 Sankar Bhattacharyya
Sankar Bhattacharyya- Vaccine and Infectious Disease Research Centre, Translational Health Science and Technology Institute, Gurgaon, India
The mammalian genome has evolved to encode a battery of mechanisms, to mitigate a progression in the life cycle of an invasive viral pathogen. Although apparently disadvantaged by their dependence on the host biosynthetic processes, an immensely faster rate of evolution provides viruses with an edge in this conflict. In this review, I have discussed the potential anti-virus activity of inositol-requiring enzyme 1 (IRE1), a well characterized effector of the cellular homeostatic response to an overloading of the endoplasmic reticulum (ER) protein-folding capacity. IRE1, an ER-membrane-resident ribonuclease (RNase), upon activation catalyses regulated cleavage of select protein-coding and non-coding host RNAs, using an RNase domain which is homologous to that of the known anti-viral effector RNaseL. The latter operates as part of the Oligoadenylate synthetase OAS/RNaseL system of anti-viral defense mechanism. Protein-coding RNA substrates are differentially treated by the IRE1 RNase to either augment, through cytoplasmic splicing of an intron in the Xbp1 transcript, or suppress gene expression. This referred suppression of gene expression is mediated through degradative cleavage of a select cohort of cellular RNA transcripts, initiating the regulated IRE1-dependent decay (RIDD) pathway. The review first discusses the anti-viral mechanism of the OAS/RNaseL system and evasion tactics employed by different viruses. This is followed by a review of the RIDD pathway and its potential effect on the stability of viral RNAs. I conclude with a comparison of the enzymatic activity of the two RNases followed by deliberations on the physiological consequences of their activation.
Introduction
Establishment prove to be inhibitory for the viral life cycle in a direct or an indirect manner. The direct mechanism involves expression of multiple anti-viral genes that have evolved to recognize, react, and thereby rid the infected host of the viral nucleic acid (Zhou et al., 1997; Thompson et al., 2011). On the other hand the pathways, e.g., those that culminate in initiating an apoptotic death for the host cell, indirectly serve to limit the spread of virus (Roulston et al., 1999). A major difference between these two mechanisms is that while the former response is transmissible to neighboring uninfected cells through interferon (IFN) signaling, the latter is observed mostly in cis. Recent reports, however, have demonstrated transmission of an apoptotic signal between cells that are in contact through gap junctions, although such a signaling from an virus infected host cell to an uninfected one is not known yet (Cusato et al., 2003; Udawatte and Ripps, 2005; Kameritsch et al., 2013). Successful viral pathogens, through a process of active selection, have evolved to replicate and simultaneously evade or block either of these host responses. The viral nucleic acids which could be the genome (positive-sense single-stranded RNA virus) or RNA derived from transcription of the genome [negative-stranded single-sense RNA or double-stranded RNA (dsRNA) or DNA virus], offer critical targets for both detection and eradication. The viral nucleic acid targeting armaments in the host arsenal include those that recognize the associated molecular patterns like toll-like receptors (TLRs), DDX58 (or RIG-1), IFIH1 (or MDA5), IFIT proteins [IFN-stimulated genes (ISG)56 and ISF54], etc. (Aoshi et al., 2011; Bowzard et al., 2011; Jensen and Thomsen, 2012). This is followed by IFN signaling and expression or activation of factors that target the inducer for degradation or modification like OAS/ribonuclease L (RNaseL) system, APOBEC3, MCPIP1, the ZC3HAV1/exosome system and RNAi pathways (Gao et al., 2002; Sheehy et al., 2002; Guo et al., 2007; Daffis et al., 2010; Sidahmed and Wilkie, 2010; Schmidt et al., 2012; Cho et al., 2013a; Lin et al., 2013). In this review we focus on two proteins containing homologous RNase domains, RNaseL with a known direct antiviral function and Inositol-requiring enzyme 1 (IRE1 or ERN1) which has an RNaseL-like RNase domain with a known role in homeostatic response to unfolded proteins in the endoplasmic reticulum (ER) and a potential to function as an antiviral (Figure 1; Tirasophon et al., 2000).
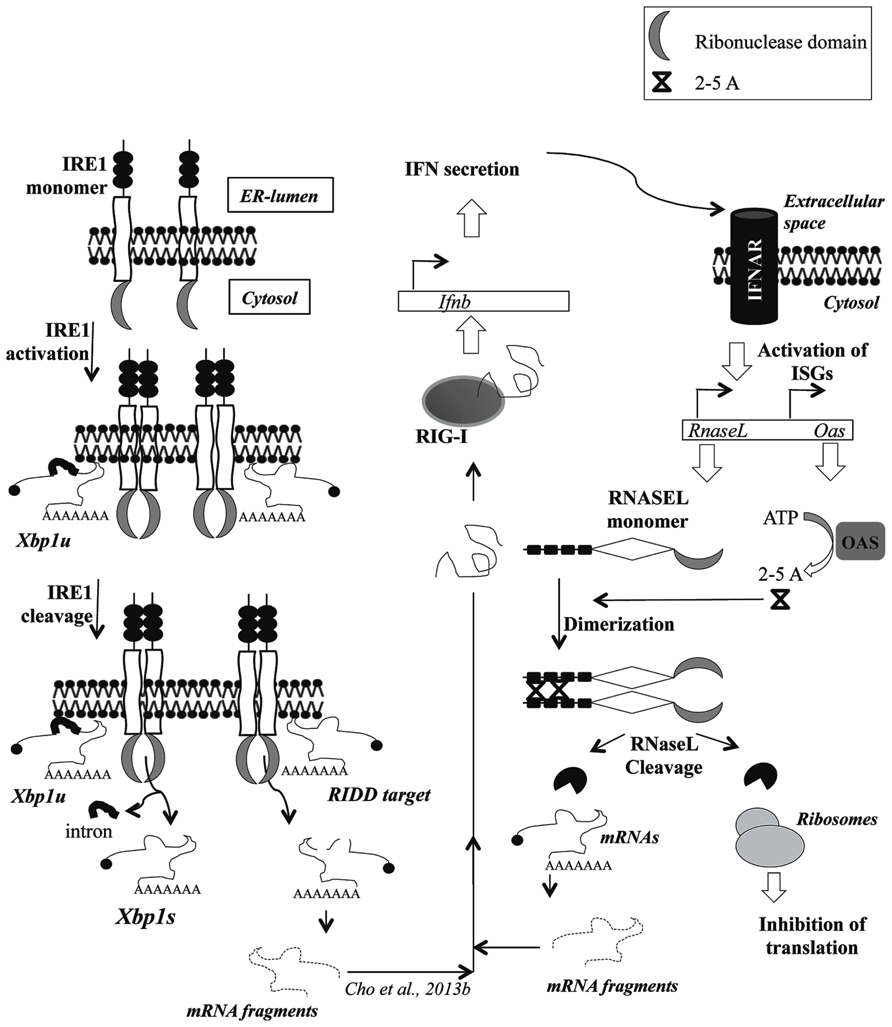
FIGURE 1. Schematic representation of the ribonuclease activity of IRE1 and RNaseL showing cross-talk between the paths catalysed by the enzymes. The figure shows activation of RNase activity following dimerization triggered by either accumulation of unfolded proteins in the ER-lumen or synthesis of 2–5A by the enzyme OAS, respectively, for IRE1 and RNaseL. The cleavage of Xbp1u by IRE1 releases an intron thus generating Xbp1s. The IRE1 targets in RIDD pathway or all RNaseL substrates are shown to undergo degradative cleavage. The cleavage products generated through degradation of the respective substrate is shown to potentially interact with RIG-I thereby leading to Interferon secretion and trans-activation of Oas genes through Interferon signaling. Abbreviations: RIG-I = retinoic acid inducible gene-I, Ifnb = interferon beta gene loci, IFN = interferons, ISG = interferon-sensitive genes, 2–5A = 2′–5′ oligoadenylates.
