 Lara Valentín
Lara Valentín Tiina Rajala
Tiina Rajala Mikko Peltoniemi
Mikko Peltoniemi Jussi Heinonsalo
Jussi Heinonsalo Taina Pennanen1
Taina Pennanen1 Raisa Mäkipää
Raisa Mäkipää- 1Vantaa Research Unit, Finnish Forest Research Institute, Vantaa, Finland
- 2Department of Chemical Engineering, Technical School of Engineering, Universitat Autònoma de Barcelona, Barcelona, Spain
- 3Department of Food and Environmental Sciences, University of Helsinki, Helsinki, Finland
Hundreds of wood-inhabiting fungal species are now threatened, principally due to a lack of dead wood in intensively managed forests, but the consequences of reduced fungal diversity on ecosystem functioning are not known. Several experiments have shown that primary productivity is negatively affected by a loss of species, but the effects of microbial diversity on decomposition are less studied. We studied the relationship between fungal diversity and the in vitro decomposition rate of slightly, moderately and heavily decayed Picea abies wood with indigenous fungal communities that were diluted to examine the influence of diversity. Respiration rate, wood-degrading hydrolytic enzymes and fungal community structure were assessed during a 16-week incubation. The number of observed OTUs in DGGE was used as a measure of fungal diversity. Respiration rate increased between early- and late-decay stages. Reduced fungal diversity was associated with lower respiration rates during intermediate stages of decay, but no effects were detected at later stages. The activity of hydrolytic enzymes varied among decay stages and fungal dilutions. Our results suggest that functioning of highly diverse communities of the late-decay stage were more resistant to the loss of diversity than less diverse communities of early decomposers. This indicates the accumulation of functional redundancy during the succession of the fungal community in decomposing substrates.
Introduction
The loss of species diversity alters ecosystem function, stability and the ability to provide goods and services to society (Loreau et al., 2001; Hooper et al., 2005; Wertz et al., 2006; Cardinale et al., 2012). Several experiments have shown that primary productivity is negatively affected by a loss of plant species (Loreau et al., 2001; Hooper et al., 2012) and ecosystem stability is reduced by decreasing functional diversity (Hooper et al., 2005). Biodiversity affects the rate of key ecosystem processes such as decomposition and nutrient cycling (Loreau et al., 2001; Hättenschwiler et al., 2005; Gessner et al., 2010). In boreal forests, where fungal communities are major decomposers (Rayner and Boddy, 1988; Lindahl and Boberg, 2008; Stenlid et al., 2008), a high diversity of soil fungi can enhance the decomposition rate (Tiunov and Scheu, 2005), especially under environmental fluctuations such as variations in temperature regimes (Toljander et al., 2011). Hundreds of fungal species in Fennoscandian forests are now threatened, principally due to a lack of dead wood in a forest landscape that is intensively managed (Siitonen, 2001). The consequences of reduced fungal diversity on decomposition are unknown. Furthermore, the stability and function of forest ecosystems might be dramatically affected if the fungal capacity to resist habitat perturbation is continuously exceeded, as is the case for many species in managed forests (Stenlid et al., 2008).
Since fungal species produce complementary enzymes, the presence of many species can improve the communities' efficiency to degrade a wide range of litter constituents and thus enhance decomposition rate (Gessner et al., 2010). Experiments with selected fungal species and their combinations revealed a strong positive relationship between fungal diversity and decomposition rate, which became asymptotic at a relatively low level of diversity (Setälä and McLean, 2004; Tiunov and Scheu, 2005). In experiments involving a few species, facilitation and resource partitioning have been observed (Tiunov and Scheu, 2005). In more diverse terrestrial fungal communities, antagonistic mechanisms might prevail (Boddy, 2000; Gessner et al., 2010) and the colonization sequence of wood-degrading fungi can further affect decomposition (Fukami et al., 2010). Studies of the decomposition rate in manipulated fungal communities are mostly performed with a few cultured species, and the results might be a poor reflection of natural communities, especially if certain community members have a greater proportional influence on the decomposition rate than the totality of the fungal community (Robinson et al., 2005).
Decaying Norway spruce logs harbor diverse fungal communities (Ovaskainen et al., 2010; Rajala et al., 2011; Kubartová et al., 2012), where the number of active species is far higher than that considered in experimental studies. Species number tends to increase with mass loss (and related changes in the substrate quality) and peaks in the most decayed logs (Rajala et al., 2012). Decomposition of cell wall polymers in wood is a complex process that is driven by a succession of species with different litter-degrading enzymes (Baldrian, 2008; Stenlid et al., 2008). During the decay process of dead wood over several decades, the fungal decomposer community changes from one dominated by ascomycetes to one of white- and brown-rot basidiomycetes, before slowly becoming one composed mainly by the mycorrhizal species found in the underlying soil (Rajala et al., 2010, 2012). Wood-degrading fungi (white-rot and brown-rot fungi) decompose polysaccharides by producing hydrolases, but only white-rot fungi efficiently degrade lignin via oxidative metalloenzymes (e.g., laccase and manganese peroxidase) (Lundell et al., 2010). Experiments with decomposing leaves and needles have linked enzyme activities (EA) with fungal communities (Šnajdr et al., 2011; Žifèáková et al., 2011), but to our knowledge, no study has yet analyzed such a relationship with the fungal community in decomposing wood. Therefore, we studied wood substrates in different stages of decay to examine the effects of manipulated fungal diversity on enzyme production and decomposition rate during the entire process.
The overall objective of this study was to examine the relationship between fungal diversity and decomposition rate by manipulating fungal communities obtained from decaying Norway spruce wood. We designed a microcosm experiment to investigate: (1) whether a reduced diversity of wood-inhabiting fungi affects the decomposition of Norway spruce wood and, if so, (2) whether functional redundancy and stability of the decomposition process varies among decay stages, and (3) whether changes in fungal diversity and rate of decomposition are related to changes in hydrolytic enzyme production. We tested the hypothesis that CO2 production and enzyme activity in the dead wood are affected by the decay stage of the substrate and diversity of the decomposing species. Furthermore, we hypothetisized that the decomposition activity (measured as CO2 production) is correlated to overall variation of the fungal community and the decomposition increases to direction that is parallel to increasing species diversity (measured as number of detected OTUs).
Materials and Methods
Wood Samples
The study material was collected from an unmanaged forest in Lapinjärvi (Southern Finland, 60°39.413′N, 26°7.352′E, altitude 50 m, temperature sum 1300°C d; further details are provided in Rajala et al., 2012). Wood samples were obtained from stem discs sawn on site from 46 fallen Norway spruce (Picea abies) logs (diameter >5 cm at breast height). Logs were classified as early, intermediate or late stages of decay (I, III, and V according to Mäkinen et al., 2006). Sample discs were sawn in May 2011 and stored in plastic bags at −18°C until processing in the laboratory. The properties of the wood (C/N ratio, lignin content and density) for individual discs were analyzed as described in Rajala et al. (2010) and the mean values of 14 discs from early-decay stages, 12 discs from intermediate-decay stages and 16 discs from late-decay stages were calculated (Table 1).
