 Ekaterina Osmekhina
Ekaterina Osmekhina Antonina Shvetsova
Antonina Shvetsova Maria Ruottinen
Maria Ruottinen Peter Neubauer
Peter Neubauer- 1Department of Process and Environmental Engineering and Biocenter Oulu, University of Oulu, Oulu, Finland
- 2Department of Biochemistry and Biocenter Oulu, University of Oulu, Oulu, Finland
- 3Laboratory of Bioprocess Engineering, Department of Biotechnology, Technische Universität Berlin, Berlin, Germany
The fast and reliable detection of bacterial spores is of great importance and still remains a challenge. Here we describe a direct RNA-based diagnostic method for the specific detection of viable bacterial spores which does not depends on an enzymatic amplification step and therefore is directly appropriate for quantification. The procedure includes the following steps: (i) heat activation of spores, (ii) germination and enrichment cultivation, (iii) cell lysis, and (iv) analysis of 16S rRNA in crude cell lysates using a sandwich hybridization assay. The sensitivity of the method is dependent on the cultivation time and the detection limit; it is possible to detect 10 spores per ml when the RNA analysis is performed after 6 h of enrichment cultivation. At spore concentrations above 106 spores per ml the cultivation time can be shortened to 30 min. Total analysis times are in the range of 2–8 h depending on the spore concentration in samples. The developed procedure is optimized at the example of Bacillus subtilis spores but should be applicable to other organisms. The new method can easily be modified for other target RNAs and is suitable for specific detection of spores from known groups of organisms.
Introduction
Detection of microorganisms in air is challenging due to the very low amount of organisms that generally are collected in air samples and the presence of organisms as dormant spores.
Various biosensors have been developed for the specific detection of harmful microbial spores in air samples (Gooding, 2006). Most methods for the detection of Bacillus spores have been designed for Bacillus anthracis, a causative factor for anthrax and a potential biological threat agent (Edwards et al., 2006; Irenge and Gala, 2012), in order to create sensors for biological warfare agents. Being a causative agent, B. anthracis is difficult to work with, and therefore the closely related species Bacillus subtilis and B. cereus are often used in the development of detection strategies (Arakawa et al., 2003; Stachowiak et al., 2007; Inami et al., 2009; Cheng et al., 2011).
The primary strategies for detection of Bacillus spores include polymerase chain reaction based techniques, immunoassays, spectrometry, chromatography, and protein profiling (Table 1). Recently also some autonomous pathogen detection systems for aerosol collection, sample preparation and detection were developed (Hindson et al., 2005; Stachowiak et al., 2007; Regan et al., 2008; Inami et al., 2009) (Table 1).
Table 1. Methods for detection of Bacillus spores.
A number of direct methods are based on DNA or protein detection and cannot distinguish between viable and dead spores. Furthermore, most of them have low sensitivity or require an amplification step such as PCR. RNA-based detection methods have the advantage to specifically analyze only viable spores, and RNA's, especially ribosomal RNAs (rRNAs), are populated in high amounts, making enzyme-based amplifications methods indispensible. Thus it is reasonable to apply RNA detection after the activation of RNA synthesis which takes place already after approximately 10 min of germination (Keijser et al., 2007).
Sandwich hybridization assays (SHA) are suitable for a rapid and quantitative RNA detection (Rautio et al., 2003). These methods are based on the hybridization of a target RNA (or denatured DNA) with two specific oligonucleotide probes. A capture probe is used to immobilize the target on a solid support, such as magnetic microbeads, which provide a large surface area for nucleic acid attachments (Walsh et al., 2001). This binding between the probes and the beads is usually performed by interaction between biotin attached to the oligonucleotide probe and streptavidin coated magnetic beads. The detection probe is labeled with a marker molecule which generates a signal proportional to the amount of target molecules. Oligonucleotide probes required for this assay can be designed for almost any RNA and can easily be modified for other targets. This means that the developed detection system can be applied for different organisms with just some small adaptations. Sandwich hybridization is relatively sensitive (10−16–10−15 moles of a specific target molecule) and can be performed with crude biological samples without any RNA purification. The method has been successfully applied for the detection of 16S rRNA from Legionella sp. in water samples (Leskelä et al., 2005), mycobacteria in soils (Nieminen et al., 2006), Lactobacillus and Pediococcus in brewery yeast slurries (Huhtamella et al., 2007), Salmonella in minced meat (Taskila et al., 2011), Bacillus cereus DNA (Gabig-Ciminska et al., 2004) and for monitoring dynamic changes of different mRNA species in microbial processes (Rautio et al., 2003; Neubauer et al., 2007; Soini et al., 2008; Thieme et al., 2008). The possibility to use various markers makes the method applicable for different read-out systems, such as fluorescence meters (Rautio et al., 2003), chip-based fluorescent biosensors (Wang et al., 2013) or electrical biochip readers (Gabig-Ciminska et al., 2004; Jürgen et al., 2005; Elsholz et al., 2006; Pioch et al., 2008a,b). The SHA coupled with capillary electrophoresis called TRAC (transcript analysis with aid of affinity capture) was developed for multiplex transcript analysis and is commercially available (Rautio et al., 2006, 2008; Rautio, 2010) (PlexPress Oy, Finland).
In the actual study a procedure was developed for detecting bacterial spores, utilizing spore activation, enrichment cultivation and an RNA-based sandwich hybridization assay. In contrast to most assays utilizing DNA and protein detection, the method developed here is specific only for viable organisms since it is based on RNA synthesis.
Materials and Methods
Spore Preparation
Bacillus subtilis spores were obtained from cells (B. subtilis 6051α, kindly provided by Prof. Dr. Thomas Schweder, Ernst-Moritz-Arndt University of Greifswald, Germany) cultured in Schaeffer's sporulation medium (8 g/l bacto-nutrient broth, 0.1% (w/v) KCl, 0.012% (w/v) MgSO4 × 7H20, 0.5 mM NaOH, 1 mM Ca(NO3)2, 0.01 mM MnCl2, 0.001 mM FeSO4) at 37°C, 200 rpm for 4 days. Cultures were carried out in 3-baffled 1 liter Erlenmeyer flasks with a liquid volume of 100 ml. After harvesting and extensive washing with water at 4°C, spores were inspected by phase-contrast microscopy to show that samples are free of vegetative cells (< 10%). Spores were lyophilized and stored at 4°C. For counting, the spores were diluted in 0.9% NaCl, activated at 70°C for 30 min and plated on nutrient agar plates (Difco, USA).
