 Pei-Ling Wang1
Pei-Ling Wang1 Yi-Ping Chiu
Yi-Ping Chiu Ting-Wen Cheng
Ting-Wen Cheng Yung-Hsin Chang
Yung-Hsin Chang Li-Hung Lin
Li-Hung Lin- 1Institute of Oceanography, National Taiwan University, Taipei, Taiwan
- 2Department of Geosciences, National Taiwan University, Taipei, Taiwan
This study analyzed cored sediments retrieved from sites distributed across a transect of the Lei-Gong-Hou mud volcanoes in eastern Taiwan to uncover the spatial distributions of biogeochemical processes and community assemblages involved in methane cycling. The profiles of methane concentration and carbon isotopic composition revealed various orders of the predominance of specific methane-related metabolisms along depth. At a site proximal to the bubbling pool, the methanogenic zone was sandwiched by the anaerobic methanotrophic zones. For two sites distributed toward the topographic depression, the methanogenic zone overlaid the anaerobic methanotrophic zone. The predominance of anaerobic methanotrophy at specific depth intervals is supported by the enhanced copy numbers of the ANME-2a 16S rRNA gene and coincides with high dissolved Fe/Mn concentrations and copy numbers of the Desulfuromonas/Pelobacter 16S rRNA gene. Assemblages of 16S rRNA and mcrA genes revealed that methanogenesis was mediated by Methanococcoides and Methanosarcina. pmoA genes and a few 16S rRNA genes related to aerobic methanotrophs were detected in limited numbers of subsurface samples. While dissolved Fe/Mn signifies the presence of anaerobic metabolisms near the surface, the correlations between geochemical characteristics and gene abundances, and the absence of aerobic methanotrophs in top sediments suggest that anaerobic methanotrophy is potentially dependent on iron/manganese reduction and dominates over aerobic methanotrophy for the removal of methane produced in situ or from a deep source. Near-surface methanogenesis contributes to the methane emissions from mud platform. The alternating arrangements of methanogenic and methanotrophic zones at different sites suggest that the interactions between mud deposition, evaporation, oxidation and fluid transport modulate the assemblages of microbial communities and methane cycling in different compartments of terrestrial mud volcanoes.
Introduction
Terrestrial mud volcanoes are ubiquitous in compressional tectonic regimes (Kopf, 2004). These peculiar cone-shaped features expel fluidized muds or breccia through either seepages or explosive discharges (Mazzini, 2009). Often, methane constitutes the major gaseous phase accompanying the release of fluids and sediments (Dimitrov, 2003). Despite focused flows channeling along the fracture network, deeply-sourced gases and fluids also percolate through pore space of sediments via diffusion and/or advection (Milkov, 2005; Mazzini et al., 2009). Such pervasive gas transport underneath mud volcanoes enables even greater summed methane emissions from surrounding mud platforms (termed micro- or mini-seepage) when compared with those from main conduits or fractures (termed macro-seepage) (Etiope et al., 2004a,b; Hong et al., 2013). Unlike marine counterparts where the overlying seawater can buffer methane released from the sediment-seawater interface (Yvon-Lewis et al., 2011), terrestrial mud volcanoes directly emit methane and gaseous hydrocarbons into the atmosphere. As the methane greenhouse effect is greater than that of CO2 by a factor of ~25, terrestrial mud volcanoes are considered a potent contributor intensifying climatic fluctuations on contemporary and geological time scales (Etiope et al., 2008).
The exact magnitude of methane released from or retained within terrestrial mud volcanoes is dependent on several factors. In addition to fluid transport, in situ microbial production and consumption appear to be the most critical factor governing methane abundances in the pore space and methane fluxes across the sediment-air interface (Alain et al., 2006; Chang et al., 2012; Cheng et al., 2012). In particular, terrestrial mud volcanoes are generally limited in sulfate. Sulfate-dependent methanotrophy commonly observed in marine counterparts (Knittel and Boetius, 2009) would be likely inhibited under such sulfate-deficient conditions, allowing more methane emitted into the atmosphere. Using geochemical profiles, incubation approaches, and 16S rRNA gene abundances and assemblages, previous studies indicated that methane originating from deep sources mixes with microbial methane produced at shallow depths (Chang et al., 2012; Cheng et al., 2012). The quantities of microbial methane produced in situ are two to three times those of deeply-sourced methane, providing abundant electron donors for methanotrophy. In addition, anaerobic methanotrophs (ANME-1 to -3 lineages) proliferate in methane-transition zones, likely being coupled with either sulfate or iron/manganese reduction fueled by mineral-derived sulfate or iron/manganese oxide generated from the atmospheric oxidation of reduced sulfur or metals (Chang et al., 2012; Cheng et al., 2012). Such subsurface-surface interactions create steep redox gradients that lead to the compartmentalization of different metabolic schemes into discrete horizons.
Metabolic stratification driven by subsurface-surface interactions in terrestrial mud volcanoes is persistent over years, effectively removing methane from the pore space (Chang et al., 2012; Cheng et al., 2012). However, these observations are primarily drawn from cores retrieved near fluid conduits. Considering that a typical mud volcano is cone-shaped, methane transport and emission may be susceptible to factors that would vary on multiple dimensions. First, advective methane fluxes originating from deep sources are expected to decrease as the distance from the major conduit or bubbling pool increases (Etiope et al., 2004a,b). The decrease in methane flux also reflects the reduced flux of reducing fluid, thereby favoring the penetration of aerobic methanotrophy and other aerobic metabolisms into deeper levels while inhibiting anaerobic methanotrophy, methanogenesis, and other anaerobic metabolisms. Second, top sediments distributed along the slope of mud volcanoes are episodically immersed by fluids expelled from bubbling pools. Solute contents in pore water could be enhanced due to the loss of pore water through evaporation (Svensen et al., 2007). Previous studies have demonstrated that evaporation could rapidly enhance solute concentrations, even for samples located close to the bubbling pool (Chang et al., 2012; Cheng et al., 2012). Therefore, increasing distance from the bubbling pool is expected to generate enrichments of solutes in the top pore water downslope the cone-shaped structure. Such a salinity increase would pose various effects on community assemblages and metabolic pathways. Determining the spatial variations of pore water geochemistry and microbial community structure would provide a basis for the assessment of total methane emission from a mud volcano.
The aim of this study was to identify the architecture of microbial methane producing and consuming zones in a sulfate-deficient terrestrial mud volcano using geochemical patterns and community assemblages based on 16S rRNA and methane-related functional genes. This study extends previous efforts (Chang et al., 2012) to a broader spatial scale by analyzing sediments across a transect of a cone-shaped feature at the Lei-Gong-Hou mud volcanoes (LGHMVs) in eastern Taiwan. Geochemical profiles, and gene assemblages and abundances were integrated with those obtained previously (Chang et al., 2012) to address the spatial variations in microbial cycling of methane.
Materials and Methods
Site Background and Sample Acquisition
Samples were collected from the LGHMVs (22°58'59.02”N, 121°12'33.85”E) in the Kuan-Shan area in eastern Taiwan (Figure 1). The stratum hosting the LGHMVs, the Li-Chi Formation, is distributed trending north-south on the east side of the Longitudinal Valley that separates the Coastal Range to the east from the metamorphic complex to the west (Chang et al., 2001). The Li-Chi Formation is composed of primarily clays mixed with rock fragments in different sizes and lithologies, and represents the intra-arc sediments that formed during the convergence between the Eurasian and Philippine Sea Plates (Chang et al., 2001).

Figure 1. Field photo of the study sites at the LGHMVs in eastern Taiwan. The investigated area is located in the topographic high where a cluster of at least six bubbling pools formed. Bubbling fluids from pool LGH02 and three cores located downslope along the gulley between four bubbling pools were collected in 2009. Black oily and yellowish patches were distributed in the mud platform near the bubbling pools and became extinct in the dry area.
The LGHMVs are composed of more than ten bubbling, cone-shaped features distributed along a nearly north-south transect potentially related to the Longitudinal Fault. Muddy fluids with abundant hydrocarbon gases were discharged in every pool during sampling in February 2009 (Chang et al., 2012). Previous studies indicated that exsolved gases from bubbling pools consist primarily of methane (90–98%) with δ13C values ranging between −51 and −49‰ [referenced to the Vienna Pee Dee Belemnite (VPDB)] (Sun et al., 2010).
In addition to the bubbling pool (LGH02-ew) and cored sediments (LGH02-c1) used for the previous study (Chang et al., 2012), two additional cores (LGH02-c2 and -c3) at a distance of 46 and 153 cm from core LGH02-c1 were also collected (Figure 1). These two cores had a total length of 20 cm and were sectioned (mostly at a 2-cm interval) for analyses of gas and aqueous geochemistry, 16S rRNA gene and methane-related functional genes. Samples collected from LGH02-ew and core LGH02-c1 were also used for analyses of methane-related functional genes. Gears for core retrieval and sample processing in the field were heat-sterilized prior to the field trip. All samples were stored either at room temperature (for gas analyses) or on dry ice (for aqueous chemistry and molecular analyses) during transportation, and transferred back to the laboratory within 6 h. The reported depth represents the mean of depth across the section interval.
Sample Processing and Analysis in the Laboratory
Upon arriving at the laboratory, samples for aqueous geochemistry and DNA analyses were immediately subject to centrifugation at ~8200 ×g for 15 min. Supernatants were decanted from the tubes, filtered using 0.22 μm pore-sized membranes, and split into two fractions; one was treated with 10% by volume of 2 M nitric acid for the preservation of cations, and the other was used for anions without any preservative. One milliliter of the anion sample was fixed with 0.1 mL of 1 M Zn-acetate for the determination of HS− concentration. Geochemical and DNA samples were stored in a 4°C refrigerator and a −80°C freezer, respectively, until further analysis.
The abundances of major anions, dissolved Fe and Mn, sulfide, and methane, and carbon isotopic compositions of methane were measured using the same methods as those described in Chang et al. (2012). The measured partial pressure of methane was converted to the dissolved concentration using the Henry's law constant (Wiesenburg and Guinasso, 1979). The isotopic compositions were reported as δ notation [δ value = (Rsample/Rstd-1) × 1000‰, where R is the 13C/12C ratio and std is the VPDB]. The uncertainties for aqueous chemistry and gas abundance were ±2 and ±5%, respectively, whereas the uncertainty for δ13C-CH4 was ±0.3‰.