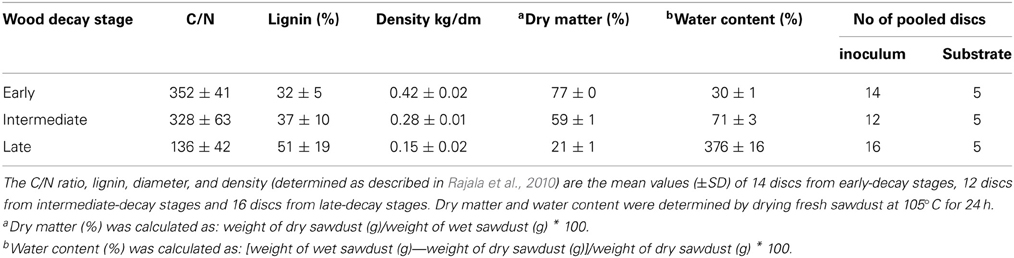
Table 1. Properties of Norway spruce (Picea abies) wood of different decay stages (early, intermediate and late) and the number of pooled discs used to prepare the inoculum (non-sterile sawdust) or the autoclaved sawdust for the microcosms.
Bark from the outermost layer of each frozen sample disc was removed with a flame-sterilized knife. Frozen discs were then drilled with a flame-sterilized and cooled bit (Ø = 10 mm) from the surface through the sapwood and heartwood. Shavings and sawdust of each decay stage were pooled into a single sample and stored in a plastic bag at −18°C prior to use as a fungal inoculum. A few active fungi might be suppressed during the processing of the samples, but species inhabiting dead wood in boreal forests are acclimated to varying temperatures and the fungal community obtained from the inocula was considered to reflect species composition in each decay phase.
The experimental growth medium (i.e., sawdust) was prepared by milling five discs from each decay stage that had been debarked and dried at 105°C for 48 h. Dry sawdust from each stage was pooled into a single sample and sterilized twice by autoclaving (121°C for 20 min) with a 3-day interval.
Experimental Set-Up
The incubation took place in sterile 100-mL glass flasks containing different amounts of inoculum and autoclaved substrate (Table 2). Sterile distilled water was added to each flask to remoisten the culture to a dry matter content similar to that at sampling, i.e., 77% for early, 59% for intermediate and 21% for late decay stages (Tables 1, 2). Flasks were then sealed with rubber septa and a one-way stopcock was connected to a 50 mm needle. The stopcock system was designed to ventilate air and prevent anoxic conditions in the microcosm. The stopcock was opened only when the ventilation was performed after measurement of CO2 (see below). The air in the flasks was ventilated by connecting the capstock to a diaphragm vacuum pump (type N 022 AN.18, KNF Neuberger GmbH, Germany) for 2 min 30 s. The capstock was then closed and reopened to flush the flasks for 5 min with moist air previously channeled through an autoclaved vent filter (0.2 μm, Millipore, USA). The evacuation-flushing system prevented oxygen depletion and drought in the microcosms.
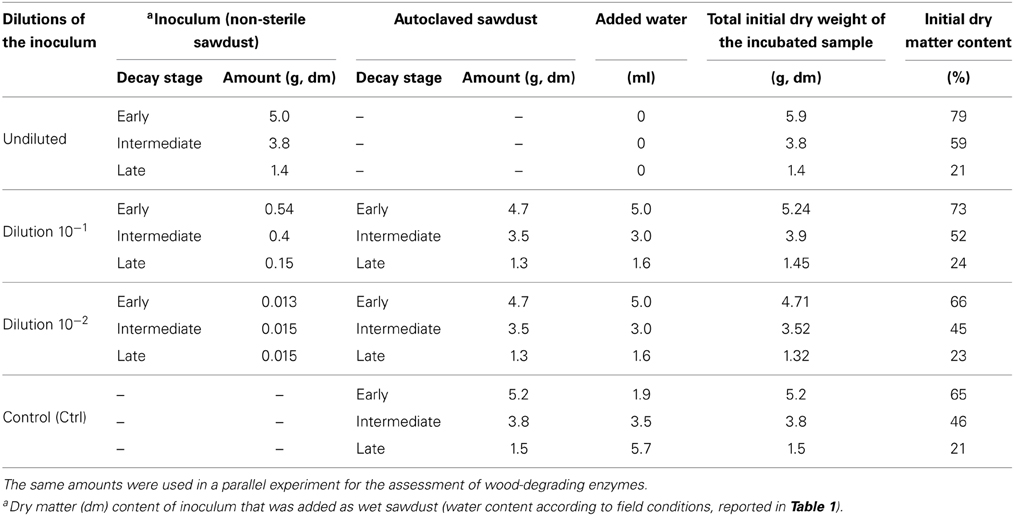
Table 2. Amount of inoculum (non-sterile sawdust) or autoclaved sawdust in dry form added to 100-mL glass flasks to assess the effect of fungal diversity on the decomposition activities of Norway spruce sawdust in early, intermediate and late stages of decay.
Preparation of Microcosms and Experimental Design of Decomposition Study (Experiment 1)
The effect of fungal diversity on respiration activity was assessed in microcosms by exposing the inoculum to a dilution procedure (Wertz et al., 2007). We tested four levels of fungal diversity (i.e., dilutions) on triplicate substrates of three different qualities (i.e., decay stages). Experimental conditions in microcosms were as follows: (1) undiluted (UD) inoculum prepared with non-sterile sawdust; (2) a dilution of approximately 1 g inoculum (dry matter, dm) per 10 g of substrate (dm) was prepared by first mixing non-sterile sawdust with sterile distilled water to create slurries with dry matter of 27% (i.e., early), 14% (i.e., intermediate), or 3% (i.e., late), then adding aliquots of 540 mg dm (early), 400 mg dm (intermediate), and 150 mg dm (late) to the corresponding dry substrate to yield a final inoculum dilution of 10−1 (Table 2); (3) a dilution of approximately 1 g inoculum (dm) per 100 g of substrate (dm) was prepared similarly, but slurry dry matter was 0.8% (early), 0.5% (intermediate) or 0.3% (late), and aliquots of 15 mg (dm) of each slurry were added to the corresponding dry substrate (Table 2); (4) autoclaved sawdust was used as a control (Ctrl). The initial fungal diversity of the microcosms was tested with the DGGE (Rajala et al., 2010 and description below) and the number of observed OTUs was used as a measure of fungal diversity. The test showed that the dilution procedure reduced the number of observed fungal OTU in the inocula (Figure 1). All treatments were performed in triplicate, yielding a total of 36 flasks (four levels of fungal diversity * three decay stages * three replicates = 36 flasks). Sealed flasks were incubated at 21°C in total darkness for 16 weeks. The cumulative CO2 was measured periodically (see below) and the airspace of the incubation flasks was ventilated immediately after CO2 measurement.
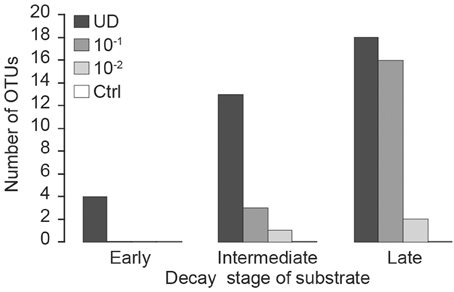
Figure 1. Number of ITS1F-GC/ITS2 fungal DNA-derived DGGE bands (Operational Taxonomic Units; OTUs) in Norway spruce pooled substrate samples at the beginning of incubation under a range of inoculum dilutions (UD, undiluted fungal inoculum; dilution 10−1 g inoculum/g substrate (dm); dilution 10−2 g inoculum/g substrate (dm); Ctrl, autoclaved sawdust) for early, intermediate and late stages of decay.
