 Hailan Piao1
Hailan Piao1 Medora Lachman2Stephanie Malfatti3
Medora Lachman2Stephanie Malfatti3 Alexander Sczyrba4
Alexander Sczyrba4 Bernhard Knierim5
Bernhard Knierim5 Manfred Auer5
Manfred Auer5 Susannah G. Tringe6Roderick I. Mackie7
Susannah G. Tringe6Roderick I. Mackie7 Carl J. Yeoman2
Carl J. Yeoman2 Matthias Hess1,6,8*
Matthias Hess1,6,8*- 1Systems Microbiology and Biotechnology Group, School of Molecular Biosciences, Washington State University, Richland, WA, USA
- 2Department of Animal and Range Sciences, Montana State University, Bozeman, MT, USA
- 3Lawrence Livermore National Laboratory, Biosciences and Biotechnology Division, Livermore, CA, USA
- 4Faculty of Technology and Center for Biotechnology, Bielefeld University, Bielefeld, Germany
- 5Lawrence Berkeley National Laboratory, Life Sciences Division, Berkeley, CA, USA
- 6Prokaryote Super Program, DOE Joint Genome Institute, Walnut Creek, CA, USA
- 7Department of Animal Sciences and Institute for Genomic Biology, University of Illinois, Urbana-Champaign, IL, USA
- 8Energy and Efficiency Division, Chemical and Biological Process Development Group, Pacific Northwest National Laboratory, Richland, WA, USA
The rumen microbial ecosystem is known for its biomass-degrading and methane-producing phenotype. Fermentation of recalcitrant plant material, comprised of a multitude of interwoven fibers, necessitates the synergistic activity of diverse microbial taxonomic groups that inhabit the anaerobic rumen ecosystem. Although interspecies hydrogen (H2) transfer, a process during which bacterially generated H2 is transferred to methanogenic Archaea, has obtained significant attention over the last decades, the temporal variation of the different taxa involved in in situ biomass-degradation, H2 transfer and the methanogenesis process remains to be established. Here we investigated the temporal succession of microbial taxa and its effect on fiber composition during rumen incubation using 16S rRNA amplicon sequencing. Switchgrass filled nylon bags were placed in the rumen of a cannulated cow and collected at nine time points for DNA extraction and 16S pyrotag profiling. The microbial community colonizing the air-dried and non-incubated (0 h) switchgrass was dominated by members of the Bacilli (recruiting 63% of the pyrotag reads). During in situ incubation of the switchgrass, two major shifts in the community composition were observed: Bacilli were replaced within 30 min by members belonging to the Bacteroidia and Clostridia, which recruited 34 and 25% of the 16S rRNA reads generated, respectively. A second significant shift was observed after 16 h of rumen incubation, when members of the Spirochaetes and Fibrobacteria classes became more abundant in the fiber-adherent community. During the first 30 min of rumen incubation ~13% of the switchgrass dry matter was degraded, whereas little biomass degradation appeared to have occurred between 30 min and 4 h after the switchgrass was placed in the rumen. Interestingly, methanogenic members of the Euryarchaeota (i.e., Methanobacteria) increased up to 3-fold during this period of reduced biomass-degradation, with peak abundance just before rates of dry matter degradation increased again. We hypothesize that during this period microbial-mediated fibrolysis was temporarily inhibited until H2 was metabolized into CH4 by methanogens. Collectively, our results demonstrate the importance of inter-species interactions for the biomass-degrading and methane-producing phenotype of the rumen microbiome—both microbially facilitated processes with global significance.
Introduction
The microbial community (microbiota) inhabiting the cow rumen has been described as “the most elegant and highly evolved cellulose-digesting system in nature” (Weimer et al., 2009). Cellulose, the most abundant natural polymer on earth (Klemm et al., 2005), and a major component of plant biomass (Heredia et al., 1995) is degraded within the rumen by various bacteria (Weimer, 1996), fungi (Bauchop and Mountfort, 1981; Theodorou et al., 1996), and protozoa (Coleman, 1992). The efficacy of cellulose-degradation, and ultimately fiber-degradation, is mediated by important microbial enzymatic and metabolic interactions and it has been shown that major cellulolytic bacteria, such as Fibrobacter succinogenes, Ruminococcus flavefaciens, and Ruminococcus albus (Weimer, 1996), exhibit suboptimal cellulolytic activity if they are not able to synergistically engage with non-cellulolytic microorganisms (Kudo et al., 1987; Fondevila and Dehority, 1996). In particular methanogenic archaea have been described to boost the carbohydrate-degrading activity of cellulolytic rumen bacteria (Joblin et al., 1989). The role of rumen methanogens is unique in that these organisms do not degrade or ferment any portion of plant biomass, but instead obtain their energy from byproducts, principally H2 and CO2, of fibrolytic organisms (Janssen and Kirs, 2008). By using hydrogen to reduce CO2, methanogens remove otherwise inhibitory levels of H2 (Janssen and Kirs, 2008), increase ATP yields by redirecting reducing equivalents toward acetate (Latham and Wolin, 1977) and thereby promote growth (Rychlik and May, 2000) and the ability to produce higher concentrations of fibrolytic enzymes. Physical co-aggregation between fibrolytic and methanogenic populations are commonly observed (i.e., Leahy et al., 2010) and this is thought to be important for both the efficient transfer of H2 and maintaining the low H2 partial pressures necessary to sustain active growth of the fermentative bacteria (Stams, 1994; Ishii et al., 2006).
To this end, the temporal dynamics of fibrolytic bacteria and methanogenic archaea remain to be systematically explored during in situ colonization and degradation of plant biomass within the rumen. Previous studies have shown temporal changes in bacterial (Edwards et al., 2007; Huws et al., 2013) and fungal (Edwards et al., 2008) populations during ruminal incubation of fresh perennial rye grass (Lolium perenne). Specifically, these studies revealed a rapid colonization of the plant material within 5 min of rumen-incubation by both bacterial (Edwards et al., 2007) and fungal populations, followed by a compositional shift in bacterial populations between 2 and 4 h following incubation (Huws et al., 2013). Compositional dynamics may vary among forage types based on observed differences in fiber disappearance and fibrolytic enzyme activities (Bowman and Firkins, 1993) and the observed temporal changes in microbiota may be affected by residual plant metabolism of fresh forages (Huws et al., 2013). Here we provide evidence that similar temporal changes occur during the in situ incubation of dried switchgrass and correspond to changes in methanogen abundance. Furthermore, these changes in microbiota also correspond to notable differences in the rate of forage degradation.
