Abstract
Like other HIV-1 auxiliary proteins, Vpr is conserved within all the human (HIV-1, HIV-2) and simian (SIV) immunodeficiency viruses. However, Vpr and homologous HIV-2, and SIV Vpx are the only viral auxiliary proteins specifically incorporated into virus particles through direct interaction with the Gag precursor, indicating that this presence in the core of the mature virions is mainly required for optimal establishment of the early steps of the virus life cycle in the newly infected cell. In spite of its small size, a plethora of effects and functions have been attributed to Vpr, including induction of cell cycle arrest and apoptosis, modulation of the fidelity of reverse transcription, nuclear import of viral DNA in macrophages and other non-dividing cells, and transcriptional modulation of viral and host cell genes. Even if some more recent studies identified a few cellular targets that HIV-1 Vpr may utilize in order to perform its different tasks, the real role and functions of Vpr during the course of natural infection are still enigmatic. In this review, we will summarize the main reported functions of HIV-1 Vpr and their significance in the context of the viral life cycle.
Introduction
The vpr gene is conserved among human (HIV-1 and HIV-2) and simian immunodeficiency viruses (SIV) and encodes the regulatory viral protein R (Vpr), a small basic protein (14 kDa) of 96 amino acids (Ogawa et al., 1989; Hattori et al., 1990; Steffy and Wong-Staal, 1991; Tristem et al., 1992). The importance of Vpr has been initially shown in macaque rhesus monkeys that were experimentally infected with a vpr-mutated SIVmac, and exhibited a decrease in virus replication and a delay in disease development progression (Lang et al., 1993; Hoch et al., 1995). In vitro, in the absence of Vpr, HIV-1 replicates less efficiently in macrophages, a cell type that represents an important viral reservoir by harboring the virus over long periods of time (Connor et al., 1995). Despite its small size, HIV-1 Vpr has been shown to have several roles during the viral life cycle. Due to its specific incorporation into the viral particle by interaction with the Pr55Gag-derived p6 protein, Vpr is readily present upon entry of the virus into the cell, which speaks in favor for enrollment during early steps of viral replication (see Figure 1). In this regard, Vpr has been shown to influence the reverse transcription of HIV-1 via the interaction and recruitment of the human uracil DNA glycosylase 2, an enzyme of the DNA repair machinery (Guenzel et al., 2012). A relationship that is not without controversy since different research reports argue whether UNG2 might rather have a negative impact or even no impact on HIV-1 replication (Schrofelbauer et al., 2005; Kaiser and Emerman, 2006; Yang et al., 2007). Furthermore, Vpr also affects the nuclear import of the viral DNA within the pre-integration complex (PIC), the cell cycle progression, the regulation of apoptosis and the transactivation of the HIV-1 LTR as well as host cell genes.
Figure 1
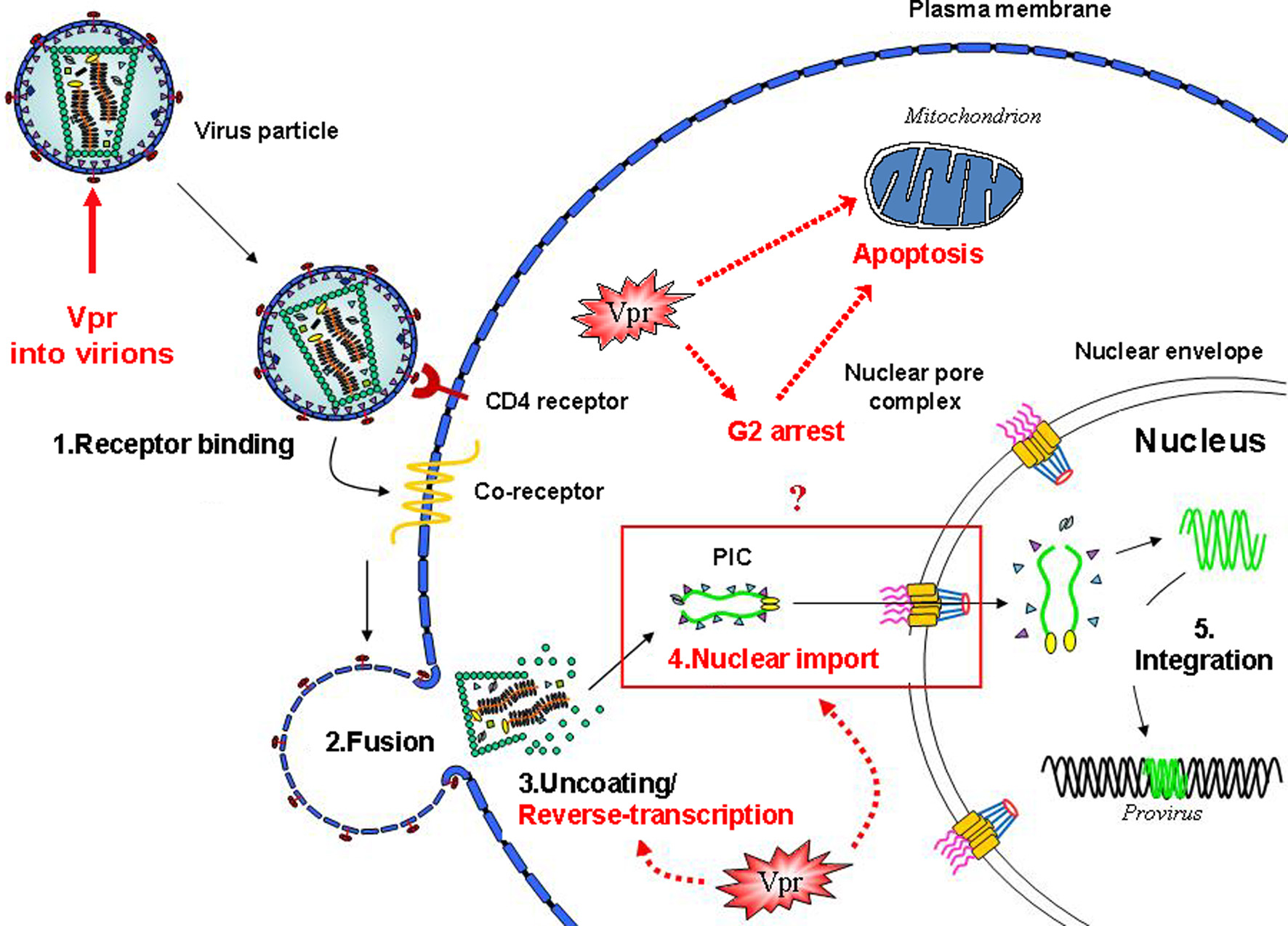
Vpr functions and early steps of the HIV-1 life cycle. Schematic view of the early steps of the HIV-1 infection of a target cell. The functional events in which the Vpr protein is involved are highlighted. Vpr has been shown to play multiple functions during the virus life cycle, including an effect on the accuracy of the reverse-transcription process, the nuclear import of the viral DNA as a component of the pre-integration complex, cell cycle progression, regulation of apoptosis, and the transactivation of the HIV-LTR as well as host cell genes.
This review will be focused on the Vpr protein of HIV-1 and will give a summary of the multifunctional nature of Vpr during the viral life cycle in order to integrate previous and more recent studies.
The structure of Vpr
HIV-1 Vpr is a relatively small protein composed of 96 amino acid residues (Figure 2A) (Checroune et al., 1995; Ramboarina et al., 2004; Kamiyama et al., 2013). The secondary and higher-order structures of Vpr have been investigated by nuclear magnetic resonance (NMR), circular dichroism (CD), and fluorescence spectroscopy (Zhao et al., 1994; Wang et al., 1996; Mahalingam et al., 1997; Kichler et al., 2000; Bruns et al., 2003; Morellet et al., 2003; Kamiyama et al., 2013). According to NMR studies on the full-length Vpr protein dissolved in acidic aqueous-organic solvents (Figure 2B) (Morellet et al., 2003), the central region of the Vpr polypeptide chain folds into three amphiphilic helices (Sherman et al., 2002; Bruns et al., 2003; Kamiyama et al., 2013). These bundled α-helices span residues 17–33, 38–50, and 55–77 and are flanked by unstructured flexible N- and C-terminal domains that are negatively or positively charged, respectively (Morellet et al., 2003). Four conserved proline residues at position 4, 10, 14, and 35 which are subjected to cis/trans isomerization are found in the N-terminal domain (reviewed in Bruns et al., 2003; Le Rouzic and Benichou, 2005). It was indeed reported that the cellular peptidyl-propyl isomerase cyclophilin A was able to interact with Vpr via prolines (position 14 and 35) for correct folding of the viral protein (Zander et al., 2003). The carboxy-terminal domain of Vpr contains six arginine residues between positions 73 and 96 (see Figure 2A), and this domain shows similarity to those of arginine-rich protein transduction domains. This might potentially explain the transducing properties of Vpr and its ability to cross the cell membrane lipid bilayer (Kichler et al., 2000; Sherman et al., 2002; Coeytaux et al., 2003). Additionally, the third helix of Vpr is rich in leucine residues (Schüler et al., 1999), where one side of the helix presents a stretch of hydrophobic side chains that can form a leucine-zipper like motif (Figure 2). This region was proposed to account for the formation of Vpr oligomers (Wang et al., 1996; Mahalingam et al., 1997; Schüler et al., 1999; Fritz et al., 2008) and for interaction with certain cellular partners (reviewed in Planelles and Benichou, 2009).
Figure 2
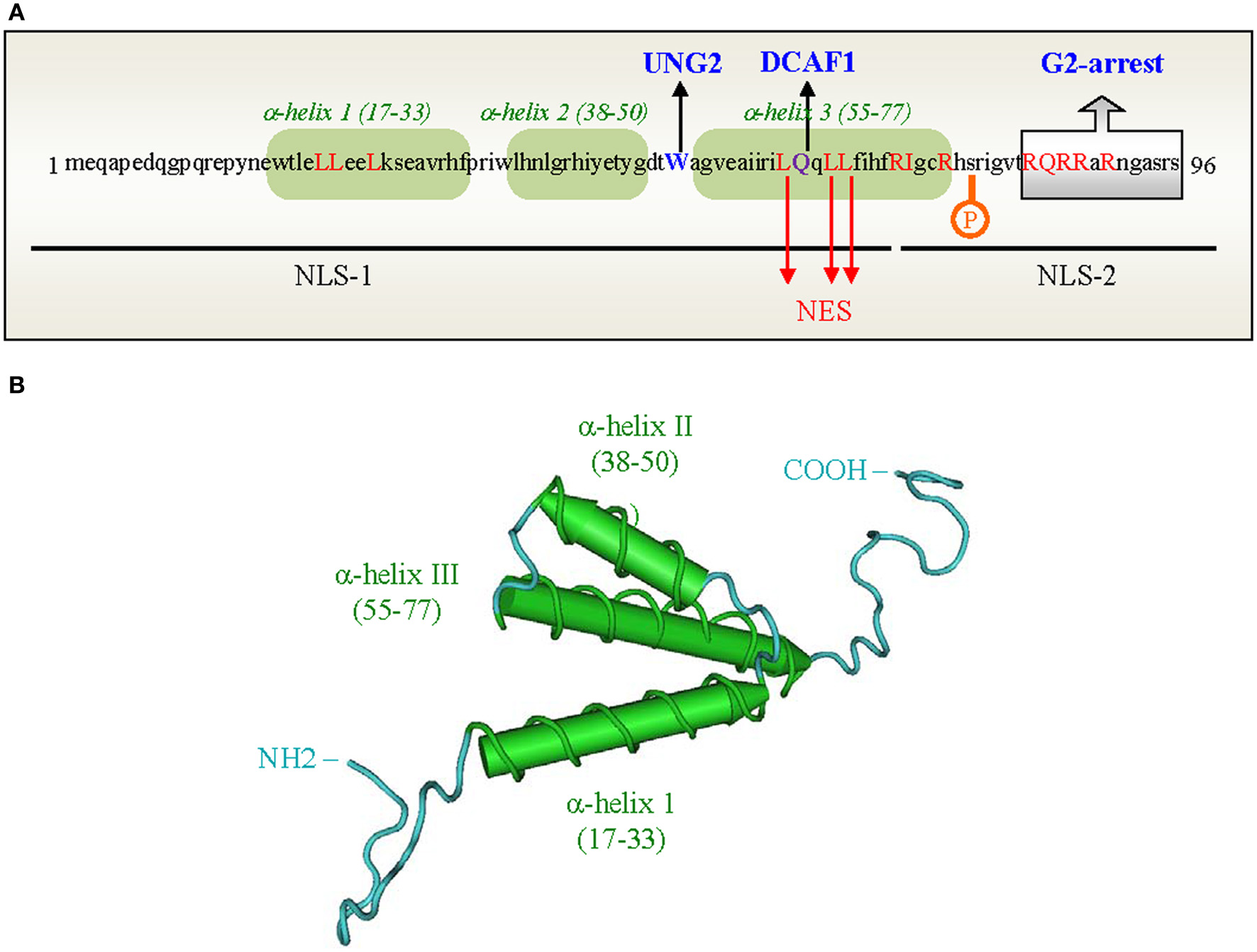
Primary sequence and three-dimensional structure of the HIV-1 Vpr protein. (A) Primary sequence of the Vpr protein from the HIV-1Lai strain. The 3 α-helices are boxed in green. Domains and Leu residues of Vpr involved in the nuclear import (black lines) and nuclear export (Leu in red) of proteins are indicated. The Trp residue in position 54 as well as the Gln residue critical for Vpr binding to UNG2 and DCAF1 are highlighted in blue and purple, respectively. (B) Three-dimensional structure of the HIV-1 Vpr protein (adapted from Morellet et al., 2003). The three α-helices (17–33, 38–50, 55–77) are colored in green, respectively; the loops and flexible domains are in blue.