At the end of the incubation, the substrate from each replicate was pooled into a single sample and stored at –18°C prior to the extraction of total DNA.
Measurement of CO2, Evacuation and Calculation of Carbon Loss
An air sample (100 μL) was collected from the sealed flask with a Hamilton Microfilter™ glass syringe, previously wiped with 95% ethanol, and injected manually into a gas chromatograph (Hewlett-Packard 6890, Finland) to analyse CO2 (assuming that most of the produced CO2 was transferred to the gas phase). The chromatograph was equipped with a capillary column (Agilent 19095P-MS6 HP-PLOT MoleSieve 5 Å, length 30 m and diameter 530 μm) and a temperature conductor detector at 250°C for detecting CO2. Helium was used as carrier gas at a split ratio of 10:1 and flow rate of 7.9 mL min−1. Injector and oven temperatures were 120 and 40°C, respectively.
The CO2 level was measured weekly at the beginning of the incubation and every 2 weeks after week five. Estimated carbon loss (C loss, %) from microcosms was calculated using cumulative CO2 at the end of the incubation following equations (1) and (2):
where Max CO2 is the maximum production of CO2 from each microcosm, mass sawdust is the amount of sawdust in the flask (in dm), C is total carbon content in the sawdust, and MCO2 and MC are the molecular masses of CO2 (44 g mol−1) and carbon (12 g mol−1), respectively.
Preparation of Microcosms and Experimental Design of the Hydrolytic Enzyme Study (Experiment 2)
The activity of several wood-degrading hydrolytic enzymes was measured from flasks incubated in parallel with the microcosms in Experiment 1. The experimental design was similar to that in Experiment 1 (i.e., four levels of fungal diversity * three decay stages * three replicates = 36 flasks). In this study, each flask was opened under sterile conditions after weeks 4, 6, 9, 11, 13, and 16 to collect about 1 g (wet weight, ww) of the substrate. Half of the sample was subjected immediately to enzyme activity assays and the other half was stored at −18°C prior to DNA extraction (samples from weeks 9 and 13).
Extraction of Enzymes and Activity Assays
Four glycoside hydrolases [β-glucuronidase (EC 3.2.1.31), β-xylosidase (EC 3.2.1.37), β-glucosidase (EC 3.2.1.21), and cellobiohydrolase (EC 3.2.1.37)] were recovered from the substrate samples following a recently described enzyme extraction method with slight modifications (Heinonsalo et al., 2012). The method is based on the recovery of extracellular enzymes by centrifuging a small amount of substrate (~155 mg, ww) placed into a centrifuge tube filter (0.45 μm pore size; 500 μL working volume; Costar® Spin-X® CLS8162; Corning Inc., NY, USA). The centrifugation speed was 15,700 g for 30 min. To obtain enough enzyme solution to run the assays, three sub-samples were prepared from each flask. The day before extraction, 200 μL sterile distilled water was added to each substrate sub-sample. After centrifugation, approximately 250 μL enzyme solution was recovered, pooled with that from the other three sub-samples, adjusted to a total volume of 2 mL with water and used directly for the activity assays. Once all the tubes were centrifuged, the inner filter tube was placed in an oven at 70°C for 48 h to measure substrate dry matter.
Hydrolytic enzymes were measured by a fluorimetric assay (Pritsch et al., 2004, 2011) and modified according to the requirements of this study. Three solutions were prepared: (1) incubation buffer (pH 4.5) containing 7.2 mM maleic acid, 7.3 mM citric acid, 10 mM boric acid, 10 mM Tris (2-amino-2(hydroxymethyl)-1,3-propanediol), and 48.8 mM sodium hydroxide; (2) different fluorogenic substrate solutions based on 4-methylumbelliferone (MU) for the detection of each glycosidic enzyme were prepared from 5 mM stock solutions in 2-methoxyethanol. The concentration of each fluorogenic substrate in the working solution was 1500 μM of MU-β-D-glucuronide (for detection of β-glucuronidase), 1500 μM of MU-β-D-xyloside (for detection of β-xylosidase), 1500 μM of MU-β-D-glucoside (for detection of β-glucosidase), and 1200 μM of MU-β-D-cellobioside (for detection of cellobiohydrolase); (3) the stop solution was 1 M Tris at pH 10-11 to enhance the fluorescence response. Triplicate assays were performed in black flat-bottom 96-well microplates, with each well-containing a reaction mixture of 50 μL enzyme solution, 50 μL incubation buffer and 50 μL of the respective fluorogenic substrate solution. Plates were incubated at 20°C on a microplate shaker for 30 min, except for β-glucosidase where 15 min was used. At the end of incubation, 150 μlL stop solution was added to all wells and the fluorescence response was measured with a Wallac 1430 Victor3 (PerkinElmer, Inc., USA) multilabel plate reader at an excitation wavelength of 364 nm and emission wavelength of 450 nm. Calibration wells were prepared by adding 100 μL stop solution, 100 μL incubation buffer and 50 μL stock solution mixed with water to give final MU concentrations of 0, 0.4, 0.8, 1.2, 1.6, and 2 μM.
Enzyme activities (EA) were expressed as picokatals per gram of substrate (in dm). A picokatal is one picomol of reacted fluorogenic substrate per second. EA were calculated using the equation (3). Total hydrolytic activity was estimated by summing that of the four measured enzymes.
where sample is the fluorescence response (counts) of the enzyme solution and negative control is the response without enzyme (fluorogenic substrate without sample), a is the slope of the regression line of the calibration curve (counts/pmol), t is the incubation time (sec), Va is the adjusted volume of the three sub-samples (2000 μL), Vs is the volume of the enzyme solution in the well (50 μL), and ms is the sum of the extracted substrate mass of the three sub-samples (g, dm).
Molecular Analysis of the Fungal Community
Total DNA was extracted from substrates (100–150 mg, ww) at the beginning, after weeks 9 and 13 (only from the microcosms subjected to enzyme analyses) and at the end of incubation (week 16) using the NucleoSpin® 96 Soil Kit (Macherey-Nagel, Germany). Extracted total DNA was amplified using polymerase chain reaction (PCR) with the GC-clamped internal transcribed spacer 1 (ITS1F) primer (Gardes and Bruns, 1993) and the ITS2 primer pair (White et al., 1990) typical of fungal ribosomal DNA. PCR was performed in a 50 μL reaction containing 25 μL MyTaq™ HS Red Mix (which included reaction buffer, dNTP, DNA Polymerase and loading dye; Bioline, Germany), 1 μL each 25 μM primer, 2.5 μL template and 20.5 μL sterile distilled water. The thermal profile for the PCR was: 1 min at 95°C for initial denaturation, 34 cycles of denaturation for 15 s at 95°C, annealing for 15 s at 58°C and extension for 10 s at 72°C, and a final extension of 72°C for 10 s. Equal concentrations of PCR products were resolved by denaturing gradient gel electrophoresis (DGGE) as described in Rajala et al. (2010). Briefly, the denaturing gradient in an acrylamide gel was 18–58% and running conditions were 75 V at 60°C for 16 h. Gels were stained with SYBR® Gold (Molecular Probes, Eugene, Oregon) and visualized with blue light on a SafeImager™ transilluminator (Invitrogen, Carlsbad, California). The DGGE gels were analyzed using GelCompar II software (Applied Maths BVBA, Belgium). The bands at the same position in the DGGE gel were considered the same operational taxonomic unit (OTU). Thus, this step generated a presence–absence matrix of fungal taxa (OTUs) in each microcosm and we used the number of the detected OTUs as a measure of fungal diversity in this experiment.