Materials and Methods
Sample Collection
To enrich for fiber-adherent rumen microorganisms, air-dried switchgrass (Panicum virgatum) was ground into 2 mm pieces using a Wiley mill and weighed into individual in situ nylon bags (50 μm pores; Ankom Technology, Macedon, NY, USA). A total of 18 nylon bags, each containing 5 g of air-dried switchgrass, were placed in the rumen of one cannulated Friesian cow, fed on a mixed diet containing 60% fiber (Hess et al., 2011). Nylon bags were retrieved from the cow's rumen at 0.5, 1, 2, 4, 6, 16, 24, 48, and 72 h and washed immediately with 2 × 50 ml PBS buffer (pH7) to remove rumen fluid containing loosely adherent microbes, placed on dry-ice and transported immediately to the laboratory where DNA extraction and fiber degradation analysis were performed. Non-incubated nylon bags filled with ground switchgrass were used as control (0 h). To account for biological variation during the fiber colonization process, nylon bags were retrieved from the cow's rumen in duplicates. All animal procedures were carried out under an approved protocol with the University of Illinois Institutional Animal Care and Use of Animals Committee (IUCAC #06081).
Fiber Degradation Analysis
Relative biomass (switchgrass) degradation during rumen-incubation was determined by dry matter, organic matter, neutral detergent fiber (NDF), acid detergent fiber (ADF), and acid detergent lignin (ADL) analysis. NDF, ADF, and ADL were determined using the procedures of Goering and Van Soest (1970). Cellulose content was estimated as ADF–ADL and hemicellulose as NDF–ADF.
DNA Extraction and 16S rRNA Gene Amplification
Total microbial genomic DNA was extracted from 100 mg of the non-incubated control sample and from each rumen-incubated biomass sample using a FastDNA SPIN Kit for Soil (MP Biomedical, Solon, OH) according to the manufacturer's instructions. Extracted DNA was quantified with a spectrophotometer (Nanodrop ND1000; Thermo Scientific, USA). The hypervariable V6 to V8 region of the 16S ribosomal RNA (rRNA) gene was amplified from the environmental DNA using the primer set 926F/1392R (926F: 5′- cct atc ccc tgt gtg cct tgg cag tct cag AAA CTY AAA KGA ATT GRC GG- 3′ and 1392R: 5′ - cca tct cat ccc tgc gtg tct ccg act cag xxxxx ACG GGC GGT GTG TRC – 3′) described by Engelbrektson et al. (2010). Primer sequences were modified by the addition of 454 A or B adapter sequences (lower case). In addition, the reverse primer included a 5 bp barcode, indicated by xxxxx in the reverse primer sequence above, for multiplexing of samples during sequencing. The barcode sequence for each sample is listed in Table 1. The generated raw reads were deposited in NCBI's Short Read Archive under the accession number SRP042121.
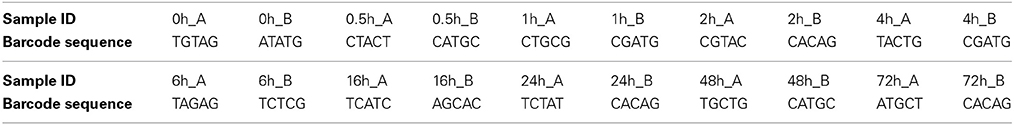
Table 1. Barcode sequences.
Twenty-microliter PCR reactions were performed in duplicate and pooled to minimize PCR bias. PCR reaction was performed using 0.4 μl Advantage GC 2 Polymerase Mix (Advantage-2 GC PCR Kit, Clontech), 4 μl 5x GC PCR buffer, 2 μl 5 M GC Melt Solution, 0.4 μl 10 mM dNTP mix (MBI Fermentas), 1.0 μl of each 25 nM primer, and 10 ng sample DNA. The thermal cycler protocol was 95°C for 3 min, 25 cycles of 95°C for 30 s, 50°C for 45 s, and 68°C for 90 s, and a final 10 min extension at 68°C. PCR amplicons were purified using Solid Phase Reversible Immobilization (SPRI) beads and quantified using a Qubit flurometer (Invitrogen). Samples were diluted to 10 ng/μl and mixed in equal concentrations. Emulsion PCR and sequencing of the PCR amplicons were performed following the Roche 454 GS FLX Titanium technology instructions provided by the manufacturer.
Pyrotag Sequence Analysis
Pyrosequencing data from the 20 samples were demultiplexed and processed using QIIME version 1.7.0 (Caporaso et al., 2010) according to the standard operating procedure described at http://qiime.org/tutorials/tutorial.html. Primers and barcodes were removed before the raw reads were quality filtered. Sequences were removed if they had long homopolymeric regions (>6 nt), were smaller than 200 nt, had quality scores lower than 25, or if they were identified as being chimeric. This resulted in a total of 198,037 high quality 16S rRNA gene sequences. Sequences generated from each of the biological duplicates were combined prior to sequence analysis to account for biological variation.
OTU-Based Sequence Analysis
Sequences were clustered into operational taxonomic units (OTUs) at a 97% sequence identity cut-off using UCLUST (Edgar, 2010). The most abundant sequence of each OTU was picked as representative sequence. Singleton and doubleton abundance, Shannon, Simpson, Simpson reciprocal, and Chao1 estimators were calculated using the QIIME software. Representative sequences were aligned using the PyNAST algorithm and the alignment was filtered to remove common gaps. Following the quality filtering and grouping steps, 7,168 unique sequences (representing 198,037 total sequences) were used for OTU analyses. QIIME scripts were used to calculate α-diversity metrics for OTU representative sequences and to generate a Principal Coordinate Analysis plot according to the standard operating procedure described at http://qiime.org/tutorials/tutorial.html.
Taxonomic Classification of Unique Representative Sequences
Taxonomic classification of the final set of representative sequences was performed using QIIME. Each sequence was normalized to contain six taxonomic levels, ranging from the domain to the genus level.