Vpr has been shown to exist as dimers, trimers, tetramers and higher order multimers (Zhao et al., 1994), however it is still not completely elucidated how the dimeric or multimeric states of the protein affect the different functions of Vpr. A real-time study using a flow cytometry fluorescence resonance energy transfer has confirmed that Vpr self-associates within live cells (Bolton and Lenardo, 2007). Self-association was dependent on the hydrophobic patch that is located on the third α-helix and mutations in this region did not impair the ability of Vpr to induce G2 arrest, suggesting that oligomerization of Vpr is not absolutely required for the functions of the protein. In addition, mutations in the arginine-rich domain, such as R80A and R87/88A, did not impair self-association but were unable to induce G2 arrest (Bolton and Lenardo, 2007). Therefore, it appears that Vpr does not require oligomerization toward induction of the cell cycle blockage but the exposed hydrophobic amino acids in the amino-terminal helix-1 are important for the cell cycle arrest and cytopathogenic functions of Vpr (Barnitz et al., 2011). A more recent study reports that oligomerization of Vpr is essential for incorporation into virus particles (Venkatachari et al., 2010). Moreover, it has been recently proposed that Vpr may assume an antiparallel helical dimer with the third α-helices of the two subunits facing each other, and the His71 and Trp54 play a crucial role in this dimer formation (Kamiyama et al., 2013).
Vpr is incorporated into virus particles
Vpr is expressed at a late stage of the virus life cycle, but it is present during the early steps of infection in target cells since it is packaged into virions that were released from the producing cells. The incorporation of Vpr occurs through a direct interaction with the carboxy-terminal p6 region of the gag-encoded Pr55Gag precursor (Bachand et al., 1999; Paillart and Göttlinger, 1999; Selig et al., 1999; Jenkins et al., 2001a,b). The integrity of the α-helices of Vpr is required for efficient packaging into virions (Singh et al., 2000), and a leucine-rich (LR) motif found in the p6 region of the Pr55Gag precursor is directly involved in the interaction with Vpr (Kondo and Göttlinger, 1996; Selig et al., 1999; Jenkins et al., 2001a,b; Fritz et al., 2010). The Pr55Gag p6 region has also been found to be phosphorylated during HIV-1 infection by atypical protein kinase C (Hemonnot et al., 2004) regulating the incorporation of Vpr into HIV-1 virions and thereby supporting virus infectivity (Kudoh et al., 2014). After assembly and proteolytic cleavage of Pr55Gag in matrix, capsid, nucleocapsid (NCp7), and p6 mature proteins, Vpr is recruited into the conical core of the virus particle (Accola et al., 2000; Welker et al., 2000) where it is tightly associated with the viral RNA (Zhang et al., 1998; De Rocquigny et al., 2000). Interestingly, Vpr displays a higher avidity for NCp7 than for the mature p6 protein (Dong et al., 1997; Selig et al., 1999; Jenkins et al., 2001a,b). Since p6 is excluded from the virion core (Accola et al., 2000; Welker et al., 2000), Vpr could switch from the p6Gag region of the precursor to the mature NCp7 protein in order to gain access to the core of the infectious virus particle budding at the cell surface. In any case, p6 has been reported to show a high affinity for membrane bilayers which substantially increases the interaction between p6 and Vpr (Salgado et al., 2009). It was estimated that Vpr is efficiently incorporated in HIV-1 virions with a Vpr/Pr55Gag ratio of ~1:7 (Knight et al., 1987; Cohen et al., 1990; Welker et al., 2000), which may represent 275 molecules of Vpr per virion. More recently it has been shown that the HIV-1 Pr55Gag precursor induces the recruitment of Vpr oligomers to the plasma membrane (Fritz et al., 2010). Vpr oligomerization has been found to be essential for binding of Vpr to Pr55Gag and for its accumulation at the plasma membrane early during Pr55Gag assembly, but the exact role of these oligomers is not certain yet (Fritz et al., 2010).
The incorporation of Vpr has also been used as a unique tool to target cargoes such as cellular and viral proteins or drugs into viral particles (Wu et al., 1995; Yao et al., 1999; Fritz et al., 2010). This property found extensive use in studies that evaluated the respective functions of integrase (IN) and reverse transcriptase (RT) during virus replication by expressing Vpr-IN and Vpr-RT fusions in trans in virus-producing cells (Wu et al., 1997, 1999; Liu et al., 1999). Furthermore, Vpr fused to the green fluorescence protein (GFP) has been used to tag HIV particles in order to follow intracellular virus behavior during the early intracellular steps of infection in target cells (Loeb et al., 2002; Steffens and Hope, 2003; Fritz et al., 2010).
Vpr and the cell cycle
Among the range of functions of the Vpr protein, the Vpr-dependent G2 arrest activity was extensively explored since it was described for the first time in 1995 (He et al., 1995; Jowett et al., 1995; Re et al., 1995; Rogel et al., 1995). The Vpr protein encapsided into HIV-1 virions is able to block proliferation of newly infected T lymphocytes. Following infection, these cells accumulate at the G2-M phase and show a 4N DNA content. The first studies proposed that the presence of Vpr leads to the accumulation of the hyperphosphorylated form of the cyclin-dependent kinase CDC2 (the complex p34 cdc2/cyclin B). This inactive form of the complex would be able to block the cell cycle before the mitosis.
This cytostatic function of Vpr is well conserved among primate lentiviruses (Planelles et al., 1996; Stivahtis et al., 1997), and could be a strategy used by HIV and SIV to improve viral replication and protein expression, and even to reactivate the virus through an epigenetic control of the LTR promoter (Yao et al., 1998; Thierry et al., 2004). The biological significance of this cell cycle arrest during the natural infection is not well understood, but the HIV-1 LTR seems to be more active in the G2 phase, implying that the G2 arrest may confer a favorable cellular environment for efficient transcription of HIV-1 (Goh et al., 1998). In agreement, the Vpr-induced G2 arrest correlates with high level of viral replication in primary human T cells. Overexpression of dominant negative mutant of the p34 cdc2 kinase shows that Vpr-induced G2 arrest correlates with HIV-1 activation (Goh et al., 1998). Vpr might also be involved in virus activation through other interactions such as the formation of a complex with p53 and the transcription factors Sp1 (Wang et al., 1995; Sawaya et al., 1998). This complex could lead to the activation of the p21/WAF promoter resulting in the transactivation of the viral LTR (Cui et al., 2006). Using a human hematopoietic stem cell-transplanted humanized mouse model, it was recently shown that Vpr causes G2 cell cycle arrest and apoptosis predominantly in proliferating CCR5+ CD4+ T cells, which mainly consist of regulatory CD4+ T cells (Tregs), resulting in Treg depletion and enhanced virus production during acute infection in vivo (Sato et al., 2013). In addition, recent results just published by Laguette et al. (2014) show that the interaction of Vpr with the structure-specific endonuclease (SSE) regulator SLX4 complex (SLX4com) is crucial for the G2-arrest activity but also for escape of HIV-1 from innate immune sensing in infected cells.
Some studies try to correlate the Vpr structure with cell cycle regulation. Historically, this function of Vpr was associated with the helix-3 and the flexible C-terminal part of the protein (Marzio et al., 1995; Mahalingam et al., 1997; Chen et al., 1999). Some key phosphorylations of the C-terminus part have also been associated with the G2 arrest, such as phosphorylation of the Ser79 residue (see Figure 2A) (Zhou and Ratner, 2000). Vpr is mainly localized in the nucleus and at the nuclear envelope where previous reports indicated it could induce herniations and burstings of the nuclear membrane and even defects in the nuclear lamina (de Noronha et al., 2001; Sörgel et al., 2012). These morphological modifications could impact several nuclear factors and redistribute a large range of proteins from the nucleus to the cytoplasm leading to alteration of the cell cycle. Indeed, the cyclins involved in the cell cycle regulation are closely regulated and their spatio-temporal distribution is primordial for the continuity of the cell cycle. More recently, interactions between Vpr and chromatin have been reported (Belzile et al., 2010; Shimura et al., 2011). Vpr can cause epigenetic disruption of heterochromatin by inducing the displacement of heterochromatin protein 1-α (HP1-α) through acetylation of the histone H3 and causes premature chromatids separation and consequently G2 arrest (Shimura et al., 2011). The interaction between Vpr and the chromatin should target and activate the ataxia telangiectasia mutated and Rad3-related kinases ATM/ATR, two of the main sensors of the cell cycle (Koundrioukoff et al., 2004). The link between ATR and the Vpr-dependent G2 arrest was initially reported by Roshal et al. (Roshal et al., 2003) (for review on ATR pathway, see Sørensen and Syljuåsen, 2012). The ATR and ATM proteins control the G2 arrest provoked by DNA damage but it is controversial if Vpr really causes DNA damage or just mimics this damage and activates some sensors involved in this process (Cliby et al., 2002). It was reported that the inhibition of ATR abrogates the Vpr-dependent G2 arrest. Following ATR activation by Vpr, Chk1 is activated through phosphorylation and required for the G2 arrest (Li et al., 2010). Clearly, Vpr acts on the cell cycle by a cascade of reversible phosphorylations. The expression of Vpr correlates with inactivation of the p34/cdc2 CDK1 kinase associated with cyclin B. Cdc2 is normally activated by the cdc5 phosphatase which is inactive in its hypophosphorylated form in Vpr-expressing cells (He et al., 1995; Re et al., 1995), whereas Wee1 inhibits the cdc2 kinases (Sørensen and Syljuåsen, 2012). Vpr seems to be able to directly activate the Wee1 protein by binding to its “N” lobe but this interaction is not sufficient for induction of the G2 arrest (Kamata et al., 2008). However, other key regulators of the cell cycle interacting with Vpr could be members of the 14-3-3 protein family (Kino et al., 2005) which bind phosphorylated serine/threonine proteins such as the cell cycle regulators Wee1, Cdc25, and Chk1. Consequently, 14-3-3 could regulate activities and distribution of these proteins (Lopez-Girona et al., 1999; Hermeking and Benzinger, 2006). These authors revealed that overexpression of 14-3-3 leads to an increase of the cell cycle arrest in the presence of Vpr while the absence of this scaffolding protein reduces the Vpr-induced activity. Another study revealed how Vpr disrupts 14-3-3θ from centrosome and increases its association with the importin β, Cyclin B1, and Cdk1 (Bolton et al., 2008).