Activation and Growth Conditions
B. subtilis spores were diluted in 0.9% NaCl and activated by temperature treatment at 70°C for 30 min. Thereafter the suspensions were transferred into 3-baffled 1 l Erlenmeyer flasks containing 100 ml of a germination medium (20 g/l tryptone, 10 g/l yeast extract, 10 g/l NaCl and 0.5 mM L-alanine) (Moeller et al., 2006). Cultures were carried out at 37°C and 200 rpm on a rotary shaker. Culture growth was monitored by measuring the optical density at 600 nm (OD600, Ultrospec Pro 2100 UV/Visible Spectrophotometer, GE Healthcare, Buckinghamshire, UK). The corresponding colony forming units (cfu/ml) were determined by plating on nutrient agar plates.
Sample Preparation
1 ml of the B. subtilis cultures were removed into pre-cooled 1.5 ml microreaction tubes containing 0.125 × sample volume of cold 95:5 ethanol:phenol (v/v) (Thieme et al., 2008). The cells were collected by centrifugation (15,000 × g, + 4°C, 5 min). The pellets were suspended in 500 μl of 1 × TEN buffer (10 mM Tris-HCl, 1 mM EDTA, 100 mM NaCl, pH 8.0) containing 1 μl/ml RNAguard® RNase inhibitor (Amersham Biosciences, New Jersey, USA) and mixed by vortexing for 1 min. Then the cell suspensions were transferred into 2 ml microcentrifuge tubes with a skirt (Greiner Bio-One GmbH, Frickenhausen, Germany) containing 100 mg glass beads (BioSpec Products Inc., Bartlesville, OK, USA) with a diameter of 0.1 mm. Cell disruption was performed with a FastPrep® FP120 Cell Disrupter (Bio-101, Thermo Savant, USA) at 4.5 m/s for 4 × 25 s. The vials were kept on ice for 1 min between the disruption cycles. Cell homogenates were centrifuged (20,000 ×g, + 4°C, 5 min) and supernatants were snap frozen in liquid nitrogen.
Generation of the DNA for the In Vitro 16S rRNA Transcript
The target gene was amplified by PCR using a colony of B. subtilis cells as a template and 10 pmol of the forward and reverse primers (Weisburg et al., 1991) (Table 2). The forward primer contained at the 5′–end the T7 promoter sequence CTA ATA CGA CTC ACT ATA GGG. The PCR conditions were as follows: 3 min at 95°C; 35 cycles of 15 s at 95°C, 30 s at 50°C and 1 min 45 s at 72°C; 10 min at 72°C and afterwards held at 4°C. PCR products were analyzed by agarose gel electrophoresis and purified with the QIAquick PCR purification kit (Qiagen, Hilden, Germany). Purified PCR products were quantified by absorbance measurement at 260 nm (GeneQuant II, Viochrom Ltd., Cambridge, UK).
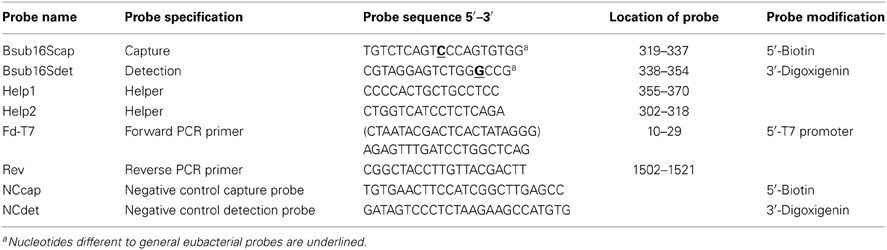
Table 2. Sequences of the oligonucleotide probes used in sandwich hybridization for the detection of B. subtilis 16S rRNA and PCR primers.
Target RNA was generated from the purified PCR product by means of T7 RNA polymerase in vitro transcription using the MAXIscript™ T7-kit (Ambion, Austin, TX, USA). The quality of the in vitro transcribed RNA was checked by the Agilent 2100 Bioanalyser and RNA 6000 Nano LabChip Kit (Agilent Technologies, Waldbronn, Germany). RNA was quantified by absorbance measurement at 260 nm (NanoDrop ND 1000, Thermo Fisher Scientific, USA).
Oligonucleotide Probes
The oligonucleotide probes for B. subtilis 16S rRNA were designed on the basis of general eubacterial probes with some modifications (Table 2). The oligonucleotide probes were purchased from Oligomer Oy (Helsinki, Finland). The detection probe was labeled with the DIG Oligonucleotide Tailing Kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's instructions.
Sandwich Hybridization Assay
A quantitative sandwich hybridization assay (SHA) was used for detecting 16S rRNA molecules. The procedure was performed as described by Rautio et al. (2003) and Thieme et al. (2008) with small modifications.