Degradation of Viral RNA by RNaseL and Viral Evasion
In mammalian cells the tell-tale signs of RNA virus infection, like the presence of cytosolic RNA having 5′-ppp or extensive (>30 bp) dsRNA segments are detected by dedicated pathogen associated molecular pattern receptors (PAMPs) or pattern recognition receptors (PRRs) in the host cell, like RIG-1, MDA5, and the IFIT family of proteins (Aoshi et al., 2011; Bowzard et al., 2011; Vabret and Blander, 2013). The transduction of a signal of this recognition results in the expression of IFN genes the products of which upon secretion outside the cell bind to cognate receptors, initiating further downstream signaling (Figure 1; Randall and Goodbourn, 2008). The genes that are regulated as a result of IFN signaling are termed as IFN-stimulated or IFN-regulated genes (ISGs or IRGs; Sen and Sarkar, 2007; Schoggins and Rice, 2011). Oligoadenylate synthetase or OAS genes are canonical ISGs that convert ATP into 2′–5′ linked oligoadenylates (2–5A) by an unique enzymatic mechanism (Figure 1; Hartmann et al., 2003). Further, they are RNA-binding proteins that function like PRRs, in a way that the 2–5A synthesizing activity needs to be induced through an interaction with dsRNA (Minks et al., 1979; Hartmann et al., 2003). In a host cell infected by an RNA virus, such dsRNA is present in the form of replication-intermediates (RI), which are synthesized by the virus-encoded RNA-dependent RNA polymerases (RdRp) and subsequently used by the same enzyme to synthesize more genomic RNA, through asymmetric transcription (Weber et al., 2006). However, the replications complexes (RCs) harboring these RI molecules are found secluded inside host-membrane derived vesicles, at least in positive-strand RNA viruses, a group which contains many human pathogens (Uchil and Satchidanandam, 2003; Denison, 2008). Reports from different groups suggest OAS proteins to be distributed both in the cytoplasm as well as in membrane-associated fractions, perhaps indicating an evolution of the host anti-viral methodologies towards detection of the membrane-associated viral dsRNAs (Marie et al., 1990; Lin et al., 2009). DNA viruses on the other hand, produce dsRNA by annealing of RNA derived from transcription of both strands in the same viral genomic loci, which are probably detected by the cytoplasmic pool of OAS proteins (Jacobs and Langland, 1996; Weber et al., 2006). Post-activation the OAS enzymes synthesize 2–5A molecules in a non-processive reaction producing oligomers which, although potentially ranging in size from dimeric to multimeric, are functionally active only in a trimeric or tetrameric form (Dong et al., 1994; Sarkar et al., 1999; Silverman, 2007). These small ligands, which bear phosphate groups (1–3) at the 5′ end and hydroxyl groups at the 2′ and 3′ positions, serve as co-factor which can specifically interact with and thereby allosterically activate, existing RNaseL molecules (Knight et al., 1980; Zhou et al., 1997, 2005; Sarkar et al., 1999). As part of a physiological control system these 2–5A oligomers are quite unstable in that they are highly susceptible to degradation by cellular 5′-phosphatases and PDE12 (2′-phosphodiesterase; Silverman et al., 1981; Johnston and Hearl, 1987; Kubota et al., 2004; Schmidt et al., 2012). Viral strategies to evade or overcome this host defense mechanism ranges from preventing IFN signaling which would hinder the induction of OAS expression or thwarting activation of expressed OAS proteins by either shielding the viral dsRNA from interacting with it or modulating the host pathway to synthesize inactive 2–5A derivatives (Cayley et al., 1984; Hersh et al., 1984; Rice et al., 1985; Maitra et al., 1994; Beattie et al., 1995; Rivas et al., 1998; Child et al., 2004; Min and Krug, 2006; Sanchez and Mohr, 2007; Sorgeloos et al., 2013). Shielding of viral RNA from interacting with OAS is possible through enclosure of dsRNA replication intermediates in membrane enclosed compartments as observed in many flaviviruses (Ahlquist, 2006; Miller and Krijnse-Locker, 2008; Miorin et al., 2013).
RNaseL is a 741 amino acid protein containing three predominantly structured region, an N-terminal ankyrin repeat domain (ARD), a middle catalytically inactive pseudo-kinase (PK) and a C-terminal RNase domain (Figure 2A; Hassel et al., 1993; Zhou et al., 1993). The activity of the RNase domain is negatively regulated by the ARD, which is relieved upon binding of 2–5A molecules to ankyrin repeats 2 and 4 followed by a conformational alteration (Figure 1; Hassel et al., 1993; Tanaka et al., 2004; Nakanishi et al., 2005). In support of this contention, deletion of the ARD has been demonstrated to produce constitutively active RNaseL, although with dramatically lower RNase activity (Dong and Silverman, 1997). However, recent reports suggest that while 2–5A links the ankyrin repeats from adjacent molecules leading to formation of dimer and higher order structures, at sufficiently high in vitro concentrations, RNaseL could oligomerize even in the absence of 2–5A (Han et al., 2012). Nonetheless, in vivo the RNaseL nuclease activity still seems to be under the sole regulation of 2–5A (Al-Saif and Khabar, 2012). In order to exploit this dependence, multiple viruses like mouse hepatitis virus (MHV) and rotavirus group A (RVA) have evolved to encode phosphodiesterases capable of hydrolysing the 2′–5′ linkages in 2–5A and thereby attenuate the RNaseL cleavage activity (Zhao et al., 2012; Zhang et al., 2013). In addition to 5′-phosphatases and 2′-phosphodiesterases to reduce the endogenous 2–5A levels, mammalian genomes encode post-transcriptional and post-translation inhibitors of RNaseL activity in the form of microRNA-29 and the protein ABCE1 (RNaseL inhibitor or RLI), respectively (Bisbal et al., 1995; Lee et al., 2013). Direct inhibition of RNaseL function is also observed upon infection by Picornaviruses through, either inducing the expression of ABCE1 or exercising a unique inhibitory property of a segment of the viral RNA (Martinand et al., 1998, 1999; Townsend et al., 2008; Sorgeloos et al., 2013).
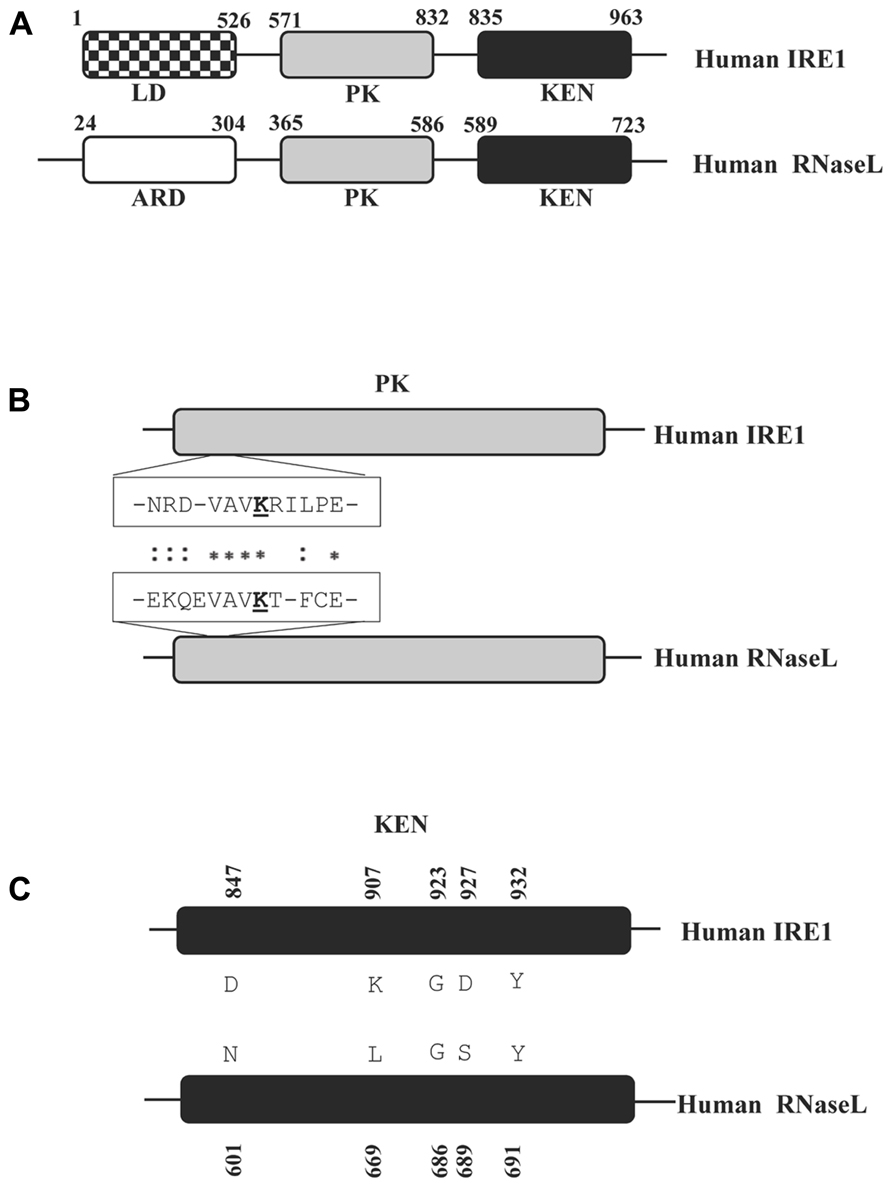
FIGURE 2. Schematic representation of distinct protein domains in human RNaseL and IRE1. (A) The domains homologous between RNaseL and IRE1 are shaded identically. The domain name abbreviations denote the following: ARD = ankyrin repeat domain; LD = luminal domain; PK = protein kinase domain; KEN = kinase extension nuclease domain. The amino acid positions bordering each domain are numbered. The schematic drawings are not according to scale. (B) ClustalW alignment of primary sequence from a segment of the PK domain indicating amino acid residues which are important for interacting with nucleotide cofactors. The conserved lysine residues, critical for this interaction (K599 for IRE1 and K392 in RNaseL) are underlined. (C) Alignment of the KEN domains in RNaseL and IRE1. The amino acids highlighted and numbered in IRE1 are critical for the IRE1 RNase activity (Tirasophon et al., 2000).