Scanning Electron Microscopy
For scanning electron microscopy (SEM) pieces of switchgrass taken from the cow rumen as described above were immediately fixed with glutaraldehyde. The samples were washed several times with phosphate buffered saline, treated with 1% OsO4 for 1 h on ice, prior to dehydration and critical point drying using a Tousimis Autosamdri-815 critical point dryer. The dried samples were then mounted on precut brass sample stubs using double sided carbon tape and sputter coated with approximately 30 Angstrom of Au/Pd. SEM imaging was performed on a Hitachi S-5000 microscope at 10 kV accelerating voltage.
Results
Temporal Changes in α-Diversity of Fiber-Adherent Rumen Microbiome
To determine α-diversity within the fiber-adherent community the quality-filtered pyrotag reads of each sample were clustered into OTUs at a sequence identify level of 97%. This resulted in a total of 7,168 distinct OTUs, with a range of 788–2,758 OTUs observed for each community. Table 2 summarizes the number of raw reads, quality filtered reads, and OTUs that were obtained for each time point. Of the observed 7,168 OTUs, 7,005 (97.7%) were determined to be unambiguously of bacterial origin, while 0.2% (17 OTUs) were assigned to be archaeal (Table 3). Table 4 summarizes the number of OTUs unique to each time point sampled. A complete list of all OTUs observed during this study and an OTU count for each sample is provided in Supplementary Table 1. Analysis of the successional appearance of OTUs suggests an initial increase in community richness within the first 30 min of rumen incubation (Tables 2, 3) and a second increase in richness of the bacterial community associated with the switchgrass samples subjected to rumen-incubation for 16, 24, and 48 h (Table 3). This finding is supported by the calculated rarefaction curves (Figure 1) and Chao 1 indices (Table 5). Likewise the Shannon and Simpson Reciprocal indices increased noticeably for all rumen-incubated samples (Table 5). An average Good's coverage estimator of 94.9% was calculated for all samples, with 92% (6 h) being the lowest and 98.6% (non-incubated switchgrass) being the highest (Table 5).
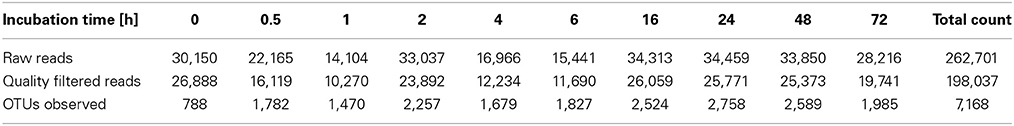
Table 2. Summary of pyrosequencing reads generated and OTUs observed for each time point sampled during this study.
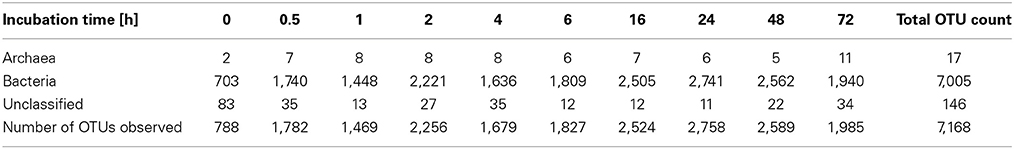
Table 3. Phylogenetic assignment of OTUs at domain level.

Table 4. Distinct OTUs observed.
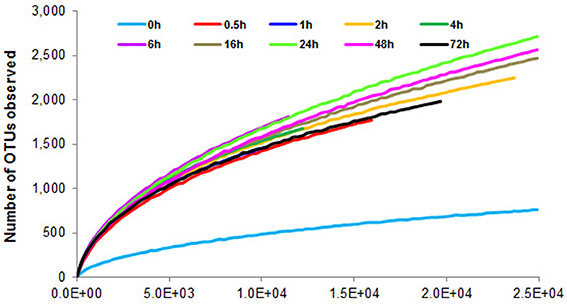
Figure 1. Rarefaction curves calculated from 16S rRNA pyrotag data of switchgrass-adherent microbiomes after rumen-incubation. 0 h indicates the non-incubated switchgrass sample (control).
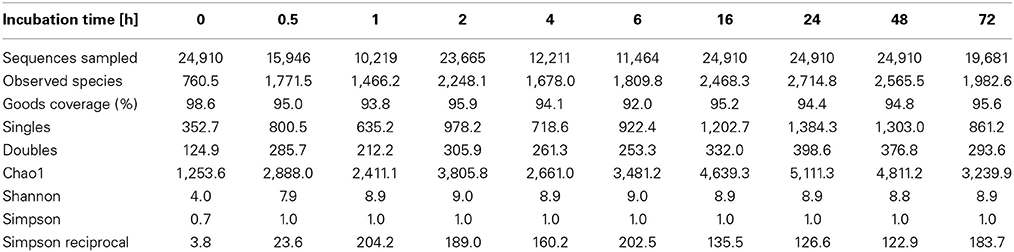
Table 5. Estimated OTU richness and diversity metrics and estimated sample coverage of rarefied bacterial and archaeal 16S rRNA sequences.
Temporal Changes in β-Diversity of Fiber-Adherent Rumen Microbiome
Principal coordinates analysis comparing the temporal dynamics of switchgrass-associated microbiota indicated that the community associated with the air-dried switchgrass sample (0 h) was distinct from the microbiomes of all rumen-incubated samples (Figure 2). After 30 min of rumen incubation the switchgrass-associated microbiota exhibited clearly noticeable changes: for example, members of the Archaea, which were essentially absent in the non-incubated microbiome, were detected in all rumen-incubated samples. In total, 17 distinct archaeal OTUs were detected (Table 3), with 14 (82%) of them categorized as Methanobacteriaceae, a family belonging to the methanogenic Euryarchaeota (Supplementary Table 1). Of the reads derived from the Archaea, the majority (≥76%) was recruited for each sample (median 88%) by two OTUs both categorized as Methanobrevibacter following rumen incubation. Abundance of members belonging to the genus Methylobacterium was notably high in the samples retrieved after 30 min of rumen incubation. The bacterial phylum Bacteroidetes, represented primarily by members of the Sphingobacteria in the 0 h sample, increased up to five fold throughout rumen incubation, mainly through large increases in Bacteroidia populations and in particular of increases in members belonging to the genus Prevotella (Tables 6–8). This overall increase in Bacteroidetes was accompanied by increased abundance of Cyanobacteria, Fibrobacteres, Spirochaetes, and Tenericutes and decreases in Actinobacteria (i.e., Kineosporiaceae and Microbacteriaceae), Firmicutes, and Proteobacteria abundance (Table 8).