SHAs were carried out in 96-well bright U-shaped microplates (Greiner-Bio-One GmbH, Frickenhausen, Germany) using a Thermomixer Comfort (Eppendorf, Hamburg, Germany). The target RNA was mixed with the 5 × SSC hybridization buffer (0.75 M sodium chloride, 0.075 M sodium citrate, pH 7.0), 20% (v/v) deionized formamide, 3% (w/v) dextran sulphate, 0.2% (v/v) TWEEN20, 0.02% (v/v) Ficoll, 0.02% (w/v) polyvinyl pyrrolidone, 0.02% (w/v) bovine serum albumin, 1% (v/v) blocking reagent (Roche, Mannheim, Germany) in 100 mM maleic acid with 150 mM NaCl (pH 7.5), 5 pmol biotin-labeled capture probe, 1 pmol DIG-labeled detection probe, and 1 pmol of each helper probe in a total volume of 100 μl. The hybridization reactions were performed in three parallels at 750 rpm and 50°C for 30 min. 15 μl of Streptavidin MagneSphere® Paramagnetic Particles (Promega, Madison, WI, USA) were washed as recommended by the manufacturer, and added to each well after the hybridization reaction and incubated at 750 rpm and 50°C for 30 min. Subsequently, beads were washed three times with 120 μl of a washing buffer (1% SSC, 0.1% TWEEN20) at 25°C and 750 rpm for 2 min. During the washing step beads were kept in the wells with a MagnaBot® 96 Magnetic Separation Device (Promega, Madison, WI, USA). 100 μl of anti-DIG alkaline phosphatase Fab-fragments (Roche Diagnostics, Mannheim, Germany) were diluted with 1 × SSC, 0.1% TWEEN20 to a final concentration of 375 U/l, and applied to each well. The 96-well plate was incubated at 450 rpm and 25°C for 30 min. Unbound enzyme was removed by washing three times as described above. Afterwards the whole solutions were transferred into new microplates and washed once more. Finally, 100 μl of 1 mM AttoPhos® fluorescent substrate (Promega, Madison, WI, USA) was added to each well and the enzymatic reaction was performed at 37°C and 750 rpm for 20 min. 90 μl of the contents of well were transferred into a Costar black 96-well assay plate (Corning Inc., Corning, NY, USA) for the fluorescence measurement with a Wallac Victor multilabel counter (PerkinElmer Life Sciences, Turku, Finland) at an excitation wavelength of 430 nm and an emission wavelength of 560 nm.
The detection limit was defined as 3 × SD added to the background fluorescence. Detection limits were calculated separately for each SHA and signals above it were considered as positive. Capture and detection probes which are not complementary to any RNA of B. subtilis (Table 2, NCcap and NCdet, respectively) were used as a negative control for each sample to estimate the background signal.
Results
The aim of this study was the development of a diagnostic method for the detection of bacterial spores at the example of Bacillus subtilis spores. The sequence of the procedure is shown in Figure 1A. First, the spores are activated by a heat treatment (30 min, 70°C) and following transfer into germination medium (Keijser et al., 2007). The duration of the enrichment cultivation was varied depending on the initial amount of spores. After the incubation phase collected samples were disrupted and the supernatant was utilized for the quantification of the 16S ribosomal RNA directly from the cell broth by the use of a sandwich hybridization assay without any further purification.
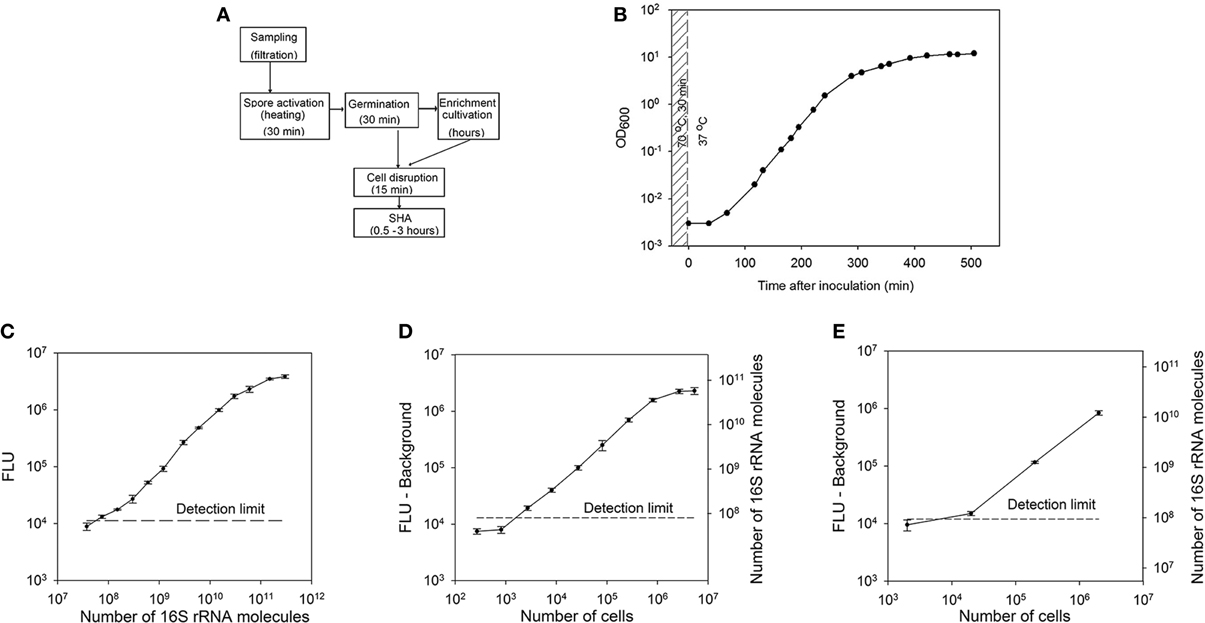
Figure 1. Spore detection using a sandwich hybridization assay. (A) Principle of airborne spore detection using a sandwich hybridization assay. Spores collected from air are activated and cultivated in a germination medium. The first sample for analysis can be collected after approximately 30 min of germination. If the amount of spores is too low to be detected at this step, the incubation can be continued to allow a multiplication of the cells. (B) A Growth curve of B. subtilis cells. After heat activation (70°C for 30 min) B. subtilis spores were inoculated into a germination medium to a final concentration of 106 spores/ml. (C) Standard curve for the B. subtilis 16S rRNA sandwich hybridization assay. (D) SHA dilution curve for B. subtilis 16S rRNA. The dilutions of the crude extracts of B. subtilis cells collected at the exponential growth phase (OD600 = 1.4) were used as a target. The corresponding numbers of 16S rRNA molecules were calculated according to the standard curve of in vitro transcribed 16SrRNA at 260 nm. (E) 16S rRNA measured with SHA in B. subtilis spores after their activation and germination for 30 min. Corresponding numbers of 16S rRNA molecules were calculated according to the standard curve (Figure 1C). The error bars show the ±SD of three parallel experiments and the detection limit is shown as a dashed horizontal line.