Once activated by 2–5A, RNaseL can degrade single-stranded RNA irrespective of its origin (virus or host) although there seems to exist a bias towards cleavage of viral RNA (Wreschner et al., 1981a; Silverman et al., 1983; Li et al., 1998). RNA sequences that are predominantly cleaved by RNaseL are U-rich with the cleavage points being typically at the 3′ end of UA or UG or UU di-nucleotides, leaving a 5′-OH and a 3′-monophosphate in the cleavage product (Floyd-Smith et al., 1981; Wreschner et al., 1981b). A recent report shows a more general consensus of 5′-UNN-3′ with the cleavage point between the second and the third nucleotide (Han et al., 2014). Cellular targets of RNaseL include both ribosomal RNA (rRNA) and mRNAs, the latter predominantly representing genes involved in protein biosynthesis (Wreschner et al., 1981a; Al-Ahmadi et al., 2009; Andersen et al., 2009). Additionally, RNaseL activity can also degrade specific ISG mRNA transcripts and thereby attenuate the effect of IFN signaling (Li et al., 2000). Probably an evolution towards insulating gene expression from RNaseL activity is observed in the coding region of mammalian genes where the UU/UA dinucleotide frequency is rarer (Bisbal et al., 2000; Khabar et al., 2003; Al-Saif and Khabar, 2012). Perhaps not surprisingly, with a much faster rate of evolution, similar observations have been made with respect to evasion of RNaseL mediated degradation by viral RNAs too (Han and Barton, 2002; Washenberger et al., 2007). Moreover, nucleoside modifications in host mRNAs, rarely observed in viral RNAs, have also been shown to confer protection from RNaseL (Anderson et al., 2011). In addition to directly targeting viral RNA, the reduction in functional ribosomes and ribosomal protein mRNA affects viral protein synthesis and replication in an indirect manner. Probably, as a reflection of these effects on cellular RNAs, RNaseL is implicated as one of the factors determining the anti-proliferative effect of IFN activity (Hassel et al., 1993). The anti-viral activity of RNaseL extends beyond direct cleavage of viral RNA, through stimulation of RIG-I by the cleavage product (Malathi et al., 2005, 2007, 2010). A global effect of RNaseL is observed in the form of autophagy induced through c-jun N-terminal kinase (JNK) signaling and apoptosis, probably as a consequence of rRNA cleavage (Li et al., 2004; Chakrabarti et al., 2012; Siddiqui and Malathi, 2012). RNaseL has also been demonstrated to play a role in apoptotic cell death initiated by pharmacological agents extending the physiological role of this pathway beyond the boundary of being only an anti-viral mechanism (Castelli et al., 1997, 1998).
IRE1 and the RIDD Pathway
The ER serves as a conduit for maturation of cellular proteins which are either secreted or destined to be associated with a membrane for its function. An exclusive microenvironment (high Calcium ion and unique ratio of reduced to oxidized glutathione) along with a battery of ER-lumen resident enzymes (foldases, chaperones, and lectins) catalyse/mediate the necessary folding, disulfide-bond formation, and glycosylation reactions (Schroder and Kaufman, 2005). A perturbation of the folding capacity, due to either physiological disturbances or virus infection, can lead to an accumulation of unfolded proteins in the ER lumen, which signals an unfolded protein response (UPR). UPR encompasses a networked transcriptional and translational gene-expression program, initiated by three ER-membrane resident sensors namely IRE1 or ERN1, PKR-like ER Kinase (PERK or EIF2AK3) and activating transcription factor 6 (ATF6; Hetz, 2012). IRE1 is a type I single-pass trans-membrane protein in which, similar to what is observed with RNaseL, the N-terminal resident in the ER lumen serves as sensor and the cytosolic C-terminal as the effector (Figure 1; Chen and Brandizzi, 2013). The IRE1 coding gene is present in genomes ranging from yeast to mammals and in the latter is ubiquitously expressed in all tissues (Tirasophon et al., 1998). Signal transduction by stimulated IRE1 initiates multiple gene regulatory pathways with either pro-survival or pro-apoptotic consequences (Kaufman, 1999). During homeostasis or unstressed conditions the sensor molecules are monomeric, a state maintained co-operatively by the “ absence” of unfolded proteins and the “presence” of HSPA5 (GRP78 or Bip, an ER-resident chaperone) molecules bound to a membrane-proximal disordered segment of the protein in the ER-lumen-resident N-terminus (Credle et al., 2005). Accumulated unfolded proteins in the lumen triggers coupling of this domain from adjacent sensor molecules through a combination of (a) titration of the bound HSPA5 chaperone molecules and (b) direct tethering by malfolded protein molecules (Shamu and Walter, 1996; Credle et al., 2005; Aragon et al., 2009; Korennykh et al., 2009). Abutting of the luminal domains juxtapose the cytosolic C-terminal segments, leading to an aggregation of the IRE1 molecules into distinct ER-membrane foci (Kimata et al., 2007; Li et al., 2010). The C-terminal segment has a serine/threonine kinase domain and a RNase domain homologous to that of RNaseL (Figure 1; Tirasophon et al., 1998, 2000). A trans-autophosphorylation by the kinase domain allosterically activates the RNase domain (Tirasophon et al., 2000; Lee et al., 2008; Korennykh et al., 2009). In fact, exogenous over-expression of IRE1 in mammalian cells lead to activation suggesting that, under homeostatic conditions, the non-juxtaposition of cytosolic domains maintains an inactive IRE1 (Tirasophon et al., 1998). Once activated, IRE1 performs cleavage of a variety of RNA substrates mediated by its RNase domain, in addition to phosphorylating and thereby activating JNK (Cox and Walter, 1996; Urano et al., 2000). Depending on the RNA substrate, the cleavage catalyzed by IRE1 RNase produces differential consequence. Although scission of the Xbp1 mRNA transcript at two internal positions is followed by splicing of the internal segment through ligation of the terminal cleavage products, that in all other known IRE1 target RNA is followed by degradation (Figure 1; Sidrauski and Walter, 1997; Calfon et al., 2002). The latter mode of negative regulation of gene expression is termed as the regulated IRE1-dependent decay (RIDD) pathway (Hollien and Weissman, 2006; Oikawa et al., 2007; Iqbal et al., 2008; Lipson et al., 2008). Gene transcripts regulated by RIDD pathway includes that from IRE1 (i.e., self-transcripts), probably in a negative feedback loop mechanism (Tirasophon et al., 2000). In addition to protein coding RNA, RIDD pathway down-regulates the level of a host of microRNA precursors (pre-miRNAs) and can potentially cleave in the anti-codon loop of tRNAPhe (Korennykh et al., 2011; Upton et al., 2012).
The IRE1 RNase domain cleaves the Xbp1u (u for unspliced) mRNA transcript at two precise internal positions within the open reading frame (ORF) generating three segments, the terminal two of which are ligated by a tRNA ligase in yeast and by an unknown ligase in mammalian cells, to produce the Xbp1s (s for spliced) mRNA transcript (Figure 1; Yoshida et al., 2001). The Xbp1s thus generated has a longer ORF, which is created by a frame-shift in the coding sequence downstream of the splice site (Cox and Walter, 1996; Calfon et al., 2002). A similar dual endonucleolytic cleavage is also observed to initiate the XRN1 and Ski2-3-8 dependent degradation of transcripts in the RIDD degradation pathway (Hollien and Weissman, 2006). The RIDD target transcript genes are predominantly those that encode membrane-associated or secretory proteins and which are not necessary for ER protein-folding reactions (Hollien and Weissman, 2006). The cleavage of Xbp1 and the RIDD-target transcripts constitute homeostatic or pro-survival response by IRE1 since XBP1S trans-activates genes encoding multiple chaperones (to fold unfolded proteins) and the ERAD pathway genes (to degrade terminally misfolded proteins) whereas RIDD reduces flux of polypeptides entering the ER lumen (Lee et al., 2003; Hollien and Weissman, 2006). On the other hand, cleavage of pre-miRNA transcripts which are processed in the cell to generate CASPASE-2 mRNA (Casp2) controlling miRNAs, constitutes the pro-apoptotic function of IRE1 (Upton et al., 2012). Another pro-apoptotic signal from IRE1 emanates from signaling through phosphorylation of JNK1 (Urano et al., 2000). Although in the initial phase RIDD activity does not cleave mRNAs encoding essential ER proteins, at later stages of chronic UPR such transcripts are rendered susceptible to degradation promoting apoptosis induction (Han et al., 2009; Bhattacharyya et al., 2014).