Statistical Analysis
We analyzed the effects of inoculum dilution (undiluted, 10−1 and 10−2) and decay stage on CO2 production and enzymes activities measured from samples during the incubation period using the methods of longitudinal data analysis (Diggle et al., 2002). We also performed some additional analyses of cumulative CO2 production during the entire incubation period and on EA at the end of the experiment (week 16) using ANOVA, where longitudinal data analysis was not needed. In addition, we tested correlation between the number of observed OTUs and the cumulative CO2 production at the end of the experiment.
In the longitudinal analyses of consecutive weekly CO2 production and log-transformed total hydrolytic EA of the samples, we used a mixed model where replicates (r) represented random effects [~ N(0, σ2r)]. The fixed part of the model consisted of the decay stage and dilution factors, and their interaction, i.e., the effects we were interested in. Serial correlation of repeated measurements in the time series of replicates was treated with a continuous autoregressive covariance structure AR(1).
Residuals of the fitted models were inspected visually for normality and homoscedasticity. In all cases, we found that the residuals of the CO2 fit were normally distributed by fitted values, and the distributions were also normal and fairly similar by explanatory variables. Random effects were distributed approximately normally as well. To fulfil these requirements for the total hydrolytic EA, we converted them to a log-scale.
Strong interactions in the fixed part of the model can make it difficult to interpret the model. Therefore, if we found significant interactions between the fixed effects, we simplified the model by either regrouping the dilution and decay stage to a single factor, or by fitting the model for dilution factor by the decay-stage factor, or the other way around.
We also investigated the trends of log-transformed total hydrolytic enzyme activity, and the ratio of hydrolytic activity to CO2 production during the incubation period. In these cases, we added a slope parameter for incubation week to the fixed part of the model. The trends for these variables were tested for each decay stage separately, so that we allowed the trend to vary by dilution. For the ratio of hydrolytic activity and CO2 production, the results are presented for the early decay stage by dilution.
Longitudinal analyses were implemented using the lme function of the nlme library of R, using the maximum likelihood method. Parameter significance was estimated by the lme-function.
When a time-series approach was not needed, we used a type II ANOVA with interaction between the decay stage. These analyses were conducted using the ANOVA function in the car-library (Fox and Weisberg, 2011) of R (R Development Core Team, 2011). The same principles of examining the residuals and splitting the model into parts if interactions were present, were applied as with the longitudinal approach. Pairwise comparisons were made with the Tukey Honestly Significant Difference (HSD) test.
Fungal community composition (i.e., the presence/absence of OTUs) was visualized by non-metric multidimensional scaling (NMDS) using metaMDS of the vegan library (Oksanen et al., 2008). We generated a dissimilarity matrix (nsample * nsample) of Bray-Curtis coefficients and used NMDS to create a graph where the distances between points (representing samples) corresponded as closely as possible to the original dissimilarity matrix. Arrows that indicate the direction of maximum correlation of cumulative CO2 and OTU number with the ordinated samples were superimposed onto the ordination graph. The significance of these correlations was assessed with a permutation test implemented in the envfit function of vegan library (Oksanen et al., 2008). The NMDS was performed separately for each incubation time. The analyses made for week 0 and 16 corresponded to the experiment where flasks were closed during incubation (Experiment 1), whereas those made for week 9 and 13 corresponded to the microcosms in which flasks were opened to collect samples (Experiment 2).
Results
Effects of Substrate and Reduced Diversity on Decomposition
The wood decay stage was an important factor to explain CO2 production during the experiment (Figure 2, Supplementary Material). When we analyzed the consecutive CO2 observations and their association with the decay stage with longitudinal analysis, we found that late- and intermediate-decay substrates were associated with significantly higher CO2 production rates than early-decay substrates (t = 8.0, df = 17, p ≤ 0.001, and t = 5.7, df = 17, p < 0.001, respectively). This effect was present when we analyzed each inoculum separately (p < 0.02 for decay-stage differences within inocula), although the 10−1 inoculum responded significantly differently from other inocula in the full model with interactions.
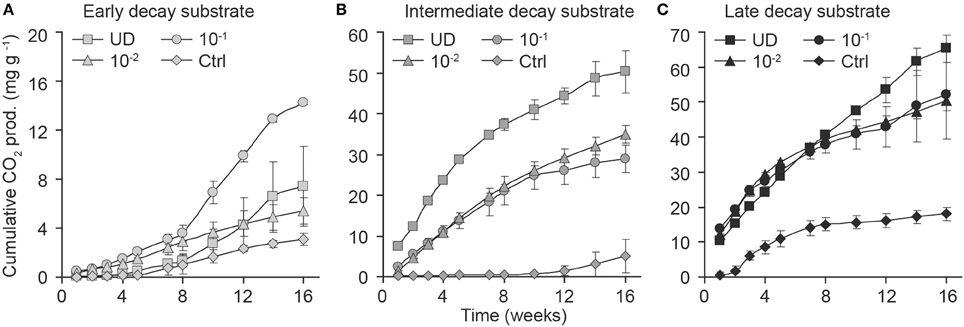
Figure 2. Effect of dilution on the production of CO2 (cumulative mg of CO2 per g of substrate; in dm) within early- (A), intermediate- (B), and late- (C) decay stages. Dilution of fungal diversity is shown by symbols as undiluted fungal inoculum (squares), dilution 10−1 g inoculum/g substrate (circles), dilution 10−2 g inoculum/g substrate (triangles), and controls (diamonds). Error bars indicate the SD of the mean for observations and were calculated for each decay stage and dilution separately.
When we analyzed the effect of inoculum (which had different number of fungal OTUs) according to decay stage with longitudinal analysis, we found that the weekly CO2 production of diluted 10−1 g and 10−2 g inocula differed from that of undiluted inoculum in intermediate-decay substrates (t = −5.5, p = 0.002 and t = −3.9, p = 0.008, respectively). The ANOVA of cumulative CO2 at week 16 supported these findings. When fungal diversity (OTU) was added to the previous model, it was also significant (F = 4.4, df = 1, p = 0.05). After 16 weeks, the cumulative production of CO2 from undiluted inoculum in late-decay substrates reached 65 mg CO2 g−1 substrate (dm), corresponding to a C loss of 3.1%, whereas C loss from undiluted intermediate- and early-decay substrates were 2.6 and 0.2%, respectively (Figures 2C, 3). At the end of the experiment, the cumulative CO2 production was correlated with the observed number of OTUs (r = 0.83, p < 0.001).
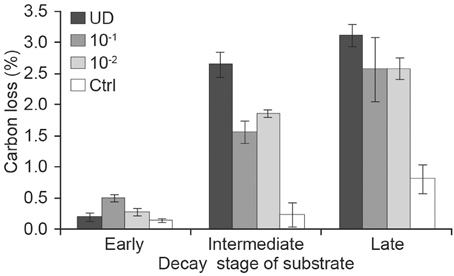
Figure 3. Cumulative carbon loss (means of three replicates) during the incubation of early, intermediate and late stages of decay and receiving various dilutions of fungal inoculum [UD, undiluted; dilution 10−1 g inoculum/g substrate (dm); dilution 10−2 g inoculum/g substrate (dm); Ctrl, autoclaved sawdust].
