 Sven Breider1†
Sven Breider1† Carmen Scheuner2†
Carmen Scheuner2† Peter Schumann2Anne Fiebig2Jörn Petersen2Silke Pradella2Hans-Peter Klenk2
Peter Schumann2Anne Fiebig2Jörn Petersen2Silke Pradella2Hans-Peter Klenk2 Thorsten Brinkhoff1
Thorsten Brinkhoff1 Markus Göker2*
Markus Göker2*- 1Department of Biology of Geological Processes - Aquatic Microbial Ecology, Institute for Chemistry and Biology of the Marine Environment (ICBM), University of Oldenburg, Oldenburg, Germany
- 2Department of Microorganisms, Leibniz Institute DSMZ - German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany
Earlier phylogenetic analyses of the marine Rhodobacteraceae (class Alphaproteobacteria) genera Leisingera and Phaeobacter indicated that neither genus might be monophyletic. We here used phylogenetic reconstruction from genome-scale data, MALDI-TOF mass-spectrometry analysis and a re-assessment of the phenotypic data from the literature to settle this matter, aiming at a reclassification of the two genera. Neither Phaeobacter nor Leisingera formed a clade in any of the phylogenetic analyses conducted. Rather, smaller monophyletic assemblages emerged, which were phenotypically more homogeneous, too. We thus propose the reclassification of Leisingera nanhaiensis as the type species of a new genus as Sedimentitalea nanhaiensis gen. nov., comb. nov., the reclassification of Phaeobacter arcticus and Phaeobacter leonis as Pseudophaeobacter arcticus gen. nov., comb. nov. and Pseudophaeobacter leonis comb. nov., and the reclassification of Phaeobacter aquaemixtae, Phaeobacter caeruleus, and Phaeobacter daeponensis as Leisingera aquaemixtae comb. nov., Leisingera caerulea comb. nov., and Leisingera daeponensis comb. nov. The genera Phaeobacter and Leisingera are accordingly emended.
Introduction
Bacteria belonging to the Roseobacter group are presumed to form a monophyletic group within the Rhodobacteraceae (Alphaproteobacteria), the great majority of them being of marine origin, which is reflected by their absolute requirement of sodium ions (Brinkhoff et al., 2008). Members of this group constitute up to 25% of the total bacterial community in a large variety of habitats (Brinkhoff et al., 2008). Because of their high abundance and their adaptive life style, they are thought to play a major role in global chemical cycles. Some of the important traits of the Roseobacter group are the use of a multitude of organic compounds, sulfur oxidation, oxidation of carbon monoxide, DMSP demethylation, the production of secondary metabolites (Buchan et al., 2005; Moran et al., 2007; Brinkhoff et al., 2008) and their affiliation with the cohort of aerobic anoxygenic phototrophic bacteria (Yurkov and Beatty, 1998).
Many representatives of the Roseobacter group can be cultivated in the lab, which is one of the reasons why the group is continuously growing. Until 2008, 38 genera were described (Brinkhoff et al., 2008), and at the time of writing the group contained at least 54 genera and 135 species with validly published names. The huge amount of genera and species reflects the physiological and genetic diversity within this group (Buchan et al., 2005) and makes it necessary to monitor the classification of previously published species and genera. Several reclassifications were already necessary within the Roseobacter group, e. g., for the genera Ruegeria (Uchino et al., 1998; Arahal et al., 2005; Martens et al., 2006) and Roseobacter (Martens et al., 2006).
The genus Leisingera was proposed by Schaefer et al. (2002) and currently consists of three species, Leisingera methylohalidivorans (Schaefer et al., 2002), Leisingera aquimarina (Vandecandelaere et al., 2008), and Leisingera nanhaiensis (Sun et al., 2010). The strains belonging to these species were isolated from sea water, a marine electro-active biofilm and marine sandy sediment, respectively. Phylogenetic trees for the trimethylamine monooxygenase (tmm) and the gamma-glutamyl-methylamine synthetase (Chen, 2012) showed that L. nanhaiensis is more distantly related to the other Leisingera species. This was confirmed by recent 16S rRNA gene sequence analyses (even though largely unresolved) and preliminary genomic analyses using the Genome-to-Genome Distance Calculator (GGDC; Auch et al., 2010a,b; Meier-Kolthoff et al., 2013) of diverse Phaeobacter and Leisingera type strains (Beyersmann et al., 2013; Buddruhs et al., 2013a; Dogs et al., 2013a,b; Freese et al., 2013; Riedel et al., 2013; Breider et al., 2014), suggesting the need for reclassification of L. nanhaiensis. Digital DNA:DNA hybridization (DDH) estimates as delivered by the GGDC were preferred over Average Nucleotide Identity (ANI) estimates because they provide higher correlations with traditional DDH results than do any of the ANI implementations (Auch et al., 2010b; Meier-Kolthoff et al., 2013).
