by hydrostatic pressure. J. Mar. Res. 11, 10–18.
Preheim, S. P., Timberlake, S., and Pols, M. F. (2011). Merging taxonomy with ecological population prediction in a case study of Vibrionaceae. Appl. Environ. Microbiol. 77, 7195–7206. doi: 10.1128/AEM.00665-11
Rameshkumar, N., Fukui, Y., Sawabe, T., and Nair, S. (2008). Vibrio porteresiae sp. nov., a diazotrophic bacterium isolated from a mangrove-associated wild rice (Porteresia coarctata Tateoka). Int. J. Syst. Evol. Microbiol. 58, 1608–1615. doi: 10.1099/ijs.0.65604-0
Richer, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Roger, F., Marchandin, H., Jumas-Bilak, E., Kodjo, A., the colBVH study group, and Lamy, B. (2012). Multilocus genetics to reconstruct aeromonad evolution. BMC Microbiol. 12:62. doi: 10.1186/1471-2180-12-62
Roh, H., Yun, E. J., Lee, S., Ko, H. J., Kim, S., Kim, B. Y., et al. (2012). Genome sequence of Vibrio sp. strain EJY3, an agarolytic marine bacterium metabolizing 3,6-anhydro-L-galactose as a sole carbon source. J. Bacteriol. 194, 2773–2774. doi: 10.1128/JB.00303-12
Sawabe, T., Kita-Tsukamoto, K., and Thompson, F. L. (2007). Inferring the evolutionary history of vibrios by means of multilocus sequence analysis. J. Bacteriol. 189, 7932–7936. doi: 10.1128/JB.00693-07
Sawabe, T., Koizumi, S., Fukui, Y., Nakagawa, S., Ivanova, E. P., Kita-Tsukamoto, K., et al. (2009). Mutation is the main driving force in the diversification of the Vibrio splendidus clade. Microbes Environ. 24, 281–285. doi: 10.1264/jsme2.ME09128
Sawabe, T., Sugimura, I., Ohtsuka, M., Nakano, K., Tajima, K., Ezura, Y., et al. (1998). Vibrio halioticoli sp. nov., a non-motile alginolytic marine bacterium isolated from the gut of abalone Haliotis discus hannai. Int. J. Syst. Bacteriol. 48, 573–580. doi: 10.1099/00207713-48-2-573
Shieh, W. Y., Chen, A.-L., and Chiu, H.-H. (2000). Vibrio aerogenes sp. nov., a facultatively anaerobic marine bacterium that ferments glucose with gas production. Int. J. Syst. Evol. Microbiol. 50, 321–329. doi: 10.1099/00207713-50-1-321
Shieh, W. Y., Chen, Y.-W., Chaw, S.-M., and Chiu, H.-H. (2003). Vibrio ruber sp. nov., a red, facultatively anaerobic, marine bacterium isolated from sea water. Int. J. Syst. Evol. Microbiol. 53, 479–484. doi: 10.1099/ijs.0.02307-0
Stackebrandt, E., Frederiksen, W., Garrity, G. M., Grimont, P. A. D., Kämpfer, P., Maiden, M. C. J., et al. (2002). Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 52, 1043–1047. doi: 10.1099/ijs.0.02360-0
Staley, J. T. (2006). The bacterial species dilemma and the genomic–phylogenetic species concept. Philos. Trans. R. Soc. B Biol. Sci. 361, 1899–1909. doi: 10.1098/rstb.2006.1914
Sugawara, H., Ohyama, A., Mori, H., and Kurokawa, K. (2009). “Microbial Genome Annotation Pipeline (MiGAP) for diverse users,” in 20th International Conference on Genome Informatics (Kanagawa), S-001, 1–2.
Tamaoka, J., and Komagata, K. (1984). Determination of DNA base composition by reversed-phase high-performance liquid chromatography. FEMS Microbiol. Lett. 25, 125–128. doi: 10.1111/j.1574-6968.1984.tb01388.x
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Thompson, C. C., Vicente, A. C., Souza, R. C., Vasconcelos, A. T., Vesth, T., Alves, N. Jr., Ussery, D. W., et al. (2009). Genomic taxonomy of Vibrios. BMC Evol. Biol. 9:258. doi: 10.1186/1471-2148-9-258
Thompson, F. L., Gevers, D., Thompson, C. C., Dawyndt, P., Naser, S., Hoste, B., et al. (2005). Phylogeny and molecular identification of Vibrios on the basis of multilocus sequence analysis. Appl. Environ. Microbiol. 71, 5107–5115. doi: 10.1128/AEM.71.9.5107-5115.2005
Thompson, F. L., Gomez-Gil, B., Vasconcelos, A. T. R., and Sawabe, T. (2007). Multilocus sequence analysis reveals that Vibrio harveyi and V. campbellii form distinct species. Appl. Environ. Microbiol. 73, 4279–4285. doi: 10.1128/AEM.00020-07
Thompson, F. L., Hoste, B., Vandemeulebroecke, K., and Swings, J. (2001). Genomic diversity amongst Vibrio isolated from different sources determined by fluorescent amplified fragment length polymorphism. Syst. Appl. Microbiol. 24, 520–538. doi: 10.1078/0723-2020-00067
Keywords: vibrios, Vibrionaceae, multilocus sequence analysis, evolution, housekeeping protein gene, Vibrio tritonius
Citation: Sawabe T, Ogura Y, Matsumura Y, Feng G, Amin AKMR, Mino S, Nakagawa S, Sawabe T, Kumar R, Fukui Y, Satomi M, Matsushima R, Thompson FL, Gomez-Gil B, Christen R, Maruyama F, Kurokawa K and Hayashi T (2013) Updating the Vibrio clades defined by multilocus sequence phylogeny: proposal of eight new clades, and the description of Vibrio tritonius sp. nov. Front. Microbiol. 4:414. doi: 10.3389/fmicb.2013.00414
Received: 27 September 2013; Paper pending published: 18 October 2013;
Accepted: 16 December 2013; Published online: 27 December 2013.
Edited by:
Daniela Ceccarelli, University of Maryland, USAReviewed by:
Xiu-Lan Chen, Shandong University, ChinaMatteo Spagnoletti, University College London, UK
Copyright © 2013 Sawabe, Ogura, Matsumura, Feng, Amin, Mino, Nakagawa, Sawabe, Kumar, Fukui, Satomi, Matsushima, Thompson, Gomez-Gil, Christen, Maruyama, Kurokawa and Hayashi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomoo Sawabe, Laboratory of Microbiology, Faculty of Fisheries Sciences, Hokkaido University, 3-1-1 Minato-cho, Hakodate 041-8611, Japan e-mail:c2F3YWJlQGZpc2guaG9rdWRhaS5hYy5qcA==