Today, almost all the new studies about the Vpr-induced G2 arrest try to identify the potential target of Vpr degraded by the proteasome machinery. Indeed, several groups clearly showed that Vpr connects the DCAF1 adaptor of the Cul4A ubiquitin ligase to a so far unidentified host target protein linked to the G2 arrest (Belzile et al., 2007; DeHart et al., 2007; Le Rouzic et al., 2007; Schrofelbauer et al., 2007). First, the interactions between Vpr and cullins 1 and 4 (Cul1, Cul4), belonging to the ubiquitin ligase complex, were reported (Schrofelbauer et al., 2007). Then, the Vpr-binding protein (VprBP) was described as a substrate specificity module in Cul4 and DDB1 (damaged-DNA specific binding protein 1)-based ubiquitine ligase E3 complexes (Angers et al., 2006; He et al., 2006; Higa et al., 2006a; Jin et al., 2006). Furthermore, other teams described a larger complex where Vpr was associated with Cul4A, DDB1, Rbx2/Roc1 and an ubiquitin-conjugating enzyme or E2. At the same time VprBP was renamed DDB1-and Cul4-associated factor (DCAF)-1 (Belzile et al., 2007; DeHart et al., 2007; Hrecka et al., 2007; Le Rouzic et al., 2007; Schrofelbauer et al., 2007; Tan et al., 2007; Wen et al., 2007). The Cul4-DDB1-E3 ligase complex can bind several DCAFs and seems involved in the maintenance and control of the genome stability, DNA replication and cell cycle check point control (Sugasawa et al., 2005; Higa et al., 2006b; Wang et al., 2006). From these studies, a model where Vpr binds the Cul4-DDB1-DCAF1 E3 ligase to trigger the degradation of a putative protein responsible for the G2 arrest has emerged (Dehart and Planelles, 2008). In this model, Vpr uses two distinct interfaces for binding, one for the attachment to VprBP/DCAF1 and the other for the putative substrate protein. Vpr binds DCAF1 through the LR motif found between amino acids 60 and 68 while the C-terminal basic flexible region binds to the substrate to be ubiquitinylated and degraded and responsible for G2 arrest (Zhao et al., 1994; DeHart et al., 2007; Le Rouzic et al., 2007). Recently, Belzile et al. (2010) proposed that Vpr is present in the nucleus and more specifically inside nuclear foci where it is associated with VprBP and the DDB1-CUL4A-E3 ubiquitine ligase. These foci colocalize with DNA repair foci containing proteins such as γH2AX and RPA2. This association may lead to the recruitment and the degradation of a chromatin-bound substrate via a K48-linked polyubiquitinylation (Belzile et al., 2010) which activates the key protein ATR and the G2 arrest. Finally, a new essential actor of the Vpr-dependent G2 arrest, the SSE regulator SLX4com has been recently identified by proteomic analysis (Laguette et al., 2014). Vpr activates SLX4com through direct interaction with SLX4 leading to the recruitment of VprBP and the kinase-active PLK1. This association would lead to the cleavage of DNA by SLX4-associated MUS81-EME1 endonucleases. Vpr activation of premature MUS81-EME1 induces accumulation of FANCD2 foci and consequently DNA intermediates cleavage and replication stress.
Vpr and apoptosis
Acute phase of AIDS is characterized by a net decrease of CD4+ T cells, and the hallmark of the chronic phase is a gradual decrease of the peripheral CD4+ T cells. While the virus mainly targets lymphocytes and macrophages, no depletion of macrophages has been reported and these terminally-differentiated cells may rather serve as virus reservoirs. The reason why infected macrophages were not susceptible to apoptosis has been recently explored. Using macrophage-like cells derived from differentiated THP1 CD4+ myeloid cells, a recent report showed that Vpr is not able to downregulate the anti-apoptotic protein cIAP1/2 (Busca et al., 2012). However, Mishra et al. (2007) previously revealed the possibility that the C-terminal part of Vpr could induce apoptosis in monocytes via a JNK-dependant pathway.
Although different HIV-1-induced pathways for apoptosis induction have been described, Vpr appears as one of the main actors of the cell death observed during HIV-1 infection. However, it is still controversial how Vpr induces apoptosis and/or necrosis. Moreover, uninfected bystander T cells can be also targeted by Vpr, since Vpr can get access to the extracellular compartment like a soluble protein (Reiss et al., 1990; Cummins and Badley, 2010; Abbas, 2013). A previous model for Vpr-induced apoptosis proposed that Vpr would be able to bind the WxxF motif of the transmembrane adenine nucleotide transporter (ANT) protein exposed in the inner membrane of mitochondria. Jacotot et al. (2000, 2001) were the first to detect this interaction and found that Vpr could also bind to another member of the permeability transition pore complex (PTPC), the voltage-dependent anion channel (VDAC). This team showed the capacity of a synthetic Vpr polypeptide to trigger permeabilization of the mitochondrial membrane resulting in the collapse of the mitochondrial transmembrane potential. Following permeabilization of both inner and outer mitochondrial membranes (Ghiotto et al., 2010), the release of pro-apoptotic proteins like the cytochrome c forms the apoptosome with the caspase 9 and Apaf-1 and allows recruitment of caspase 3. Bax, another pore forming complex protein should also be involved in the Vpr-induced cell death since a conformational change and activation of Bax was detected in apoptotic cells expressing Vpr (Andersen et al., 2006). In this study, the authors characterized cell death in mice, and described that ANT may promote a necrotic cell death rather than apoptosis.
It was indeed discussed whether the Vpr-induced G2 arrest was linked to the observed apoptosis in Vpr expressing cells. Earlier, some studies concluded that Vpr-induced apoptosis was independent of the G2 arrest activity (Nishizawa et al., 2000a,b) showing that a C-terminal truncated form of Vpr still induced apoptosis but did not induce G2 arrest. However, others and more recent studies found a correlation between both Vpr activities and suggested that apoptosis was a consequence of the prolonged G2 arrest (Andersen et al., 2006). According to Stewart and colleagues, apoptosis would happen in cells after the G2 arrest as a consequence of the blockage, and this was observed in human fibroblasts, T cell lines, as well as primary peripheral blood lymphocytes (Stewart et al., 1997, 1999). Accordingly, Zhu et al. (2001) showed that treating cells with caffeine, an inhibitor of both ATM and ATR, which are key proteins involved in cell cycle control, abrogated both G2 arrest and apoptosis. Trying to understand this ATR-dependent mechanism, subsequent studies from the same team described an activation of the DNA repair enzyme BRCA1 leading to the regulation of the growth arrest and DNA damage protein 45α (GADD45α) involved in the cell death process (Zimmerman et al., 2004; Andersen et al., 2005). Moreover, some cell cycle regulators such as Wee1 and Chk1 could also be involved in the Vpr-dependent apoptosis pathway. The Vpr-dependent phosphorylation of Chk1, an event that begins during S phase of the cell cycle, could also trigger apoptosis (Li et al., 2010).
Furthermore, it was reported that Vpr may impact the immune system homeostasis by stimulating the secretion of TNF-α by dendritic cells, resulting in apoptosis of CD8+ T cells (Majumder et al., 2007). Vpr could also increase expression of the NKG2D stress ligand in CD4+ T cells promoting their destruction by the Natural Killer (NK) cells (Ward et al., 2009; Richard et al., 2010). According to Ward et al. (2009), this mechanism causes a link between the G2 arrest and the apoptosis since they showed NKG2D expression is dependent of ATR activation by Vpr.
Finally, some studies revealed that 40–50% of HIV-1 seropositive patients have neurocognitive disorders (Ances and Ellis, 2007; Jones et al., 2007; Harezlak et al., 2011), and different theories have been proposed to explain these neurological disorders. Among the Vpr effects, some hypothesized that extracellular Vpr might be able to enter into neurons (Rom et al., 2009) where it can cause apoptosis. Jones et al. (2007) tested the effect of soluble Vpr in neurons and detected apoptosis involving cytochrome c release, p53 induction, and activation of caspase-9. This study also suggested that Vpr triggers the release of the inflammatory IL-6 cytokine by astrocytes which could affect neuron survival. More recently, it was shown that Vpr could also act on the glycolytic pathway of astrocytes leading to secretion of stress molecules (Ferrucci et al., 2013).
Vpr and the reverse transcription
After virus entry, the viral core is released into the cytoplasm of the target cell where the reverse transcription of the viral RNA takes place within a large nucleoprotein complex (Farnet and Haseltine, 1991; Bukrinsky et al., 1993; Miller et al., 1997; Fassati and Goff, 2001; Nermut and Fassati, 2003; Lyonnais et al., 2013). This reverse transcription complex (RTC) contains the two copies of viral RNA and the viral RT, IN, NCp7, Vpr and a few molecules of the matrix protein. It is generally believed that the reverse transcription process is initiated in virus particles and then completed in the cytoplasm after the virus has entered into the target cell (Figure 1). The reverse transcription process is likely to take place in parallel during both virus uncoating and trafficking through the cytoplasm (for reviews, see Goff, 2001; and Pomerantz, 2000). Several studies confirmed that Vpr co-localizes with the viral nucleic acids and IN within purified HIV-1 RTCs (Fassati et al., 2003; Nermut and Fassati, 2003; Steffens and Hope, 2003), and remains associated with the viral DNA within 4–16 h after infection (Fassati and Goff, 2001). Interestingly, Vpr has recently been reported to be essential for unintegrated HIV-1 gene expression and de-novo virus production in a virus replication pathway utilizing RT DNA products that failed to integrate (Poon and Chen, 2003; Trinité et al., 2013).
In addition to a potential role in the initiation step of the reverse transcription process (Stark and Hay, 1998), it has been shown that Vpr modulated the in vivo mutation rate of HIV-1 by influencing the accuracy of the reverse transcription. The HIV-1 RT is an error-prone RNA-dependent DNA polymerase, and quantification of the in vivo rate of forward virus mutations per replication cycle revealed that the mutation rate was 4-fold higher in the absence of Vpr expression when measured in dividing cells using a genetically engineered system (Mansky and Yemin, 1995; Mansky, 1996). Furthermore, analysis in non-dividing cells showed that this phenotype is even more pronounced in primary monocyte-derived macrophages (MDMs) leading to a 16-fold increase of the HIV-1 mutation frequency (Chen et al., 2004). Strikingly, this activity correlates with the interaction of Vpr with the nuclear form of uracil DNA glycosylase (UNG2) (Mansky et al., 2000), an enzyme of the base excision repair system that specifically removes the RNA base uracil from DNA. The inclusion of uracil in DNA can occur either by misincorporation of dUTP or by cytosine deamination. While the Trp residue in position 54 located in the exposed loop connecting the second and the third α-helix of HIV-1 Vpr has been shown to be critical to maintain the interaction with UNG2, the Vpr-binding site was mapped within the C-terminal part of UNG2, and occurs through a WxxF motif. So far, three distinct cellular partners of Vpr were known to contain a WxxF motif: the TFIIB transcription factor, the adenosine-nucleotide translocator (ANT) and UNG2 (as reviewed in Planelles and Benichou, 2009). However, the WxxF motif is not sufficient for Vpr binding, since other cellular Vpr-interacting proteins, such as DCAF1 or DICER for example, still bind to Vpr independently of the presence of a WxxF motif within their primary sequence (Belzile et al., 2007; DeHart et al., 2007; Le Rouzic et al., 2007; Schrofelbauer et al., 2007; Casey Klockow et al., 2013).
Some authors suggested that the association of Vpr with UNG2 in virus-producing cells allows the incorporation of a catalytically active enzyme into HIV-1 particles where UNG2 may directly influence the reverse transcription accuracy (Mansky et al., 2000; Chen et al., 2004), and this plays a specific role in the modulation of the virus mutation rate. The model supporting the direct contribution of incorporated UNG2 in the reverse transcription process was demonstrated by using an experimental system in which UNG2 was recruited into virions independently of Vpr. UNG2 was expressed as a chimeric protein fused to the C-terminal extremity of the VprW54R mutant, a Vpr variant that fails to recruit UNG2 into virions and to influence the virus mutation rate, even though it is incorporated as efficiently as the wild-type Vpr protein. The VprW54R-UNG2 fusion is also efficiently packaged into HIV-1 virions and can restore a mutation rate equivalent to that observed with wild-type Vpr, both in actively dividing cells and in MDMs. In agreement with this phenotype on the virus mutation frequency, it was finally documented that the Vpr-mediated incorporation of UNG2 into virus particles contributed to the ability of HIV-1 to replicate in primary macrophages. When the VprW54R variant was introduced into an infectious HIV-1 molecular clone, virus replication in macrophages was both reduced and delayed. Although it was proposed that the viral integrase was also able to mediate interaction with UNG2 (Priet et al., 2003), Vpr seems to be the main viral determinant that allows for the incorporation of UNG2 into virus particles. However, further analyses are required to document the nature of interactions between UNG2, Vpr, IN as well as RT both in virus-producing cells and in target cells.