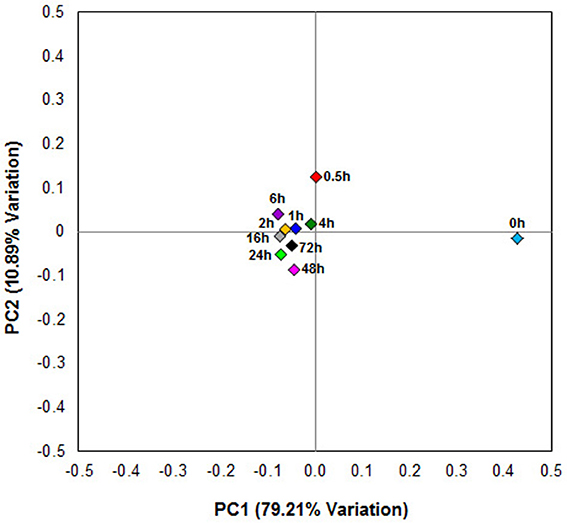
Figure 2. Principal coordinate analysis of switchgrass-adherent microbiomes. Each point corresponds to one of nine rumen-incubated samples (0.5, 1, 2, 4, 6, 16, 24, 48, and 72 h) and one control sample (0 h). The percentage of variation explained by the plotted principal coordinates is indicated on the axes.
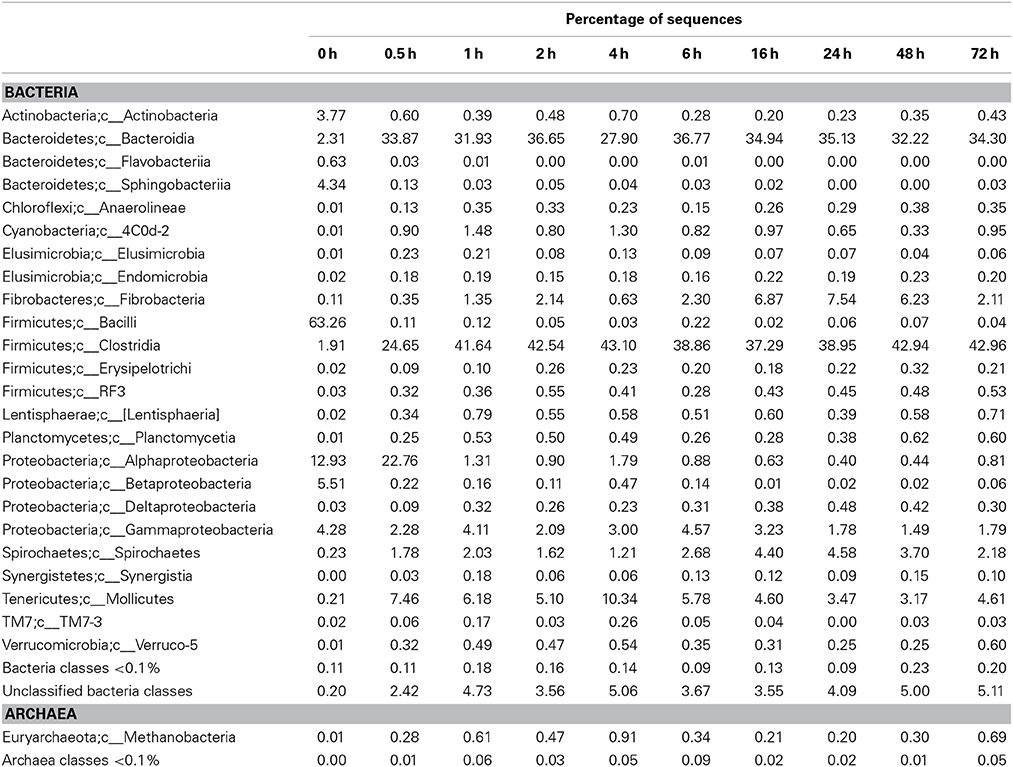
Table 6. Community composition of switchgrass-adherent microbiome at the class level based on 16S rRNA pyrotag data.
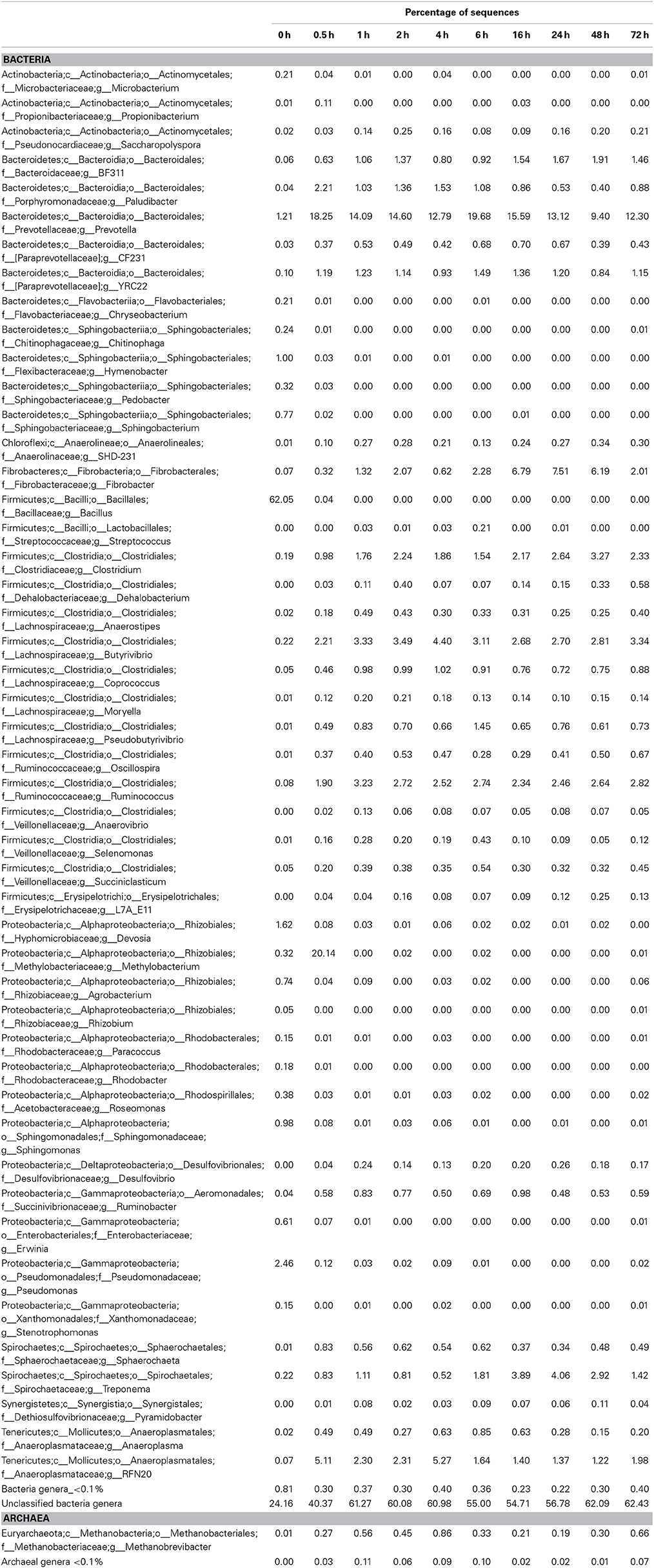
Table 7. Community composition of switchgrass-adherent microbiome at the genus level based on 16S rRNA pyrotag data.