The genus Phaeobacter was introduced by Martens et al. (2006) and currently comprises the species Phaeobacter aquaemixtae (Park et al., 2014), Phaeobacter arcticus (Zhang et al., 2008), Phaeobacter caeruleus (Vandecandelaere et al., 2009), Phaeobacter daeponensis (Yoon et al., 2007), Phaeobacter inhibens (Martens et al., 2006), Phaeobacter leonis (Gaboyer et al., 2013), and Phaeobacter gallaeciensis (Ruiz-Ponte et al., 1998; Martens et al., 2006), which is the type species, its type strain being BS107T = CIP 105210T = DSM 26640T but not DSM 17395 (Buddruhs et al., 2013b). The Phaeobacter species were isolated from a mixing zone of the ocean and a freshwater spring, marine arctic sediment, a marine electro-active biofilm, tidal flat sediment, a tidal mud flat, marine surface sediment and rearings and collectors of the scallop Pecten maximus, respectively. Recently, Phaeobacter strains retrieved a lot of interest because of their production of various secondary metabolites (e.g., Berger et al., 2011). Analyses of the 16S rRNA gene and in some publications also preliminary genomic analyses were in conflict with the current classification (Jin et al., 2011; Beyersmann et al., 2013; Buddruhs et al., 2013a; Dogs et al., 2013a,b; Freese et al., 2013; Gaboyer et al., 2013; Riedel et al., 2013; Breider et al., 2014; Liu et al., 2014; Park et al., 2014). These analyses mostly showed that P. aquaemixtae, P. caeruleus, P. daeponensis, Leisingera methylohalodivorans, and L. aquimarina form a clade, P. arcticus and P. leonis comprise a distinct monophyletic group, and P. gallaeciensis and P. inhibens form a third clade.
Thus, the two genera Leisingera and Phaeobacter appear intermixed. When 16S rRNA genes are insufficiently resolved, it is necessary to conduct phylogenetic analyses with additional genes. In many respects, using genome-scale data is the most promising approach (Klenk and Göker, 2010). As shown, e.g., in a series of studies using the DSMZ phylogenomics pipeline (Spring et al., 2010; Anderson et al., 2011; Abt et al., 2012, 2013; Frank et al., 2014; Stackebrandt et al., 2014; Verbarg et al., 2014), the more characters are assembled, the better statistically supported are the resulting phylogenies. Thus, phylogenomics has the potential to yield a more stable taxonomy, given the general goal that the taxonomic classification should summarize the phylogeny of the organisms (Klenk and Göker, 2010; Wiley and Lieberman, 2011; Göker and Klenk, 2013).
According to the aforementioned preliminary genomic analyses, we here re-investigated the phylogenetic relationships of Phaeobacter and Leisingera spp. using a variety of methods applied to genome-scale data, for determining monophyletic groups that are stable under a broad range of conditions. We also analyzed the relevant type strains using Matrix-assisted laser desorption/ionization time-of-flight mass spectronomy (MALDI-TOF MS) technology and re-assessed the published phenotypic information for providing descriptions of new or redefined taxa, including a recalculation of the G+C content from the genome data (Meier-Kolthoff et al., 2014).
Materials and Methods
Comprehensive samples of 16S rRNA gene data available from the Living Tree Project (LTP; Munoz et al., 2011), version s111, were used to determine the range of other genera that should be compared with the genera Phaeobacter and Leisingera. Because as yet the LTP phylogeny does not contain branch-support values, it has only limited use for directly assessing evolutionary relationships. We thus extracted the Rhodobacteraceae part of the LTP alignment, deleted all resulting gap-only alignment columns and phylogenetically analyzed the resulting matrix including bootstrapping as described below. Taxon sampling for all further, more detailed analyses was based on this initial assessment.
Protein sequences from the 14 available type-strain genomes of Leisingera, Phaeobacter, Ruegeria and outgroup (Oceanibulbus, Roseobacter, and Sediminimonas) species (Beyersmann et al., 2013; Buddruhs et al., 2013a; Dogs et al., 2013a,b; Freese et al., 2013; Riedel et al., 2013; Breider et al., 2014) were retrieved from the IMG website (http://img.jgi.doe.gov/cgi-bin/w/main.cgi) (L. aquimarina DSM 24565T, ID 2516653083 = AXBE00000000; L. methylohalidivorans MB2T, ID 2512564009 = CP006773/CP006774/CP006775; L. nanhaiensis NH52FT, ID 2512047090 = AXBG00000000; P. arcticus DSM 23566T; ID 2516653081 = AXBF00000000; P. caeruleus 13T, ID 2512047087 = AXBI00000000; P. daeponensis TF-218T, ID 2516493020 = AXBD00000000; P. inhibens T5T, ID 2516653078 = AXBB00000000; Sediminimonas qiaohouensis DSM 21189T, ID 2523533612 = AUIJ00000000) or from NCBI (Oceanibulbus indolifex HEL-45T, ABID00000000; P. gallaeciensis CIP105210T, AOQA01000000; Roseobacter denitrificans Och 114T, CP000362, CP000464, CP000465, CP000466, CP000467; Roseobacter litoralis Och 149T, CP002623, CP002624, CP002625, CP002626; Ruegeria lacuscaerulensis ITI-1157T, ACNX00000000; ACNX00000000; Ruegeria pomeroyi DSS-3T, CP000031, CP000032).