Effects of Substrate and Reduced Diversity on Enzyme Activity
Differences in total hydrolytic EA depended on the substrate, therefore, we performed the analyses by substrate and by dilution (Figure 4).
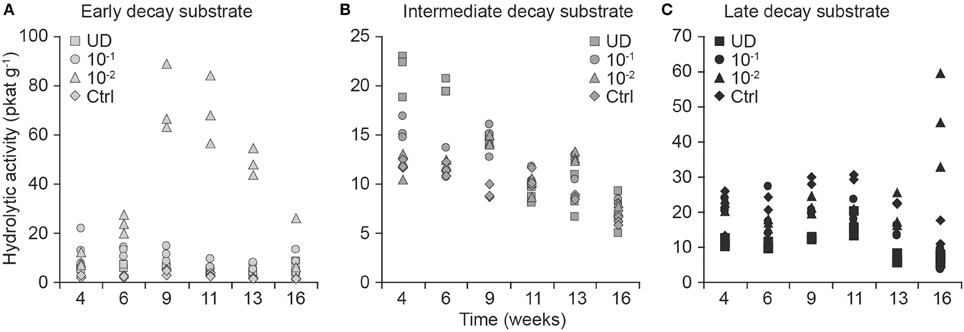
Figure 4. Effect of fungal community dilution on total hydrolytic activity (pkat/g of substrate; in dm) as the sum of 1,4-β-glucuronidase, 1,4-β-xylosidase, cellobiohydrolase, and 1,4-β-glucosidase from early-decayed (A), intermediate-decayed (B), and late-decayed substrate (C). The dilution of fungal diversity is shown by symbols as undiluted fungal inoculum (squares), dilution 10−1 g inoculum/g substrate (circles), dilution 10−2 g inoculum/g substrate (triangles), and controls (diamonds).
When we analyzed the consecutive total hydrolytic activity of replicates with longitudinal analysis, we found that it was higher in preparations containing the 10−2 dilution than in the undiluted inoculum in early- (t = 3.6, df = 5, p = 0.02) and late-decay substrates (t = 6.0, df = 6, p = 0.001). Moreover, the hydrolytic activities were also significantly different between early decayed and other substrates in undiluted inocula (both t > 4.0, df = 6, p < 0.005) and in the 10−1 dilution (both t > 3.2, df = 5, p < 0.03).
When we analyzed the total hydrolytic enzyme activity measurements at the end of the experiment with ANOVA, we also found that the activities depended on the dilution-substrate combination (interaction F = 10.2, df = 6, p < 0.001). We then grouped the inoculum and substrate into a compounded factor, performed an ANOVA and conducted a pair-wise comparison of factors, which showed that the mean hydrolytic activity of the most diluted (10−2) inoculum of the late-decayed substrate was greater than that in any of the other inoculum decay-stage combinations (p < 0.0001 for all pairs).
Furthermore, the number of fungal OTUs appeared to influence the enzyme activity at the end of the experiment (F = 14.2, df = 1, p = 0.09).
In all dilutions of the intermediate-decay inoculum, a negative trend in total hydrolytic activity was recorded toward the end of the incubation (Figure 4B). According to the longitudinal analysis, the higher intercept and stronger negative trend of hydrolytic activity in undiluted inoculum than in either of the diluted inocula (the model intercept differences in comparison to the undiluted inoculum both had t < −3.0, df = 6, p < 0.02 and for the trend coefficient, they had t > 3.4, df = 42, p < 0.001, respectively) were coincident with the measured respiration rates that started at the highest values (t < −3.9, df = 6, p < 0.01) and then decreased the quickest (t > 2.3, df = 42, p < 0.02) in the undiluted inoculum in comparison with other dilutions in the intermediate-decay substrate (Figures 2B, 4B).
The ratio of hydrolytic activity to respiration (Hydrolytic activity/CO2) clearly decreased toward the end of the incubation in the early decay substrate that received undiluted (t = −3, 4, df = 13, p = 0.005), as well as diluted inocula (10−1 dilution: t = −2.9, df = 9, p < 0.02) (Figure 5). The same phenomenon did not occur in more decayed substrates (data not shown).
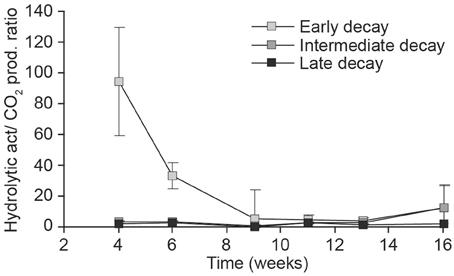
Figure 5. Relationship of carbohydrate degradation (pkat/g) and weekly CO2 production (mg/g substrate, dm) in the treatments receiving undiluted fungal inoculum of early- (light gray), intermediate- (dark gray), and late- (black) decay substrate during the 16-week incubation.
Fungal Community Structure and Respiration Activity in Different Decay Stages
Fungal communities of different decay stages differed at the start of the experiment (Figure 6A) and community structures changed during the incubation period (Figure 6). In conjunction with observed differences between undiluted and diluted fungal communities in the intermediate decay substrate (Figure 6B), we measured higher respiration activity in the microcosm prepared with the undiluted inoculum, especially during the first 6 weeks (Figure 2B). In the early decay substrate, the slightly diluted (10−1) fungal community, which had the highest respiration rate (Figure 2A), appeared to deviate from all other communities after 13 weeks of incubation (Figure 6C).
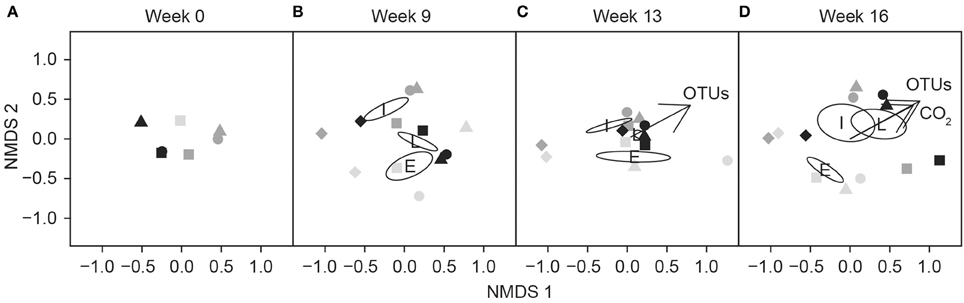
Figure 6. Non-metric multidimensional scaling (NMDS) ordination showing the separation of fungal communities at the beginning (A), after 9 weeks (B), 13 weeks (C), and 16 weeks (D) in the different dilutions or controls (autoclaved sawdust) according to the decay stage of substrate. Symbols represent the dilution of the fungal community: undiluted (squares), dilution 10−1 g inoculum/g substrate (circles), dilution 10−2 g inoculum/g substrate (triangles) and controls (diamonds). The intensity of the symbol indicates the decay stage. Vectors indicate the direction of maximum correlation of CO2 production and number of OTUs with ordinated samples (p < 0.01). Ellipses indicate the mean location of each decay stage: E, early; I, intermediate; and L, late.