Infection of mammalian cells by a multitude of viruses induce an UPR which is sometimes characterized by suppression of signaling by one or more of the three sensor(s; Su et al., 2002; Tardif et al., 2002; He, 2006; Yu et al., 2006, 2013; Medigeshi et al., 2007; Zhang et al., 2010; Merquiol et al., 2011). Among these at least two viruses from diverse families, HCMV (a DNA virus) and hepatitis C virus (a hepacivirus), interfere with IRE1 signaling by different mechanism (Tardif et al., 2004; Stahl et al., 2013). An observed inhibition of any cellular function by a virus infection could suggest a potential anti-virus function for it, which the virus has evolved to evade through blocking some critical step(s). In both the cases mentioned above, stability of the viral proteins seems to be affected by ERAD-mediated degradation, although other potential anti-viral effect of IRE1 activation are not clear yet (Isler et al., 2005; Saeed et al., 2011). Interestingly, host mRNA fragments produced following IRE1 activation during bacterial infection, has been shown to activate RIG-I signaling (Figure 1; Cho et al., 2013b). Theoretically, other functions of IRE1 can also have anti-viral effect necessitating its inhibition for uninhibited viral replication. It is, however, still not clear whether IRE1 is able to cleave any viral RNA (or mRNA) in a manner similar to that of other RIDD targets (Figure 1). The possibilities of such a direct anti-viral function are encouraged by the fact that all these viruses encode at least one protein which, as part of its maturation process, requires glycosylation and disulfide-bond formation. Such a necessity would entail translation of the mRNA encoding such a protein, which in case of positive-sense single-stranded RNA viruses would mean the genome, in association with the ER-membrane (Figure 1; Lerner et al., 2003). Additionally for many RNA viruses, replication complexes are housed in ER-derived vesicular structures (Denison, 2008; den Boon et al., 2010). Considering the proximity of IRE1 and these virus-derived RNAs it is tempting to speculate that probably at some point of time in the viral life cycle one or more virus-associated RNA would be susceptible to cleavage by IRE1. However, studies with at least two viruses have shown that instead of increasing viral titre, inhibiting the RNase activity of activated IRE1 has an opposite effect (Hassan et al., 2012; Bhattacharyya et al., 2014). This implies potential benefits of IRE1 activation through one or more of the following, (a) expression of chaperones or other pro-viral molecules downstream of XBP1S-upregulation or JNK-activation, (b) cleavage of potential anti-viral gene mRNA transcripts by RIDD activity. However, the mode of protection for the viral RNA from RIDD activity is still not clear. It is possible that the viral proteins create a subdomain within the ER membrane, which through some mechanism excludes IRE1 from diffusing near the genomic RNA, thereby protecting the replication complexes (Denison, 2008). It is therefore probably not surprising that single-stranded plus-sense RNA viruses encode a polyprotein, which produces replication complexes in cis, promoting formation of such subdomains (Egger et al., 2000). The fact that IRE1 forms bulky oligomers of higher order probably aggravates such an exclusion of the activated sensor molecules from vicinity of the viral replication complexes. The UPR signaling eventually attenuate during chronic ER-stress and since that is what a virus-induced UPR mimics, probably the viral RNA needs protection only during the initial phase of UPR activation (Lin et al., 2007). Since the choice of RIDD target seems to be grossly driven towards mRNAs that encode ER-transitory but are not ER-essential proteins, it is also possible that one or more viral protein have evolved to mimic a host protein the transcript of which is RIDD-resistant (Hollien and Weissman, 2006). Most of the RIDD target mRNA are observed to be ER-membrane associated, the proximity to IRE1 facilitating association and cleavage (Figure 1; Hollien and Weissman, 2006). Although ER-association for an mRNA is possible without the mediation of ribosomes, Gaddam and co-workers reported that continued association with polysomes for a membrane-bound mRNA can confer protection from IRE1 cleavage (Cui et al., 2012; Gaddam et al., 2013). This would suggest important implications for the observed refractory nature of Japanese encephalitis virus (JEV) and influenza virus RNA to RIDD cleavage (Hassan et al., 2012; Bhattacharyya et al., 2014). In contrast to Influenza virus, flaviviruses (which include JEV) do not suppress host protein synthesis implying the absence of a global inhibition on translation as would be expected during UPR (Clyde et al., 2006; Edgil et al., 2006). Therefore, a continued translation of viral RNA in spite of UPR activation can in principle confer protection from the pattern of RNA cleavage observed in the RIDD pathway.
Comparison of IRE1 and RNaseL
IRE1 and RNaseL, in addition to biochemical similarities in protein kinase domain and structural similarities in their RNase domain, share the functional consequences of their activation in initiating cellular apoptosis through JNK signaling (Table 1 and Figure 2; Liu and Lin, 2005; Dhanasekaran and Reddy, 2008). Though initial discoveries were made in the context of homeostatic and anti-viral role for the former and latter, differences between the pathways are narrowed by further advances in research. In the same vein, while inhibition of IRE1 signaling in virus infected cells indicates a potential anti-viral role, association of RNaseL mutations with generation of prostate cancer extends the ambit of influence of this anti-viral effector to more non-infectious physiological disorders (Silverman, 2003). Biochemically, the similarity in their RNase domains does not extend to the choice of either substrates or cleavage point, which are downstream of UU or UA in RNaseL and downstream of G (predominantly) for IRE1 (Figure 2C; Yoshida et al., 2001; Hollien and Weissman, 2006; Upton et al., 2012). Further, while RNaseL cleaves pre-dominantly in single-stranded region, IRE1 seems to cleave equally well in single- and double-stranded region (Upton et al., 2012). However, a recent report suggested a consensus cleavage site with the sequence UN/N, in RNaseL targets and in those mRNAs that are cleaved by IRE1 as part of the RIDD pathway (Han et al., 2014). Access to potential cleavage substrate for RNaseL is conjectured to be facilitated through its association with polyribosomes, while no such association is known for IRE1 (Salehzada et al., 1991). Possibilities exist that IRE1 would have preferential distribution in the rough ER which, upon activation, would give it ready access to mRNAs for initiating the RIDD pathway.
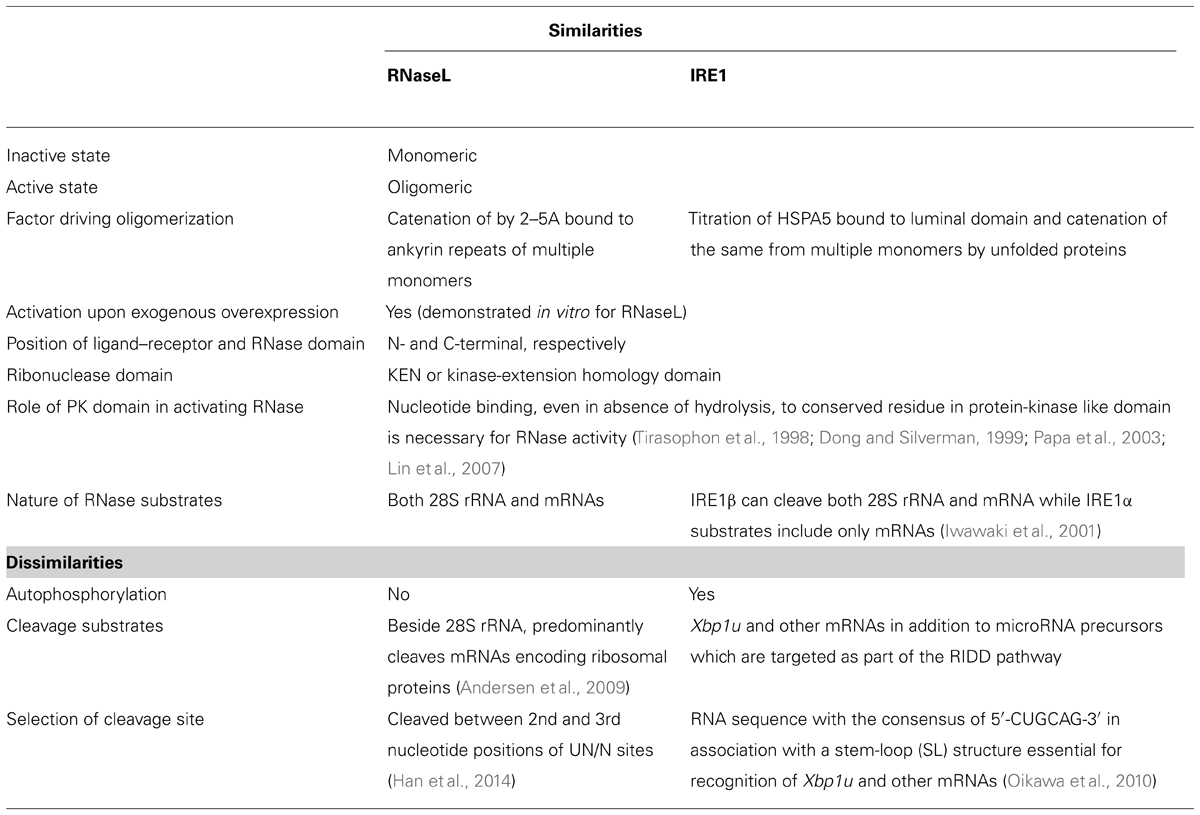
TABLE 1. A comparison of the structural and biochemical properties of RNaseL and IRE1, showing similarities and differences.