RNA-based detection of spores is feasible only after the activation of RNA synthesis, that takes place during the spore germination. In order to activate B. subtilis spores and initiate the germination process, dormant spores were exposed to heat (70°C for 30 min) and afterwards cultured in the germination medium at 37°C. Cell germination and growth were monitored by measuring the OD600 (Figure 1B). An increase of the cell density after 1 h of cultivation indicated that the germination and outgrowth were completed and proliferation began.
For this feasibility study oligonucleotide probes for the detection of B. subtilis 16S rRNA with SHA were designed on the basis of general eubacterial probes with some modifications to detect the sequences specific for B. subtilis (Table 2). The probes were tested using an in vitro transcribed fragment of B. subtilis 16S rRNA as target molecule, which also is applied as a quantitative standard. 6 × 107 (0.1 fmol) target molecules in the hybridization solution gave a signal that was significantly above the detection limit. The reaction was linear over a range of nearly 3 orders of magnitude up to approximately 3 × 1010 i.e., 50 fmol of target molecules (Figure 1C).
In order to determine the detection limit and the range of the SHA for crude cell extracts, different dilutions of B. subtilis cell extracts were analyzed with the sandwich hybridization assay. The cells were collected at the end of the exponential growth phase (OD600 = 1.4), disrupted, and 2.7 × 102 to 5.4 × 106 cells were used as targets for a dilution curve (Figure 1D). The detection limit was about 1.5 × 103 cells per assay for exponentially growing B. subtilis cells (7.5 × 104 cells per ml of culture medium). The calculated amount of 16S rRNA was 4.8 (±0.6) × 104 molecules per cell at this growth phase. Consequently, the detection limit in crude cell extracts corresponded to about 7.2 × 107 16S rRNA molecules. The linear range of the assay was between 103 and 106 cells in a hybridization solution.
In order to establish the analysis of cells from enrichment cultures the germination medium was inoculated with 105–108 activated B. subtilis spores per mL. After 30 min of cultivation (before the first proliferation), the cells were disrupted and analyzed with SHA (Figure 1E). It was possible to detect about 20,000 cells per assay (106 cells per ml of culture medium). This number of cells corresponds to approximately 108 16S rRNA molecules. According to the measurements at this stage of germination one B. subtilis cell contained 5.6 (±0.9) × 103 16S rRNA molecules.
Next, the sensitivity of the method including activation, enrichment cultivation, and 16S rRNA analysis of B. subtilis spores was studied. Therefore the germination medium was inoculated with 101–105 activated B. subtilis spores per ml. Germination and growth of the cells were monitored by measuring the OD600 (Figure 2A). The cells were collected, disrupted and analyzed with SHA using the probes against the16S rRNA (Figure 2B). Detectable signals were observed after 110, 140, 245, 340, and 370 min of cultivation for the samples with initial number of 105, 104, 103, 102, and 101 spores per ml of culture medium, respectively.
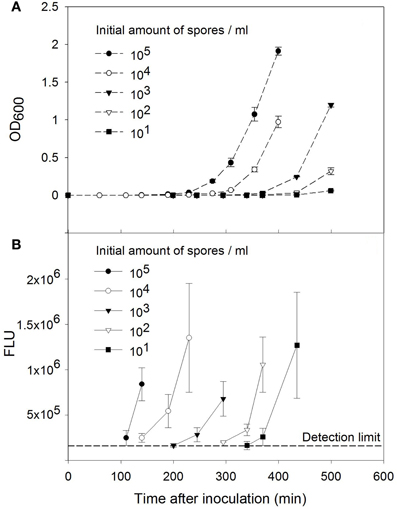
Figure 2. Sensitivity of the B. subtilis spore detection method. (A) Growth curves of B. subtilis cells after spore activation. The initial number of the spores was 101–105 spores per ml of germination medium. (B) Detection of B. subtilis 16S rRNA by sandwich hybridization after enrichment cultivation. The error bars show ±SD of three independent cultivations and measurements.
Since the SHA sensitivity depends on the number of target RNA molecules in one cell, the amount of 16S rRNA was measured for different growth phases of B. subtilis cells (Figure 3). As it was shown earlier one B. subtilis cell contained 5.6 (±0.9) × 103 16S rRNA molecules already after 30 min of germination. At the early exponential growth phase the amount of 16S rRNA molecules per cell was maximal (about 9 × 104 rRNA molecules per cell for an OD600 of 0.27) and the 16S rRNA content decreased in later growth stages. Therefore the analysis is most sensitive if the OD600 is approximately 0.3.
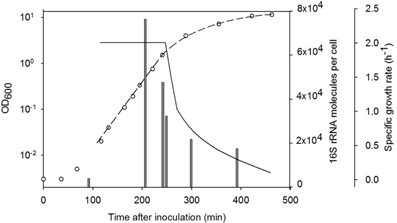
Figure 3. Level of 16S rRNA molecules per cell (gray bars) in different growth phases of B. subtilis. Quantity of RNA was measured with SHA. The growth was followed by measuring the optical density at 600 nm (◦) and the specific growth rate was calculated (black line). The dashed line shows fitting of the OD curve.
Discussion
Detection of microorganisms in bioaerosols is an important issue in determining bio-warfare agents during biological attacks and for the monitoring of indoor air quality. A fast and sensitive method for the detection of bacterial spores was developed in this study. The procedure includes activation of spores, their germination, enrichment cultivation and RNA detection using a sandwich hybridization assay, thus only viable spores are detected, which is advantageous over standard detection methods as listed in Table 1. There are no sensitivity limitations as the method is adaptable by the enrichment cultivation time. The RNA analysis can be performed directly with crude cell extracts avoiding laborious RNA purification. The method was developed for B. subtilis, a model organism capable of spore formation and quite abundant in aerosols.