At the end of the experiment, cumulative CO2 (r = 0.91, p < 0.001) and the number of OTUs (r = 0.87, p < 0.001) increased in the communities composed of intermediate- and late-decay stage species, as shown by the linear fit superimposed on the NMDS graph (Figure 6D).
Discussion
Effects of Fungal Diversity on Wood Decomposition
The main findings of the experiment confirmed our hypothesis that fungal diversity affects decomposition rate and that resistance of the decomposition process to the loss of diversity depends on the decomposition stage of woody substrates. We used inocula that originated from actively decaying wood, where the number of OTUs is higher than in previous experiments using selected species from pure cultures (Setälä and McLean, 2004; Tiunov and Scheu, 2005). These studies with communities created by combining cultured fungal species have shown how the decomposition rate becomes asymptotic with relatively few decomposer species, and our results on late-decay communities with initially high number of fungal OTUs support this finding. However, we observed that the functioning of the decomposers of the intermediate-decay stage, where the initial number of OTUs was lower than in the late-decay wood, was less resistant to the loss of diversity. In the intermediate-decay stage, decomposition activity was highest in non-diluted communities and the experimental dilution of the fungal community was associated with a lower respiration rate.
This study showed that CO2 production, as an indicator of decomposition, increased in relation to the decay stage of Norway spruce wood and peaked during the late-decay stage, where fungal diversity (measured as the number of observed OTUs) was also maximal. The observed increase in decomposition activity with increasing OTU diversity is in agreement with our hypothesis. Earlier studies, where decay models were fitted to empirical field data, predicted that the rate of decay is highest during intermediate phases of decomposition (Harmon et al., 2000; Mäkinen et al., 2006; Tuomi et al., 2011), which coincides with the period when white- and brown-rot fungi are at their peak prevalence (Rajala et al., 2011, 2012). However, we suggest that decomposition activity is also maintained in the late-decay stage wood that might be under-represented in previous studies (i.e., they used a relatively small number of late-decay samples, which are difficult to date). In addition, our results indicate that lowering fungal diversity, especially in the intermediate-decay stage, has a direct impact on fungal community function, and thereby on the decomposition of woody litter.
We showed that in the most diverse fungal community inhabiting the late-decay substrate, lowering the diversity with dilution of the inocula had only a slight impact on decomposition rate. This suggests that the community that remained after dilution was sufficiently diverse to provide the same ecological function as that of the undiluted system, which agrees well with earlier studies of the diversity-function relationship in soil microbes (Wertz et al., 2006), whereas the less diverse communities were more affected by random selection. Consequently, we predict a stronger resilience of the diverse fungal community found in advanced stages of wood decay.
In the less diverse community inhabiting the early decay substrate, a 10−1 dilution caused an unexpected result in that CO2 production increased. It appears that loss of diversity in a low-density community affects competitive interactions and decomposition in unpredictable ways. The response is highly sensitive to the activity of the remaining species and the functional redundancy in the fungal community of early-stage decomposers is lower than that found in later stages.
The use of indigenous fungal communities as inocula to study the relationship between diversity and function of terrestrial fungal communities is novel and was applied for the first time to wood-inhabiting fungi in this study. Previous laboratory studies have assessed the ability of cultured fungi to degrade woody substrates (Boddy et al., 1989; Tiunov and Scheu, 2005; Toljander et al., 2011) and compete with natural wood-inhabiting fungi (Holmer and Stenlid, 1997; Holmer et al., 1997). According to the method proposed by Wertz et al. (2006), the dilution procedure reduced the number of fungal taxa detected in the inoculum. Prior to our experiment, we tested different ratios of dilutions and found that a relatively high (1/100) dilution is needed for a remarkable reduction of OTUs in highly diverse inocula. However, for a moderate reduction in the OTU diversity, we chose to apply a dilution ratio (1/10) that was optimal for the intermediate decay stage, but compromised late decay, where the resulting reduction in OTU diversity was only slight.
As expected, the number of detected fungal species increased in all treatments with the progression of the incubation, since the applied PCR-DGGE method cannot identify infrequent fungi and only after replication of DNA during the incubation they became visible. Our sterilization procedure of the control substrate was apparently sufficient to destroy most of the fungal DNA, as OTUs were not detectable via DGGE at the start of the experiment (Figure 1). However, slight CO2 production during the incubation indicated that some microbes survived autoclaving (Figure 2), as it is known that some spores might be resistant to this form of sterilization (Cheng et al., 2008). This was the reason to detect a few fungal OTUs in the controls after 9 weeks (data not shown). However, NMDS showed the community structure in controls to be distinct from that of other microcosms. This suggests that the key species involved in decomposition did not survive autoclaving, although we cannot totally deny that some species originated from autoclaved sawdust. However, CO2 production in controls was far lower than in any of the treatments and therefore we believe that effect of contaminant species in dilution treatments is minor. The fact that sterilization of wood, soil or other substrate by autoclaving is practically impossible should still be considered in this kind of dilution procedures.
Enzyme Activity During Wood Decomposition in Response to Fungal Diversity
Contrary to our hypothesis, the most diluted (10−2) inoculum was associated with a higher activity of hydrolytic enzymes than treatments of the same substrate receiving inoculum with a higher fungal richness (Figure 4). This might be explained by less competition in the diluted communities involving species specializing in the production of carbohydrate-degrading enzymes (e.g., brown- and soft-rot fungi). We also found that the ratio of hydrolytic activity and production of CO2 per week in early-decay substrates was initially high and gradually decreased. The negative slope of the relationship between hydrolytic activity and weekly CO2 production suggests that fungal taxa that are active during the early phases of decomposition must produce hydrolytic enzymes to release the wide variety of carbohydrates that are metabolized in later stages.
The enzyme recovery method, based on a filter centrifugation approach, has been validated in agar media supplemented with different organic products (Heinonsalo et al., 2012), but has never been applied to a more complex substrate such as one composed of decomposing spruce wood. A standard and universal protocol for extracting and measuring enzymes from environmental samples does not exist (Baldrian, 2009). Common practice is to soak the samples in buffer (e.g., acetate or sulfate buffers) and extract the enzymes by agitating the slurry, but such treatment might negatively affect EA (Vepsäläinen, 2001). In our study, the level of enzyme activity was in the range 10–100 pkat g−1 dry substrate. Previous studies that reported activities as high as 1000 pkat g−1 (dm) in Norway spruce needles used pure fungal inoculum from a cultured isolate (Žifèáková et al., 2011). Our results indicate that extracellular EA in spruce wood that has not received a pure and concentrated inoculum are much lower. It is known that spruce wood is a complex substrate for the recovery of wood-degrading enzymes, since water-soluble constituents such as phenols, sugars, organic acids, xylo-oligosaccharides and proteins all act as inhibitors of hydrolytic enzymes (e.g., Lagaert et al., 2009; Kim et al., 2011). Nevertheless, the effect of wood extracts on enzyme recovery in this study was expected to be minimal as was previously reported by Valentín et al. (2010).