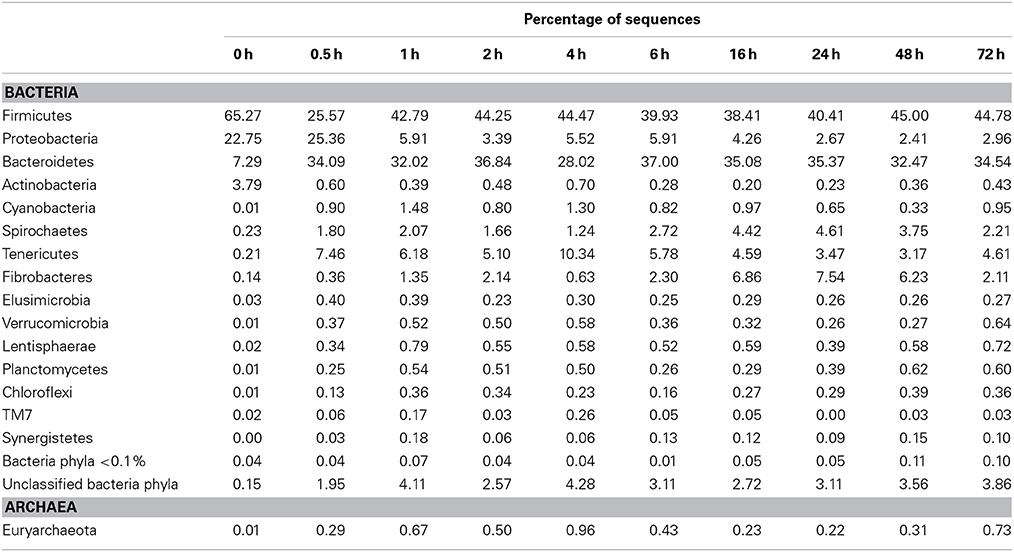
Table 8. Community composition of switchgrass-adherent microbiome at the phylum level based on 16S rRNA pyrotag data.
Observed decreases in Firmicutes were driven by a 460-fold reduction in the Bacilli (mostly Bacillus spp.), which initially accounted for 97% of the Firmicutes phylum and 63% of all bacteria prior to rumen incubation. Decreases in Bacilli were partially offset by large increases in the Clostridia including the Lachnospiraceae (particularly Butyrivibrio spp., Pseudobutyrivibrio spp., and Clostridium spp.) and the Ruminococcaceae (almost entirely Ruminococcus spp.) families. The Clostridia continued to increase to become the predominant class and made Firmicutes the predominant phylum after a brief peak in Proteobacteria abundance within the initial 30 min of rumen-incubation (Figure 3, Tables 6, 8). The switchgrass-associated microbiota stabilized at the phylum level after 1 h of rumen incubation (Figure 3), and only modest phylum- and class-level changes were observed for most taxonomic groups between 1 and 72 h of rumen incubation (Tables 6, 8). The only exceptions to this observation were the Fibrobacteres and Spirochaetes, which both increased considerably in abundance in samples collected between 6 and 16 h, peaking around 24 h before returning to their 1 h levels after a total of 72 h of rumen-incubation (Figure 3, Table 8). Each of these phylum-level changes was driven by a single genus: the Fibrobacteres driven exclusively by Fibrobacter, while changes in the Spirochaetes were driven by Treponema (Table 7).
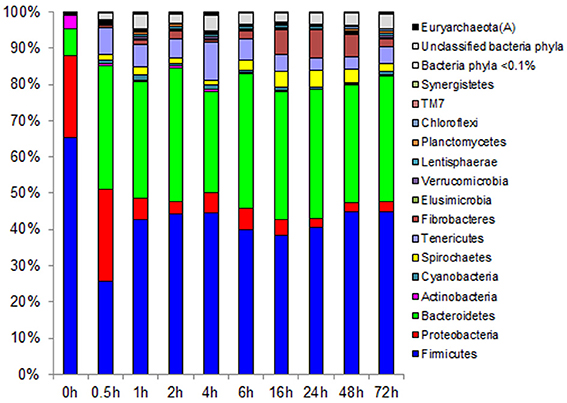
Figure 3. Effect of rumen incubation on relative abundance of bacterial and archaeal phyla of switchgrass-adherent microbiome. Relative OTU abundance of rarefied 16S rRNA gene 454-pyrotag data. Taxonomy was assigned based on Greengenes (McDonald et al., 2012).