In the context of a virus infection, the pathway leading from both these proteins have the potential to lead to cell death. Notwithstanding the fact that this might be an efficient way of virus clearance, it also portends pathological outcomes for the infected organism. Future research would probably lead to design of drugs targeting these proteins based on the structural homology of their effector domains, regulating the pathological denouement of their activation without compromising their anti-viral or potential anti-viral functions.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The research carried out was supported by intramural funding from THSTI. I would like to thank Dr. Manjula Kalia for careful reading of the manuscript.
References
Ahlquist, P. (2006). Parallels among positive-strand RNA viruses, reverse-transcribing viruses and double-stranded RNA viruses. Nat. Rev. Microbiol. 4, 371–382. doi: 10.1038/nrmicro1389
Al-Ahmadi, W., Al-Haj, L., Al-Mohanna, F. A., Silverman, R. H., and Khabar, K. S. (2009). Downmodulation of the RNA-binding protein, HuR, and cellular growth. Oncogene 28, 1782–1791. doi: 10.1038/onc.2009.16
Al-Saif, M., and Khabar, K. S. (2012). UU/UA dinucleotide frequency reduction in coding regions results in increased mRNA stability and protein expression. Mol. Ther. 20, 954–959. doi: 10.1038/mt.2012.29
Andersen, J. B., Mazan-Mamczarz, K., Zhan, M., Gorospe, M., and Hassel, B. A. (2009). Ribosomal protein mRNAs are primary targets of regulation in RNase-L-induced senescence. RNA Biol. 6, 305–315. doi: 10.4161/rna.6.3.8526
Anderson, B. R., Muramatsu, H., Jha, B. K., Silverman, R. H., Weissman, D., and Kariko, K. (2011). Nucleoside modifications in RNA limit activation of 2′-5′-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res. 39, 9329–9338. doi: 10.1093/nar/gkr586
Aoshi, T., Koyama, S., Kobiyama, K., Akira, S., and Ishii, K. J. (2011). Innate and adaptive immune responses to viral infection and vaccination. Curr. Opin. Virol. 1, 226–232. doi: 10.1016/j.coviro.2011.07.002
Aragon, T., Van Anken, E., Pincus, D., Serafimova, I. M., Korennykh, A. V., Rubio, C. A., et al. (2009). Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature 457, 736–740. doi: 10.1038/nature07641
Beattie, E., Denzler, K. L., Tartaglia, J., Perkus, M. E., Paoletti, E., and Jacobs, B. L. (1995). Reversal of the interferon-sensitive phenotype of a vaccinia virus lacking E3L by expression of the reovirus S4 gene. J. Virol. 69, 499–505.
Bhattacharyya, S., Sen, U., and Vrati, S. (2014). Regulated IRE1-dependent decay pathway is activated during Japanese encephalitis virus-induced unfolded protein response and benefits viral replication. J. Gen. Virol. 95(Pt 1), 71–79. doi: 10.1099/vir.0.057265-0
Bisbal, C., Martinand, C., Silhol, M., Lebleu, B., and Salehzada, T. (1995). Cloning and characterization of a RNAse L inhibitor. A new component of the interferon-regulated 2–5A pathway. J. Biol. Chem. 270, 13308–13317. doi: 10.1074/jbc.270.22.13308
Bisbal, C., Silhol, M., Laubenthal, H., Kaluza, T., Carnac, G., Milligan, L., et al. (2000). The 2′–5′ oligoadenylate/RNase L/RNase L inhibitor pathway regulates both MyoD mRNA stability and muscle cell differentiation. Mol. Cell. Biol. 20, 4959–4969. doi: 10.1128/MCB.20.14.4959-4969.2000
Bowzard, J. B., Davis, W. G., Jeisy-Scott, V., Ranjan, P., Gangappa, S., Fujita, T., et al. (2011). PAMPer and tRIGer: ligand-induced activation of RIG-I. Trends Biochem. Sci. 36, 314–319. doi: 10.1016/j.tibs.2011.03.003
Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., et al. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96. doi: 10.1038/415092a
Castelli, J. C., Hassel, B. A., Maran, A., Paranjape, J., Hewitt, J. A., Li, X. L., et al. (1998). The role of 2′–5′ oligoadenylate-activated ribonuclease L in apoptosis. Cell Death Differ. 5, 313–320. doi: 10.1038/sj.cdd.4400352
Castelli, J. C., Hassel, B. A., Wood, K. A., Li, X. L., Amemiya, K., Dalakas, M. C., et al. (1997). A study of the interferon antiviral mechanism: apoptosis activation by the 2–5A system. J. Exp. Med. 186, 967–972. doi: 10.1084/jem.186.6.967
Cayley, P. J., Davies, J. A., Mccullagh, K. G., and Kerr, I. M. (1984). Activation of the ppp(A2′p)nA system in interferon-treated, herpes simplex virus-infected cells and evidence for novel inhibitors of the ppp(A2′p)nA-dependent RNase. Eur. J. Biochem. 143, 165–174. doi: 10.1111/j.1432-1033.1984.tb08355.x
Chakrabarti, A., Ghosh, P. K., Banerjee, S., Gaughan, C., and Silverman, R. H. (2012). RNase L triggers autophagy in response to viral infections. J. Virol. 86, 11311–11321. doi: 10.1128/JVI.00270-12
Chen, Y., and Brandizzi, F. (2013). IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 23, 547–555. doi: 10.1016/j.tcb.2013.06.005
Child, S. J., Hakki, M., De Niro, K. L., and Geballe, A. P. (2004). Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J. Virol. 78, 197–205. doi: 10.1128/JVI.78.1.197-205.2004
Cho, H., Shrestha, B., Sen, G. C., and Diamond, M. S. (2013a). A role for Ifit2 in restricting West Nile virus infection in the brain. J. Virol. 87, 8363–8371. doi: 10.1128/JVI.01097-13
Cho, J. A., Lee, A. H., Platzer, B., Cross, B. C., Gardner, B. M., De Luca, H., et al. (2013b). The unfolded protein response element IRE1alpha senses bacterial proteins invading the ER to activate RIG-I and innate immune signaling. Cell Host Microbe 13, 558–569. doi: 10.1016/j.chom.2013.03.011
Clyde, K., Kyle, J. L., and Harris, E. (2006). Recent advances in deciphering viral and host determinants of dengue virus replication and pathogenesis. J. Virol. 80, 11418–11431. doi: 10.1128/JVI.01257-06
Cox, J. S., and Walter, P. (1996). A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 87, 391–404. doi: 10.1016/S0092-8674(00)81360-4
Credle, J. J., Finer-Moore, J. S., Papa, F. R., Stroud, R. M., and Walter, P. (2005). On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 102, 18773–18784. doi: 10.1073/pnas.0509487102
Cui, X. A., Zhang, H., and Palazzo, A. F. (2012). p180 promotes the ribosome-independent localization of a subset of mRNA to the endoplasmic reticulum. PLoS Biol. 10:e1001336. doi: 10.1371/journal.pbio.1001336
Cusato, K., Bosco, A., Rozental, R., Guimaraes, C. A., Reese, B. E., Linden, R., et al. (2003). Gap junctions mediate bystander cell death in developing retina. J. Neurosci. 23, 6413–6422.
Daffis, S., Szretter, K. J., Schriewer, J., Li, J., Youn, S., Errett, J., et al. (2010). 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468, 452–456. doi: 10.1038/nature09489
den Boon, J. A., Diaz, A., and Ahlquist, P. (2010). Cytoplasmic viral replication complexes. Cell Host Microbe 8, 77–85. doi: 10.1016/j.chom.2010.06.010
Denison, M. R. (2008). Seeking membranes: positive-strand RNA virus replication complexes. PLoS Biol. 6:e270. doi: 10.1371/journal.pbio.0060270
Dhanasekaran, D. N., and Reddy, E. P. (2008). JNK signaling in apoptosis. Oncogene 27, 6245–6251. doi: 10.1038/onc.2008.301
Dong, B., and Silverman, R. H. (1997). A bipartite model of 2–5A-dependent RNase L. J. Biol. Chem. 272, 22236–22242. doi: 10.1074/jbc.272.35.22236
Dong, B., and Silverman, R. H. (1999). Alternative function of a protein kinase homology domain in 2′, 5′-oligoadenylate dependent RNase L. Nucleic Acids Res. 27, 439–445. doi: 10.1093/nar/27.2.439
Dong, B., Xu, L., Zhou, A., Hassel, B. A., Lee, X., Torrence, P. F., et al. (1994). Intrinsic molecular activities of the interferon-induced 2-5A-dependent RNase. J. Biol. Chem. 269, 14153–14158.