Since spores only contain low amounts of RNA molecules, the idea of this study was to initiate RNA synthesis to increase the detection system sensitivity. At first, the spores are activated at 70°C for 30 min and placed into a medium containing L-alanine as a germination factor. During germination and outgrowth the activated spores synthesize RNA, proteins, and ATP. Chromosomal replication is initiated after 0.5 h (Garrick-Silversmith and Torriani, 1973) and the first cell division takes place after approximately 70 min (Keijser et al., 2007). Depending on the initial amount of spores, cells are disrupted either directly after the germination or after the enrichment cultivation required for the accumulation of necessary amount of RNA molecules. The presented method allows the use of crude cells lysates as sample material for the RNA detection, and no further RNA purification is needed (Rautio et al., 2003; Thieme et al., 2008). We believe that this is a strong advantage for using the sandwich hybridization assay for the RNA detection compared to reverse transcriptase real time PCR.
The assay is based on the hybridization event of the target RNA with two oligonucleotide probes. Alkaline phosphatase attached to one of the probes generates a fluorescent signal used for quantification. The SHA has a potential to be automated, e.g., by the use of an electrical chip reader (Gabig-Ciminska et al., 2004).
After the activation the spores germinated very quickly. The first increase of the OD600 was noticed already after 1 h for the cultures with an initial amount of 106 spores per ml. The detectable SHA signal for these cultures was observed after 0.5 h of germination. Since the RNA synthesis in germinating B. subtilis spores starts within about 5 min after spore activation (Matsuda and Kameyama, 1986), the cells have accumulated a detectable amount of RNA molecules already after 30 min. Furthermore, at this moment the spore core is rehydrated making cells more accessible to disruption (Sloma and Smith, 1979; Moeller et al., 2006).
The sensitivity of the developed method is not limited due to enrichment cultivation for which the time can be adapted. Very low initial spore amounts in the sample could be detected after about 5 h of enrichment cultivation. Consequently, the maximum duration of the assay for a very low amount of spores including spore activation, enrichment cultivation, and detection with the SHA is about 8 h. The detection limit of the SHA was estimated as 6 × 107 target molecules, which in case of using rRNA as a target corresponds to about 103–104 cells in a sample depending on their growth stage.
By the use of in vitro synthesized RNA probes as a standard with the SHA it was possible to estimate the amount of 16S rRNA molecules per cell. As expected, the number of molecules increased during the germination and the outgrowth, reached the maximum at the beginning of the exponential phase of growth, and then slowly decreased. After 0.5 h of germination one cell contained approximately 5.6 (±0.9) × 103 16S rRNA molecules. Exponentially growing cells contained about 9 × 104 16S rRNA molecules. These numbers correlate well with the known amount of ribosomes in Escherichia coli cells, that is 6700–71,000 depending on the specific growth rate (Bremer and Dennis, 1996).
The 16S rRNA oligonucleotide probes used in this study are capable of detecting most bacteria and are relevant only for method development. Specific probes for the SHA can be designed for almost any organism or groups of organisms if the gene sequence is available. Furthermore, it would be possible to apply the procedure for the analysis of mRNAs and their dynamics during germination. However, due to numerical advantage of ribosomes compared to a specific mRNA species, the method sensitivity would be lower when mRNA is used as a target for the SHA instead of rRNA and additionally the analytical error eventually would be higher due to the low stability of mRNA molecules. For selecting target mRNAs for specific identification of B. subtilis a transcription profile of germinated spores (Keijser et al., 2007) and the most abundant proteins of growing Bacillus cells have to be reviewed. Protein abundance during the exponential growth of Bacillus subtilis has been studied in great detail (Büttner et al., 2001; Eymann et al., 2004). The most abundant proteins in B. subtilis cytosolic extracts perform mainly housekeeping functions as components of the translational apparatus (e.g., translation elongation factors, ribosomal proteins), the glycolytic pathways, the tricarboxylic acid cycle, the metabolism of amino and nucleic acids, and protein quality control (e.g., chaperons). The mRNAs of these proteins can be used as a target for the specific detection of B. subtilis spores using the presented method.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Dr. Pekka Belt, Dr. Janne Härkönen, and Dr. Matti Möttönen are acknowledged for critical editing of this manuscript. This study was supported by the Finnish Funding Agency for Technology and Innovation (TEKES) project ENRICH (40254/07).
Abbreviations
SHA, sandwich hybridization assay.
References
Arakawa, E. T., Lavrik, N. V., Rajic, S., and Datskos, P. G. (2003). Detection and differentiation of biological species using microcalorimetric spectroscopy. Ultramicroscopy 97, 459–465. doi: 10.1016/S0304-3991(03)00074-3
Baeumner, A. J., Leonard, B., McElwee, J., and Montagna, R. A. (2004). A rapid biosensor for viable B. anthracis spores. Anal. Bioanal. Chem. 380, 15–23. doi: 10.1007/s00216-004-2726-7
Bhatta, D., Michel, A. A., Marti Villalba, M., Emmerson, G. D., Sparrow, I. L. G., Perkins, E. A., et al. (2011). Optical microchip array biosensor for multiplexed detection of bio-hazardous agents. Biosens. Bioelectron. 30, 78–86. doi: 10.1016/j.bios.2011.08.031
Bremer, H., and Dennis, P. P. (1996). “Modulation of chemical composition and other parameters of the cell by growth rate,” in Escherichia coli and Salmonella typhimurium Cellular and Molecular Biology, ed F. C. Neidhardt (Washington, DC: American Society of Microbiology), 1553–1569.