Biomass-Degradation Coordinated with Microbial Colonization
Biomass degradation following rumen incubation appeared to be biphasic; each phase corresponding to notable changes in microbial communities. Biomass degradation began rapidly following rumen incubation with 13% of the total biomass being degraded within 30 min (Figure 4). Biomass-degradation then appeared to stall between 30 min and 4 h, with no significant change to biomass during this period. Corresponding to this period, we observed noticeable increases in Methanobrevibacter spp. and changes in some bacterial taxa. The Butyrivibrio spp. increased, while Prevotella spp. decreased between 30 min and 4 h. Following 4 h, biomass degradation proceeded somewhat linearly at a rate of ~0.81% biomass h−1 for the next 20 h of incubation, before gradually slowing over the following two measurements (~0.41% biomass h−1 between 24 and 48 h and 0.17% biomass h−1 between 48 and 72 h). The increase in rate of biomass degradation between 4 and 24 h occurred simultaneously with increased numbers of reads assigned to Fibrobacter and Treponema species (Figure 4, Table 7). Biomass-degradation and colonization of switchgrass was also detectable by SEM with significant fiber degradation being visible for samples incubated within the rumen for 24 h and longer (Figure 5).
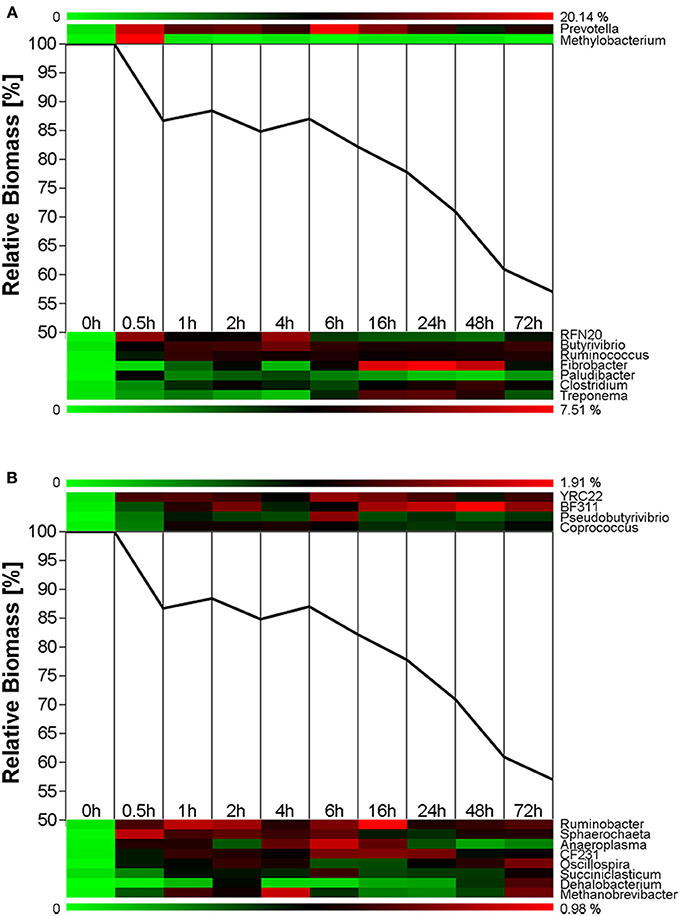
Figure 4. Microbial succession and biomass-degradation during rumen-incubation. Heat map show succession of genera recruiting >0.5% of the generated sequences. Line graphs show the relative dry mater change during rumen-incubation. (A) Upper panel shows genera with >10% relative abundance, lower panel shows genera with a relative abundance between 2 and 10%; (B) Upper panel shows genera with relative abundance between 1 and 2%, lower panel shows genera with a relative abundance between 0.5 and 1%.
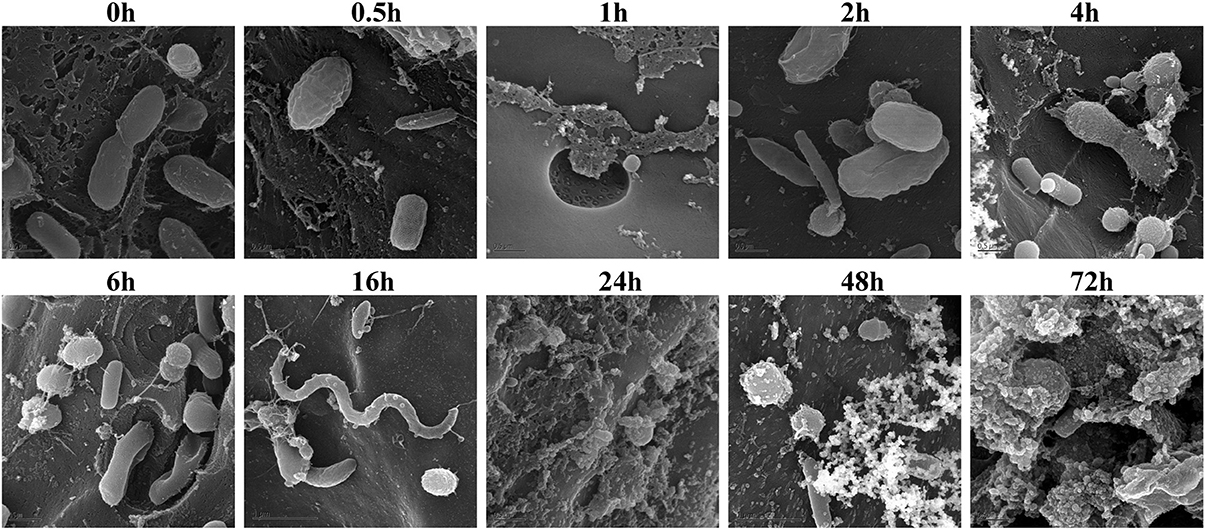
Figure 5. Scanning Electron Microscopy of air-dried and rumen-incubated switchgrass samples and adherent microorganisms. Scale bars indicate 0.5 μm.