Genomic DNA was extracted from 10 g of sediments at 0.5, 2, 4, 6, 11, 15, and 19 cm of core LGH02-c2 and 2, 7, 11, and 19 cm of core LGH02-c3 using the MoBio Ultraclean Mega DNA Prep Soil Kit (MoBio, USA) according to the manufacturer's instruction and stored in water at −20°C. Nearly complete 16S rRNA gene sequences were amplified from the crude extracts from 0.5, 2, 4, 6, and 15 cm of core LGH02-c2 and 2, 11, and 19 cm of core LGH02-c3 using the primer pairs B27f/U1492r for bacteria (Lane, 1991) and A8f/U1513r for archaea (Huber et al., 2002) by polymerase chain reaction (PCR). The amplification scheme was the same as that described in Chang et al. (2012). For the gene mcrA, which encodes methyl coenzyme M reductase subunit A (a key enzyme for methanogenesis and anaerobic methanotrophy), only the crude extracts obtained from 3 and 7 cm of core LGH02-c1, 6 and 15 cm of core LGH02-c2, and 2 and 11 cm of core LGH02-c3 were amplified using the primer pair ME1/ME2 (Scholten et al., 2005). Thermal cycling was performed with an initial melting step at 94°C for 2 min, followed by 30 cycles of denaturation at 94°C for 30 s, annealing at 48°C for 45 s, and elongation at 72°C for 1 min, and a final elongation step at 72°C for 7 min. The gene pmoA, encoding particulate methane monooxygenase subunit A (a key enzyme for aerobic methane oxidation), was amplified from every crude extract obtained from LGH02-ew and cores LGH02-c1 to -c3 using the primer pair of pmoA189f/pmoA682r or pmoA189f/mb661r (Costello and Lidstrom, 1999). Thermal cycling was performed with an initial melting step at 94°C for 2 min, followed by 40 cycles of denaturation at 94°C for 30 s, annealing at 56°C for 45 s, and elongation at 72°C for 1 min, and a final elongation step at 72°C for 7 min. Three to five positive PCR products were pooled, purified using the StrataPrep PCR purification kit (Stratagene, USA) and cloned using the pGEM-T cloning kit (Promega, USA) following the manufacturers' protocols. Positive inserts from each clone library were sequenced using the primers described above for 16S rRNA genes, and the primer pair M13f/M13r for methane-related functional genes. Parallel negative control was performed to assess the potential contamination introduced during the laboratory manipulation.
The obtained sequences were checked for chimera formation using the Pintail (Ashelford et al., 2005), and aligned to the closely related sequences retrieved from the GenBank and RDP databases using the Greengenes (http://greengenes.lbl.gov) (Desantis et al., 2006). Sequences were categorized into the operational taxonomic units (OTUs) on the basis of 98% similarity cutoff. Taxonomic assignments for representative sequences were based on the results obtained from the SILVA tools (Quast et al., 2013). The UniFrac analyses was based on the phylogenetic trees constructed by the neighbor-joining algorithm and the substitution model of the maximum composite likelihood embedded within the MEGA 4.0 program (Tamura et al., 2007), and employed the environmental cluster algorithm for unweighted OTUs (Lozupone and Knight, 2005). Phylogenetic trees for pmoA and mcrA genes were constructed with the neighbor-joining algorithm on amino acid sequences deduced from nucleotide sequences using the MEGA 4.0 program. The bootstrap number was computed on the basis of 1000 iterations. The accession numbers of the unique sequences deposited in the GenBank are HQ916477-HQ916659, JN811694-JN811725, JQ407233-JQ407300, JX648556-JX648564, and JX648584- JX648591 for 16S rRNA genes, JX648565-JX648573 for mcrA genes, and JX648574-JX648583 for pmoA genes.
Quantitative PCR (qPCR) analyses of 16S rRNA gene copy number of bacteria, archaea, ANME-2a and Desulfuromonas/Pelobacter were performed for every crude extract from cores LGH02-c2 and -c3 on a MyiQ Real Time PCR Detection system (Bio-Rad, USA). In brief, each qPCR reaction contained 1X of SsoFast EvaGreen Supermix (Bio-Rad, USA), 100 nM of each primer, and 2 μ L of template. Primers and PCR conditions were the same as those described in Chang et al. (2012). Primer specificity was first confirmed through sequencing cloned amplicons generated from qPCR assays and performing taxonomic analyses of obtained sequences. For routine qPCR analyses, primer specificity was checked by both melting curve analysis and gel electrophoresis. The annealing was set at the temperature generating the greatest yield [the lowest threshold cycle (CT)] while primer specificity was maintained. The standards were prepared from the cloned amplicons of these target groups obtained in the clone libraries. The concentrations of standard DNA were determined using a Qubit spectrophotometer (Invitrogen, USA). The standard DNA templates were then diluted in a 10-fold series and qPCR-amplified in order to measure the CT for a known concentration of standard DNA. The plots of CT vs. standard DNA concentration yielded the linear relationship with R2 values greater than 0.99. The 16S rRNA gene copy number of a specific group was then calculated assuming 650 g mole−1 of one base pair of DNA.
Results
Geochemical Characteristics
Analyses of pore water extracted from sediments yielded distinct geochemical characteristics between cores LGH02-c2 and -c3 (Figure 2). Chloride concentrations in core LGH02-c2 decreased from 709 mM at the top to 490 mM at 4 cm, followed by an increase to a level between 567 and 634 mM at depth. In core LGH02-c3, chloride concentrations fluctuated between 637 and 708 mM. Sulfate remained at a low level (<40 μM) in core LGH02-c2, whereas a significant increase from 18 to 165 μM with depth was observed in core LGH02-c3. Dissolved Mn concentrations in core LGH02-c2 increased from 0.13 mM in the top sediments to 0.23 mM at 4 cm and varied between 0.13 and 0.23 mM at depth. In contrast, dissolved Mn concentrations in core LGH02-c3 were at a nearly constant level of 0.20 ± 0.02 mM (average with one standard deviation). Dissolved Fe concentrations in core LGH02-c2 exhibited a peak at 0.62 mM at 6 cm followed by a decrease toward shallower and greater depths. For comparison, dissolved Fe concentrations in core LGH02-c3 decreased from 0.23 to 0.015 mM with depth. Methane concentrations in core LGH02-c2 increased from 0.58 mM at top to 1.6 mM at 2 cm and dropped to a level of 0.52–0.76 mM at depth, whereas methane concentrations in core LGH02-c3 decreased from 1.65 to 0.15 mM with depth. Patterns of carbon isotopic compositions of methane from both cores were in general comparable to each other with 13C-enriched methane at 5–11 cm and 13C-depleted methane at shallower and greater depths. Sulfide and nitrate concentrations were below the detection limit.
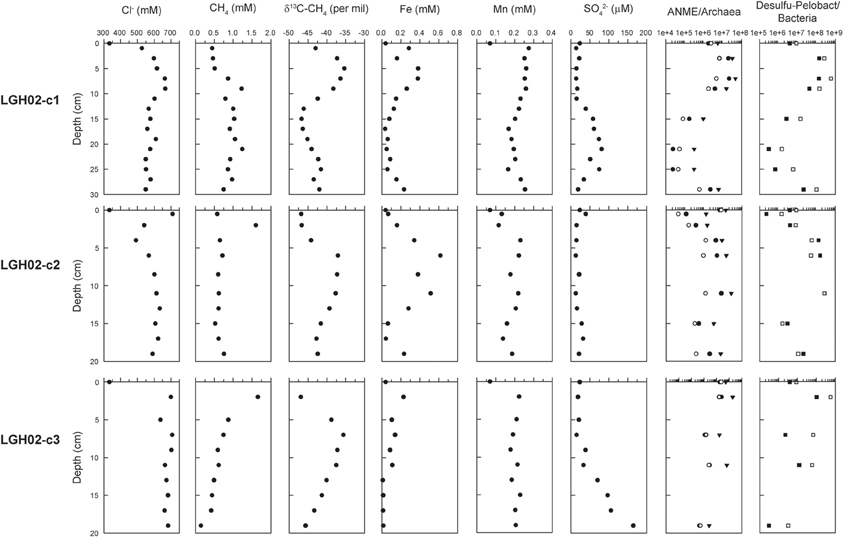
Figure 2. Pore water geochemistry and 16S rRNA gene abundances along depth. Target taxonomic units for qPCR analyses include archaea (solid triangles), ANME-2a amplified by primers A426f (solid circles) and A426fm (open circles), bacteria (open squares) and Desulfuromonas/Pelobacter (solid squares). Data points at 0 cm represent the characteristics of the bubbling fluids. The results obtained from core LGH02-c1 are adopted from Chang et al. (2012).
Assemblages of 16S rRNA Genes
PCR amplification yielded 16S rRNA gene amplicons from all crude extracts. A total of 617 bacterial and 525 archaeal 16S rRNA gene sequences were obtained (Supplementary Tables 1, 2). With a 98% similarity cutoff, 107 bacterial and 47 archaeal OTUs were identified. About 46% of bacterial OTUs and 40% of archaeal OTUs appeared to be singletons (appearing only once in any library).
Bacterial communities were diverse and complexly structured (Figure 3 and Supplementary Table 1). Taxonomic analyses revealed that Proteobacteria, Bacteroidetes, Firmicutes, Cyanobacteria, and Spirochaetes were the most abundant phyla identified (summed up to be more than 80% of the clones in each clone library). The proportion of each phylum/division varied among the samples. In particular, Cyanobacteria outnumbered other phyla/divisions in the top sediments of core LGH02-c2. Bacteroidetes constituted the most abundant phylum/division at 2, 4, and 6 cm of core LGH02-c2, and at 2 and 11 cm of core LGH02-c3, whereas Deltaproteobacteria and Gammaproteobacteria outnumbered others at 15 cm of core LGH02-c2 and 19 cm of core LGH02-c3, respectively. Clone sequences belonging to Acidobacteria, Actinobacteria, Chloroflexi, Deferribacteres, Lentisphaerae, Nitrospirae, OP9, OP11, Planctomycetes, Alphaproteobacteria, Betaproteobacteria, Epsilonproteobacteria, and Zetaproteobacteria were observed in a minor proportion.

Figure 3. Community structures along depth on the basis of 16S rRNA and mcrA gene sequences. Profiles of methane concentrations and δ13C-CH4 values are shown for constraining methanogenic and methanotrophic zones. Abundances of bacterial phyla/divisions less than 1% of the community size are summed and categorized as “others.” The “Methanosarcinales” represents the sequences affiliated with the ANME-2-excluded Methanosarcinales. Abbreviations: ANME, ANaerobic Methanotrophic Euryarchaeota; AAA, AOM Associated Archaea; MBG-B, Marine Benthic Group B; SCG, Soil Crenarchaeaota Group; MCG, Miscellaneous Crenarchaeota Group; SAMEG, South Africa Gold Mine Euryarchaeota Group; DSEG, Deep Sea Euryarchaeota Group. Detailed taxonomic categorization and abundances of individual OTUs in individual samples are shown in Supplementary Tables 1, 2.