Other studies also confirmed that UNG2 was efficiently recruited into virus particles (Priet et al., 2005; Kaiser and Emerman, 2006; Yang et al., 2007; Jones et al., 2010), indicating that this recruitment might influence the accuracy of the reverse transcription process and has a positive influence on viral replication (Chen et al., 2004; Priet et al., 2005; Jones et al., 2010). Interestingly, it has been recently reported that HIV-1 DNA generated in infected macrophages and CD4-positive T cells is heavily uracilated (Yan et al., 2011). However, the specific role of UNG2 incorporation into virions was challenged by other studies (Schrofelbauer et al., 2005; Kaiser and Emerman, 2006; Yang et al., 2007). While the specificity of the interaction between Vpr and UNG2 was not questioned, these studies suggested a detrimental (Schrofelbauer et al., 2005; Yang et al., 2007; Eldin et al., 2013) or dispensable (Kaiser and Emerman, 2006) effect of UNG2 on virus replication. In the model suggesting a detrimental effect on UNG2 on virus replication, Vpr was shown to induce the proteasomal degradation of UNG2 in virus-producing cells in order to prevent its recruitment into virus particles (Schrofelbauer et al., 2005, 2007; Eldin et al., 2013). It has also been reported that the Vpr-UNG2 interaction temporarily impairs the uracil excision activity of UNG2 in infected cells (Eldin et al., 2013). However, other data have indicated that the Vpr-induced reduction of endogenous UNG2 observed in HIV-1 infected cells was not solely related to proteasomal degradation (Langevin et al., 2009; Nekorchuk et al., 2013) and that UNG2 might not be responsible for the degradation of HIV-1 DNA containing misincorporated dUTP which prevents viral integration (Weil et al., 2013). More recently, it has been argued that incorporation of UNG2 into HIV-1 particles may not be detrimental for virus infection in target cells but rather has a positive impact on virus replication and virus infectivity achieved through a non-enzymatic mechanism mapping within a 60-amino-acid long domain located in the N-terminal region of UNG2 (Guenzel et al., 2012). Interestingly, this domain is also required for interaction of UNG2 with the p32 subunit (RPA2) of the replication protein A complex (Nagelhus et al., 1997; Otterlei et al., 1999; Mer et al., 2000; De Silva and Moss, 2008). It was observed that enforced virion recruitment of UNG2, through UNG2 overexpression in virus producing cells, similarly influenced infectivity of X4 and R5 HIV-1 strains in transformed cell lines and MDMs, respectively (Guenzel et al., 2012), which stands in contrast to another report suggesting that UNG2 was exclusively required for efficient infection of primary cells by R5-tropic viruses (Jones et al., 2010). Strikingly, viruses produced from cells depleted of endogenous UNG2 and RPA2 resulted in significantly reduced infectivity and replication, the latter evidenced by a reduced amount of viral transcripts measured during the reverse transcription process (Guenzel et al., 2012). These new intriguing findings are not yet completely understood and further investigations are needed to clarify the mechanism.
HIV-1 and other lentiviruses are unusual among retroviruses in their ability to infect resting or terminally differentiated cells. While Vpr has been shown to facilitate the nuclear import of viral DNA in non-dividing cells (see below), the virion incorporation of UNG2 via Vpr may also contribute to the ability of HIV-1 to replicate in primary macrophages. This implies that UNG2 is a cellular factor that plays an important role in the early steps of the HIV-1 replication cycle (i.e., viral DNA synthesis). This observation is in agreement with a report showing that the misincorporation of uracil into minus strand viral DNA affects the initiation of the plus strand DNA synthesis in vitro (Klarmann et al., 2003). This observation suggests that UNG2 is likely to be recruited into HIV-1 particles to subsequently minimize the detrimental accumulation of uracil into the newly synthesized proviral DNA. While further works are needed to explain the precise mechanism for how the UNG2 catalytic activity may specifically influence HIV-1 replication in macrophages, it is worth noting that non-dividing cells express low levels of UNG and contain relatively high levels of dUTP (Chen et al., 2002). Similarly, most non-primate lentiviruses, such as feline immunodeficiency virus (FIV), caprine-arthritis-encephalitis virus (CAEV) and equine infectious anemia virus (EIAV), have also developed an efficient strategy to reduce accumulation of uracil into viral DNA. These lentiviruses encode and package a viral-encoded dUTP pyropshophatase (dUTPase) into virus particles, an enzyme that hydrolyzed dUTP to dUMP, and thus maintains a low level of dUTP. Interestingly, replication of FIV, CAEV, or EIAV that lack functional dUTPase activity is severely affected in non-dividing host cells (e.g., primary macrophages). Taken together, these results indicate that uracil misincorporation in viral DNA strands during reverse transcription is deleterious for the ongoing steps of the virus life cycle. The presence of a viral dUTPase or a cellular UNG will prevent these detrimental effects for replication of non-primate and primate lentiviruses in macrophages, respectively.
Finally, it is intriguing to note that two viral auxiliary proteins from HIV-1, Vpr and Vif, can both influence the fidelity of viral DNA synthesis. The Vif protein forms a complex with the cellular deaminase APOBEC3G thereby preventing its encapsidation into virions (Sheehy et al., 2002; Lecossier et al., 2003; Mangeat et al., 2003; Mariani et al., 2003; Stopak et al., 2003), while Vpr binds the DNA repair enzyme UNG2. In this context it was suggested that incorporation of UNG2 into viral particles would have a detrimental effect on reverse transcription by introducing a basic sites into viral DNA in regards to uracil residues resulting from cytosine deamination by the cytidine deaminase APOBEC3G (Schrofelbauer et al., 2005; Yang et al., 2007). While the specific role of UNG2 in the antiviral activity of APOBEC3G was not directly questioned (Schrofelbauer et al., 2005), others reported data indicating that the antiviral activity of overexpressed APOBEC3G was partially affected when viruses were produced in UNG2-depleted 293T cells (Yang et al., 2007). However, these data are in apparent contradiction with results from other reports in which viruses were produced in UNG2-depleted cells which expressed or did not express APOBEC3G (Priet et al., 2005; Jones et al., 2010; Guenzel et al., 2012), but also from reports showing that APOBEC3G-mediated restriction of HIV-1 was independent of UNG2 (Kaiser and Emerman, 2006; Langlois and Neuberger, 2008). More recently, and in favor for a correlative positive impact of UNG2, it has been shown that the detrimental hypermutation of Hepatitis B virus DNA induced by either APOBEC3G or interferon treatment was enhanced in a human hepatocyte cell lines when UNG2 activity was inhibited (Kitamura et al., 2013; Liang et al., 2013). Additional investigations are thus required to further understand this apparent contradiction regarding the role of UNG2 for the antiviral activity of APOBEC restriction factors. However, it is tempting to speculate that the action of both viral proteins may influence the mutation rate during the course of HIV-1 infection, and their balance may play a key role during disease progression and antiretroviral treatment susceptibility in infected individuals (Fourati et al., 2010, 2012).
Vpr and the viral DNA nuclear import
Like other retroviruses, HIV-1 has the capacity to infect and integrate its genomic DNA into dividing cells like T lymphocytes, but lentiviruses are also remarkable by their capacity to infect non-dividing cells, in contrast to onco-retroviruses which need the disintegration of the nuclear envelope to allow access of their genome for integration in the host genome (Greber and Fassati, 2003). Indeed, HIV-1 can infect terminally-differentiated macrophages and produces new virions after integration of its DNA into the cell genome. The Vpr protein has been described as a potential enhancer of HIV-1 replication especially in macrophages whereas it does not impact on virus replication in proliferating T cells (Balliet et al., 1994; Connor et al., 1995; Eckstein et al., 2001). In macrophages, the viral DNA needs to be transported into the interphasic nucleus by an active mechanism (Vodicka et al., 1998). After virus entry into the cell, the viral genome is reverse-transcribed in full length viral double-strand DNA which is associated with viral and host cell proteins into the so-called PIC. Among the protein components of the PIC, four viral proteins have been detected (e.g., the reverse-transcriptase and integrase enzymes, the matrix protein and Vpr) (Heinzinger et al., 1994; Jenkins et al., 1998; Eckstein et al., 2001; Le Rouzic et al., 2002; Schang, 2003; Suzuki et al., 2009).
Despite the absence of a basic canonical or a M9-dependant nuclear localization signal (NLS) in the protein sequence, Vpr shows evident karyophilic properties (Gallay et al., 1996; Jenkins et al., 1998; Depienne et al., 2000). Finally, Vpr seems to use a non-classical pathway to be transferred in the nucleus through direct binding to importin-α (Gallay et al., 1996; Nitahara-Kasahara et al., 2007). However, it was largely shown that Vpr is able to shuttle between the cytoplasm and the nuclear compartments and could play a potential role in transport of the viral DNA (Jenkins et al., 2001a,b; Sherman et al., 2001, 2003; Le Rouzic et al., 2002). By using a photobleaching strategy on living cells, Le Rouzic and colleagues revealed that Vpr-GFP has shuttling properties (Le Rouzic et al., 2002). This activity was linked to the distal LR region, a classical nuclear export signal (NES) recognized by the CRM1-dependent pathway (Sherman et al., 2001, 2003). This NES could be involved in the release of Vpr back into the cytoplasm, making it available for virion packaging through interaction with the Pr55Gag precursor (Jenkins et al., 2001a,b).
The role of Vpr within the PIC has been studied in living cells through tracking of a GFP-tagged form of Vpr (McDonald et al., 2002). These authors evidenced a tight association between the PIC and the cytoplasmic microtubules, targeting the viral DNA toward the nucleus. The PIC moves along the cytoskeletal microtubule filaments using the dynein/dynactin complex as a motor, leading to its accumulation in the perinuclear region close to the centrosome. So far, it is not known if Vpr plays an active role in the intracytoplasmic transport of the PIC; it may be only associated with the complex and play a role later for nuclear membrane anchoring and translocation of the viral DNA into the nucleus (for review, Le Rouzic and Benichou, 2005).
The nuclear envelope contains two concentric membranes with nuclear pore complexes (NPC) consisting of aqueous channels which allow for selective transport between the cytoplasmic and nuclear compartments. The NPC corresponds to a 125 MDa structure consisting of 30 distinct nuclear pore proteins, named nucleoporins (Nups) (Cronshaw et al., 2002). Most of these Nups have filamentous structures containing FG or FxFG motif repeats emanating from both sides of the NPC and able to dock transport factors (Rout and Aitchison, 2001). It was initially reported that Vpr was able to recognize these FG motifs in Nups such as p54 and p58 leading to the docking of Vpr to the nuclear membrane (Fouchier et al., 1998; Popov et al., 1998). Another interaction with the human CG1 (hCG1) nucleoporin has been described by Le Rouzic et al. (2002). However, Vpr associated with the N-terminal region of hCG1 while the FG repeats of this Nup were located in the C-terminal part of the protein. This interaction results in Vpr accumulation at the nuclear envelope, which is believed to be involved in active nuclear import of the PIC in non-dividing cells, such as macrophages (Jacquot et al., 2007). Through these interactions with components of NPC, Vpr may be responsible for the first step of viral DNA import by targeting the PIC to the nuclear pore complex while other components of the PIC could trigger the next step of the nuclear translocation. As mentioned above, it was also reported that Vpr can induce herniations and the dissociation of lamina and nuclear envelope which provoke a blend of nuclear and cytoplasmic proteins (de Noronha et al., 2001). The exact mechanism inducing these membrane perturbations is not understood, but some authors hypothesize that the interaction of Vpr with the NPC proteins could impact nuclear membrane stability. Consequently it may also facilitate the entry of the PIC through a non-conventional pathway (Segura-Totten and Wilson, 2001).