Edgil, D., Polacek, C., and Harris, E. (2006). Dengue virus utilizes a novel strategy for translation initiation when cap-dependent translation is inhibited. J. Virol. 80, 2976–2986. doi: 10.1128/JVI.80.6.2976-2986.2006
Egger, D., Teterina, N., Ehrenfeld, E., and Bienz, K. (2000). Formation of the poliovirus replication complex requires coupled viral translation, vesicle production, and viral RNA synthesis. J. Virol. 74, 6570–6580. doi: 10.1128/JVI.74.14.6570-6580.2000
Floyd-Smith, G., Slattery, E., and Lengyel, P. (1981). Interferon action: RNA cleavage pattern of a (2′–5′)oligoadenylate-dependent endonuclease. Science 212, 1030–1032. doi: 10.1126/science.6165080
Gaddam, D., Stevens, N., and Hollien, J. (2013). Comparison of mRNA localization and regulation during endoplasmic reticulum stress in Drosophila cells. Mol. Biol. Cell 24, 14–20. doi: 10.1091/mbc.E12-06-0491
Gao, G., Guo, X., and Goff, S. P. (2002). Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science 297, 1703–1706. doi: 10.1126/science.1074276
Guo, X., Ma, J., Sun, J., and Gao, G. (2007). The zinc-finger antiviral protein recruits the RNA processing exosome to degrade the target mRNA. Proc. Natl. Acad. Sci. U.S.A. 104, 151–156. doi: 10.1073/pnas.0607063104
Han, D., Lerner, A. G., Vande Walle, L., Upton, J. P., Xu, W., Hagen, A., et al. (2009). IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138, 562–575. doi: 10.1016/j.cell.2009.07.017
Han, J. Q., and Barton, D. J. (2002). Activation and evasion of the antiviral 2′–5′ oligoadenylate synthetase/ribonuclease L pathway by hepatitis C virus mRNA. RNA 8, 512–525. doi: 10.1017/S1355838202020617
Han, Y., Donovan, J., Rath, S., Whitney, G., Chitrakar, A., and Korennykh, A. (2014). Structure of human RNase L reveals the basis for regulated RNA decay in the IFN response. Science 343, 1244–1248. doi: 10.1126/science.1249845
Han, Y., Whitney, G., Donovan, J., and Korennykh, A. (2012). Innate immune messenger 2–5A tethers human RNase L into active high-order complexes. Cell Rep. 2, 902–913. doi: 10.1016/j.celrep.2012.09.004
Hartmann, R., Justesen, J., Sarkar, S. N., Sen, G. C., and Yee, V. C. (2003). Crystal structure of the 2′-specific and double-stranded RNA-activated interferon-induced antiviral protein 2′–5′-oligoadenylate synthetase. Mol. Cell 12, 1173–1185. doi: 10.1016/S1097-2765(03)00433-7
Hassan, I. H., Zhang, M. S., Powers, L. S., Shao, J. Q., Baltrusaitis, J., Rutkowski, D. T., et al. (2012). Influenza A viral replication is blocked by inhibition of the inositol-requiring enzyme 1 (IRE1) stress pathway. J. Biol. Chem. 287, 4679–4689. doi: 10.1074/jbc.M111.284695
Hassel, B. A., Zhou, A., Sotomayor, C., Maran, A., and Silverman, R. H. (1993). A dominant negative mutant of 2–5A-dependent RNase suppresses antiproliferative and antiviral effects of interferon. EMBO J. 12, 3297–3304.
He, B. (2006). Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 13, 393–403. doi: 10.1038/sj.cdd.4401833
Hersh, C. L., Brown, R. E., Roberts, W. K., Swyryd, E. A., Kerr, I. M., and Stark, G. R. (1984). Simian virus 40-infected, interferon-treated cells contain 2′,5′-oligoadenylates which do not activate cleavage of RNA. J. Biol. Chem. 259, 1731–1737.
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102.
Hollien, J., and Weissman, J. S. (2006). Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313, 104–107. doi: 10.1126/science.1129631
Iqbal, J., Dai, K., Seimon, T., Jungreis, R., Oyadomari, M., Kuriakose, G., et al. (2008). IRE1beta inhibits chylomicron production by selectively degrading MTP mRNA. Cell Metab. 7, 445–455. doi: 10.1016/j.cmet.2008.03.005
Isler, J. A., Skalet, A. H., and Alwine, J. C. (2005). Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 79, 6890–6899. doi: 10.1128/JVI.79.11.6890-6899.2005
Iwawaki, T., Hosoda, A., Okuda, T., Kamigori, Y., Nomura-Furuwatari, C., Kimata, Y., et al. (2001). Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nat. Cell Biol. 3, 158–164. doi: 10.1038/35055065
Jacobs, B. L., and Langland, J. O. (1996). When two strands are better than one: the mediators and modulators of the cellular responses to double-stranded RNA. Virology 219, 339–349. doi: 10.1006/viro.1996.0259
Jensen, S., and Thomsen, A. R. (2012). Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 86, 2900–2910. doi: 10.1128/JVI.05738-11
Johnston, M. I., and Hearl, W. G. (1987). Purification and characterization of a 2′-phosphodiesterase from bovine spleen. J. Biol. Chem. 262, 8377–8382.
Kameritsch, P., Khandoga, N., Pohl, U., and Pogoda, K. (2013). Gap junctional communication promotes apoptosis in a connexin-type-dependent manner. Cell Death Dis. 4:e584. doi: 10.1038/cddis.2013.105
Kaufman, R. J. (1999). Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 13, 1211–1233. doi: 10.1101/gad.13.10.1211
Khabar, K. S., Siddiqui, Y. M., Al-Zoghaibi, F., Al-Haj, L., Dhalla, M., Zhou, A., et al. (2003). RNase L mediates transient control of the interferon response through modulation of the double-stranded RNA-dependent protein kinase PKR. J. Biol. Chem. 278, 20124–20132. doi: 10.1074/jbc.M208766200
Kimata, Y., Ishiwata-Kimata, Y., Ito, T., Hirata, A., Suzuki, T., Oikawa, D., et al. (2007). Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J. Cell Biol. 179, 75–86. doi: 10.1083/jcb.200704166
Knight, M., Cayley, P. J., Silverman, R. H., Wreschner, D. H., Gilbert, C. S., Brown, R. E., et al. (1980). Radioimmune, radiobinding and HPLC analysis of 2–5A and related oligonucleotides from intact cells. Nature 288, 189–192. doi: 10.1038/288189a0
Korennykh, A. V., Egea, P. F., Korostelev, A. A., Finer-Moore, J., Zhang, C., Shokat, K. M., et al. (2009). The unfolded protein response signals through high-order assembly of Ire1. Nature 457, 687–693. doi: 10.1038/nature07661
Korennykh, A. V., Korostelev, A. A., Egea, P. F., Finer-Moore, J., Stroud, R. M., Zhang, C., et al. (2011). Structural and functional basis for RNA cleavage by Ire1. BMC Biol. 9:47. doi: 10.1186/1741-7007-9-47
Kubota, K., Nakahara, K., Ohtsuka, T., Yoshida, S., Kawaguchi, J., Fujita, Y., et al. (2004). Identification of 2′-phosphodiesterase, which plays a role in the 2–5A system regulated by interferon. J. Biol. Chem. 279, 37832–37841. doi: 10.1074/jbc.M400089200
Lee, A. H., Iwakoshi, N. N., and Glimcher, L. H. (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003
Lee, K. P., Dey, M., Neculai, D., Cao, C., Dever, T. E., and Sicheri, F. (2008). Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell 132, 89–100. doi: 10.1016/j.cell.2007.10.057
Lee, T. Y., Ezelle, H. J., Venkataraman, T., Lapidus, R. G., Scheibner, K. A., and Hassel, B. A. (2013). Regulation of human RNase-L by the miR-29 family reveals a novel oncogenic role in chronic myelogenous leukemia. J. Interferon Cytokine Res. 33, 34–42. doi: 10.1089/jir.2012.0062
Lerner, R. S., Seiser, R. M., Zheng, T., Lager, P. J., Reedy, M. C., Keene, J. D., et al. (2003). Partitioning and translation of mRNAs encoding soluble proteins on membrane-bound ribosomes. RNA 9, 1123–1137. doi: 10.1261/rna.5610403
Li, G., Xiang, Y., Sabapathy, K., and Silverman, R. H. (2004). An apoptotic signaling pathway in the interferon antiviral response mediated by RNase L and c-Jun NH2-terminal kinase. J. Biol. Chem. 279, 1123–1131. doi: 10.1074/jbc.M305893200
Li, H., Korennykh, A. V., Behrman, S. L., and Walter, P. (2010). Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. U.S.A. 107, 16113–16118. doi: 10.1073/pnas.1010580107
Li, X. L., Blackford, J. A., and Hassel, B. A. (1998). RNase L mediates the antiviral effect of interferon through a selective reduction in viral RNA during encephalomyocarditis virus infection. J. Virol. 72, 2752–2759.