Sequences of dominant bacterial OTUs (>10% of the clones in any individual library) were affiliated with Bacteroidetes-related clones recovered from hydrocarbon degradation enrichments (Knight et al., 1999; Alain et al., 2012) (LGH02-B-052 and -053), an uncultured Cyanobacteria-related clone from a wetland (LGH02-B-090), uncultured Deltaproteobacteria-related clones obtained from the Kazan mud volcano (Pachiadaki et al., 2010) (LGH02-B-027), an uncultured Desulfuromonadales-related clone (LGH02-B-030), Desulfuromonas spp. (Coates et al., 1995) (LGH02-B-22), an unclassified clone (LGH02-B-178), Thiohalophilus thiocyanatoxydans (Sorokin et al., 2007) (LGH02-B-012), and an uncultured Clostridiales-related clone from a coral (Sunagawa et al., 2009) (LGH02-B-174). The proportions of these OTUs in each library varied substantially. In particular, OTUs LGH02-B-052 and -053 dominated over others at 2, 4, and 6 cm of core LGH02-c2, and at 2 and 11 cm of core LGH02-c3. OTUs LGH02-B-022 and -030 were abundant at 15 cm of core LGH02-c2, and at 2 and 11 cm of core LGH02-c3. OTUs LGH02-B-090, -174 and -178, and OTU LGH02-B-012 were abundant at shallow intervals of core LGH02-c2 and the deepest interval of core LGH02-c3, respectively. OTU LGH02-B-027 was only detected in core LGH02-c2.
In addition to the major OTUs described above, the OTUs with an overall frequency greater than 1% possessed sequences affiliated with culture sequences belonging to Desulfovibrio alkaliphilus (Sorokin et al., 2008) (LGH02-B-136), Pelobacter acetylenicus (Schink, 1985) (LGH02-B-024), Caenispirillum spp. (LGH02-B-002), Marinobacter spp. (Gu et al., 2007) (LGH02-B-011 and -013), Desulfopila aestuarii (Suzuki et al., 2007) (LGH02-B-025) and Spirochaeta spp. (LGH02-B-150), and with uncultured sequences related to Bacteroidetes (Isenbarger et al., 2008) (LGH02-B-054), Firmicutes (Hubert et al., 2009; Orcutt et al., 2010) (LGH02-B-066), Spirochaetes (LGH02-B-097 and -150), and Lentisphaerae (Bano and Hollibaugh, 2002) (LGH02-B-100).
Archaeal communities were essentially composed of ANME members and methanogens although a few crenarchaeal and thaumarchaeal clones were detected (Figure 3 and Supplementary Table 2). Of archaeal OTUs, ANME members constituted 23–55 and 3–60% of clones in individual libraries of cores LGH02-c2 and -c3, respectively. Most detected ANME-related sequences were phylogenetically assigned to the ANME-2a group and shared ≥98% similarity with ANME-2a sequences recovered from core LGH02-c1 (Chang et al., 2012) or 92–97% similarity with sequences from the Nankai Trough (Miyashita et al., 2009), the Baltic Sea (Jagersma et al., 2009), the Okinawa Trough (Inagaki et al., 2006) and other onshore and offshore methane-rich sediments (Hinrichs et al., 1999; Knittel et al., 2005; Alain et al., 2006; Kendall et al., 2007). Few ANME-1 sequences affiliated with those obtained from the Kazan mud volcano (Pachiadaki et al., 2010) and freshwater sediments in the Kanto Plain of Japan (Takeuchi et al., 2009) were recovered from 0.5, 2 and 15 cm of core LGH02-c2 and 19 cm of core LGH02-03. A total of 30 clone sequences from cores LGH02-c2 and -c3 were assigned to the AAA (AOM Associated Archaea) lineage (Supplementary Table 2) that has been proposed to be capable of oxidizing methane anaerobically (Knittel and Boetius, 2009). Most of the AAA-related sequences were clustered into one OTU and particularly enriched at 19 cm of core LGH02-c3 (20% of the clones in the library).
The proportions of Methanosarcinales-related clones resembled those of Methanomicrobiales-related clones at 2, 4, and 6 cm of core LGH02-c2 (Figure 3). However, Methanosarcinales-related clones outnumbered Methanomicrobiales-related clones in other samples. The majority of these Methanosarcinales-related sequences were affiliated with Methanosarcina semesiae (Lyimo et al., 2000, 2009) and Methanococcoides methylutens (Sowers and Ferry, 1983), both of which are obligatorily methylotrophic methanogens. Other Methanosarcinales-related sequences were affiliated with uncultured sequences recovered from a wetland and from the Kazan mud volcanoes (Pachiadaki et al., 2010), Methanosaeta spp. (Zengler et al., 1999), Methanococcoides spp. (Asakawa et al., 1998), and Methanolobus profundi (Mochimaru et al., 2009). Within Methanomicrobiales, sequences related to Methanoplanus petrolearius (Ollivier et al., 1997), Methanocalculus pumilus (Mori et al., 2000), and uncultured sequences retrieved from the Pearl River estuary (Jiang et al., 2011) were detected. A few sequences affiliated with DSEG and MBG-B from Mediterranean mud volcanoes (Heijs et al., 2007; Kormas et al., 2008) and a hypersaline mat (Robertson et al., 2009), MCG from various environments (Huang et al., 2003; Nelson et al., 2009; Kubo et al., 2012; Lin et al., 2012), SAGMEG from South African gold mine groundwater and a hypersaline mat (Takai et al., 2001; Robertson et al., 2009), and SCG from groundwater in a Japanese gold mine (Nunoura et al., 2005) were also obtained.
Assemblages of mcrA Genes
PCR amplification yielded amplicons of mcrA genes for two samples selected from each core. A total of 356 sequences were clustered into nine OTUs (98% similarity) (Figures 3, 4). About 92 and 68% of mcrA gene sequences from cores LGH02-c1 and -c2, respectively, were represented by OTU LGH02-M-01 and affiliated with the ANME-2 related sequences recovered from coastal (Roussel et al., 2009) and estuarine sediments (Jiang et al., 2011). In contrast, only 20% of mcrA gene sequences from core LGH02-c3 were related to the ANME-2 lineage. The second largest OTU (LGH02-M-02) constituted about 20% of the total clones and possessed sequences affiliated with environmental sequences recovered from estuarine sediments (Jiang et al., 2011) and Methanosarcina spp. known to being capable of using acetate and methyl-compounds for methane production (Whitman et al., 2006). About 3–10% of sequences were related to clone sequences from estuarine sediments (Jiang et al., 2011; Xie et al., 2013) and classified into lineages belonging to Methanospirillum (LGH02-M-03) Methanococcoides (LGH02-M-04), Methanoplanus (LGH02-M-05) and Methanosarcina (LGH02-M-06). A few sequences were related to the ANME-1 (LGH02-M-07) and Methanomicrobiales (LGH-M-09).
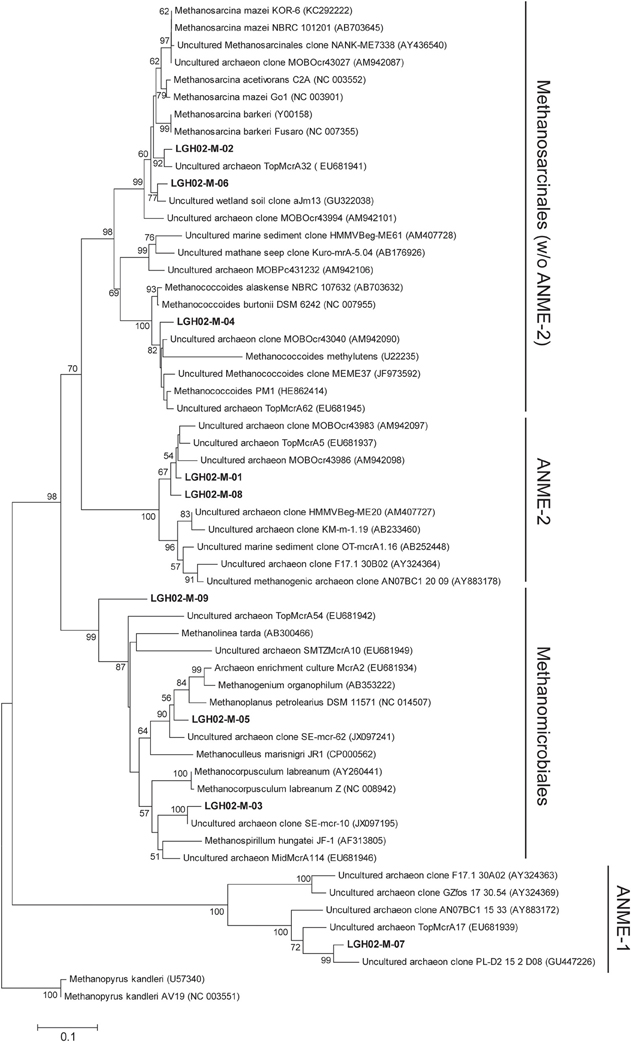
Figure 4. Phylogenetic tree based on the deduced partial amino acid sequences of mcrA genes. The tree was constructed on the basis of ~263 amino acids. Sequences obtained from this study (in boldface) were categorized into different OTUs on the basis of 98% identity. The scale bar represents the substitution for every base pair. The bootstrap number is shown where greater than 50%.
Assemblages of pmoA Genes
PCR amplification using the primer pair A189f/A682r did not yield any amplicon. Instead, pmoA genes were only detected from sediments at 4, 6, and 11 cm of core LGH02-c2 using the primer pair A189f/mb661. A total of 74 sequences were categorized into ten OTUs at a 98% similarity cutoff (Figure 5). These sequences were classified as type I methanotrophs within the Gammaproteobacteria. The majority of these sequences (~64% of the total clones) represented by OTUs LGH02-pmoA-1, −3, and −6 formed a monophyletic lineage that is distinct from all the other reported pmoA sequences. The exact topology between this lineage and other methanotrophs or environmental sequences could not be constrained confidently as the bootstrap value was less than 50% on both the amino acid (Figure 5) and nucleic acid levels (data not shown). Comparisons with sequences deposited in the GenBank showed that these sequences were related to uncultured Methylococcales-related clones obtained from lake sediments (Lin et al., 2004) or to strain M200 (Kip et al., 2011), Methylosoma spp. (Bussmann et al., 2006) and Methylovurum spp. (Iguchi et al., 2011), all of which rely on methane and methanol as carbon and energy sources. The other numerically abundant sequences represented by OTUs LGH02-pmoA-2, −5, and −9 were related to Methylomicrobium pelagicum (Sieburth et al., 1987; Bowman et al., 1995) or uncultured Methylococcales-related clones obtained from landfill soils (Chang et al., 2010). This closest strain also relies on methane and methanol as carbon and energy sources (Sieburth et al., 1987). The proportions of the largest two clusters did not vary systematically with depth. A few sequences were affiliated with Methylohalobius spp. isolated from an alkaline lake (Heyer et al., 2005) or with uncultured clones recovered from a hydrothermal field (Kato et al., 2009).