Büttner, K., Bernhardt, J., Scharf, C., Schmid, R., Mäder, U., Eymann, C., et al. (2001). A comprehensive two-dimensional map of cytosolic proteins of Bacillus subtilis. Electrophoresis 22, 2908–2935. doi: 10.1002/1522-2683(200108)22:14<2908::AID-ELPS2908>3.0.CO;2-M
Campbell, G. A., and Mutharasan, R. (2007). Method of measuring Bacillus anthracis spores in the presence of copious amounts of Bacillus thuringiensis and Bacillus cereus. Anal. Chem. 79, 1145–1152. doi: 10.1021/ac060982b
Chenau, J., Fenaille, F., Ezan, E., Morel, N., Lamourette, P., Goossens, P. L., et al. (2011). Sensitive detection of Bacillus anthracis spores by immunocapture and liquid chromatography-tandem mass spectrometry. Anal. Chem. 83, 8675–8682. doi: 10.1021/ac2020992
Cheng, H. W., Chen, Y. Y., Lin, X. X., Huan, S. Y., Wu, H. L., Shen, G. L., et al. (2011). Surface-enhanced Raman spectroscopic detection of Bacillus subtilis spores using gold nanoparticle based substrates. Anal. Chim. Acta 707, 155–163. doi: 10.1016/j.aca.2011.09.007
Cheng, H. W., Huan, S. Y., and Yu, R. Q. (2012). Nanoparticle-based substrates for surface-enhanced Raman scattering detection of bacterial spores. Analyst 137, 3601–3608. doi: 10.1039/c2an35448a
Cowcher, D. P., Xu, Y., and Goodacre, R. (2013). Portable, quantitative detection of Bacillus bacterial spores using surface-enhanced Raman scattering. Anal. Chem. 85, 3297–3302. doi: 10.1021/ac303657k
Dhayal, B., Henne, W. A., Doorneweerd, D. D., Reifenberger, R. G., and Low, P. S. (2006). Detection of Bacillus subtilis spores using peptide-functionalized cantilever arrays. J. Am. Chem. Soc. 128, 3716–3721. doi: 10.1021/ja0570887
Edwards, K. A., Clancy, H. A., and Baeumner, A. J. (2006). Bacillus anthracis: toxicology, epidemiology and current rapid-detection methods. Anal. Bioanal. Chem. 384, 73–84. doi: 10.1007/s00216-005-0090-x
Elsholz, B., Worl, R., Blohm, L., Albers, J., Feucht, H., Grunwald, T., et al. (2006). Automated detection and quantitation of bacterial RNA by using electrical microarrays. Anal. Chem. 78, 4794–4802. doi: 10.1021/ac0600914
Eymann, C., Dreisbach, A., Albrecht, D., Bernhardt, J., Becher, D., Gentner, S., et al. (2004). A comprehensive proteome map of growing Bacillus subtilis cells. Proteomics 4, 2849–2876. doi: 10.1002/pmic.200400907
Gabig-Ciminska, M., Holmgren, A., Andresen, H., Bundvig, B. K., Wümpelmann, M., Albers, J., et al. (2004). Electric chips for rapid detection and quantification of nucleic acids. Biosens. Bioelectron. 19, 537–546. doi: 10.1016/S0956-5663(03)00273-2
Garrick-Silversmith, L., and Torriani, A. (1973). Macromolecular syntheses during germination and outgrowth of Bacillus subtilis spores. J. Bacteriol. 114, 507–516.
Gooding, J. J. (2006). Biosensor technology for detecting biological warfare agents: recent progress and future trends. Anal. Chim. Acta 559, 137–151. doi: 10.1016/j.aca.2005.12.020
Hadfield, T., Ryan, V., Spaulding, U. K., Clemens, K. M., Ota, I. M., and Brunelle, S. L. (2013). RAZOR EX Anthrax Air Detection System for detection of Bacillus anthracis spores from aerosol collection samples: collaborative study. J. AOAC Int. 96, 392–398. doi: 10.5740/jaoacint.CS2012-06
Hindson, B. J., Brown, S. B., Marshall, G. D., McBride, M. T., Makarewicz, A. J., Gutierrez, D. M., et al. (2004). Development of an automated sample preparation module for environmental monitoring of biowarfare agents. Anal. Chem. 76, 3492–3497. doi: 10.1021/ac035365r
Hindson, B. J., McBride, M. T., Makarewicz, A. J., Henderer, B. D., Setlur, U. S., Smith, S. M., et al. (2005). Autonomous detection of aerosolized biological agents by multiplexed immunoassay with polymerase chain reaction confirmation. Anal. Chem. 77, 284–289. doi: 10.1021/ac0489014
Huhtamella, S., Leinonen, M., Nieminen, T., Fahnert, B., Myllykoski, L., Breitenstein, A., et al. (2007). RNA-based sandwich hybridisation method for detection of lactic acid bacteria in brewery samples. J. Microbiol. Methods 68, 543–553. doi: 10.1016/j.mimet.2006.10.009
Hybl, J. D., Lithgow, G. A., and Buckley, S. G. (2003). Laser-induced breakdown spectroscopy detection and classification of biological aerosols. Appl. Spectrosc. 57, 1207–1215. doi: 10.1366/000370203769699054
Hybl, J. D., Tysk, S. M., Berry, S. R., and Jordan, M. P. (2006). Laser-induced fluorescence-cued, laser-induced breakdown spectroscopy biological-agent detection. Appl. Opt. 45, 8806–8814. doi: 10.1364/AO.45.008806
Inami, H., Tsuge, K., Matsuzawa, M., Sasaki, Y., Togashi, S., Komano, A., et al. (2009). Semi-automated bacterial spore detection system with micro-fluidic chips for aerosol collection, spore treatment and ICAN DNA detection. Biosens. Bioelectron. 24, 3299–3305. doi: 10.1016/j.bios.2009.04.025
Irenge, L. M., Durant, J. F., Tomaso, H., Pilo, P., Olsen, J. S., Ramisse, V., et al. (2010). Development and validation of a real-time quantitative PCR assay for rapid identification of Bacillus anthracis in environmental samples. Appl. Microbiol. Biotechnol. 88, 1179–1192. doi: 10.1007/s00253-010-2848-0