Conclusions and future directions
Like other HIV-1 auxiliary proteins, Vpr is a small but multifunctional protein which is potentially able to interact with plenty of cellular partners. During the last two decades, several groups looked for such partners but the importance of such interactions often needs to be better documented to support their real impact on HIV-1 propagation, immune and antiretroviral treatment evasion and disease progression. While major efforts have been made during the last years to better define the molecular mechanisms and cellular targets of Vpr, additional works are needed for the complete understanding of its wide range of activities in key processes during the early steps of the viral life cycle (i.e., reverse transcription, intra-cytoplasmic routing and nuclear import of the viral DNA). However, precise characterization of Vpr interactions leading to the proteasomal degradation of some host cell factors is certainly the main challenge for a better understanding of the Vpr contribution to the overall pathogenesis of HIV-1 infection.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Statements
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AbbasW. (2013). T-Cell Signaling in HIV-1 Infection. Open Virol. J. 7, 57–71. 10.2174/1874357920130621001
2
AccolaM. A.OhagenA.GöttlingerH. G. (2000). Isolation of human immunodeficiency virus type 1 cores: retention of vpr in the absence of P6(gag). J. Virol. 74, 6198–6202. 10.1128/JVI.74.13.6198-6202.2000
3
AncesB. M.EllisR. J. (2007). Dementia and neurocognitive disorders due to HIV-1 infection. Semin. Neurol. 27, 86–92. 10.1055/s-2006-956759
4
AndersenJ. L.DeHartJ. L.ZimmermanE. S.ArdonO.KimB.JacquotG.et al. (2006). HIV-1 Vpr-induced apoptosis is cell cycle dependent and requires Bax but not ANT. PLoS Pathog. 2:e127. 10.1371/journal.ppat.0020127
5
AndersenJ. L.ZimmermanE. S.DeHartJ. L.MuralaS.ArdonO.BlackettJ.et al. (2005). ATR and GADD45alpha mediate HIV-1 Vpr-induced apoptosis. Cell Death Differ. 12, 326–334. 10.1038/sj.cdd.4401565
6
AngersS.LiT.YiX.MacCossM. J.MoonR. T.ZhengN. (2006). Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature443, 590–593. 10.1038/nature05175
7
BachandF.YaoX. J.HrimechM.RougeauN.CohenE. A. (1999). Incorporation of Vpr into human immunodeficiency virus type 1 requires a direct interaction with the P6 domain of the P55 gag precursor. J. Biol. Chem. 274, 9083–9091. 10.1074/jbc.274.13.9083
8
BallietJ. W.KolsonD. L.EigerG.KimF. M.McGannK. A.SrinivasanA.et al. (1994) Distinct effects in primary macrophages and lymphocytes of the human immunodeficiency virus type 1 accessory genes vpr, vpu, and nef: mutational analysis of a primary HIV-1 isolate. Virology200, 623–631. 10.1006/viro.1994.1225
9
BarnitzR. A.Chaigne-DelalandeB.BoltonD. L.LenardoM. J. (2011). Exposed Hydrophobic residues in human immunodeficiency virus type 1 Vpr Helix-1 are important for cell cycle arrest and cell death. PLoS ONE6:e24924. 10.1371/journal.pone.0024924
10
BelzileJ.-P.DuisitG.RougeauN.MercierJ.FinziA.CohenE. A. (2007). HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase. PLoS Pathog. 3:e85. 10.1371/journal.ppat.0030085
11
BelzileJ.-P.RichardJ.RougeauN.XiaoY.CohenE. A. (2010). HIV-1 Vpr induces the K48-linked polyubiquitination and proteasomal degradation of target cellular proteins to activate ATR and promote G2 arrest. J. Virol. 84, 3320–3330. 10.1128/JVI.02590-09
12
BoltonD. L.BarnitzR. A.SakaiK.LenardoM. J. (2008). 14-3-3 theta binding to cell cycle regulatory factors is enhanced by HIV-1 Vpr. Biol. Direct3, 17. 10.1186/1745-6150-3-17
13
BoltonD. L.LenardoM. J. (2007). Vpr cytopathicity independent of G2/M cell cycle arrest in human immunodeficiency virus type 1-infected CD4+ T Cells. J. Virol. 81, 8878–8890. 10.1128/JVI.00122-07
14
BrunsK.FossenT.WrayV.HenkleinP.TessmerU.SchubertU. (2003). Structural characterization of the HIV-1 Vpr N terminus: evidence of Cis/trans-proline Isomerism. J. Biol. Chem. 278, 43188–43201. 10.1074/jbc.M305413200
15
BukrinskyM. I.SharovaN.McDonaldT. L.PushkarskayaT.TarpleyW. G.StevensonM. (1993). Association of integrase, matrix, and reverse transcriptase antigens of human immunodeficiency virus type 1 with viral nucleic acids following acute infection. Proc. Natl. Acad. Sci. U.S.A. 90, 6125–6129. 10.1073/pnas.90.13.6125
16
BuscaA.SaxenaM.KumarA. (2012). Critical role for antiapoptotic Bcl-xL and Mcl-1 in human macrophage survival and cellular IAP1/2 (cIAP1/2) in resistance to HIV-Vpr-induced apoptosis. J. Biol. Chem. 287, 15118–15133. 10.1074/jbc.M111.312660
17
Casey KlockowL.SharifiH. J.WenX.FlaggM.FuruyaA. K.NekorchukM.et al. (2013) The HIV-1 protein Vpr targets the endoribonuclease Dicer for proteasomal degradation to boost macrophage infection. Virology444, 191–202. 10.1016/j.virol.2013.06.010
18
ChecrouneF.YaoX. J.GöttlingerH. G.BergeronD.CohenE. A. (1995) Incorporation of Vpr into human immunodeficiency virus type 1: role of conserved regions within the P6 domain of Pr55gag. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 10, 1–7. 10.1097/00042560-199509000-00001
19
ChenM.ElderR. T.YuM.O'GormanM. G.SeligL.BenarousR.et al. (1999). Mutational analysis of Vpr-induced G2Arrest, nuclear localization, and cell death in fission yeast. J. Virol. 73, 3236–3245.
20
ChenR.Le RouzicE.KearneyJ. A.ManskyL. M.BenichouS. (2004). Vpr-mediated incorporation of UNG2 into HIV-1 particles is required to modulate the virus mutation rate and for replication in macrophages. J. Biol. Chem. 279, 28419–28425. 10.1074/jbc.M403875200
21
ChenR.WangH.ManskyL. M. (2002). Roles of uracil-DNA glycosylase and dUTPase in virus replication. J. Gen. Virol. 83, 2339–2345.
22
ClibyW. A.LewisK. A.LillyK. K.KaufmannS. H. (2002). S Phase and G2 Arrests induced by topoisomerase i poisons are dependent on ATR kinase function. J. Biol. Chem. 277, 1599–1606. 10.1074/jbc.M106287200
23
CoeytauxE.CoulaudD.Le CamE.DanosO.KichlerA. (2003). The cationic amphipathic alpha-helix of HIV-1 viral protein R (Vpr) binds to nucleic acids, permeabilizes membranes, and efficiently transfects cells. J. Biol. Chem. 278, 18110–18116. 10.1074/jbc.M300248200
24
CohenE. A.DehniG.SodroskiJ. G.HaseltineW. A. (1990). Human immunodeficiency virus Vpr product is a virion-associated regulatory protein. J. Virol. 64, 3097–3099.
25
ConnorR. I.ChenB. K.ChoeS.LandauN. R. (1995). Vpr is required for efficient replication of human immunodeficiency virus Type-1 in mononuclear phagocytes. Virology206, 935–944. 10.1006/viro.1995.1016
26
CronshawJ. M.KrutchinskyA. N.ZhangW.ChaitB. T.MatunisM. J. (2002). Proteomic analysis of the mammalian nuclear pore complex. J. Cell Biol. 158, 915–927. 10.1083/jcb.200206106
27
CuiJ.TungaturthiP. K.AyyavooV.GhafouriM.ArigaH.KhaliliK.et al. (2006). The role of Vpr in the regulation of HIV-1 gene expression. Cell Cycle5, 2626–2638. 10.4161/cc.5.22.3442
28
CumminsN. W.BadleyA. D. (2010). Mechanisms of HIV-associated lymphocyte apoptosis: 2010. Cell Death Dis. 1, e99. 10.1038/cddis.2010.77
29
DehartJ. L.PlanellesV. (2008). Human immunodeficiency virus type 1 Vpr links proteasomal degradation and checkpoint activation. J. Virol. 82, 1066–1072. 10.1128/JVI.01628-07
30
DeHartJ. L.ZimmermanE. S.ArdonO.Monteiro-FilhoC. M.ArgañarazE. R.PlanellesV. (2007). HIV-1 Vpr activates the G2 checkpoint through manipulation of the ubiquitin proteasome system. Virol. J. 4, 57. 10.1186/1743-422X-4-57
31
de NoronhaC. M. C.ShermanM. P.LinH. W.CavroisM. V.MoirR. D.GoldmanR. D.GreeneW. C. (2001). Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr. Science294, 1105–1108. 10.1126/science.1063957
32
DepienneC.RoquesP.CréminonC.FritschL.CasseronR.DormontD.et al. (2000). Cellular distribution and karyophilic properties of matrix, integrase, and Vpr proteins from the human and simian immunodeficiency viruses. Exp. Cell Res. 260, 387–395. 10.1006/excr.2000.5016
33
De RocquignyH.CaneparoA.DelaunayT.BischerourJ.MouscadetJ. F.RoquesB. P. (2000). Interactions of the C-terminus of viral protein R with nucleic acids are modulated by its N-terminus. Eur. J. Biochem. 267, 3654–3660. 10.1046/j.1432-1327.2000.01397.x
34
De SilvaF. S.MossB. (2008). Effects of vaccinia virus uracil DNA glycosylase catalytic site and deoxyuridine triphosphatase deletion mutations individually and together on replication in active and quiescent cells and pathogenesis in mice. Virol. J. 5, 145. 10.1186/1743-422X-5-145
35
DongC. Z.De RocquignyH.RémyE.MellacS.Fournié-ZaluskiM. C.RoquesB. P. (1997). Synthesis and biological activities of fluorescent acridine-containing HIV-1 nucleocapsid proteins for investigation of nucleic acid-NCp7 interactions. J. Pept. Res. 50, 269–278. 10.1111/j.1399-3011.1997.tb01468.x
36
EcksteinD. A.ShermanM. P.PennM. L.ChinP. S.De NoronhaC. M.GreeneW. C.et al. (2001). HIV-1 Vpr enhances viral burden by facilitating infection of tissue macrophages but not nondividing CD4+ T cells. J. Exp. Med. 194, 1407–1419. 10.1084/jem.194.10.1407
37
EldinP.ChazalN.FenardD.BernardE.GuichouJ.-F.BriantL. (2013). Vpr expression abolishes the capacity of HIV-1 infected cells to repair uracilated DNA. Nucleic Acids Res. 42, 1698–1710. 10.1093/nar/gkt974
38
FarnetC. M.HaseltineW. A. (1991). Determination of viral proteins present in the human immunodeficiency virus type 1 preintegration complex. J. Virol. 65, 1910–1915.
39
FassatiA.GoffS. P. (2001). Characterization of intracellular reverse transcription complexes of human immunodeficiency virus type 1. J. Virol. 75, 3626–3635. 10.1128/JVI.75.8.3626-3635.2001
40
FassatiA.GörlichD.HarrisonI.ZaytsevaL.MingotJ.-M. (2003). Nuclear import of HIV-1 intracellular reverse transcription complexes is mediated by importin 7. EMBO J. 22, 3675–3685. 10.1093/emboj/cdg357
41
FerrucciA.NonnemacherM. R.WigdahlB. (2013). Extracellular HIV-1 viral protein R affects astrocytic glyceraldehyde 3-phosphate dehydrogenase activity and neuronal survival. J. Neurovirol. 19, 239–253. 10.1007/s13365-013-0170-1
42
FouchierR. A.MeyerB. E.SimonJ. H.FischerU.AlbrightA. V.González-ScaranoF.et al. (1998). Interaction of the human immunodeficiency virus type 1 Vpr protein with the nuclear pore complex. J. Virol. 72, 6004–6013.
43
FouratiS.MaletI.BinkaM.BoukobzaS.WirdenM.SayonS.et al. (2010). Partially active HIV-1 Vif alleles facilitate viral escape from specific antiretrovirals. AIDS24, 2313–2321. 10.1097/QAD.0b013e32833e515a
44
FouratiS.MaletI.GuenzelC. A.SoulieC.Maidou-PeindaraP.Morand-JoubertL.et al. (2012). E17A mutation in HIV-1 Vpr confers resistance to didanosine in association with thymidine analog mutations. Antiviral Res. 93, 167–174. 10.1016/j.antiviral.2011.11.008
45
FritzJ. V.DidierP.ClammeJ.-P.SchaubE.MuriauxD.CabanneC.et al. (2008). Direct Vpr-Vpr interaction in cells monitored by two photon fluorescence correlation spectroscopy and fluorescence lifetime imaging. Retrovirology5, 87. 10.1186/1742-4690-5-87
46
FritzJ. V.DujardinD.GodetJ.DidierP.De MeyJ.DarlixJ.-L.et al. (2010). HIV-1 Vpr oligomerization but not that of gag directs the interaction between Vpr and Gag. J. Virol. 84, 1585–1596. 10.1128/JVI.01691-09
47
GallayP.StittV.MundyC.OettingerM.TronoD. (1996). Role of the karyopherin pathway in human immunodeficiency virus type 1 nuclear import. J. Virol. 70, 1027–1032.