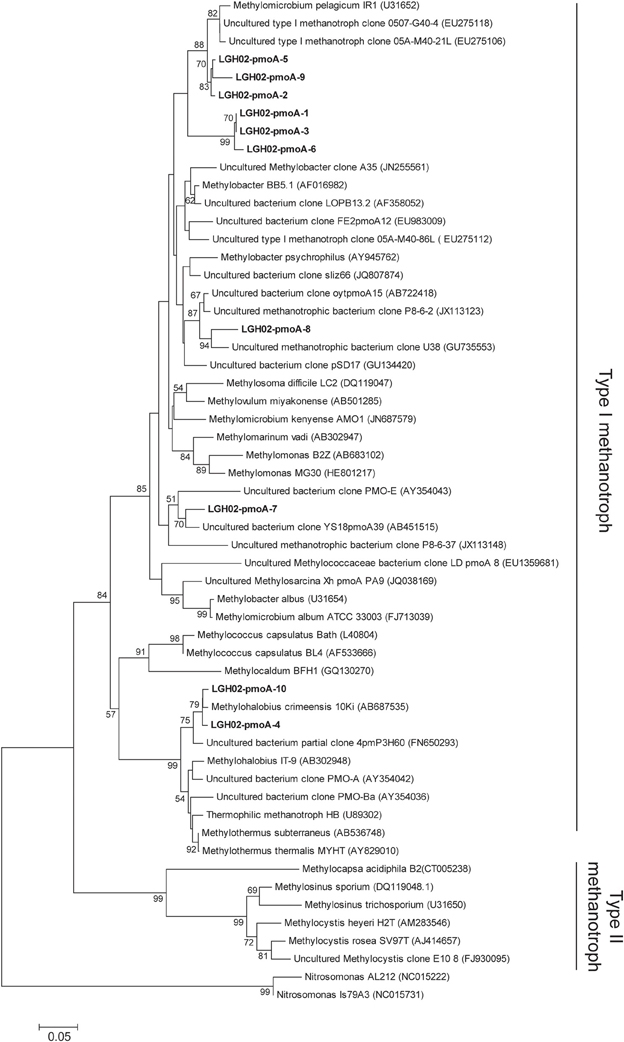
Figure 5. Phylogenetic tree based on the deduced partial amino acid sequences of pmoA genes. The tree was constructed on the basis of ~152 amino acids. Sequences obtained from this study (in boldface) were categorized into different OTUs on the basis of 98% identity. The scale bar represents the substitution for every base pair. The bootstrap number is shown where greater than 50%.
Abundances of 16S rRNA Genes
The qPCR analyses revealed that the gene abundances of all investigated taxonomic groups varied in accordance with the dissolved Fe profiles (Figure 2). The bacterial 16S rRNA gene abundances in core LGH02-c2 ranged between 1.46 × 106 and 2.70 × 108 copies (g−1 sediments). Along the depth profile, an increase from 1.46 × 106 to 2.70 × 108 copies (g−1 sediments) at ≤11 cm was followed by a decrease of one to two orders of magnitude at 11–19 cm. The Desulfuromonas/Pelobacter members made up 25 to >100% of the bacterial abundances. Their abundances varied in a fashion similar to those of bacteria. The archaeal 16S rRNA gene abundances ranged between 1.48 × 105 and 1.49 × 107 copies (g−1 sediments), whereas the ANME-2a 16S rRNA gene abundances detected by two different ANME-2a specific primers varied from 3.80 × 103 to 2.39 × 106copies (g−1 sediments) and from 9.02 × 101 to 1.34 × 104 copies (g−1 sediments), respectively. The sum of these two ANME-2a subgroups constituted 3–39% of the archaeal abundances. The gene abundance patterns for archaea and ANME-2a were comparable with those for bacteria and Desulfuromonas/Pelobacter. In core LGH02-c3, the bacterial 16S rRNA gene abundances decreased from 1.05 × 108 copies (g−1 sediments) at 2 cm to 2.29 × 106 copies (g−1 sediments) at 7 cm, increased to 1.26 × 107 copies (g−1 sediments) at 11 cm, and decreased to 3.10 × 105 copies (g−1 sediments) at 19 cm. The abundances of the other investigated taxonomic groups exhibited a pattern similar to that of bacteria.
Principle Component Analyses
Principle component analyses (PCA) revealed that PC1 and PC2 could explain ~29 and ~45% the variance in the bacterial and archaeal communities, respectively (Figure 6). For bacteria, communities at 3, 7, 9, and 29 cm of core LGH02-c1, 15 cm of core LGH02-c2, and 2, 11, and 19 cm of core LGH02-c3 clustered near the origin of the plot (Figure 6A). Three other clusters were formed by two samples from similar depths of the same core (groups composed of communities at 15 and 21 cm of core LGH02-c1, of communities at 0.5 and 2 cm of core LGH02-c2, and of communities at 4 and 6 cm of core LGH02-c2). The community associated with the bubbling fluid was distinguished from the groups described above. For archaea, communities at 3, 7, and 9 cm of core LGH02-c1 and 11 cm of core LGH02-c3 formed a cluster in the third quadrant of the plot, whereas communities at 15, 21, and 29 cm of core LGH02-c1 and from the bubbling fluids clustered in the fourth quadrant (Figure 6B). Communities in core LGH02-c2 and at the top and bottom of core LGH02-c3 were distributed over the first and second quadrants.
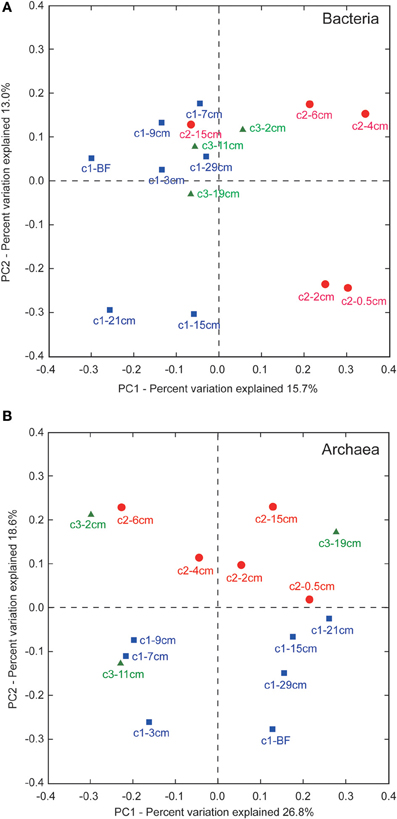
Figure 6. Unweighted principle component analyses for (A) bacterial and (B) archaeal community assemblages. Labels for data points consist of core name (c1–c3) and average depth interval. “BF” stands for bubbling fluids.
Discussion
Surface and Fluid Processes
Previous studies have suggested that subsurface-surface interactions are essential to charge the deeply-sourced fluids with substantial amounts of electron acceptors for anaerobic metabolisms in terrestrial mud volcanoes (Chang et al., 2012; Cheng et al., 2012). To assess the role of surface processes in shaping community assemblages at different sites of the LGHMVs, the field observations were examined. The sites for cores LGH02-c1, -c2, and -c3 were distributed along a gulley between four cone-shaped structures covered by patchy yellowish and oily materials (Figure 1). As evidenced by the mud hardness and the distribution of mud crack, the site proximal to the bubbling pool (e.g., core LGH02-c1) was immersed by the expelled fluids at a frequency much greater than the distant site (e.g., core LGH02-c3). The presence of a thick soft mud pile and simultaneous release of mud with fluids also suggest high rates of mud accumulation at sites near the bubbling pool. Therefore, the site for core LGH02-c1 would experience cycled wetting and drying as well as mud deposition at a frequency greater than the other two sites. In contrast, the site for core LGH02-c3 would experience less frequent fluid immersion and mud accumulation than the other two sites, and be subject to more intensive drying and evaporation.
The progressive enhancement of evaporation with increasing distance from the bubbling pool is supported by chloride concentrations in pore water extracted from the cored sediments. When compared with that in the bubbling fluid (333 mM), the chloride concentrations in the top pore water increased from 527 mM for core LGH02-c1 to ~700 mM for the other two cores (Figure 2). The averaged chloride concentration (± one standard deviation) in pore water also increased from 586 ± 41 mM for core LGH02-c1, to 598 ± 58 mM for core LGH02-c2, and 679 ± 23 mM for core LGH02-c3. Considering that the detected chloride concentrations are far less than the halite solubility at the in situ temperature, the enhancement in the chloride concentration of the top pore water reflects the effects of short-term evaporation on the overflowing bubbling fluid. Compounds susceptible to microbial transformation (e.g., methane) exhibit drastically different patterns and variation magnitudes from those (e.g., chloride) inert to abiotic and biotic reactions (Figure 2). Such a difference is essentially accounted for by physiological capabilities and activities that would transiently surpass the effect of fluid processes as well as mineral formation, dissolution and sorption (Chang et al., 2012; Cheng et al., 2012). Therefore, potential microbial metabolisms could be determined using the observed geochemical abundances while excluding the signals inherited from abiotic processes.
Microbial Methane Cycling
Metabolisms catalyzing methane cycling in the cored sediments were first assessed using the geochemical characteristics of the pore water (Figure 2). Like those interpreted in the previous study (Chang et al., 2012), the variations in concentration and δ13C value of methane for core LGH02-c2 suggest the compartmentalization of methanogenesis at <4 cm and anaerobic oxidation of methane (AOM) at 4–13 cm. Aerobic methanotrophy is likely not significant at shallow depths. This is because culture experiments have shown that aerobic methanotrophy preferentially utilizes 12CH4 over 13CH4 with an isotopic fractionation magnitude of 2–35‰ (Templeton et al., 2006). If aerobic methane oxidizers are active in consuming methane, the δ13C-CH4 values would have been shifted toward a higher range. The virtually identical δ13C-CH4 values at the two shallowest intervals do not corroborate this assertion. In addition, the high concentration of 13C-depleted methane and the presence of dissolved Fe/Mn at shallower depths signify methanogenesis and metal reduction, both of which are obligatorily anaerobic metabolisms. The most plausible explanation for the depletion in abundance and isotopic composition near the surface would be the loss of subsurface methane to the atmosphere when taking into account that methane levels in the atmosphere are low (~2 ppmv) and molecular diffusion at the centimeter scale would not cause significant isotopic fractionation. The possibility that aerobic methanotrophy is confined within 0.5 cm could not be completely excluded. Determination of the fine-scaled variations in dissolved methane and oxygen concentration is warranted to address the exact distribution of aerobic methanotrophy.