Irenge, L. M., and Gala, J. L. (2012). Rapid detection methods for Bacillus anthracis in environmental samples: a review. Appl. Microbiol. Biotechnol. 93, 1411–1422. doi: 10.1007/s00253-011-3845-7
Jürgen, B., Pioch, D., Hoi, L. T., Albers, J., Hintsche, R., and Schweder, T. (2005). At-line monitoring of bioprocess-relevant marker genes using an electric DNA-chip. J. Biotechnol. 118:S36. doi: 10.1002/bit.20578
Kane, S. R., Létant, S. E., Murphy, G. A., Alfaro, T. M., Krauter, P. W., Mahnke, R., et al. (2009). Rapid, high-throughput, culture-based PCR methods to analyze samples for viable spores of Bacillus anthracis and its surrogates. J. Microbiol. Methods 76, 278–284. doi: 10.1016/j.mimet.2008.12.005
Keijser, B. J., Ter Beek, A., Rauwerda, H., Schuren, F., Montijn, R., van der Spek, H., et al. (2007). Analysis of temporal gene expression during Bacillus subtilis spore germination and outgrowth. J. Bacteriol. 189, 3624–3634. doi: 10.1128/JB.01736-06
Krebs, M. D., Mansfield, B., Yip, P., Cohen, S. J., Sonenshein, A. L., Hitt, B. A., et al. (2006). Novel technology for rapid species-specific detection of Bacillus spores. Biomol. Eng. 23, 119–127. doi: 10.1016/j.bioeng.2005.12.003
Lasch, P., Beyer, W., Nattermann, H., Stämmler, M., Siegbrecht, E., Grunow, R., et al. (2009). Identification of Bacillus anthracis by using matrix-assisted laser desorption ionization-time of flight mass spectrometry and artificial neural networks. Appl. Environ. Microbiol. 75, 7229–7242. doi: 10.1128/AEM.00857-09
Lee, M. A., Brightwell, G., Leslie, D., Bird, H., and Hamilton, A. (1999). Fluorescent detection techniques for real-time multiplex strand specific detection of Bacillus anthracis using rapid PCR. J. Appl. Microbiol. 87, 218–223. doi: 10.1046/j.1365-2672.1999.00908.x
Leskelä, T., Tilsala-Timisjärvi, A., Kusnetsov, J., Neubauer, P., and Breitenstein, A. (2005). Sensitive genus-specific detection of Legionella by a 16S rRNA based sandwich hybridization assay. J. Microbiol. Methods 62, 167–179. doi: 10.1016/j.mimet.2005.02.008
Li, D., Truong, T. V., Bills, T. M., Holt, B. C., VanDerwerken, D. N., Williams, J. R., et al. (2012). GC/MS method for positive detection of Bacillus anthracis endospores. Anal. Chem. 84, 1637–1644. doi: 10.1021/ac202606x
Makino, S., and Cheun, H. (2003). Application of the real-time PCR for the detection of airborne microbial pathogens in reference to the anthrax spores. J. Microbiol. Methods 53, 141–147. doi: 10.1016/S0167-7012(03)00019-8
Matsuda, M., and Kameyama, T. (1986). Fractionation of ribonucleic-acid transcripts synthesized during spore germination in Bacillus subtilis. J. Basic Microbiol. 26, 91–99. doi: 10.1002/jobm.3620260209
McBride, M. T., Masquelier, D., Hindson, B. J., Makarewicz, A. J., Brown, S., Burris, K., et al. (2003). Autonomous detection of aerosolized Bacillus anthracis and Yersinia pestis. Anal. Chem. 75, 5293–5299. doi: 10.1021/ac034722v
Moeller, R., Horneck, G., Rettberg, P., Mollenkopf, H. J., Stackebrandt, E., and Nicholson, W. L. (2006). A method for extracting RNA from dormant and germinating Bacillus subtilis strain 168 endospores. Curr. Microbiol. 53, 227–231. doi: 10.1007/s00284-006-0099-1
Morel, N., Volland, H., Dano, J., Lamourette, P., Sylvestre, P., Mock, M., et al. (2012). Fast and sensitive detection of Bacillus anthracis spores by immunoassay. Appl. Environ. Microbiol. 78, 6491–6498. doi: 10.1128/AEM.01282-12
Neubauer, A., Soini, J., Bollok, M., Zenker, M., Sandqvist, J., Myllyharju, J., et al. (2007). Fermentation process for tetrameric human collagen prolyl 4-hydroxylase in Escherichia coli: improvement by gene optimisation of the PDI/beta subunit and repeated addition of the inducer anhydrotetracycline. J. Biotechnol. 128, 308–321. doi: 10.1016/j.jbiotec.2006.10.017
Nieminen, T., Pakarinen, J., Tsitko, I., Salkinoja-Salonen, M., Breitenstein, A., Ali-Vehmas, T., et al. (2006). 16S rRNA targeted sandwich hybridization method for direct quantification of mycobacteria in soils. J. Microbiol. Methods 67, 44–55. doi: 10.1016/j.mimet.2006.02.015
Pioch, D., Jürgen, B., Evers, S., Maurer, K. H., Hecker, M., and Schweder, T. (2008a). Improved sandwich-hybridization assay for an electrical DNA-chip-based monitoring of bioprocess-relevant marker genes. Appl. Microbiol. Biotechnol. 78, 719–728. doi: 10.1007/s00253-008-1347-z
Pioch, D., Schweder, T., and Jürgen, B. (2008b). Novel developments for improved detection of specific mRNAs by DNA chips. Appl. Microbiol. Biotechnol. 80, 953–963. doi: 10.1007/s00253-008-1680-2
Pizarro, S. A., Lane, P., Lane, T. W., Cruz, E., Haroldsen, B., and VanderNoot, V. A. (2007). Bacterial characterization using protein profiling in a microchip separations platform. Electrophoresis 28, 4697–4704. doi: 10.1002/elps.200700005
Rautio, J., Barken, K. B., Lahdenperä, J., Breitenstein, A., Molin, S., and Neubauer, P. (2003). Sandwich hybridisation assay for quantitative detection of yeast RNAs in crude cell lysates. Microb. Cell Fact. 2, 4. doi: 10.1186/1475-2859-2-4