48
GhiottoF.FaisF.BrunoS. (2010). BH3-only proteins: the death-puppeteer's wires. Cytometry A77, 11–21. 10.1002/cyto.a.20819
49
GoffS. P. (2001). Intracellular trafficking of retroviral genomes during the early phase of infection: viral exploitation of cellular pathways. J. Gene. Med. 3, 517–528. 10.1002/1521-2254(200111)3:6<517::AID-JGM234>3.0.CO;2-E
50
GohW. C.RogelM. E.KinseyC. M.MichaelS. F.FultzP. N.NowakM. A.et al. (1998). HIV-1 Vpr increases viral expression by manipulation of the cell cycle: a mechanism for selection of Vpr in vivo. Nat. Med. 4, 65–71. 10.1038/nm0198-065
51
GreberU. F.FassatiA. (2003). Nuclear import of viral DNA genomes. Traffic4, 136–143. 10.1034/j.1600-0854.2003.00114.x
52
GuenzelC. A.HérateC.Le RouzicE.Maidou-PeindaraP.SadlerH. A.RouyezM.-C.et al. (2012). Recruitment of the nuclear form of uracil DNA glycosylase into virus particles participates in the full infectivity of HIV-1. J. Virol. 86, 2533–2544. 10.1128/JVI.05163-11
53
HarezlakJ.BuchthalS.TaylorM.SchifittoG.ZhongJ.DaarE.et al. (2011). Persistence of HIV-associated cognitive impairment, inflammation, and neuronal injury in era of highly active antiretroviral treatment. AIDS25, 625–633. 10.1097/QAD.0b013e3283427da7
54
HattoriN.MichaelsF.FargnoliK.MarconL.GalloR. C.FranchiniG. (1990). The human immunodeficiency virus type 2 Vpr gene is essential for productive infection of human macrophages. Proc. Natl. Acad. Sci. U.S.A. 87, 8080–8084. 10.1073/pnas.87.20.8080
55
HeJ.ChoeS.WalkerR.Di MarzioP.MorganD. O.LandauN. R. (1995). Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 69, 6705–6711.
56
HeY. J.McCallC. M.HuJ.ZengY.XiongY. (2006). DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev. 20, 2949–2954. 10.1101/gad.1483206
57
HeinzingerN. K.BukinskyM. I.HaggertyS. A.RaglandA. M.KewalramaniV.LeeM. A.et al. (1994). The Vpr protein of human immunodeficiency virus type 1 influences nuclear localization of viral nucleic acids in nondividing host cells. Proc. Natl. Acad. Sci. U.S.A. 91, 7311–7315. 10.1073/pnas.91.15.7311
58
HemonnotB.CartierC.GayB.RebuffatS.BardyM.DevauxC.et al. (2004). The host cell MAP kinase ERK-2 regulates viral assembly and release by phosphorylating the P6gag protein of HIV-1. J. Biol. Chem279, 32426–32434. 10.1074/jbc.M313137200
59
HermekingH.BenzingerA. (2006). 14-3-3 proteins in cell cycle regulation. Semin. Cancer Biol. 16, 183–192. 10.1016/j.semcancer.2006.03.002
60
HigaL. A.WuM.YeT.KobayashiR.SunH.ZhangH. (2006a). CUL4–DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat. Cell Biol. 8, 1277–1283. 10.1038/ncb1490
61
HigaL. A.YangX.ZhengJ.BanksD.WuM.GhoshP.et al. (2006b). Involvement of CUL4 ubiquitin E3 ligases in regulating CDK inhibitors Dacapo/p27Kip1 and cyclin E degradation. Cell Cycle5, 71–77. 10.4161/cc.5.1.2266
62
HochJ.LangS. M.WeegerM.Stahl-HennigC.CoulibalyC.DittmerU.et al. (1995). Vpr deletion mutant of simian immunodeficiency virus induces AIDS in rhesus monkeys. J. Virol. 69, 4807–4813.
63
HreckaK.GierszewskaM.SrivastavaS.KozaczkiewiczL.SwansonS. K.FlorensL.et al. (2007). Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc. Natl. Acad. Sci. U.S.A. 104, 11778–11783. 10.1073/pnas.0702102104
64
JacototE.FerriK. F.El HamelC.BrennerC.DruillennecS.HoebekeJ.et al. (2001). Control of mitochondrial membrane permeabilization by adenine nucleotide translocator interacting with HIV-1 viral protein rR and Bcl-2. J. Exp. Med. 193, 509–519. 10.1084/jem.193.4.509
65
JacototE.RavagnanL.LoefflerM.FerriK. F.VieiraH. L. A.ZamzamiN.et al. (2000). The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J. Exp. Med. 191, 33. 10.1084/jem.191.1.33
66
JacquotG.Le RouzicE.DavidA.MazzoliniJ.BouchetJ.BouazizS.et al. (2007). Localization of HIV-1 Vpr to the nuclear envelope: impact on Vpr functions and virus replication in macrophages. Retrovirology4, 84. 10.1186/1742-4690-4-84
67
JenkinsY.McEnteeM.WeisK.GreeneW. C. (1998). Characterization of HIV-1 vpr nuclear import: analysis of signals and pathways. J. Cell Biol. 143, 875–885. 10.1083/jcb.143.4.875
68
JenkinsY.PornillosO.RichR. L.MyszkaD. G.SundquistW. I.MalimM. H. (2001a). Biochemical analyses of the interactions between human immunodeficiency virus type 1 Vpr and p6(Gag). J. Virol. 75, 10537–10542. 10.1128/JVI.75.21.10537-10542.2001
69
JenkinsY.SanchezP. V.MeyerB. E.MalimM. H. (2001b). Nuclear export of human immunodeficiency virus type 1 Vpr is not required for virion packaging. J. Virol. 75, 8348–8352. 10.1128/JVI.75.17.8348-8352.2001
70
JinJ.AriasE. E.ChenJ.HarperJ. W.WalterJ. C. (2006). A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell23, 709–721. 10.1016/j.molcel.2006.08.010
71
JonesG. J.BarsbyN. L.CohenE. A.HoldenJ.HarrisK.DickieP.et al. (2007). HIV-1 Vpr causes neuronal apoptosis and in vivo neurodegeneration. J. Neurosci. 27, 3703–3711. 10.1523/JNEUROSCI.5522-06.2007
72
JonesK. L.RocheM.GantierM. P.BegumN. A.HonjoT.CaradonnaS.et al. (2010). X4 and R5 HIV-1 have distinct post-entry requirements for uracil DNA glycosylase during infection of primary cells. J. Biol. Chem. 285, 18603–18614. 10.1074/jbc.M109.090126
73
JowettJ. B.PlanellesV.PoonB.ShahN. P.ChenM. L.ChenI. S. (1995). The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2 + M phase of the cell cycle. J. Virol. 69, 6304–6313.
74
KaiserS. M.EmermanM. (2006). Uracil DNA glycosylase is dispensable for human immunodeficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidine deaminase apobec3G. J. Virol. 80, 875–882. 10.1128/JVI.80.2.875-882.2006
75
KamataM.WatanabeN.NagaokaY.ChenI. S. Y. (2008). Human immunodeficiency virus type 1 Vpr binds to the N lobe of the Wee1 kinase domain and enhances kinase activity for CDC2. J. Virol. 82, 5672–5682. 10.1128/JVI.01330-07
76
KamiyamaT.MiuraT.TakeuchiH. (2013). His-Trp cation-π interaction and its structural role in an A-helical dimer of HIV-1 Vpr protein. Biophys. Chem. 173–174, 8–14. 10.1016/j.bpc.2013.01.004
77
KichlerA.PagesJ. C.LeborgneC.DruillennecS.LenoirC.CoulaudD.et al. (2000). Efficient DNA transfection mediated by the C-terminal domain of human immunodeficiency virus type 1 viral protein R. J. Virol. 74, 5424–5431. 10.1128/JVI.74.12.5424-5431.2000
78
KinoT.GragerovA.ValentinA.TsopanomihalouM.Ilyina-GragerovaG.Erwin-CohenR.et al. (2005). Vpr protein of human immunodeficiency virus type 1 binds to 14-3-3 proteins and facilitates complex formation with Cdc25C: implications for cell cycle arrest. J. Virol. 79, 2780–2787. 10.1128/JVI.79.5.2780-2787.2005
79
KitamuraK.WangZ.ChowdhuryS.SimaduM.KouraM.MuramatsuM. (2013) Uracil DNA glycosylase counteracts APOBEC3G-induced hypermutation of hepatitis B viral genomes: excision repair of covalently closed circular DNA. PLoS Pathog. 9:e1003361. 10.1371/journal.ppat.1003361
80
KlarmannG. J.ChenX.NorthT. W.PrestonB. D. (2003). Incorporation of uracil into minus strand DNA affects the specificity of plus strand synthesis initiation during lentiviral reverse transcription. J. Biol. Chem. 278, 7902–7909. 10.1074/jbc.M207223200
81
KnightD. M.FlomerfeltF. A.GhrayebJ. (1987). Expression of the art/trs protein of HIV and study of its role in viral envelope synthesis. Science236, 837–840. 10.1126/science.3033827
82
KondoE.GöttlingerH. G. (1996). A conserved LXXLF sequence is the major determinant in P6gag required for the incorporation of human immunodeficiency virus type 1 Vpr. J. Virol. 70, 159–164.
83
KoundrioukoffS.PoloS.AlmouzniG. (2004). Interplay between chromatin and cell cycle checkpoints in the context of ATR/ATM-dependent checkpoints. DNA Repair (Amst.)3, 969–978. 10.1016/j.dnarep.2004.03.010
84
KudohA.TakahamaS.SawasakiT.OdeH.YokoyamaM.OkayamaA.et al. (2014). The phosphorylation of HIV-1 gag by atypical protein kinase C facilitates viral infectivity by promoting Vpr incorporation into virions. Retrovirology11, 9. 10.1186/1742-4690-11-9
85
LaguetteN.BrégnardC.HueP.BasbousJ.YatimA.LarroqueM.et al. (2014). Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell156, 134–145. 10.1016/j.cell.2013.12.011
86
LangS. M.WeegerM.Stahl-HennigC.CoulibalyC.HunsmannG.MüllerJ.et al. (1993). Importance of Vpr for infection of rhesus monkeys with simian immunodeficiency virus. J. Virol. 67, 902–912.
87
LangevinC.Maidou-PeindaraP.AasP. A.JacquotG.OtterleiM.SlupphaugG.et al. (2009). Human immunodeficiency virus type 1 Vpr modulates cellular expression of UNG2 via a negative transcriptional effect. J. Virol. 83, 10256–10263. 10.1128/JVI.02654-08
88
LangloisM.-A.NeubergerM. S. (2008). Human APOBEC3G can restrict retroviral infection in avian cells and acts independently of both UNG and SMUG1. J. Virol. 82, 4660–4664. 10.1128/JVI.02469-07
89
LecossierD.BouchonnetF.ClavelF.HanceA. J. (2003). Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science300, 1112. 10.1126/science.1083338
90
Le RouzicE.BelaïdouniN.EstrabaudE.MorelM.RainJ.-C.TransyC.et al. (2007). HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4-DDB1 ubiquitin ligase. Cell Cycle6, 182–188. 10.4161/cc.6.2.3732
91
Le RouzicE.BenichouS. (2005). The Vpr protein from HIV-1: distinct roles along the viral life cycle. Retrovirology2, 11. 10.1186/1742-4690-2-11
92
Le RouzicE.MousnierA.RustumC.StutzF.HallbergE.DargemontC.et al. (2002). Docking of HIV-1 Vpr to the nuclear envelope is mediated by the interaction with the nucleoporin hCG1. J. Biol. Chem. 277, 45091–45098. 10.1074/jbc.M207439200
93
LiG.ParkH. U.LiangD.ZhaoR. Y. (2010). Cell cycle G2/M arrest through an S phase-dependent mechanism by HIV-1 viral protein R. Retrovirology7, 59. 10.1186/1742-4690-7-59
94
LiangG.KitamuraK.WangZ.LiuG.ChowdhuryS.FuW.et al. (2013). RNA editing of hepatitis B virus transcripts by activation-induced cytidine deaminase. Proc. Natl. Acad. Sci. U.S.A. 110, 2246–2251. 10.1073/pnas.1221921110
95
LiuH.WuX.XiaoH.KappesJ. C. (1999). Targeting human immunodeficiency virus (HIV) type 2 integrase protein into HIV type 1. J. Virol. 73, 8831–8836.