In addition to the depletion of 13C-enriched methane, the AOM zone is also characterized by the enhancement of dissolved Fe and Mn concentrations (up to 0.62 mM Fe and 0.23 mM Mn) (Figure 2). Therefore, high correlations between δ13C-CH4 values and dissolved Fe/Mn concentrations [Pearson correlation coefficients are 0.73 for Fe and 0.66 for Mn (p < 0.05)] are consistent with the presence of metal-dependent anaerobic methanotrophy (Beal et al., 2009; Sivan et al., 2011; Chang et al., 2012). At depths greater than 13 cm of core LGH02-c2, δ13C-CH4 values decreased to -42‰ while methane concentrations remained at a nearly constant level of 0.6 mM. The depletion of 13C in methane was less significant at >13 cm than that at shallow intervals. This combined with a nearly constant concentration of methane suggests that AOM prevails to this depth interval and is sustained by in situ methanogenesis and/or deeply-sourced methane, with δ13C-CH4 values ranging between -51 and -49‰ (Sun et al., 2010).
The profile of methane concentration in core LGH02-c3 was different from that in core LGH02-c2 (Figure 2). While methane concentrations decreased gradually with depth, δ13C-CH4 values were at a maximum of −36‰ at 7 cm and decreased toward shallower and deeper regions. The geochemical pattern suggests that methanogenesis and AOM were active at <5 and 5–11 cm, respectively. For intervals deeper than 11 cm, the depletion in abundance and isotopic composition of methane contradicts the expression of methanogenesis. Instead, the mixing between residual methane derived from the methanotrophic zone at 5–11 cm and a deep component characterized by the depleted isotopic composition (between −51 and −49‰; Sun et al., 2010) could account for the observed variations. With the exclusion of the data from the depth of 2 cm, δ13C-CH4 values are correlated with dissolved Fe and sulfate concentrations, respectively [Pearson correlation coefficients are 0.84 for Fe and −0.96 for sulfate (p < 0.05)] but irrelevant with dissolved Mn concentrations (p > 0.05). These lines of evidence suggest that AOM is potentially dependent on iron and sulfate reduction processes and manganese reduction is driven by organic mineralization.
The geochemical inference for methanotrophy and methanogenesis is supported by the molecular results (Figures 2, 3). Like those observed for core LGH02-c1 (Chang et al., 2012), the gene copy numbers of ANME-2a and Desulfuromonas/Pelobacter populations were high at 4–11 cm of core LGH02-c2 (constituting 17–38% of the archaeal populations and >100% of the bacterial populations, respectively). The log-scaled ANME-2a abundances are correlated with the δ13C-CH4 values [Pearson correlation coefficient is 0.78 (p < 0.05)], a pattern consistent with the geochemical interpretation that AOM is active in the methane transition zone. Furthermore, since Desulfuromonas/Pelobacter members are potential iron/manganese reducers (Lovley, 2006), positive correlation between the gene abundances of Desulfuromonas/Pelobacter and ANME-2a lineages [Pearson correlation coefficient is 0.99 (p < 0.05)] again suggests a likely metabolic interdependence between AOM and iron/manganese reduction. The ANME-2a abundances in core LGH02-c3 exhibited a pattern different from those in the other two cores. The gene copy numbers generally decreased with depth, with an exception that an increase by a factor of ~2 was observed for samples at 7 and 11 cm of core LGH02-c3. The summed ANME-2a abundance at 11 cm (5.24 × 105 copies g−1 sediments) was, however, 4–40 times less than those in the methane transition zones of the other two cores. While the high δ13C-CH4 values indicate the presence of AOM, the low abundance of ANME-2a members together with limited methane depletion at ≥7 cm suggests that AOM might not be as active as in the other two cores.
Methanogens represent the other major component of archaeal communities at the LGHMVs (Figure 3). Their relative abundances in the 16S rRNA or mcrA gene clone libraries were inversely correlated with those of the ANME-2a members. Most of these methanogen-related sequences were affiliated with obligatorily methylotrophic methanogens (Methanococcoides spp. and Methanosarcina spp.; Figures 3, 4) (Asakawa et al., 1998; Lyimo et al., 2000, 2009). These methanogens can proliferate under saline or hypersaline conditions by metabolizing methyl-compounds produced from the fermentation of osmoprotectants (e.g., choline, glycine betaine) that are used to counterbalance osmotic stress (Whitman et al., 2006). The prevalence of Methanosarcina and Methanococcoides over other methanogens has been constantly observed in hypersaline environments, such as microbial mats in Baja California (Orphan et al., 2008; Smith et al., 2008). However, surface evaporation at the LGHMVs does not generate salinities falling into the hypersaline range. In addition, neither the proportions nor the abundances of these methanogens were correlated with the variations in pore water salinity (p > 0.05). As stated previously, the high methane concentrations and low δ13C-CH4 values near the surface in cores LGH02-c2 and -c3 suggest active methanogenesis. Therefore, the prevalence of these obligatorily methylotrophic methanogens near the surface seems to be best explained by their physiological durability against transient desiccation and oxygen perfusion associated with surface evaporation.
The pmoA gene, which is responsible for aerobic methane oxidation, was only detected at 4, 6, and 11 cm of core LGH02-c2. The detected pmoA gene sequences were related to type I methanotrophs belonging to the Gammaproteobacteria (Figure 5). Most of these sequences were clustered into two OTUs (90% of the total clones) and affiliated with aerobic methanotrophs relying solely on methane and methanol (Sieburth et al., 1987; Bussmann et al., 2006; Iguchi et al., 2011). The presence of these sequences is partly consistent with abundant methane but does not corroborate with the anoxic conditions at these depth intervals geochemically and molecularly interpreted as the AOM zone. Since no culture evidence has attested that these Gammaproteobacteria could use nitrate, nitrite, or metal oxides as electron acceptors for methanotrophy, these sequences probably represent the relics of aerobic methanotrophs that proliferate upon exposure to the atmosphere and survive being buried in the anoxic environment. The rare detection of 16S rRNA genes affiliated with known aerobic methanotrophs (7 out of 1305 sequences in Supplementary Table 1) from all three cores and the absence of these sequences in the top sediments further suggest that aerobic methanotrophy might not contribute significantly to the overall methane cycling at the LGHMVs.
Combining geochemical and molecular data reported in this and previous studies (Chang et al., 2012) reveals that methanogenesis and AOM are prevalent regardless of the investigated site. However, their distribution along depth varied from site to site. At the site proximal to the bubbling pool (LGH02-c1), the AOM zone was underlain by the methanogenic and another AOM zones. In contrast, the methanogenic zone overlaid the AOM zone in two other cores (LGH02-c2 and -c3) distributed away from the bubbling pool. The reversible and alternating order of AOM and methanogenic zones along depth is not uncommon at the LGHMVs. For example, the AOM zone was either sandwiched or underlain by the methanogenic zone in other cores retrieved 18 months later from the other sites near bubbling pools (Chang et al., 2012). The alternating distribution of AOM and methanogenic zones is, however, not consistent with the successive predominance of sulfate reduction, AOM, and methanogenesis in marine sediments or terrestrial aquifers, a metabolic zonation pattern controlled by the competition for limited organic matters and H2 (Hoehler et al., 1998). Instead, the pattern seems to reflect the development and superposition of one specific metabolic zone over one developed previously, a scenario analogous to mud deposition over time.
The enhanced methane concentrations, low δ13C-CH4 values, and predominance of sequences affiliated with obligatorily methylotrophic methanogens near the surface also suggest that the mud platform could be an effective source of methane emission. Oxygen penetration is apparently attenuated by active oxygen consumption through biological respiration, and is thereby limited at very shallow intervals. Therefore, oxygen-sensitive methanogenesis could occur close to the surface. Previous studies have revealed that the summed flux of methane emission from the mud platform could exceed that from the corresponding macro-seepages by up to two orders of magnitude and attributed this to the gas transport through either pore space or micro-/mini-seepages (Etiope et al., 2004a,b; Hong et al., 2013). The results observed in this study provide an alternative mechanistic explanation that methanogenesis in near-surface environments can account for enhanced methane emissions from the mud platform.
Sulfur Cycling and Other Metabolisms
Sulfate concentrations increased from nearly below the limit of detection at 2 cm to 165 μM at the deepest interval in core LGH02-c3 (Figure 2). In addition, sulfate depletion occurred at depth intervals (5–10 cm) coinciding with the high δ13C-CH4 values, the decrease in methane concentration, and the high dissolved Fe concentrations. Considering that the bubbling fluid with 23 μM sulfate represents a deeply-sourced fluid (Chang et al., 2012), the high sulfate level at depth would require an additional source and could be accounted for by anaerobic sulfur disproportionation or sulfur oxidation potentially driven by the reduction of iron oxyhydroxide (Holmkvist et al., 2011). Such geochemical characteristics also indicate that AOM might be dependent on iron and sulfate reduction.
Analyses of 16S rRNA gene libraries revealed that a great proportion of bacterial sequences at 15 and 21 cm of core LGH02-c1 (18.4 and 8.0%) and 19 cm of core LGH02-c3 (29.3%) were affiliated with sequences retrieved from bubbling fluids and surface sediments of terrestrial and marine mud volcanoes (Yakimov et al., 2002; Omoregie et al., 2008, 2009), and with Thiohalophilus thiocyanatoxydans strain HRhD 2 (Sorokin et al., 2007) at ≥96% similarity (Supplementary Table 1). This strain is known to grow using thiosulfate and nitrate as energy sources under saline conditions (1–4 M NaCl) (Sorokin et al., 2007). As sulfate represents the end product from the oxidation of thiosulfate, the observed increase in the sulfate concentration is likely accounted for by the Thiohalophilus-related microorganisms. The clone library analyses also indicated that some bacterial sequences were related to Desulfovibrio alkaliphilus, Desulfobacterium spp. and unclassified Desulfobacterales and Desulfarculales sequences (Supplementary Table 1). Although the abundances of these potential sulfate reducers were low at these or adjacent intervals, their presence is presumably linked to the observed decrease in sulfate concentration. Whether these potential sulfate reducers are involved in the AOM remains to be investigated.