Li, X. L., Blackford, J. A., Judge, C. S., Liu, M., Xiao, W., Kalvakolanu, D. V., et al. (2000). RNase-L-dependent destabilization of interferon-induced mRNAs. A role for the 2–5A system in attenuation of the interferon response. J. Biol. Chem. 275, 8880–8888. doi: 10.1074/jbc.275.12.8880
Lin, J. H., Li, H., Yasumura, D., Cohen, H. R., Zhang, C., Panning, B., et al. (2007). IRE1 signaling affects cell fate during the unfolded protein response. Science 318, 944–949. doi: 10.1126/science.1146361
Lin, R. J., Chien, H. L., Lin, S. Y., Chang, B. L., Yu, H. P., Tang, W. C., et al. (2013). MCPIP1 ribonuclease exhibits broad-spectrum antiviral effects through viral RNA binding and degradation. Nucleic Acids Res. 41, 3314–3326. doi: 10.1093/nar/gkt019
Lin, R. J., Yu, H. P., Chang, B. L., Tang, W. C., Liao, C. L., and Lin, Y. L. (2009). Distinct antiviral roles for human 2′,5′-oligoadenylate synthetase family members against dengue virus infection. J. Immunol. 183, 8035–8043. doi: 10.4049/jimmunol.0902728
Lipson, K. L., Ghosh, R., and Urano, F. (2008). The role of IRE1alpha in the degradation of insulin mRNA in pancreatic beta-cells. PLoS ONE 3:e1648. doi: 10.1371/journal.pone.0001648
Liu, J., and Lin, A. (2005). Role of JNK activation in apoptosis: a double-edged sword. Cell Res. 15, 36–42. doi: 10.1038/sj.cr.7290262
Maitra, R. K., Mcmillan, N. A., Desai, S., Mcswiggen, J., Hovanessian, A. G., Sen, G., et al. (1994). HIV-1 TAR RNA has an intrinsic ability to activate interferon-inducible enzymes. Virology 204, 823–827. doi: 10.1006/viro.1994.1601
Malathi, K., Dong, B., Gale, M. Jr., and Silverman, R. H. (2007). Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448, 816–819. doi: 10.1038/nature06042
Malathi, K., Paranjape, J. M., Bulanova, E., Shim, M., Guenther-Johnson, J. M., Faber, P. W., et al. (2005). A transcriptional signaling pathway in the IFN system mediated by 2′–5′-oligoadenylate activation of RNase L. Proc. Natl. Acad. Sci. U.S.A. 102, 14533–14538. doi: 10.1073/pnas.0507551102
Malathi, K., Saito, T., Crochet, N., Barton, D. J., Gale, M. Jr., and Silverman, R. H. (2010). RNase L releases a small RNA from HCV RNA that refolds into a potent PAMP. RNA 16, 2108–2119. doi: 10.1261/rna.2244210
Marie, I., Svab, J., Robert, N., Galabru, J., and Hovanessian, A. G. (1990). Differential expression and distinct structure of 69- and 100-kDa forms of 2–5A synthetase in human cells treated with interferon. J. Biol. Chem. 265, 18601–18607.
Martinand, C., Montavon, C., Salehzada, T., Silhol, M., Lebleu, B., and Bisbal, C. (1999). RNase L inhibitor is induced during human immunodeficiency virus type 1 infection and down regulates the 2-5A/RNase L pathway in human T cells. J. Virol. 73, 290–296.
Martinand, C., Salehzada, T., Silhol, M., Lebleu, B., and Bisbal, C. (1998). RNase L inhibitor (RLI) antisense constructions block partially the down regulation of the 2–5A/RNase L pathway in encephalomyocarditis-virus-(EMCV)-infected cells. Eur. J. Biochem. 254, 248–255. doi: 10.1046/j.1432-1327.1998.2540248.x
Medigeshi, G. R., Lancaster, A. M., Hirsch, A. J., Briese, T., Lipkin, W. I., Defilippis, V., et al. (2007). West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J. Virol. 81, 10849–10860. doi: 10.1128/JVI.01151-07
Merquiol, E., Uzi, D., Mueller, T., Goldenberg, D., Nahmias, Y., Xavier, R. J., et al. (2011). HCV causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response. PLoS ONE 6:e24660. doi: 10.1371/journal.pone.0024660
Miller, S., and Krijnse-Locker, J. (2008). Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 6, 363–374. doi: 10.1038/nrmicro1890
Min, J. Y., and Krug, R. M. (2006). The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2′–5′ oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. U.S.A. 103, 7100–7105. doi: 10.1073/pnas.0602184103
Minks, M. A., West, D. K., Benvin, S., and Baglioni, C. (1979). Structural requirements of double-stranded RNA for the activation of 2′,5′-oligo(A) polymerase and protein kinase of interferon-treated HeLa cells. J. Biol. Chem. 254, 10180–10183.
Miorin, L., Romero-Brey, I., Maiuri, P., Hoppe, S., Krijnse-Locker, J., Bartenschlager, R., et al. (2013). Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. J. Virol. 87, 6469–6481. doi: 10.1128/JVI.03456-12
Nakanishi, M., Goto, Y., and Kitade, Y. (2005). 2–5A induces a conformational change in the ankyrin-repeat domain of RNase L. Proteins 60, 131–138. doi: 10.1002/prot.20474
Oikawa, D., Tokuda, M., Hosoda, A., and Iwawaki, T. (2010). Identification of a consensus element recognized and cleaved by IRE1 alpha. Nucleic Acids Res. 38, 6265–6273. doi: 10.1093/nar/gkq452
Oikawa, D., Tokuda, M., and Iwawaki, T. (2007). Site-specific cleavage of CD59 mRNA by endoplasmic reticulum-localized ribonuclease, IRE1. Biochem. Biophys. Res. Commun. 360, 122–127. doi: 10.1016/j.bbrc.2007.06.020
Papa, F. R., Zhang, C., Shokat, K., and Walter, P. (2003). Bypassing a kinase activity with an ATP-competitive drug. Science 302, 1533–1537. doi: 10.1126/science.1090031
Randall, R. E., and Goodbourn, S. (2008). Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89, 1–47. doi: 10.1099/vir.0.83391-0
Rice, A. P., Kerr, S. M., Roberts, W. K., Brown, R. E., and Kerr, I. M. (1985). Novel 2′,5′-oligoadenylates synthesized in interferon-treated, vaccinia virus-infected cells. J. Virol. 56, 1041–1044.
Rivas, C., Gil, J., Melkova, Z., Esteban, M., and Diaz-Guerra, M. (1998). Vaccinia virus E3L protein is an inhibitor of the interferon (i.f.n.)-induced 2–5A synthetase enzyme. Virology 243, 406–414. doi: 10.1006/viro.1998.9072
Roulston, A., Marcellus, R. C., and Branton, P. E. (1999). Viruses and apoptosis. Annu. Rev. Microbiol. 53, 577–628. doi: 10.1146/annurev.micro.53.1.577
Saeed, M., Suzuki, R., Watanabe, N., Masaki, T., Tomonaga, M., Muhammad, A., et al. (2011). Role of the endoplasmic reticulum-associated degradation (ERAD) pathway in degradation of hepatitis C virus envelope proteins and production of virus particles. J. Biol. Chem. 286, 37264–37273. doi: 10.1074/jbc.M111.259085
Salehzada, T., Silhol, M., Lebleu, B., and Bisbal, C. (1991). Polyclonal antibodies against RNase L. Subcellular localization of this enzyme in mouse cells. J. Biol. Chem. 266, 5808–5813.
Sanchez, R., and Mohr, I. (2007). Inhibition of cellular 2′–5′ oligoadenylate synthetase by the herpes simplex virus type 1 Us11 protein. J. Virol. 81, 3455–3464. doi: 10.1128/JVI.02520-06
Sarkar, S. N., Bandyopadhyay, S., Ghosh, A., and Sen, G. C. (1999). Enzymatic characteristics of recombinant medium isozyme of 2′–5′ oligoadenylate synthetase. J. Biol. Chem. 274, 1848–1855. doi: 10.1074/jbc.274.3.1848
Schmidt, A., Rothenfusser, S., and Hopfner, K. P. (2012). Sensing of viral nucleic acids by RIG-I: from translocation to translation. Eur. J. Cell Biol. 91, 78–85. doi: 10.1016/j.ejcb.2011.01.015
Schoggins, J. W., and Rice, C. M. (2011). Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 1, 519–525. doi: 10.1016/j.coviro.2011.10.008
Schroder, M., and Kaufman, R. J. (2005). ER stress and the unfolded protein response. Mutat. Res. 569, 29–63. doi: 10.1016/j.mrfmmm.2004.06.056
Sen, G. C., and Sarkar, S. N. (2007). The interferon-stimulated genes: targets of direct signaling by interferons, double-stranded RNA, and viruses. Curr. Top. Microbiol. Immunol. 316, 233–250. doi: 10.1007/978-3-540-71329-6_12
Shamu, C. E., and Walter, P. (1996). Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 15, 3028–3039.