Conclusions
Dilution of fungal inocula recovered from Picea abies logs in three different decay stages revealed a stage-dependent response in decomposition rate. Reduced fungal diversity was associated with lower respiration rates during the intermediate stages of decay, but no diversity effects were detected in later stages, where initial number of detected species was high. This suggested that the highly diverse community of the late-decay stage was more resistant to the loss of diversity than less diverse communities of early decomposers. In early-decay communities, dilution caused unexpected changes to the decomposition process, probably due to the strong stochastic effect of dilution on less diverse communities. The results of this study also showed that the decomposition rate and the fungal diversity increased as decay advanced. We suggest that fungal activity and the functional redundancy of the fungal community in decaying wood increase during the fungal succession from intermediate- to late-decay stages.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the forest and laboratory personnel of the Finnish Forest Research Institute for their assistance in field work and the preparation of samples for chemical, enzymatic and molecular analyses. The study was funded by the Academy of Finland (project numbers 121630 and 257701).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2014.00230/abstract
References
Baldrian, P. (2008). “Enzymes of saprotrophic basidiomycetes,” in Ecology of Saprotrophic Basidiomycetes, eds L. Boddy, J. C. Frankland, and P. van West (London: Academic Press), 19–41.
Baldrian, P. (2009). Microbial enzyme-catalyzed processes in soils and their analysis. Plant Soil Environ. 55, 370–378. Available online at: http://www.agriculturejournals.cz/web/pse.htm?volume=55&firstPage=370&type=publishedArticle
Boddy, L. (2000). Interspecific combative interactions between wood-decaying basidiomycetes. FEMS Microbiol. Ecol. 31, 185–194. doi: 10.1111/j.1574-6941.2000.tb00683.x
Boddy, L., Owens, E., and Chapela, I. (1989). Small scale variation in decay rate witin logs one year after felling: effect of fungal community structure and moisture content. FEMS Microbiol. Lett. 62, 173–184. doi: 10.1111/j.1574-6968.1989.tb03691.x
Cardinale, B. J., Duffy, J. E., Gonzalez, A., Hooper, D. U., Perrings, C., Venail, P., et al. (2012). Biodiversity loss and its impact on humanity. Nature 486, 59–67. doi: 10.1038/nature11148
Cheng, J. Y. W., Lau, A. P. S., and Fang, M. (2008). Assessment of the atmospheric fungal prevalence through field ergosterol measurement I - Determination of the specific ergosterol content in common ambient fungal spores and yeast cells. Atmos. Environ. 42, 5526–5533. doi: 10.1016/j.atmosenv.2008.03.021
Diggle, P. J., Heagerty, P., Liang, K. Y., and Zeger, S. L. (2002). Analysis of Longitudinal Data, 2nd Edn. Oxford: Oxford University Press.
Fox, J., and Weisberg, S. (2011). An R Companion to Applied Regression, 2nd Edn. Sage Publications. Available online at: http://socserv.socsci.mcmaster.ca/jfox/Books/Companion/ (Accessed December 19, 2013).
Fukami, T., Dickie, I. A., Paula Wilkie, J., Paulus, B. C., Park, D., et al. (2010). Assembly history dictates ecosystem functioning: evidence from wood decomposer communities. Ecol. Lett. 13, 675–684. doi: 10.1111/j.1461-0248.2010.01465.x
Gardes, M., and Bruns, T. D. (1993). ITS primers with enhanced specificity for basidiomycetes - application to the identification of mycorrhizae and rusts. Mol. Ecol. 2, 113–118. doi: 10.1111/j.1365-294X.1993.tb00005.x
Gessner, M. O., Swan, C. M., Dang, C. K., McKie, B. G., Bardgett, R. D., Wall, D. H., et al. (2010). Diversity meets decomposition. Trends Ecol. Evol. 25, 372–380. doi: 10.1016/j.tree.2010.01.010
Harmon, M., Krankina, O., and J, S. (2000). Decomposition vectors: a new approach to estimating woody detritus decomposition dynamics. Can. J. For. Res. 30, 76–84. doi: 10.1139/cjfr-30-1-76
Hättenschwiler, S., Tiunov, A. V., and Scheu, S. (2005). Biodiversity and litter decomposition in terrestrial ecosystems. Annu. Rev. Ecol. Evol. Syst. 36, 191–218. doi: 10.1146/annurev.ecolsys.36.112904.151932
Heinonsalo, J., Kabiersch, G., Niemi, R. M., Simpanen, S., Ilvesniemi, H., Hofrichter, M., et al. (2012). Filter centrifugation as a sampling method for miniaturization of extracellular fungal enzyme activity measurements in solid media. Fungal Ecol. 5, 261–269. doi: 10.1016/j.funeco.2011.07.008
Holmer, L., Renvall, P., and Stenlid, J. (1997). Selective replacement between species of wood-rotting basidiomycetes, a laboratory study. Mycol. Res. 101, 714–720. doi: 10.1017/S0953756296003243
Holmer, L., and Stenlid, J. (1997). Competitive hierarchies of wood decomposing basidiomycetes in artificial systems based on variable inoculum sizes. Oikos 79, 77–84. doi: 10.2307/3546092
Hooper, D., Chapin, F. III., Ewel, J., Hector, A., Inchausti, P., Lavorel, S., et al. (2005). Effects of biodiversity on ecosystem functioning a consensus of current knowledge. Ecol. Monogr. 75, 3–35. doi: 10.1890/04-0922
Hooper, D. U., Adair, E. C., Cardinale, B. J., Byrnes, J. E. K., Hungate, B. A., Matulich, K. L., et al. (2012). A global synthesis reveals biodiversity loss as a major driver of ecosystem change. Nature 486, 105–108. doi: 10.1038/nature11118
Kim, Y., Ximenes, E., Mosier, N. S., and Ladisch, M. R. (2011). Soluble inhibitors/deactivators of cellulase enzymes from lignocellulosic biomass. Enzyme Microb. Technol. 48, 408–415. doi: 10.1016/j.enzmictec.2011.01.007
Kubartová, A., Ottosson, E., Dahlberg, A., and Stenlid, J. (2012). Patterns of fungal communities among and within decaying logs revealed by 454 sequencing. Mol. Ecol. 21, 4514–4532. doi: 10.1111/j.1365-294X.2012.05723.x
Lagaert, S., Beliën, T., and Volckaert, G. (2009). Plant cell walls: protecting the barrier from degradation by microbial enzymes. Semin. Cell Dev. Biol. 20, 1064–1073. doi: 10.1016/j.semcdb.2009.05.008
Lindahl, B., and Boberg, J. (2008). “Distribution and function of litter basidiomycetes in coniferous forests,” in Ecology of Saprotrophic Basidiomycetes, eds L. Boddy, J. C. Frankland, and P. van West (London: Academic Press), 183–196.
Loreau, M., Naeem, S., Inchausti, P., Bengtsson, J., Grime, J. P., Hector, A., et al. (2001). Biodiversity and ecosystem functioning: current knowledge and future challenges. Science 294, 804–808. doi: 10.1126/science.1064088
Lundell, T. K., Mäkelä, M. R., and Hildén, K. (2010). Lignin-modifying enzymes in filamentous basidiomycetes - ecological, functional and phylogenetic review. J. Basic Microbiol. 50, 5–20. doi: 10.1002/jobm.200900338
Mäkinen, H., Hynynen, J., Siitonen, J., and Sievänen, R. (2006). Predicting the decomposition of Scots pine, Norway spruce, and birch stems in Finland. Ecol. Appl. 16, 1865–1879. doi: 10.1890/1051-0761(2006)016[1865:PTDOSP]2.0.CO;2
Oksanen, J., Kindt, R., Legendre, P., Simpson, G., Sólymos, P., Stevens, M. H., et al. (2008). Vegan: Community ecology package - R package version 1.15-2. Available online at: http://cran.r-project.org (Accessed December 19, 2013).