OTUs LGH02-B-052 and -53 with sequences classified into Bacteroidetes were represented by 17.7% of the total clones (Supplementary Table 1). Their proportions in clone libraries were enhanced at depth intervals geochemically and molecularly interpreted as the AOM zone (Figure 2). Most related sequences are derived from mixed populations of polluted sediments and enrichment cultures capable of anaerobically degrading complex organic compounds (e.g., benzene, phenol) (Knight et al., 1999; Alain et al., 2012). Whether and how microorganisms represented by the detected sequences are capable of degrading organic compounds or involved in the metabolic network related to the AOM warrants more investigation. Finally, OTU LGH02-B-90, represented by sequences related to Cyanobacteria, constituted 45.0% of the clones recovered from 0.5 cm of core LGH02-c2. Its relative abundance in this core decreased significantly with depth (from 45.0% at 0.5 cm to 1.1% at 15 cm). The decreasing trend of cyanobacteria is consistent with the phototrophic and oxygen producing natures of cyanobacteria, and further suggests that the proliferation of cyanobacteria in the top sediments would intensify the redox gradient and provide an additional pathway of primary production and carbon sequestration in terrestrial mud volcanoes.
Community Patterns
The unweighted Unifrac PCA revealed that bacterial communities in a specific metabolic zone of a core were more similar to each other than those in a different metabolic zone (Figure 6A). For example, samples from 3, 7, 9, and 29 cm and from 15 to 21 cm of core LGH02-c1 were geochemically and molecularly interpreted as the AOM and methanogenic zones, respectively. Communities in these samples were categorized into two clusters in accordance with the interpretations. A similar clustering pattern was observed for core LGH02-c2 (0.5 and 2 cm for methanogenesis, and 4 and 6 cm for AOM) but not for core LGH02-c3. If all communities are pooled together, communities recovered from individual metabolic zones in different cores are not coherently clustered. For archaea, communities from either core LGH02-c1 or -c2 could be differentiated from each other mostly by PC1 (Figure 6B). Furthermore, communities from core LGH02-c1 could be differentiated from those from core LGH02-c2 mostly by PC2. Archaeal communities from core LGH02-c3 were distributed over the entire plot. Compared to bacterial communities, archaeal communities exhibited no systematic variation with respect to depth or metabolic characteristics.
The clustering pattern for bacterial communities from cores LGH02-c1 and -c2 suggests that a specific community function could be mediated by different community members. As the imposed geochemical context varies from site to site, different community members possessing similar metabolic capabilities might be stimulated or inhibited by varying degrees. Therefore, their role in the metabolic network could be possibly swapped among different members whose physiological limitations could be accommodated under a specific geochemical context. In contrast, the lack of correlation between the archaeal assemblage and specific methane-related metabolism suggests that factors other than the observed geochemical characteristics are required to account for community variance. Since most detected archaeal members are affiliated with ANME-2a and Methanosarcinales, the community variance could be controlled by the presence or absence of minor community members. Overall, the discordance between community clustering and metabolic interpretation might reflect the heterogeneous nature of geochemical characteristics over various spatial scales in terrestrial mud volcanoes. We note that the community pattern could be biased by PCR or sequencing depth and DNA signatures may not be necessarily translated into metabolic activity or population size.
Conclusions
The geochemical patterns and assemblages of 16S rRNA and methane-related genes suggest the prevalence and stratified distribution of AOM and methanogenesis at different sites of the LGHMVs. The order of predominant methane-related metabolisms along depth, however, varied from site to site. At the site near the fringe of the bubbling pool, the AOM zone overlaid the methanogenic zone, providing an effective biological filtration for the removal of methane produced in situ or migrating from a deep source. For sites away from the bubbling pool, the methanogenic zone overlaid the AOM zone, highlighting a potential contribution of near-surface methanogenesis from the mud platform to the overall methane emission in the area. In situ methanogenesis catalyzed by methylotrophic methanogens produced methane at quantities sufficient to fuel anaerobic methanotrophy. Finally, the downcore alternating sequences of the methanogenic and AOM zones at different sites provide evidence that the pattern of metabolic and geochemical zonation is modulated by the frequency and magnitude of mud deposition on the surface as well as the fluid transport and mixing processes in the subsurface. Such subsurface-surface interactions would enable the transient development of various organizations of methane cycling in sulfate-deficient terrestrial mud volcanoes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the late Chih-Hsien Sun for his assistance with the analyses of the carbon isotopic compositions of methane. We acknowledge the support of the Taiwanese National Science Council (100-2627-M-002-003, 102-2116-M-002-017, and 102-3113-P-002 -011).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2014.00121/abstract
References
Alain, K., Harder, J., Widdel, F., and Zengler, K. (2012). Anaerobic utilization of toluene by marine alpha- and gammaproteobacteria reducing nitrate. Microbiology 158, 2946–2957. doi: 10.1099/mic.0.061598-0
Alain, K., Holler, T., Musat, F., Elvert, M., Treude, T., and Kruger, M. (2006). Microbiological investigation of methane- and hydrocarbon-discharging mud volcanoes in the Carpathian Mountains, Romania. Environ. Microbiol. 8, 574–590. doi: 10.1111/j.1462-2920.2005.00922.x
Asakawa, S., Sauer, K., Liesack, W., and Thauer, R. K. (1998). Tetramethylammonium:coenzyme M methyltransferase system from Methanococcoides sp. Arch. Microbiol. 170, 220–226. doi: 10.1007/s002030050636
Ashelford, K. E., Chuzhanova, N. A., Fry, J. C., Jones, A. J., and Weightman, A. J. (2005). At least 1 in 20 16S rRNA sequence record currently held in public repositories is estimated to contain substantial anomalies. Appl. Environ. Microbiol. 71, 7724–7736. doi: 10.1128/AEM.71.12.7724-7736.2005
Bano, N., and Hollibaugh, J. T. (2002). Phylogenetic composition of bacterioplankton assemblages from the Arctic Ocean. Appl. Environ. Microbiol. 68, 505–518. doi: 10.1128/AEM.68.2.505-518.2002
Beal, E. J., House, C. H., and Orpan, V. J. (2009). Manganese- and iron-dependent marine methane oxidation. Science 325, 184–187. doi: 10.1126/science.1169984
Bowman, J. P., Sly, L. I., and Stackerbrandt, E. (1995). The phylogenetic position of the family Methylococcaceae. Int. J. Syst. Bacteriol. 45, 182–185. doi: 10.1099/00207713-45-1-182
Bussmann, I., Rahalkar, M., and Schink, B. (2006). Cultivation of methanotrophic bacteria in opposing gradients of methane and oxygen. FEMS Microbiol. Ecol. 56, 331–344. doi: 10.1111/j.1574-6941.2006.00076.x
Chang, C. P., Angelier, J., Huang, C. Y., and Liu, C. S. (2001). Structural evolution and significance of a mélange in a collision belt: the Lichi Mélange and the Taiwan arc–continent collision. Geol. Mag. 138, 633–651. doi: 10.1017/S0016756801005970
Chang, C.-Y., Tung, H.-H., Tseng, I.-C., Wu, J.-H., Liu, Y.-F., and Lin, H.-M. (2010). Dynamics of methanotrophic communities in tropical alkaline landfill upland soil. Appl. Soil Ecol. 46, 192–199. doi: 10.1016/j.apsoil.2010.08.009
Chang, Y.-H., Cheng, T.-W., Lai, W.-J., Tsai, W.-Y., Sun, C.-H., Lin, L.-H., et al. (2012). Microbial methane cycling in a terrestrial mud volcano in eastern Taiwan. Environ. Microbiol. 14, 895–908. doi: 10.1111/j.1462-2920.2011.02658.x
Cheng, T.-W., Chang, Y.-H., Tang, S.-L., Tseng, C.-H., Chiang, P.-W., Chang, K.-T., et al. (2012). Subsurface and surface interactions drive metabolic stratification in terrestrial mud volcanoes. ISME J. 6, 2280–2290. doi: 10.1038/ismej.2012.61
Coates, J. D., Lonergan, D. J., Philips, E. J., Jenter, H., and Lovley, D. R. (1995). Desulfuromonas palmitatis sp. nov., a marine dissimilatory Fe(III) reducer that can oxidize long-chain fatty acids. Arch. Microbiol. 164, 406–413. doi: 10.1007/BF02529738
Costello, A. M., and Lidstrom, M. E. (1999). Molecular characterization of functional and phylogenetic genes from natural populations of methanotrophs in lake sediments. Appl. Environ. Microbiol. 65, 5066–5074.