96
LoebJ. E.WeitzmanM. D.HopeT. J. (2002). Enhancement of green fluorescent protein expression in adeno-associated virus with the woodchuck hepatitis virus post-transcriptional regulatory element. Methods Mol. Biol. 183, 331–340. 10.1089/10430349950016942
97
Lopez-GironaA.FurnariB.MondesertO.RussellP. (1999). Nuclear localization of Cdc25 is regulated by DNA damage and a 14-3-3 protein. Nature397, 172–175. 10.1038/16488
98
LyonnaisS.GorelickR. J.Heniche-BoukhalfaF.BouazizS.ParissiV.MouscadetJ.-F.et al. (2013). A protein ballet around the viral genome orchestrated by HIV-1 reverse transcriptase leads to an architectural switch: from nucleocapsid-condensed RNA to Vpr-bridged DNA. Virus Res. 171, 287–303. 10.1016/j.virusres.2012.09.008
99
MahalingamS.AyyavooV.PatelM.Kieber-EmmonsT.WeinerD. B. (1997). Nuclear import, virion incorporation, and cell cycle arrest/differentiation are mediated by distinct functional domains of human immunodeficiency virus type 1 Vpr. J. Virol. 71, 6339–6347.
100
MajumderB.VenkatachariN. J.SchaferE. A.JanketM. L.AyyavooV. (2007). Dendritic cells infected with vpr-positive human immunodeficiency virus type 1 induce CD8+ T-cell apoptosis via upregulation of tumor necrosis factor alpha. J. Virol. 81, 7388–7399. 10.1128/JVI.00893-06
101
MangeatB.TurelliP.CaronG.FriedliM.PerrinL.TronoD. (2003). Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature424, 99–103. 10.1038/nature01709
102
ManskyL. M. (1996). The mutation rate of human immunodeficiency virus type 1 is influenced by the Vpr Gene. Virology222, 391–400. 10.1006/viro.1996.0436
103
ManskyL. M.PreveralS.SeligL.BenarousR.BenichouS. (2000). The interaction of Vpr with uracil DNA glycosylase modulates the human immunodeficiency virus type 1 in vivo mutation rate. J. Virol. 74, 7039–7047. 10.1128/JVI.74.15.7039-7047.2000
104
ManskyL. M.YeminH. M. (1995). Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 69, 5087–5094.
105
MarianiR.ChenD.SchröfelbauerB.NavarroF.KönigR.BollmannB.et al. (2003). Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell114, 21–31. 10.1016/S0092-8674(03)00515-4
106
MarzioP. D.ChoeS.EbrightM.KnoblauchR.LandauN. R. (1995). Mutational analysis of cell cycle arrest, nuclear localization and virion packaging of human immunodeficiency virus type 1 Vpr. J. Virol. 69, 7909–7916.
107
McDonaldD.VodickaM. A.LuceroG.SvitkinaT. M.BorisyG. G.EmermanM.et al. (2002). Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 159, 441–452. 10.1083/jcb.200203150
108
MerG.BochkarevA.ChazinW. J.EdwardsA. M. (2000). Three-dimensional structure and function of replication protein A. Cold Spring Harb. Symp. Quant. Biol. 65, 193–200. 10.1101/sqb.2000.65.193
109
MillerM. D.FarnetC. M.BushmanF. D. (1997). Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. J. Virol. 71, 5382–5390.
110
MishraS.MishraJ. P.KumarA. (2007). Activation of JNK-dependent pathway is required for HIV viral protein R-induced apoptosis in human monocytic cells: involvement of antiapoptotic BCL2 and c-IAP1 genes. J. Biol. Chem. 282, 4288–4300. 10.1074/jbc.M608307200
111
MorelletN.BouazizS.PetitjeanP.RoquesB. P. (2003). NMR structure of the HIV-1 regulatory protein VPR. J. Mol. Biol. 327, 215–227. 10.1016/S0022-2836(03)00060-3
112
NagelhusT. A.HaugT.SinghK. K.KeshavK. F.SkorpenF.OtterleiM.et al. (1997). A sequence in the N-terminal region of human uracil-DNA glycosylase with homology to XPA interacts with the C-terminal part of the 34-kDa subunit of replication protein A. J. Biol. Chem. 272, 6561–6566. 10.1074/jbc.272.10.6561
113
NekorchukM. D.SharifiH. J.FuruyaA. K.JellingerR.de NoronhaC. M. (2013) HIV relies on neddylation for ubiquitin ligase-mediated functions. Retrovirology10, 138. 10.1186/1742-4690-10-138
114
NermutM. V.FassatiA. (2003). Structural analyses of purified human immunodeficiency virus type 1 intracellular reverse transcription complexes. J. Virol. 77, 8196–8206. 10.1128/JVI.77.15.8196-8206.2003
115
NishizawaM.KamataM.KatsumataR.AidaY. (2000a). A carboxy-terminally truncated form of the human immunodeficiency virus type 1 Vpr protein induces apoptosis via G(1) cell cycle arrest. J. Virol. 74, 6058–6067. 10.1128/JVI.74.13.6058-6067.2000
116
NishizawaM.KamataM.MojinT.NakaiY.AidaY. (2000b). Induction of apoptosis by the Vpr protein of human immunodeficiency virus type 1 occurs independently of G(2) arrest of the cell cycle. Virology276, 16–26. 10.1006/viro.2000.0534
117
Nitahara-KasaharaY.KamataM.YamamotoT.ZhangX.MiyamotoY.MunetaK.et al. (2007). Novel nuclear import of Vpr promoted by importin alpha is crucial for human immunodeficiency virus type 1 replication in macrophages. J. Virol. 81, 5284–5293. 10.1128/JVI.01928-06
118
OgawaK.ShibataR.KiyomasuT.HiguchiI.KishidaY.IshimotoA.et al. (1989). Mutational analysis of the human immunodeficiency virus Vpr open reading frame. J. Virol. 63, 4110–4114.
119
OtterleiM.WarbrickE.NagelhusT. A.HaugT.SlupphaugG.AkbariM.et al. (1999). Post-replicative base excision repair in replication foci. EMBO J. 18, 3834–3844. 10.1093/emboj/18.13.3834
120
PaillartJ. C.GöttlingerH. G. (1999). Opposing effects of human immunodeficiency virus type 1 matrix mutations support a myristyl switch model of gag membrane targeting. J. Virol. 73, 2604–2612.
121
PlanellesV.BenichouS. (2009). Vpr and its interactions with cellular proteins. Curr. Top. Microbiol. Immunol. 339, 177–200. 10.1007/978-3-642-02175-6_9
122
PlanellesV.JowettJ. B.LiQ. X.XieY.HahnB.ChenI. S. (1996). Vpr-induced cell cycle arrest is conserved among primate lentiviruses. J. Virol. 70, 2516–2524.
123
PomerantzR. J. (2000). Residual HIV-1 replication. Biomed. Pharmacother. 54, 13–15. 10.1016/S0753-3322(00)88635-7
124
PoonB.ChenI. S. (2003) Human immunodeficiency virus type 1 (HIV-1) Vpr enhances expression from unintegrated HIV-1 DNA. J. Virol. 77, 3962–3972. 10.1128/JVI.77.7.3962-3972.2003
125
PopovS.RexachM.RatnerL.BlobelG.BukrinskyM. (1998). Viral protein R regulates docking of the HIV-1 preintegration complex to the nuclear pore complex. J. Biol. Chem. 273, 13347–13352. 10.1074/jbc.273.21.13347
126
PrietS.GrosN.NavarroJ.-M.BorettoJ.CanardB.QuératG.et al. (2005). HIV-1-associated uracil DNA glycosylase activity controls dUTP misincorporation in viral DNA and is essential to the HIV-1 life cycle. Mol. Cell17, 479–490. 10.1016/j.molcel.2005.01.016
127
PrietS.NavarroJ. M.GrosN.QueratG.SireJ. (2003) Functional role of HIV-1 virion-associated uracil DNA glycosylase 2 in the correction of G:U mispairs to G:C pairs. J. Biol. Chem. 278, 4566–4571. 10.1074/jbc.M209311200
128
RamboarinaS.DruillennecS.MorelletN.BouazizS.RoquesB. P. (2004). Target Specificity of Human Immunodeficiency Virus Type 1 NCp7 Requires an Intact Conformation of Its CCHC N-terminal Zinc Finger. J. Virol. 78, 6682–6687. 10.1128/JVI.78.12.6682-6687.2004
129
ReF.BraatenD.FrankeE. K.LubanJ. (1995). Human immunodeficiency virus type 1 Vpr arrests the cell cycle in G2 by inhibiting the activation of p34cdc2-cyclin B. J. Virol. 69, 6859–6864.
130
ReissP.LangeJ. M.de RondeA.de WolfF.DekkerJ.DannerS. A.et al. (1990). Antibody response to viral proteins U (vpu) and R (vpr) in HIV-1-infected individuals. J. Acquir. Immune Defic. Syndr. 3, 115–122.
131
RichardJ.SindhuS.PhamT. N. Q.BelzileJ.-P.CohenE. A. (2010). HIV-1 Vpr up-regulates expression of ligands for the activating NKG2D receptor and promotes NK cell-mediated killing. Blood115, 1354–1363. 10.1182/blood-2009-08-237370
132
RogelM. E.WuL. I.EmermanM. (1995). The human immunodeficiency virus type 1 vpr gene prevents cell proliferation during chronic infection. J. Virol. 69, 882–888.
133
RomI.DeshmaneS. L.MukerjeeR.KhaliliK.AminiS.SawayaB. E. (2009). HIV-1 Vpr deregulates calcium secretion in neural cells. Brain Res. 1275, 81–86. 10.1016/j.brainres.2009.03.024
134
RomaniB.CohenE. A. (2012). Lentivirus Vpr and Vpx accessory proteins usurp the cullin4-DDB1 (DCAF1) E3 ubiquitin ligase. Curr. Opin. Virol. 2, 755–763. 10.1016/j.coviro.2012.09.010
135
RoshalM.KimB.ZhuY.NghiemP.PlanellesV. (2003). Activation of the ATR-mediated DNA damage response by the HIV-1 viral protein R. J. Biol. Chem. 278, 25879–25886. 10.1074/jbc.M303948200
136
RoutM. P.AitchisonJ. D. (2001). The nuclear pore complex as a transport machine. J. Biol. Chem. 276, 16593–16596. 10.1074/jbc.R100015200
137
SalgadoG. F.VogelA.MarquantR.FellerS. E.BouazizS.AlvesI. D. (2009). The Role of membranes in the organization of HIV-1 Gag P6 and Vpr: P6 shows high affinity for membrane bilayers which substantially increases the interaction between P6 and Vpr. J. Med. Chem. 52, 7157–7162. 10.1021/jm901106t
138
SatoK.MisawaN.IwamiS.SatouY.MatsuokaM.IshizakaY.et al. (2013) HIV-1 Vpr accelerates viral replication during acute infection by exploitation of proliferating CD4(+) T Cells in vivo. PLoS Pathog. 9:e1003812. 10.1371/journal.ppat.1003812
139
SawayaB. E.KhaliliK.MercerW. E.DenisovaL.AminiS. (1998). Cooperative actions of HIV-1 Vpr and p53 modulate viral gene transcription. J. Biol. Chem. 273, 20052–20057. 10.1074/jbc.273.32.20052
140
SchangL. M. (2003). The cell cycle, cyclin-dependent kinases, and viral infections: new horizons and unexpected connections. Prog. Cell Cycle Res. 5, 103–124.
141
SchrofelbauerB.HakataY.LandauN. R. (2007). HIV-1 Vpr function is mediated by interaction with the damage-specific DNA-binding protein DDB1. Proc. Natl. Acad. Sci. U.S.A. 104, 4130–4135. 10.1073/pnas.0610167104
142
SchrofelbauerB.YuQ.ZeitlinS. G.LandauN. R. (2005). Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J. Virol. 79, 10978–10987. 10.1128/JVI.79.17.10978-10987.2005
143
SchülerW.WeckerL.De RocquignyH.BaudatY.SireJ.RoquesB. P. (1999). NMR structure of the (52-96) C-terminal domain of the HIV-1 regulatory protein Vpr: molecular insights into its biological functions. J. Mol. Biol. 285, 2105–2117. 10.1006/jmbi.1998.2381
144
Segura-TottenM.WilsonK. L. (2001). Virology. HIV–breaking the rules for nuclear entry. Science294, 1016–1017. 10.1126/science.1066729
145
SeligL.PagesJ. C.TanchouV.PrévéralS.Berlioz-TorrentC.LiuL. X.et al. (1999). Interaction with the P6 domain of the gag precursor mediates incorporation into virions of Vpr and Vpx proteins from primate lentiviruses. J. Virol. 73, 592–600.