Discussion
In the present study we observed temporal shifts in the plant biomass-associated microbiota and in the rates of biomass degradation during the in situ rumen-incubation of dried switchgrass. Changes in the microbiome were in particular prevailing immediately within the first 30 min and after 4 h of rumen incubation. These observations are consistent with analogous trials on rumen-incubated fresh perennial ryegrass, where denaturing gradient gel electrophoresis (DGGE)-based analyses identified discrete microbial profiles at 0–2 h and after 4 h onwards (Huws et al., 2013). Notable similarities and differences were observed in several findings between these two studies. Consistent with our findings, Huws and colleagues reported increases in richness and diversity following rumen incubation (Huws et al., 2013). The authors of the former study also found the major ruminal genus Prevotella (Stevenson and Weimer, 2007) increased after 4 h. In our study, Prevotella spp. growth was biphasic, increasing dramatically within the first 30 min following rumen incubation and decreasing between 30 min and 4 h, before increasing again. In our study, Prevotella spp. were most abundant at 6 h and did not return to 4 h levels until after 24 h of incubation. In contrast to our observations, Huws et al. reported a loss of a band corresponding to Treponema spp. after 4 h rumen incubation (Huws et al., 2013), whereas we saw great increases in this genus between 6 and 16 h. This discrepancy likely reflects differences in analytical techniques (DGGE vs. next-generation sequencing), where loss of a single Treponema band may not reflect all members of that taxonomic group. Direct sequencing of 16S rRNA amplicons also facilitated the detection of a significant increase of Methylobacterium in switchgrass samples retrieved after 30 min of rumen incubation (Figure 4). Members of the genus Methylobacterium are strictly aerobic, can utilize C1 compounds such as formate, methanol and methaylamine and a variety of C2 (including acetate), C3, and C4 compounds (Green, 2006) as sole carbon source, and have been found in association with a variety of plants (Corpe, 1985). We hypothesize that members of the Methylobacterium were associated with the switchgrass fibers and introduced into the rumen, where they metabolized common fermentation intermediates, such as formate and acetate (Sabine and Johnson, 1964; Hungate et al., 1970), until all oxygen that might have been introduced with the ground switchgrass was depleted. Overall many similarities exist between the two studies and may indicate that the temporal changes in microbiota during the first 30 min following rumen-incubation and then after 4 h may be common features of rumen-microbial digestion among multiple plant species in various states (fresh vs. dried). Changes in the microbiota corresponded with differences in the rates of switchgrass degradation. The rate of switchgrass degradation was greatest within the first 30 min of rumen-incubation during which 13% of the total biomass was lost. This may reflect rapid utilization of soluble sugars and other easily fermentable nutrients that were unabated by proximal H2 partial pressures. However, the rate of degradation stalled between 30 min and 4 h. During this period we observed a dramatic increase in methanogen density such that the total abundance of methanogens at 4 h post-incubation increased 3.3-fold compared to 30 min incubation. We hypothesize that these two observations were linked and related to a dramatic increase in proximal H2 partial pressure caused by the rapid initial fermentation of easily accessible plant polysaccharides. Methanogen numbers swelled, most likely in response to the hypothesized high H2 partial pressures and were eventually able to overcome these and create an environment that was favorable to the energetically-efficient growth of the fibrolytic microbial consortia, as has been proposed based on in vitro evidence (Stams, 1994; Ishii et al., 2006). Although the community was largely established by 1 h, as evidenced by less dramatic changes in the overall microbiota 16S rRNA profile following this period, it was not until after 4 h before their collective metabolism was operating efficiently.
In summary the findings presented here support the hypothesis that the hydrogenotrophic metabolism of methanogens is an essential part of the fibrolytic activity of the rumen ecosystem and is essential for the degradation of the more recalcitrant plant polysaccharides by rumen microbes. Additional in situ work during which methane emission, fiber composition and community are monitored more stringently will be essential for obtaining a better holistic understanding of the rumen microbiome and the contribution of particular community members to the fibrolytic and methanogenic rumen phenotype.
Author Contributions
Conceived and designed the experiments: Matthias Hess, Susannah G. Tringe, and Roderick I. Mackie. Performed the experiments: Matthias Hess, Hailan Piao, Roderick I. Mackie. Generated and analyzed the data: Matthias Hess, Hailan Piao, Medora Lachman, Stephanie Malfatti, Alexander Sczyrba, Bernhard Knierim, Manfred Auer, Susannah G. Tringe, and Carl J. Yeoman. Wrote the paper: Matthias Hess, Hailan Piao, Medora Lachman, and Carl J. Yeoman.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2014.00307/abstract
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work conducted by the US Department of Energy Joint Genome Institute is supported by the Office of Science of the US Department of Energy under Contract No. DE-AC02-05CH11231. MH was partially funded by the Energy Biosciences Institute at the University of California, Berkeley. We would like to thank Dr. Edward Rubin at the DOE Joint Genome Institute and Dr. Philip Hugenholtz at the University of Queensland for their help and guidance during this project as well as Dr. Anthony Yannarell at the University of Illinois Urbana-Champaign and Anna Engelbrektson for helping us with the DNA extraction and sample preparation.
References
Bauchop, T., and Mountfort, D. O. (1981). Cellulose fermentation by a rumen anaerobic fungus in both the absence and presence of rumen methanogens. Appl. Environ. Microbiol. 42, 1103–1110.
Bowman, J. G., and Firkins, J. L. (1993). Effects of forage species and particle size on bacterial cellulolytic activity and colonization in situ. J. Anim. Sci. 71, 1623–1633.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Coleman, G. S. (1992). The rate of uptake and metabolism of starch grains and cellulose particles by Entodinium species, Eudiplodinium maggii, some other entodiniomorphid protozoa and natural protozoal populations taken from the ovine rumen. J. Appl. Bacteriol. 73, 507–513. doi: 10.1111/j.1365-2672.1992.tb05013.x
Corpe, W. A. (1985). A method for detecting methylotrophic bacteria on solid-surfaces. J. Microbiol. Methods 3, 215–221. doi: 10.1016/0167-7012(85)90049-1
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edwards, J. E., Huws, S. A., Kim, E. J., and Kingston-Smith, A. H. (2007). Characterization of the dynamics of initial bacterial colonization of nonconserved forage in the bovine rumen. FEMS Microbiol. Ecol. 62, 323–335. doi: 10.1111/j.1574-6941.2007.00392.x
Edwards, J. E., Kingston-Smith, A. H., Jimenez, H. R., Huws, S. A., Skot, K. P., Griffith, G. W., et al. (2008). Dynamics of initial colonization of nonconserved perennial ryegrass by anaerobic fungi in the bovine rumen. FEMS Microbiol. Ecol. 66, 537–545. doi: 10.1111/j.1574-6941.2008.00563.x
Engelbrektson, A., Kunin, V., Wrighton, K. C., Zvenigorodsky, N., Chen, F., Ochman, H., et al. (2010). Experimental factors affecting PCR-based estimates of microbial species richness and evenness. ISME J. 4, 642–647. doi: 10.1038/ismej.2009.153
Fondevila, M., and Dehority, B. A. (1996). Interactions between Fibrobacter succinogenes, Prevotella ruminicola, and Ruminococcus flavefaciens in the digestion of cellulose from forages. J. Anim. Sci. 74, 678–684.