Desantis, T. Z., Hugenholtz, P., Keller, K., Brodie, E. L., Larsen, N., Piceno, Y. M., et al. (2006). NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 34, W394–W399. doi: 10.1093/nar/gkl244
Dimitrov, L. I. (2003). Mud volcanoes - a significant source of atmospheric methane. Geo-Mar. Lett. 23, 155–161. doi: 10.1007/s00367-003-0140-3
Etiope, G., Baciu, C., Caracausi, A., Italiano, F., and Cosma, C. (2004a). Gas flux to the atmosphere from mud volcanoes in eastern Romania. Terra Nova 16, 179–184. doi: 10.1111/j.1365-3121.2004.00542.x
Etiope, G., Feyzullayev, A., Baciu, C., and Milkov, A. V. (2004b). Methane emission from mud volcanoes in eastern Azerbaijan. Geology 32, 465–468. doi: 10.1130/G20320.1
Etiope, G., Lassey, K. R., Klusman, R. W., and Boschi, E. (2008). Reappraisal of the fossil methane budget and related emission from geologic sources. Geophys. Res. Lett. 35:L09307. doi: 10.1029/2008GL033623
Gu, J., Cai, H., Yu, S. L., Qu, R., Yin, B., Guo, Y. F., et al. (2007). Marinobacter gudaonensis sp. nov., isolated from an oil-polluted saline soil in a Chinese oilfield. Int. J. Syst. Evol. Microbiol. 57, 250–254. doi: 10.1099/ijs.0.64522-0
Heijs, S. K., Haese, R. R., van der Wielen, P. W. J. J., Forney, L. J., and van Elsas, J. D. (2007). Use of 16S rRNA gene based clone libraries to assess microbial communities potentially involved in anaerobic methane oxidation in a Mediterranean cold seep. Microb. Ecol. 53, 384–398. doi: 10.1007/s00248-006-9172-3
Heyer, J., Berger, U., Hardt, M., and Dunfield, P. F. (2005). Methylohalobius crimeensis gen. nov., sp. nov., a moderately halophilic, methanotrophic bacterium isolated from hypersaline lakes of Crimea. Int. J. Syst. Evol. Microbiol. 55, 1817–1826. doi: 10.1099/ijs.0.63213-0
Hinrichs, K.-U., Hayes, J. M., Sylva, S. P., Brewer, P. G., and Delong, E. F. (1999). Methane-consuming archaebacteria in marine sediments. Nature 398, 802–805. doi: 10.1038/19751
Hoehler, T. M., Alperin, M. J., Albert, D. B., and Martens, C. S. (1998). Thermodynamic control on hydrogen concentrations in anoxic sediments. Geochim. Cosmochim. Acta 62, 1745–1756. doi: 10.1016/S0016-7037(98)00106-9
Holmkvist, L., Ferdelman, T. G., and Jorgensen, B. B. (2011). A cryptic sulfur cycle driven by iron in the methane zone of marine sediment (Aarhus Bay, Denmark). Geochim. Cosmochim. Acta 75, 3581–3599. doi: 10.1016/j.gca.2011.03.033
Hong, W. L., Etiope, G., Yang, T. F., and Chang, P. Y. (2013). Methane flux from miniseepage in mud volcanoes of SW Taiwan: comparison with the data from Italy, Romania, and Azerbaijan. J. Asian Earth Sci. 65, 3–12. doi: 10.1016/j.jseaes.2012.02.005
Huang, L. N., Chen, Y. Q., Zhou, H., Luo, S., Lan, C. Y., and Qu, L. H. (2003). Characterization of methanogenic Archaea in the leachate of a closed municipal solid waste landfill. FEMS Microbiol. Ecol. 46, 171–177. doi: 10.1016/S0168-6496(03)00218-6
Huber, H., Hohn, M. J., Rachel, R., Fuchs, T., Wimmer, V. C., and Stetter, K. O. (2002). A new phylum of Archaea represented by a nanosized hyperthermophilic symbiont. Nature 417, 63–67. doi: 10.1038/417063a
Hubert, C., Loy, A., Nickel, M., Arnosti, C., Baranyi, C., Bruchert, V., et al. (2009). A constant flux of diverse thermophilic bacteria into the cold Arctic seabed. Science 325, 1541–1544. doi: 10.1126/science.1174012
Iguchi, H., Yurimoto, H., and Sakai, Y. (2011). Methylovulum miyakonense gen. nov., sp. nov., a type I methanotroph isolated from forest soil. Int. J. Syst. Evol. Microbiol. 61, 810–815. doi: 10.1099/ijs.0.019604-0
Inagaki, F., Kuypers, M. N. M., Tsunogai, U., Ishibashi, J.-I., Nakamura, K.-I., Treude, T., et al. (2006). Microbial communities in a sediment-hosted CO2 lake of the Southern Okinawa Trough hydrothermal system. Proc. Natl. Acad. Sci. U.S.A. 103, 14164–14169. doi: 10.1073/pnas.0606083103
Isenbarger, T. A., Finney, M., Rios-Velazquez, C., Handelsman, J., and Ruvkun, G. (2008). Miniprimer PCR, a new lens for viewing the microbial world. Appl. Environ. Microbiol. 74, 840–849. doi: 10.1128/AEM.01933-07
Jagersma, G. C., Meulepas, R. J. W., Jong, I. H., Gieteling, J., Klimiuk, A., Schouten, S., et al. (2009). Microbial diversity and community structure of a highly active anaerobic methane-oxidizing sulfate-reducing enrichment. Environ. Microbiol. 11, 3223–3232. doi: 10.1111/j.1462-2920.2009.02036.x
Jiang, L., Zheng, Y., Chen, J., Xiao, X., and Wang, F. (2011). Stratification of archaeal communities in shallow sediments of the Pearl River estuary, Southern China. Antonie Van Leeuwenhoek 99, 739–751. doi: 10.1007/s10482-011-9548-3
Kato, S., Kobayashi, C., Kakegawa, T., and Yamagishi, A. (2009). Microbial communities in iron-silica-rich microbial mats at deep-sea hydrothermal fields of the Southern Mariana Trough. Environ. Microbiol. 11, 2094–2111. doi: 10.1111/j.1462-2920.2009.01930.x
Kendall, M. M., Wardlaw, G. D., Tang, C. F., Bonin, A. S., Liu, Y., and Valentine, D. L. (2007). Diversity of Archaea in marine sediments from Skan Bay, Alaska, including cultivated methanogens, and description of Methanogenium boonei sp. nov. Appl. Environ. Microbiol. 73, 407–414. doi: 10.1128/AEM.01154-06
Kip, N., Ouyang, W., van Winden, J., Raghoebarsing, A., van Niftrik, L., Pol, A., et al. (2011). Detection, isolation, and characterization of acidophilic methanotrophs from Sphagnum mosses. Appl. Environ. Microbiol. 77, 5643–5654. doi: 10.1128/AEM.05017-11
Knight, V. K., Kerkhof, L. J., and Haggblom, M. M. (1999). Community analyses of sulfidogenic 2-bromophenol-dehalogenating and phenol-degrading microbial consortia. FEMS Microbiol. Ecol. 29, 137–147. doi: 10.1111/j.1574-6941.1999.tb00605.x
Knittel, K., and Boetius, A. (2009). Anaerobic oxidation of methane: progress with an unknown process. Annu. Rev. Microbiol. 63, 311–334. doi: 10.1146/annurev.micro.61.080706.093130
Knittel, K., Losekann, T., Boetius, A., Kort, R., and Amann, R. (2005). Diversity and distribution of methanotrophic Archaea at cold seeps. Appl. Environ. Microbiol. 71, 467–479. doi: 10.1128/AEM.71.1.467-479.2005
Kopf, A. J. (2004). Significance of mud volcanism. Rev. Geophys. 40, 1–52. doi: 10.1029/2000RG000093
Kormas, K. A., Meziti, A., Dahlmann, A., de Lange, G. J., and Lykousis, V. (2008). Characterization of methanogenic and prokaryotic assemblages based on mcrA and 16S rRNA gene diversity in sediments of the Kazan mud volcano (Mediterranean Sea). Geobiology 6, 450–460. doi: 10.1111/j.1472-4669.2008.00172.x
Kubo, K., Lloyd, K. G., Biddle, J. F., Amann, R., Teske, A. P., and Knittel, K. (2012). Archaea of the Miscellaneous Crenarchaeotal Group are abundant, diverse and widespread in marine sediments. ISME J. 6, 1949–1965. doi: 10.1038/ismej.2012.37
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackenbrandt and M. Goodfellow (New York, NY: Wiley), 115–175.
Lin, J. L., Radajewski, S., Eshinimaev, B. T., Trotsenko, Y. A., McDonald, I. R., and Murrell, J. C. (2004). Molecular diversity of methanotrophs in Transbaikal soda lake sediments and identification of potentially active populations by stable isotope probing. Environ. Microbiol. 6, 1049–1060. doi: 10.1111/j.1462-2920.2004.00635.x
Lin, X., Kennedy, D., Fredrickson, J., Bjornstad, B., and Konopka, A. (2012). Vertical stratification of subsurface microbial community composition across geological formations at the Hanford Site. Environ. Microbiol. 14, 414–425. doi: 10.1111/j.1462-2920.2011.02659.x
Lovley, D. R. (2006). Dissimilatory Fe(III)- and Mn(IV)-reducing prokaryotes. Prokaryotes 2, 635–658. doi: 10.1007/0-387-30742-7_21
Lozupone, C., and Knight, R. (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005
Lyimo, T. J., Pol, A., Jetten, M. S., and Op den Camp, H. J. M. (2009). Diversity of methanogenic archaea in a mangrove sediment and isolation of a new Methanococcoides strain. FEMS Microbiol. Lett. 291, 247–253. doi: 10.1111/j.1574-6968.2008.01464.x
Lyimo, T. J., Pol, A., Op den Camp, H. J. M., Harhangi, H. R., and Vogels, G. D. (2000). Methanosarcina semesiae sp. nov., a dimethylsulfide-utilizing methanogen from mangrove sediment. Int. J. Syst. Evol. Microbiol. 50, 171–178. doi: 10.1099/00207713-50-1-171
Mazzini, A. (2009). Mud volcanism: processes and implications. Mar. Petrol. Geol. 26, 1677–1680. doi: 10.1016/j.marpetgeo.2009.05.003
Mazzini, A., Svensen, H., Planke, S., Guliyev, I., Akhmanov, G. G., Fallik, T., et al. (2009). When mud volcanoes sleep: insight from seep geochemistry at the Dashgil mud volcano, Azerbaijan. Mar. Petrol. Geol. 26, 1704–1715. doi: 10.1016/j.marpetgeo.2008.11.003
Milkov, A. V. (2005). “Global distribution of mud volcanoes and their significance in petroleum exploration as a source of methane in the atmosphere and hydrosphere and as a geohazard,” in Mud Volcanoes, Geodynamics and Seismicity, eds G. Martinelli and B. Panahi (Dordrecht: Springer), 29–34.