Rautio, J. J. (2010). Multiplex gene expression analysis by TRAC in fungal cultures. Methods Mol. Biol. 638, 165–173. doi: 10.1007/978-1-60761-611-5_12
Rautio, J. J., Kataja, K., Satokani, R., Penttilä, M., Söderlund, H., and Saloheimo, M. (2006). Rapid and multiplexed transcript analysis of microbial cultures using capillary electophoresis-detectable oligonucleotide probe pools. J. Microbiol. Methods 65, 404–416. doi: 10.1016/j.mimet.2005.08.010
Rautio, J. J., Satokari, R., Vehmaan-Kreula, P., Serkkola, E., and Söderlund, H. (2008). TRAC in high-content gene expression analysis: applications in microbial population studies, process biotechnology and biomedical research. Expert Rev. Mol. Diagn. 8, 379–385. doi: 10.1586/14737159.8.4.379
Regan, J. F., Makarewicz, A. J., Hindson, B. J., Metz, T. R., Gutierrez, D. M., Corzett, T. H., et al. (2008). Environmental monitoring for biological threat agents using the autonomous pathogen detection system with multiplexed polymerase chain reaction. Anal. Chem. 80, 7422–7429. doi: 10.1021/ac801125x
Sloma, A., and Smith, I. (1979). RNA synthesis during spore germination in Bacillus subtilis. Mol. Gen. Genet. 175, 113–120. doi: 10.1007/BF00425526
Soini J., Falschlehner C., Liedert C., Bernhard J., Vuoristo J., and Neubauer, P. (2008). Norvaline is accumulated after a down-shift of oxygen in Escherichia coli W3110. Microb. Cell Fact. 7:30. doi: 10.1186/1475-2859-7-30
Spaulding, U. K., Christensen, C. J., Crisp, R. J., Vaughn, M. B., Trauscht, R. C., Gardner, J. R., et al. (2012). RAZOR EX anthrax air detection system. J. AOAC Int. 95, 860–891. doi: 10.5740/jaoacint.11-521
Stachowiak, J. C., Shugard, E. E., Mosier, B. P., Renzi, R. F., Caton, P. F., Ferko, S. M., et al. (2007). Autonomous microfluidic sample preparation system for protein profile-based detection of aerosolized bacterial cells and spores. Anal. Chem. 79, 5763–5770. doi: 10.1021/ac070567z
Stratis-Cullum, D. N., Griffin, G. D., Mobley, J., Vass, A. A., and Vo-Dinh, T. (2003). A miniature biochip system for detection of aerosolized Bacillus globigii spores. Anal. Chem. 75, 275–280. doi: 10.1021/ac026068+
Tamborrini, M., Holzer, M., Seeberger, P. H., Schürch, N., and Pluschke, G. (2010). Anthrax spore detection by a luminex assay based on monoclonal antibodies that recognize anthrose-containing oligosaccharides. Clin. Vaccine Immunol. 17, 1446–1451. doi: 10.1128/CVI.00205-10
Taskila, S., Osmekhina, E., Tuomola, M., Ruuska, J., and Neubauer, P. (2011). Modification of buffered peptone water for improved recovery of heat-injured Salmonella Typhimurium. J. Food Sci. 76, M157–M162. doi: 10.1111/j.1750-3841.2010.02050.x
Thieme, D., Neubauer, P., Nies, D. H., and Grass, G. (2008). Sandwich hybridization assay for sensitive detection of dynamic changes in mRNA transcript levels in crude Escherichia coli cell extracts in response to copper ions. Appl. Environ. Microbiol. 74, 7463–7470. doi: 10.1128/AEM.01370-08
Walsh, M. K., Wang, X., and Weimer, B. C. (2001). Optimizing the immobilization of single-stranded DNA onto glass beads. J. Biochem. Biophys. Methods 47, 221–231. doi: 10.1016/S0165-022X(00)00146-9
Wang, Z., Fan, Y., Chen, J., Guo, Y., Wu, W., He, Y., et al. (2013). A micro-fluidic chip-based fluorescent biosensor for the sensitive and specific detection of label-free single-base mismatch via magnetic beads-based “sandwich” hybridization strategy. Electrophoresis 34, 2177–2184. doi: 10.1002/elps.201300131
Weerasekara, M. L., Ryuda, N., Miyamoto, H., Okumura, T., Ueno, D., Inoue, K., et al. (2013). Double-color fluorescence in situ hybridization (FISH) for the detection of Bacillus anthracis spores in environmental samples with a novel permeabilization protocol. J. Microbiol. Methods 93, 177–184. doi: 10.1016/j.mimet.2013.03.007
Weisburg, W. G., Barns, S. M., Pelletier, D. A., and Lane, D. J. (1991). 16S Ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173, 697–703.
Wielinga, P. R., Hamidjaja, R. A., Agren, J., Knutsson, R., Segerman, B., Fricker, M., et al. (2011). A multiplex real-time PCR for identifying and differentiating B. anthracis virulent types. Int. J. Food Microbiol. 145 (Suppl. 1), S137–S144. doi: 10.1016/j.ijfoodmicro.2010.07.039
Keywords: spore detection, Bacillus subtilis, RNA hybridization
Citation: Osmekhina E, Shvetsova A, Ruottinen M and Neubauer P (2014) Quantitative and sensitive RNA based detection of Bacillus spores. Front. Microbiol. 5:92. doi: 10.3389/fmicb.2014.00092
Received: 21 January 2014; Accepted: 19 February 2014;
Published online: 11 March 2014.
Edited by:
Eric Altermann, AgResearch Ltd, New ZealandReviewed by:
Grzegorz Wegrzyn, University of Gdansk, PolandJohannes Kabisch, Ernst-Moritz-Arndt Universität Greifswald, Germany
Copyright © 2014 Osmekhina, Shvetsova, Ruottinen and Neubauer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Neubauer, Laboratory of Bioprocess Engineering, Department of Biotechnology, Technische Universität Berlin, Ackerstraße 76, D-13355 Berlin, Germany e-mail:cGV0ZXIubmV1YmF1ZXJAdHUtYmVybGluLmRl