Goering, H. K., and Van Soest, P. J. (1970). Forage Fiber Analysis. Vol. 379. Washington, DC: Agricultural Research Service, US Department of Agriculture.
Heredia, A., Jimenez, A., and Guillen, R. (1995). Composition of plant-cell walls. Zeitschrift für Lebensmittel-Untersuchung -Forschung 200, 24–31. doi: 10.1007/BF01192903
Hess, M., Sczyrba, A., Egan, R., Kim, T. W., Chokhawala, H., Schroth, G., et al. (2011). Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331, 463–467. doi: 10.1126/science.1200387
Hungate, R. E., Smith, W., Bauchop, T., Yu, I., and Rabinowitz, J. C. (1970). Formate as an intermediate in the bovine rumen fermentation. J. Bacteriol. 102, 389–397.
Huws, S. A., Mayorga, O. L., Theodorou, M. K., Onime, L. A., Kim, E. J., Cookson, A. H., et al. (2013). Successional colonization of perennial ryegrass by rumen bacteria. Lett. Appl. Microbiol. 56, 186–196. doi: 10.1111/lam.12033
Ishii, S., Kosaka, T., Hotta, Y., and Watanabe, K. (2006). Simulating the contribution of coaggregation to interspecies hydrogen fluxes in syntrophic methanogenic consortia. Appl. Environ. Microbiol. 72, 5093–5096. doi: 10.1128/AEM.00333-06
Janssen, P. H., and Kirs, M. (2008). Structure of the archaeal community of the rumen. Appl. Environ. Microbiol. 74, 3619–3625. doi: 10.1128/AEM.02812-07
Joblin, K. N., Campbell, G. P., Richardson, A. J., and Stewart, C. S. (1989). Fermentation of barley straw by anaerobic rumen bacteria and fungi in axenic culture and in co-culture with methanogens. Lett. Appl. Microbiol. 9, 195–197. doi: 10.1111/j.1472-765X.1989.tb00323.x
Klemm, D., Heublein, B., Fink, H.-P., and Bohn, A. (2005). Cellulose: fascinating biopolymer and sustainable raw material. Angew. Chem. Int. Ed. 44, 3358–3393. doi: 10.1002/anie.200460587
Kudo, H., Cheng, K. J., and Costerton, J. W. (1987). Interactions between Treponema bryantii and cellulolytic bacteria in the in vitro degradation of straw cellulose. Can. J. Microbiol. 33, 244–248. doi: 10.1139/m87-041
Latham, M. J., and Wolin, M. J. (1977). Fermentation of cellulose by Ruminococcus flavefaciens in the presence and absence of Methanobacterium ruminantium. Appl. Environ. Microbiol. 34, 297–301.
Leahy, S. C., Kelly, W. J., Altermann, E., Ronimus, R. S., Yeoman, C. J., Pacheco, D. M., et al. (2010). The genome sequence of the rumen methanogen Methanobrevibacter ruminantium reveals new possibilities for controlling ruminant methane emissions. PLoS ONE 5:e8926. doi: 10.1371/journal.pone.0008926
McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., DeSantis, T. Z., Probst, A., et al. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. doi: 10.1038/ismej.2011.139
Rychlik, J. L., and May, T. (2000). The effect of a methanogen, Methanobrevibacter smithii, on the growth rate, organic acid production, and specific ATP activity of three predominant ruminal cellulolytic bacteria. Curr. Microbiol. 40, 176–180. doi: 10.1007/s002849910035
Sabine, J. R., and Johnson, B. C. (1964). Acetate metabolism in the ruminant. J. Biol. Chem. 239, 89–93.
Stams, A. J. M. (1994). Metabolic interactions between anaerobic-bacteria in methanogenic environments. Antonie van Leeuwenhoek 66, 271–294. doi: 10.1007/BF00871644
Stevenson, D. M., and Weimer, P. J. (2007). Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR. Appl. Microbiol. Biotechnol. 75, 165–174. doi: 10.1007/s00253-006-0802-y
Theodorou, M. K., Mennim, G., Davies, D. R., Zhu, W. Y., Trinci, A. P., and Brookman, J. L. (1996). Anaerobic fungi in the digestive tract of mammalian herbivores and their potential for exploitation. Proc. Nutr. Soc. 55, 913–926. doi: 10.1079/PNS19960088
Weimer, P. J. (1996). Why don't ruminal bacteria digest cellulose faster? J. Dairy Sci. 79, 1496–1502. doi: 10.3168/jds.S0022-0302(96)76509-8
Keywords: rumen microbiology, microbe-microbe interactions, cellulolytic bacteria, methanogenic archaea, interspecies H2 transfer
Citation: Piao H, Lachman M, Malfatti S, Sczyrba A, Knierim B, Auer M, Tringe SG, Mackie RI, Yeoman CJ and Hess M (2014) Temporal dynamics of fibrolytic and methanogenic rumen microorganisms during in situ incubation of switchgrass determined by 16S rRNA gene profiling. Front. Microbiol. 5:307. doi: 10.3389/fmicb.2014.00307
Received: 09 May 2014; Accepted: 03 June 2014;
Published online: 22 July 2014.
Edited by:
Luis Raul Comolli, Lawrence Berkeley National Laboratory, USAReviewed by:
William John Kelly, AgResearch Ltd., New ZealandEtinosa Igbinosa, University of Benin, Nigeria
Copyright © 2014 Piao, Lachman, Malfatti, Sczyrba, Knierim, Auer, Tringe, Mackie, Yeoman and Hess. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthias Hess, Systems Microbiology and Biotechnology Group, School of Molecular Biosciences, Washington State University, 2710 Crimson Way, Richland, WA 99354-1671, USA e-mail:bWhlc3NAbGJsLmdvdg==