Ovaskainen, O., Nokso-Koivisto, J., Hottola, J., Rajala, T., Pennanen, T., Ali-Kovero, H., et al. (2010). Identifying wood-inhabiting fungi with 454 sequencing – what is the probability that BLAST gives the correct species? Fungal Ecol. 3, 274–283. doi: 10.1016/j.funeco.2010.01.001
Pritsch, K., Courty, P. E., Churin, J.-L., Cloutier-Hurteau, B., Ali, M. A., Damon, C., et al. (2011). Optimized assay and storage conditions for enzyme activity profiling of ectomycorrhizae. Mycorrhiza 21, 589–600. doi: 10.1007/s00572-011-0364-4
Pritsch, K., Raidl, S., Marksteiner, E., Blaschke, H., Agerer, R., Schloter, M., et al. (2004). A rapid and highly sensitive method for measuring enzyme activities in single mycorrhizal tips using 4-methylumbelliferone-labelled fluorogenic substrates in a microplate system. J. Microbiol. Methods 58, 233–241. doi: 10.1016/j.mimet.2004.04.001
R Development Core Team. (2011). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Available online at: http://www.r-project.org (Accessed December 19, 2013).
Rajala, T., Peltoniemi, M., Hantula, J., Mäkipää, R., and Pennanen, T. (2011). RNA reveals a succession of active fungi during the decay of Norway spruce logs. Fungal Ecol. 4, 437–448. doi: 10.1016/j.funeco.2011.05.005
Rajala, T., Peltoniemi, M., Pennanen, T., and Mäkipää, R. (2010). Relationship between wood-inhabiting fungi determined by molecular analysis (denaturing gradient gel electrophoresis) and quality of decaying logs. Can. J. For. Res. 40, 2384–2397. doi: 10.1139/X10-176
Rajala, T., Peltoniemi, M., Pennanen, T., and Mäkipää, R. (2012). Fungal community dynamics in relation to substrate quality of decaying Norway spruce (Picea abies [L.] Karst.) logs in boreal forests. FEMS Microbiol. Ecol. 81, 494–505. doi: 10.1111/j.1574-6941.2012.01376.x
Rayner, A. D. M., and Boddy, L. (1988). Fungal Decomposition of Wood. Its Biology and Ecology. Chichester: John Wiley & Sons Ltd.
Robinson, C. H., Miller, E. J. P., and Deacon, L. J. (2005). “Biodiversity of saprotrophic fungi in relation to their function: Do fungi obey the rules?,” in Biological Diversity and Function in Soils, eds R. Bardgett, M. Usher, and D. Hopkins (Cambridge: Cambridge University Press), 189–215. doi: 10.1017/CBO9780511541926
Setälä, H., and McLean, M. A. (2004). Decomposition rate of organic substrates in relation to the species diversity of soil saprophytic fungi. Oecologia 139, 98–107. doi: 10.1007/s00442-003-1478-y
Siitonen, J. (2001). Forest management, coarse woody debris and saproxylic organisms: Fennoscandian boreal forests as an example. Ecol. Bull. 49, 11–41.
Šnajdr, J., Cajthaml, T., Valášková, V., Merhautová, V., Petránková, M., Spetz, P., et al. (2011). Transformation of Quercus petraea litter: successive changes in litter chemistry are reflected in differential enzyme activity and changes in the microbial community composition. FEMS Microbiol. Ecol. 75, 291–303. doi: 10.1111/j.1574-6941.2010.00999.x
Stenlid, J., Penttilä, R., and Dahlberg, A. (2008). “Wood-decay basidiomycetes in boreal forests: Distribution and community development,” in Ecology of Saprotrophic Basidiomycetes, eds L. Boddy, J. C. Frankland, and P. van West (London: Academic Press), 239–262.
Tiunov, A. V., and Scheu, S. (2005). Facilitative interactions rather than resource partitioning drive diversity-functioning relationships in laboratory fungal communities. Ecol. Lett. 8, 618–625. doi: 10.1111/j.1461-0248.2005.00757.x
Toljander, Y. K., Lindahl, B. J. D., Holmer, L., and Högberg, N. O. S. (2011). Environmental fluctuations facilitate species co-existence and increase decomposition. Oecologia 148, 625–631. doi: 10.1007/s00442-006-0406-3
Tuomi, M., Laiho, R., Repo, A., and Liski, J. (2011). Wood decomposition model for boreal forests. Ecol. Model. 222, 709–718. doi: 10.1016/j.ecolmodel.2010.10.025
Valentín, L., Kluczek-Turpeinen, B., Willför, S., Hemming, J., Hatakka, A., Steffen, K., et al. (2010). Scots pine (Pinus sylvestris) bark composition and degradation by fungi: potential substrate for bioremediation. Bioresour. Technol. 101, 2203–2209. doi: 10.1016/j.biortech.2009.11.052
Vepsäläinen, M. (2001). Poor enzyme recovery by extraction from soils. Soil Biol. Biochem. 33, 1131–1135. doi: 10.1016/S0038-0717(00)00240-6
Wertz, S., Degrange, V., Prosser, J. I., Poly, F., Commeaux, C., Freitag, T., et al. (2006). Maintenance of soil functioning following erosion of microbial diversity. Environ. Microbiol. 8, 2162–2169. doi: 10.1111/j.1462-2920.2006.01098.x
Wertz, S., Degrange, V., Prosser, J. I., Poly, F., Commeaux, C., Guillaumaud, N., et al. (2007). Decline of soil microbial diversity does not influence the resistance and resilience of key soil microbial functional groups following a model disturbance. Environ. Microbiol. 9, 2211–2219. doi: 10.1111/j.1462-2920.2007.01335.x
White, T. J., Bruns, S., Lee, S., and Taylor, J. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR Protocols: A Guide to Methods and Applications, eds M. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (San Diego, CA: Academic Press), 315–322.
Keywords: biodiversity, woody debris, respiration activity, functional redundancy, enzymes
Citation: Valentín L, Rajala T, Peltoniemi M, Heinonsalo J, Pennanen T and Mäkipää R (2014) Loss of diversity in wood-inhabiting fungal communities affects decomposition activity in Norway spruce wood. Front. Microbiol. 5:230. doi: 10.3389/fmicb.2014.00230
Received: 24 January 2014; Accepted: 29 April 2014;
Published online: 20 May 2014.
Edited by:
John J. Kelly, Loyola University Chicago, USAReviewed by:
Stefan Bertilsson, Uppsala University, SwedenPetra Fransson, Swedish University of Agricultral Sciences, Sweden
Copyright © 2014 Valentín, Rajala, Peltoniemi, Heinonsalo, Pennanen and Mäkipää. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raisa Mäkipää, Vantaa Research Unit, Finnish Forest Research Institute, PO Box 18, Jokiniemenkuja 1, FI-01301 Vantaa, Finland e-mail:cmFpc2EubWFraXBhYUBtZXRsYS5maQ==