Sheehy, A. M., Gaddis, N. C., Choi, J. D., and Malim, M. H. (2002). Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418, 646–650. doi: 10.1038/nature00939
Sidahmed, A. M., and Wilkie, B. (2010). Endogenous antiviral mechanisms of RNA interference: a comparative biology perspective. Methods Mol. Biol. 623, 3–19. doi: 10.1007/978-1-60761-588-0_1
Siddiqui, M. A., and Malathi, K. (2012). RNase L induces autophagy via c-Jun N-terminal kinase and double-stranded RNA-dependent protein kinase signaling pathways. J. Biol. Chem. 287, 43651–43664. doi: 10.1074/jbc.M112.399964
Sidrauski, C., and Walter, P. (1997). The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90, 1031–1039. doi: 10.1016/S0092-8674(00)80369-4
Silverman, R. H. (2003). Implications for RNase L in prostate cancer biology. Biochemistry 42, 1805–1812. doi: 10.1021/bi027147i
Silverman, R. H. (2007). Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 81, 12720–12729. doi: 10.1128/JVI.01471-07
Silverman, R. H., Skehel, J. J., James, T. C., Wreschner, D. H., and Kerr, I. M. (1983). rRNA cleavage as an index of ppp(A2′p)nA activity in interferon-treated encephalomyocarditis virus-infected cells. J. Virol. 46, 1051–1055.
Silverman, R. H., Wreschner, D. H., Gilbert, C. S., and Kerr, I. M. (1981). Synthesis, characterization and properties of ppp(A2′p)nApCp and related high-specific-activity 32P-labelled derivatives of ppp(A2′p)nA. Eur. J. Biochem. 115, 79–85. doi: 10.1111/j.1432-1033.1981.tb06200.x
Sorgeloos, F., Jha, B. K., Silverman, R. H., and Michiels, T. (2013). Evasion of antiviral innate immunity by Theiler’s virus L* protein through direct inhibition of RNase L. PLoS Pathog. 9:e1003474. doi: 10.1371/journal.ppat.1003474
Stahl, S., Burkhart, J. M., Hinte, F., Tirosh, B., Mohr, H., Zahedi, R. P., et al. (2013). Cytomegalovirus downregulates IRE1 to repress the unfolded protein response. PLoS Pathog. 9:e1003544. doi: 10.1371/journal.ppat.1003544
Su, H. L., Liao, C. L., and Lin, Y. L. (2002). Japanese encephalitis virus infection initiates endoplasmic reticulum stress and an unfolded protein response. J. Virol. 76, 4162–4171. doi: 10.1128/JVI.76.9.4162-4171.2002
Tanaka, N., Nakanishi, M., Kusakabe, Y., Goto, Y., Kitade, Y., and Nakamura, K. T. (2004). Structural basis for recognition of 2′,5′-linked oligoadenylates by human ribonuclease L. EMBO J. 23, 3929–3938. doi: 10.1038/sj.emboj.7600420
Tardif, K. D., Mori, K., Kaufman, R. J., and Siddiqui, A. (2004). Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 279, 17158–17164. doi: 10.1074/jbc.M312144200
Tardif, K. D., Mori, K., and Siddiqui, A. (2002). Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. J. Virol. 76, 7453–7459. doi: 10.1128/JVI.76.15.7453-7459.2002
Thompson, M. R., Kaminski, J. J., Kurt-Jones, E. A., and Fitzgerald, K. A. (2011). Pattern recognition receptors and the innate immune response to viral infection. Viruses 3, 920–940. doi: 10.3390/v3060920
Tirasophon, W., Lee, K., Callaghan, B., Welihinda, A., and Kaufman, R. J. (2000). The endoribonuclease activity of mammalian IRE1 autoregulates its mRNA and is required for the unfolded protein response. Genes Dev. 14, 2725–2736. doi: 10.1101/gad.839400
Tirasophon, W., Welihinda, A. A., and Kaufman, R. J. (1998). A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812–1824. doi: 10.1101/gad.12.12.1812
Townsend, H. L., Jha, B. K., Han, J. Q., Maluf, N. K., Silverman, R. H., and Barton, D. J. (2008). A viral RNA competitively inhibits the antiviral endoribonuclease domain of RNase L. RNA 14, 1026–1036. doi: 10.1261/rna.958908
Uchil, P. D., and Satchidanandam, V. (2003). Architecture of the flaviviral replication complex. Protease, nuclease, and detergents reveal encasement within double-layered membrane compartments. J. Biol. Chem. 278, 24388–24398. doi: 10.1074/jbc.M301717200
Udawatte, C., and Ripps, H. (2005). The spread of apoptosis through gap-junctional channels in BHK cells transfected with Cx32. Apoptosis 10, 1019–1029. doi: 10.1007/s10495-005-0776-8
Upton, J. P., Wang, L., Han, D., Wang, E. S., Huskey, N. E., Lim, L., et al. (2012). IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 338, 818–822. doi: 10.1126/science.1226191
Urano, F., Wang, X., Bertolotti, A., Zhang, Y., Chung, P., Harding, H. P., et al. (2000). Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666. doi: 10.1126/science.287.5453.664
Vabret, N., and Blander, J. M. (2013). Sensing microbial RNA in the cytosol. Front. Immunol. 4:468. doi: 10.3389/fimmu.2013.00468
Washenberger, C. L., Han, J. Q., Kechris, K. J., Jha, B. K., Silverman, R. H., and Barton, D. J. (2007). Hepatitis C virus RNA: dinucleotide frequencies and cleavage by RNase L. Virus Res. 130, 85–95. doi: 10.1016/j.virusres.2007.05.020
Weber, F., Wagner, V., Rasmussen, S. B., Hartmann, R., and Paludan, S. R. (2006). Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 80, 5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006
Wreschner, D. H., James, T. C., Silverman, R. H., and Kerr, I. M. (1981a). Ribosomal RNA cleavage, nuclease activation and 2–5A(ppp(A2′p)nA) in interferon-treated cells. Nucleic Acids Res. 9, 1571–1581. doi: 10.1093/nar/9.7.1571
Wreschner, D. H., Mccauley, J. W., Skehel, J. J., and Kerr, I. M. (1981b). Interferon action–sequence specificity of the ppp(A2′p)nA-dependent ribonuclease. Nature 289, 414–417. doi: 10.1038/289414a0
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. doi: 10.1016/S0092-8674(01)00611-0
Yu, C., Achazi, K., and Niedrig, M. (2013). Tick-borne encephalitis virus triggers inositol-requiring enzyme 1 (IRE1) and transcription factor 6 (ATF6) pathways of unfolded protein response. Virus Res. 178, 471–477. doi: 10.1016/j.virusres.2013.10.012
Yu, C. Y., Hsu, Y. W., Liao, C. L., and Lin, Y. L. (2006). Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J. Virol. 80, 11868–11880. doi: 10.1128/JVI.00879-06
Zhang, H. M., Ye, X., Su, Y., Yuan, J., Liu, Z., Stein, D. A., et al. (2010). Coxsackievirus B3 infection activates the unfolded protein response and induces apoptosis through downregulation of p58IPK and activation of CHOP and SREBP1. J. Virol. 84, 8446–8459. doi: 10.1128/JVI.01416-09
Zhang, R., Jha, B. K., Ogden, K. M., Dong, B., Zhao, L., Elliott, R., et al. (2013). Homologous 2′,5′-phosphodiesterases from disparate RNA viruses antagonize antiviral innate immunity. Proc. Natl. Acad. Sci. U.S.A. 110, 13114–13119. doi: 10.1073/pnas.1306917110
Zhao, L., Jha, B. K., Wu, A., Elliott, R., Ziebuhr, J., Gorbalenya, A. E., et al. (2012). Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe 11, 607–616. doi: 10.1016/j.chom.2012.04.011
Zhou, A., Hassel, B. A., and Silverman, R. H. (1993). Expression cloning of 2–5A-dependent RNAase: a uniquely regulated mediator of interferon action. Cell 72, 753–765. doi: 10.1016/0092-8674(93)90403-D
Zhou, A., Molinaro, R. J., Malathi, K., and Silverman, R. H. (2005). Mapping of the human RNASEL promoter and expression in cancer and normal cells. J. Interferon Cytokine Res. 25, 595–603. doi: 10.1089/jir.2005.25.595
Keywords: unfolded protein response, UPR, RNaseL, OAS, IRE1, Xbp1, RIDD pathway
Citation: Bhattacharyya S (2014) Can’t RIDD off viruses. Front. Microbiol. 5:292. doi: 10.3389/fmicb.2014.00292
Received: 19 February 2014; Accepted: 27 May 2014;
Published online: 18 June 2014.
Edited by:
Shiu-Wan Chan, The University of Manchester, UKReviewed by:
Masa Shuda, University of Pittsburgh, USAHidekatsu Iha, Oita University, Japan
Fouzia Sadiq, Imperial College London, UK
Copyright © 2014 Bhattacharyya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sankar Bhattacharyya, Vaccine and Infectious Disease Research Centre, Translational Health Science and Technology Institute, Plot# 496, Phase-III, Udyog Vihar, Gurgaon, Haryana, India e-mail:c2Fua2FyQHRoc3RpLnJlcy5pbg==