Miyashita, A., Mochimaru, H., Kazama, H., Ohashi, A., Yamaguchi, T., Nunoura, T., et al. (2009). Development of 16S rRNA gene-targeted primers for detection of archaeal anaerobic methanotrophs (ANMEs). FEMS Microbiol. Lett. 297, 31–37. doi: 10.1111/j.1574-6968.2009.01648.x
Mochimaru, H., Tamaki, H., Hanada, S., Imachi, H., Nakamura, K., Sakata, S., et al. (2009). Methanolobus profundi sp. nov., a methylotrophic methanogen isolated from deep subsurface sediments in a natural gas field. Int. J. Syst. Evol. Microbiol. 59, 714–718. doi: 10.1099/ijs.0.001677-0
Mori, K., Yamamoto, H., Kamagata, Y., Hatsu, M., and Takamizawa, K. (2000). Methanocalculus pumilus sp. nov., a heavy-metal-tolerant methanogen isolated from a waste-disposal site. Int. J. Syst. Evol. Microbiol. 50, 1723–1729. doi: 10.1099/00207713-50-5-1723
Nelson, K. A., Moin, N. S., and Bernhard, A. E. (2009). Archaeal diversity and the prevalence of Crenarchaeota in salt marsh sediments. Appl. Environ. Microbiol. 75, 4211–4215. doi: 10.1128/AEM.00201-09
Nunoura, T., Hirayama, H., Takami, H., Oida, H., Nishi, S., Shimamura, S., et al. (2005). Genetic and functional properties of uncultivated thermophilic crenarchaeotes from a subsurface gold mine as revealed by analysis of genome fragments. Environ. Microbiol. 7, 1967–1984. doi: 10.1111/j.1462-2920.2005.00881.x
Ollivier, B., Cayol, J. L., Patel, B. K., Magot, M., Fardeau, M. L., and Garcia, J. L. (1997). Methanoplanus petrolearius sp. nov., a novel methanogenic bacterium from an oil-producing well. FEMS Microbiol. Lett. 147, 51–56. doi: 10.1111/j.1574-6968.1997.tb10219.x
Omoregie, E. O., Mastalerz, V., de Lange, G., Straub, K. L., Kappler, A., Roy, H., et al. (2008). Biogeochemistry and community composition of iron- and sulfur-precipitating microbial mats at the Chefren mud volcano (Nile Deep Sea fan, Eastern Mediterranean). Appl. Environ. Microbiol. 74, 3198–3215. doi: 10.1128/AEM.01751-07
Omoregie, E. O., Niemann, H., Mastalerz, V., de Lange, G. J., Stadnitskaia, A., Mascle, J., et al. (2009). Microbial methane oxidation and sulfate reduction at cold seeps of the deep Eastern Mediterranean Sea. Mar. Geol. 261, 114–127. doi: 10.1016/j.margeo.2009.02.001
Orcutt, B., Joye, S. B., Kleindienst, S., Knittel, K., Ramette, A., Rietz, A., et al. (2010). Impact of natural oil and higher hydrocarbons on microbial diversity, distribution, and activity in Gulf of Mexico cold-seep sediments. Deep Sea Res. II Top. Stud. Oceanogr. 57, 2008–2021. doi: 10.1016/j.dsr2.2010.05.014
Orphan, V. J., Jahnke, L. L., Embaye, T., Turk, K. A., Pernthaler, A., Summons, R. E., et al. (2008). Characterization and spatial distribution of methanogens and methanogenic biosignatures in hypersaline microbial mats of Baja California. Geobiology 6, 376–393. doi: 10.1111/j.1472-4669.2008.00166.x
Pachiadaki, M. G., Lykousis, V., Stefanou, E. G., and Kormas, K. A. (2010). Prokaryotic community structure and diversity in the sediments of an active submarine mud volcano (Kazan mud volcano, East Mediterranean Sea). FEMS Microbiol. Ecol. 72, 429–444. doi: 10.1111/j.1574-6941.2010.00857.x
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Robertson, C. E., Spear, J. R., Harris, J. K., and Pace, N. R. (2009). Diversity and stratification of Archaea in a hypersaline microbial mat. Appl. Environ. Microbiol. 75, 1801–1810. doi: 10.1128/AEM.01811-08
Roussel, E. G., Sauvadet, A. L., Allard, J., Chaduteau, C., Richard, P., Bonavita, M.-A. C., et al. (2009). Archaeal methane cycling communities associated with gassy subsurface sediments of Marennes-Oleron Bay (France). Geomicrobiol. J. 26, 31–43. doi: 10.1080/01490450802599284
Schink, B. (1985). Fermentation of acetylene by an obligate anaerobe, Pelobacter acetylenicus sp. nov. Arch. Microbiol. 142, 295–301. doi: 10.1007/BF00693407
Scholten, J. C. M., Joye, S. B., Hollibaugh, J. T., and Murrell, J. C. (2005). Molecular analysis of the sulfate reducing and archaeal community in a meromictic soda lake (Mono Lake, California) by targeting 16S rRNA, mcrA, aspA, and dsrAB genes. Microb. Ecol. 50, 29–39. doi: 10.1007/s00248-004-0085-8
Sieburth, J. M., Johnson, P. W., Eberhardt, M. A., Sieracki, M. E., Lidstrom, M., and Laux, D. (1987). The first methane-oxidizing bacterium from the upper mixing layer of the deep ocean: Methylomonas pelagica sp. nov. Curr. Microbiol. 14, 285–293. doi: 10.1007/BF01568138
Sivan, O., Adler, M., Pearson, A., Gelman, F., Bar-Or, I., John, S. G., et al. (2011). Geochemical evidence for iron-mediated anaerobic oxidation of methane. Limnol. Oceanogr. 56, 1536–1544. doi: 10.4319/lo.2011.56.4.1536
Smith, J. M., Green, S. J., Kelley, C. A., Prufert-Bebout, L., and Bebout, B. M. (2008). Shifts in methanogen community structure and function associated with long-term manipulation of sulfate and salinity in a hypersaline microbial mat. Environ. Microbiol. 10, 386–394. doi: 10.1111/j.1462-2920.2007.01459.x
Sorokin, D. Y., Tourova, T. P., Bezsoudnova, E. Y., Poli, A., and Muyzer, G. (2007). Denitrification in a binary culture and thiocyanate metabolism in Thiohalophilus thiocyanoxidans gen. nov. sp. nov. - a moderately halophilic chemolithoautotrophic sulfur-oxidizing Gammaproteobacterium from hypersaline lakes. Arch. Microbiol. 187, 441–450. doi: 10.1007/s00203-006-0208-3
Sorokin, D. Y., Tourova, T. P., Mussmann, M., and Muyzer, G. (2008). Dethiobacter alkaliphilus gen. nov. sp. nov., and Desulfurivibrio alkaliphilus gen. nov. sp. nov.: two novel representatives of reductive sulfur cycle from soda lakes. Extremophiles 12, 431–439. doi: 10.1007/s00792-008-0148-8
Sowers, K. R., and Ferry, J. G. (1983). Isolation and characterization of a methylotrophic marine methanogen, Methanococcoides methylutens gen. nov., sp. nov. Appl. Environ. Microbiol. 45, 684–690.
Sunagawa, S., Desantis, T. Z., Piceno, Y. M., Brodie, E. L., Desalvo, M. K., Voolstra, C. R., et al. (2009). Bacterial diversity and white plague disease-associated community changes in the Caribbean coral Montastraea faveolata. ISME J. 3, 512–521. doi: 10.1038/ismej.2008.131
Sun, C.-H., Chang, S.-C., Kuo, C.-L., Wu, J.-C., and Shao, P.-H. (2010). Origins of Taiwan's mud volcanoes: evidence from geochemistry. J. Asian Earth Sci. 37, 105–110. doi: 10.1016/j.jseaes.2009.02.007
Suzuki, D., Ueki, A., Amaishi, A., and Ueki, K. (2007). Desulfopila aestuarii gen. nov., sp. nov., a Gram-negative, rod-like, sulfate-reducing bacterium isolated from an estuarine sediment in Japan. Int. J. Syst. Evol. Microbiol. 57, 520–526. doi: 10.1099/ijs.0.64600-0
Svensen, H., Karlsen, D. A., Sturz, A., Backer-Owe, K., Banks, D. A., and Planke, S. (2007). Processes controlling water and hydrocarbon compositions in seeps from the Salton Sea geothermal system, California, USA. Geology 35, 85–88. doi: 10.1130/G23101A.1
Takai, K., Moser, D. P., Deflaun, M., Onstott, T. C., and Fredrickson, J. K. (2001). Archaeal diversity in waters from deep South African gold mines. Appl. Environ. Microbiol. 67, 5750–5760. doi: 10.1128/AEM.67.21.5750-5760.2001
Takeuchi, M., Komai, T., Hanada, S., Tamaki, H., Tanabe, S., Miyachi, Y., et al. (2009). Bacterial and archaeal 16S rRNA genes in Late Pleistocene to Holocene muddy sediments from the Kanto Plain of Japan. Geomicrobiol. J. 26, 104–118. doi: 10.1080/01490450802662355
Tamura, K., Dudley, J., Nei, M., and Kumar, S. (2007). MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599. doi: 10.1093/molbev/msm092
Templeton, A. S., Chu, K.-H., Alvarez-Cohen, L., and Conrad, M. E. (2006). Variable carbon isotope fractionation expressed by aerobic CH4-oxidizing bacteria. Geochim. Cosmochim. Acta 70, 1739–1752. doi: 10.1016/j.gca.2005.12.002
Whitman, W. B., Bowen, T. L., and Boone, D. R. (2006). The methanogenic bacteria. Prokaryotes 3, 165–207. doi: 10.1007/0-387-30743-5_9
Wiesenburg, D. A., and Guinasso, N. L. (1979). Equilibrium solubilities of methane, carbon monoxide, and hydrogen in water and seawater. J. Chem. Eng. Data 24, 356–360. doi: 10.1021/je60083a006
Xie, S., Lazar, C. S., Lin, Y.-S., Teske, A., and Hinrichs, K.-U. (2013). Ethane- and propane-producing potential and molecular characterization of an ethanogenic enrichment in an anoxic estuarine sediment. Org. Geochem. 59, 37–48. doi: 10.1016/j.orggeochem.2013.03.001
Yakimov, M. M., Giuliano, L., Crisafi, E., Chernikova, T. N., Timmis, K. N., and Golyshin, P. N. (2002). Microbial community of a saline mud volcano at San Biagio-Belpasso, Mt. Etna (Italy). Environ. Microbiol. 4, 249–256. doi: 10.1046/j.1462-2920.2002.00293.x
Yvon-Lewis, S. A., Hu, L., and Kessler, J. (2011). Methane flux to the atmosphere from the Deepwater Horizon oil disaster. Geophys. Res. Lett. 38:L01602. doi: 10.1029/2010GL045928
Keywords: mud volcano, methanogenesis, methanotrophy, metal reduction, ANME group, Taiwan
Citation: Wang P-L, Chiu Y-P, Cheng T-W, Chang Y-H, Tu W-X and Lin L-H (2014) Spatial variations of community structures and methane cycling across a transect of Lei-Gong-Hou mud volcanoes in eastern Taiwan. Front. Microbiol. 5:121. doi: 10.3389/fmicb.2014.00121
Received: 09 January 2014; Accepted: 10 March 2014;
Published online: 25 March 2014.
Edited by:
Mark Alexander Lever, Aarhus University, DenmarkReviewed by:
William D. Orsi, Woods Hole Oceanographic Institution, USAKevin R. Sowers, University of Maryland, USA
Copyright © 2014 Wang, Chiu, Cheng, Chang, Tu and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li-Hung Lin, Department of Geosciences, National Taiwan University, No. 1, Sec. 4, Roosevelt Road, Taipei 10617, Taiwan e-mail:bGhsaW5AbnR1LmVkdS50dw==