146
SheehyA. M.GaddisN. C.ChoiJ. D.MalimM. H. (2002). Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature418, 646–650. 10.1038/nature00939
147
ShermanM. P.de NoronhaC. M. C.EcksteinL. A.HatayeJ.MundtP.WilliamsS. A. F.et al. (2003). Nuclear export of Vpr is required for efficient replication of human immunodeficiency virus type 1 in tissue macrophages. J. Virol. 77, 7582–7589. 10.1128/JVI.77.13.7582-7589.2003
148
ShermanM. P.de NoronhaC. M. C.HeuschM. I.GreeneS.GreeneW. C. (2001). Nucleocytoplasmic shuttling by human immunodeficiency virus type 1 Vpr. J. Virol. 75, 1522–1532. 10.1128/JVI.75.3.1522-1532.2001
149
ShermanM. P.de NoronhaC. M. C.WilliamsS. A.GreeneW. C. (2002). Insights into the biology of HIV-1 viral protein R. DNA Cell Biol. 21, 679–688. 10.1089/104454902760330228
150
ShimuraM.ToyodaY.IijimaK.KinomotoM.TokunagaK.YodaK.et al. (2011). Epigenetic displacement of HP1 from heterochromatin by HIV-1 Vpr causes premature sister chromatid separation. J. Cell Biol. 194, 721–735. 10.1083/jcb.201010118
151
SinghS. P.TomkowiczB.LaiD.CartasM.MahalingamS.KalyanaramanV. S.et al. (2000). Functional role of residues corresponding to helical domain II (amino Acids 35 to 46) of human immunodeficiency virus type 1 Vpr. J. Virol. 74, 10650–10657. 10.1128/JVI.74.22.10650-10657.2000
152
SørensenC. S.SyljuåsenR. G. (2012). Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 40, 477–486. 10.1093/nar/gkr697
153
SörgelS.FraedrichK.VottelerJ.ThomasM.StammingerT.SchubertU. (2012). Perinuclear localization of the HIV-1 regulatory protein Vpr is important for induction of G2-arrest. Virology432, 444–451. 10.1016/j.virol.2012.06.027
154
StarkL. A.HayR. T. (1998). Human immunodeficiency virus type 1 (HIV-1) viral protein R (Vpr) interacts with Lys-tRNA synthetase: implications for priming of HIV-1 reverse transcription. J. Virol. 72, 3037–3044.
155
SteffensC. M.HopeT. J. (2003). Localization of CD4 and CCR5 in living cells. J. Virol. 77, 4985–4991. 10.1128/JVI.77.8.4985-4991.2003
156
SteffyK.Wong-StaalF. (1991). Genetic regulation of human immunodeficiency virus. Microbiol. Rev. 55, 193–205.
157
StewartS. A.PoonB.JowettJ. B.ChenI. S. (1997). Human immunodeficiency virus type 1 Vpr induces apoptosis following cell cycle arrest. J. Virol. 71, 5579–5592.
158
StewartS. A.PoonB.JowettJ. B.XieY.ChenI. S. (1999). Lentiviral delivery of HIV-1 Vpr protein induces apoptosis in transformed cells. Proc. Natl. Acad. Sci. U.S.A. 96, 12039–12043. 10.1073/pnas.96.21.12039
159
StivahtisG. L.SoaresM. A.VodickaM. A.HahnB. H.EmermanM. (1997). Conservation and host specificity of Vpr-mediated cell cycle arrest suggest a fundamental role in primate lentivirus evolution and biology. J. Virol. 71, 4331–4338.
160
StopakK.de NoronhaC.YonemotoW.GreeneW. C. (2003) HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell. 12, 591–601. 10.1016/S1097-2765(03)00353-8
161
SugasawaK.OkudaY.SaijoM.NishiR.MatsudaN.ChuG.et al. (2005). UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell121, 387–400. 10.1016/j.cell.2005.02.035
162
SuzukiT.YamamotoN.NonakaM.HashimotoY.MatsudaG.TakeshimaS.et al. (2009). Inhibition of human immunodeficiency virus type 1 (HIV-1) nuclear import via Vpr-importin alpha interactions as a novel HIV-1 therapy. Biochem. Biophys. Res. Commun. 380, 838–843. 10.1016/j.bbrc.2009.01.180
163
TanL.EhrlichE.YuX.-F. (2007). DDB1 and Cul4A are required for human immunodeficiency virus type 1 Vpr-induced G2 arrest. J. Virol. 81, 10822–10830. 10.1128/JVI.01380-07
164
ThierryS.MarechalV.RosenzwajgM.SabbahM.RedeuilhG.NicolasJ.-C.et al. (2004). Cell cycle arrest in G2 induces human immunodeficiency virus type 1 transcriptional activation through histone acetylation and recruitment of CBP, NF-κ B, and c-Jun to the long terminal repeat promoter. J. Virol. 78, 12198–12206. 10.1128/JVI.78.22.12198-12206.2004
165
TrinitéB.OhlsonE. C.VoznesenskyI.RanaS. P.ChanC. N.MahajanS.et al. (2013). An HIV-1 replication pathway utilizing reverse transcription products that fail to integrate. J. Virol. 87, 12701–12720. 10.1128/JVI.01939-13
166
TristemM.MarshallC.KarpasA.HillF. (1992). Evolution of the primate lentiviruses: evidence from Vpx and Vpr. EMBO J. 11, 3405–3012.
167
VenkatachariN. J.WalkerL. A.TastanO.LeT.DempseyT. M.LiY.et al. (2010). Human immunodeficiency virus type 1 Vpr: oligomerization is an essential feature for its incorporation into virus particles. Virology7, 119. 10.1186/1743-422X-7-119
168
VodickaM. A.KoeppD. M.SilverP. A.EmermanM. (1998). HIV-1 Vpr interacts with the nuclear transport pathway to promote macrophage infection. Genes Dev. 12, 175–185. 10.1101/gad.12.2.175
169
WangH.ZhaiL.XuJ.JooH.-Y.JacksonS.Erdjument-BromageH.et al. (2006). Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol. Cell22, 383–394. 10.1016/j.molcel.2006.03.035
170
WangL.MukherjeeS.JiaF.NarayanO.ZhaoL. J. (1995). Interaction of virion protein Vpr of human immunodeficiency virus type 1 with cellular transcription factor Sp1 and trans-activation of viral long terminal repeat. J. Biol. Chem. 270, 25564–25569. 10.1074/jbc.270.43.25564
171
WangL.MukherjeeS.NarayanO.ZhaoL. J. (1996). Characterization of a Leucine-zipper-like domain in Vpr protein of human immunodeficiency virus type 1. Gene178, 7–13. 10.1016/0378-1119(96)00312-5
172
WardJ.DavisZ.DeHartJ.ZimmermanE.BosqueA.BrunettaE.et al. (2009). HIV-1 Vpr triggers natural killer cell-mediated lysis of infected cells through activation of the ATR-mediated DNA damage response. PLoS Pathog. 5:e1000613. 10.1371/journal.ppat.1000613
173
WeilA. F.GhoshD.ZhouY.SeipleL.McMahonM. A.SpivakA. M.et al. (2013). Uracil DNA glycosylase initiates degradation of HIV-1 cDNA containing misincorporated dUTP and prevents viral integration. Proc. Natl. Acad. Sci. U.S.A. 110, E448–E457. 10.1073/pnas.1219702110
174
WelkerR.HohenbergH.TessmerU.HuckhagelC.KräusslichH. G. (2000). Biochemical and structural analysis of isolated mature cores of human immunodeficiency virus type 1. J. Virol. 74, 1168–1177. 10.1128/JVI.74.3.1168-1177.2000
175
WenX.DuusK. M.FriedrichT. D.de NoronhaC. M. C. (2007). The HIV1 protein Vpr acts to promote G2 cell cycle arrest by engaging a DDB1 and Cullin4A-containing ubiquitin ligase complex using VprBP/DCAF1 as an adaptor. J. Biol. Chem. 282, 27046–27057. 10.1074/jbc.M703955200
176
WuX.LiuH.XiaoH.ConwayJ. A.HehlE.KalpanaG. V.et al. (1999). Human immunodeficiency virus type 1 integrase protein promotes reverse transcription through specific interactions with the nucleoprotein reverse transcription complex. J. Virol. 73, 2126–2135.
177
WuX.LiuH.XiaoH.ConwayJ. A.HunterE.KappesJ. C. (1997). Functional RT and IN incorporated into HIV-1 particles independently of the gag/pol precursor protein. EMBO J. 16, 5113–5122. 10.1093/emboj/16.16.5113
178
WuX.LiuH.XiaoH.KimJ.SeshaiahP.NatsoulisG.et al. (1995). Targeting foreign proteins to human immunodeficiency virus particles via fusion with Vpr and Vpx. J. Virol. 69, 3389–3398
179
YanN.O'DayE.WheelerL. A.EngelmanA.LiebermanJ. (2011). HIV DNA is heavily uracilated, which protects it from autointegration. Proc. Natl. Acad. Sci. U.S.A. 108, 9244–9249. 10.1073/pnas.1102943108
180
YangB.ChenK.ZhangC.HuangS.ZhangH. (2007). Virion-associated uracil DNA glycosylase-2 and apurinic/apyrimidinic endonuclease are involved in the degradation of APOBEC3G-edited nascent HIV-1 DNA. J. Biol. Chem. 282, 11667–11675. 10.1074/jbc.M606864200
181
YaoX. J.KobingerG.DandacheS.RougeauN.CohenE. (1999). HIV-1 Vpr-chloramphenicol acetyltransferase fusion proteins: sequence requirement for virion incorporation and analysis of antiviral effect. Gene Ther. 6, 1590–1599. 10.1038/sj.gt.3300988
182
YaoX.-J.MoulandA. J.SubbramanianR. A.ForgetJ.RougeauN.BergeronD.et al. (1998). Vpr stimulates viral expression and induces cell killing in human immunodeficiency virus type 1-infected dividing jurkat T cells. J. Virol. 72, 4686–4693.
183
ZanderK.ShermanM. P.TessmerU.BrunsK.WrayV.PrechtelA. T.et al. (2003). Cyclophilin a interacts with HIV-1 Vpr and is required for its functional expression. J. Biol. Chem. 278, 43202–43213. 10.1074/jbc.M305414200
184
ZhangS.PointerD.SingerG.FengY.ParkK.ZhaoL. J. (1998). Direct binding to nucleic acids by Vpr of human immunodeficiency virus type 1. Gene212, 157–166. 10.1016/S0378-1119(98)00178-4
185
ZhaoL. J.WangL.MukherjeeS.NarayanO. (1994). Biochemical mechanism of HIV-1 Vpr function. oligomerization mediated by the N-terminal domain. J. Biol. Chem. 269, 32131–32137.
186
ZhouY.RatnerL. (2000). Phosphorylation of human immunodeficiency virus type 1 Vpr regulates cell cycle arrest. J. Virol. 74, 6520–6527. 10.1128/JVI.74.14.6520-6527.2000
187
ZhuY.GelbardH. A.RoshalM.PursellS.JamiesonB. D.PlanellesV. (2001). Comparison of cell cycle arrest, transactivation, and apoptosis induced by the simian immunodeficiency virus SIVagm and human immunodeficiency virus type 1 vpr genes. J. Virol. 75, 3791–3801. 10.1128/JVI.75.8.3791-3801.2001
188
ZimmermanE. S.ChenJ.AndersenJ. L.ArdonO.DehartJ. L.BlackettJ.et al. (2004). Human immunodeficiency virus type 1 Vpr-mediated G2 arrest requires Rad17 and Hus1 and induces nuclear BRCA1 and gamma-H2AX focus formation. Mol. Cell. Biol. 24, 9286–9294. 10.1128/MCB.24.21.9286-9294.2004
Summary
Keywords
HIV-1 Vpr, reverse transcription, cell cycle, apoptosis, nuclear import
Citation
Guenzel CA, Hérate C and Benichou S (2014) HIV-1 Vpr—a still “enigmatic multitasker”. Front. Microbiol. 5:127. doi: 10.3389/fmicb.2014.00127
Received
07 February 2013
Accepted
12 March 2014
Published
31 March 2014
Volume
5 - 2014
Edited by
Nadine Laguette, Centre National de la Recherche Scientifique, France
Reviewed by
Ryuta Sakuma, Tokyo Medical and Dental University, Japan; Carlos M. De Noronha, Albany Medical College, USA
Copyright
© 2014 Guenzel, Hérate and Benichou.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Serge Benichou, Cochin Institute, INSERM U1016, Centre National de la Recherche Scientifique UMR8104, Université Paris-Descartes, 22 rue Méchain, 75014 Paris, France e-mail: serge.benichou@inserm.fr
This article was submitted to Virology, a section of the journal Frontiers in Microbiology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.