The genome sequences were phylogenetically investigated using the DSMZ phylogenomics pipeline as previously described (Spring et al., 2010; Anderson et al., 2011; Göker et al., 2011; Abt et al., 2012, 2013; Frank et al., 2014; Stackebrandt et al., 2014; Verbarg et al., 2014). In brief, clusters of orthologs were determined with a re-implementation of the OrthoMCL algorithm (Li et al., 2003) using NCBI BLAST version 2.2.25 (Altschul et al., 1997) and in conjunction with MCL version 11-294 (http://micans.org/mcl/) under default settings. OrthoMCL clusters containing inparalogs were reduced by selecting the most “central” sequence from each genome (the one with highest sum of BLAST scores), aligned using MUSCLE version 3.8.31 (Edgar, 2004), and the alignments filtered with the program scan_orphanerrs from the RASCAL package version 1.3.4 (Thompson et al., 2003) to remove orphan sequences as well as GBLOCKS version 0.91b (Castresana, 2000) to remove poorly aligned columns. Here, three distinct supermatrices (concatenated alignments) were generated: (i) using the “core genes” only, i.e., those alignments containing sequences from all genomes, (ii) a “full” matrix using all alignments comprising at least four sequences, (iii) the same matrix but filtered with MARE (http://mare.zfmk.de) (Meusemann et al., 2010) without removing organisms. Additionally, two smaller matrices of preselected genes were analyzed, using the distinct sets of 31 genes, respectively, suggested by Ciccarelli et al. (2006) and Wu and Eisen (2008). The OrthoMCL clusters were also converted to an ortholog-content matrix representing the presence or absence of a gene within a certain genome and clusters of orthologs. Further, clusters of homologous sequences were determined using a re-implementation of the TribeMCL algorithm (Enright et al., 2002), applying an e-value threshold of 10−5 and an MCL inflation parameter of 2.0. The clusters of homologs were converted to a gene-content matrix in analogy to the ortholog-content matrix.
As no genomic data were available for the other organisms of interest (further Ruegeria species, Litorimicrobium taeanense (Jin et al., 2011), Phaeobacter aquamixtae (Park et al., 2014), P. leonis (Gaboyer et al., 2013), Puniceibacterium antarcticum (Liu et al., 2014), and Seohaeicola saemankumensis (Yoon et al., 2009), their position was assessed by 16S rRNA gene sequences only. These were analyzed unconstrained as well as constrained by enforcing the monophyly of the maximally supported groups from the supermatrix analysis. As the 16S rRNA gene analysis contained more species than the phylogenomic analyses, the supermatrix tree yields a backbone constraint, which enforces only the relative positioning of the species contained in all data matrices. The 16S rRNA gene alignment used was again the one from the LTP version s111, from which taxa not of interest were removed (with deletion of all resulting gap-only alignment columns) and to which the sequences of P. aquamixtae (KF554505), P. leonis (HE661585), and P. antarcticum (JX070673) were aligned using POA version 2.0 (Lee et al., 2002).
Maximum likelihood (ML) (Felsenstein, 1981) and maximum-parsimony (MP) (Fitch, 1977; Goloboff, 2003) phylogenetic trees were inferred from the data matrices as previously described (Spring et al., 2010; Anderson et al., 2011; Göker et al., 2011; Abt et al., 2012, 2013; Frank et al., 2014; Stackebrandt et al., 2014; Verbarg et al., 2014). The Pthreads-parallelized RAxML package version 7.2.8 (Stamatakis, 2006) was used for ML, applying fast bootstrapping in conjunction with the autoMRE bootstopping criterion (Pattengale et al., 2010) and subsequent search for the best tree (Stamatakis et al., 2008). Tree searches under the MP criterion were conducted with PAUP* version 4b10 (Swofford, 2002) using 100 rounds of random sequence addition and subsequent TBR branch swapping, saving no more than 10 best trees per round and collapsing potential zero-length branches during tree search. MP bootstrap support was calculated with PAUP* using 1000 replicates with 10 rounds of heuristic search per replicate. For each supermatrix, the best ML amino-acid substitution model was determined beforehand by comparing the resulting log likelihoods on a MP starting tree. For the ortholog-content and the gene-content matrices, the BINGAMMA model as implemented in RAxML was used, and for the rRNA gene matrices the GTRGAMMA model. The phylogenomic trees were checked for long-branch attraction artifacts (Felsenstein, 2004; Bergsten, 2006) using selected long-branch extraction (Siddall and Whiting, 1999) experiments. To assess the significance of phylogenetic conflict, if any, between data matrices, paired-site tests as implemented in RAxML and PAUP* were conducted, comparing the best tree(s) from unconstrained search with the best ones from a search (backbone-) constrained for the well-supported parts of the topology obtained via one to several other analyses.
The G+C content of all species was determined from the genome sequences, allowing for higher precision than the wet-lab methods. The values were taken from previous studies (Beyersmann et al., 2013; Buddruhs et al., 2013a; Dogs et al., 2013a,b; Freese et al., 2013; Riedel et al., 2013; Breider et al., 2014; Frank et al., 2014).
Whole-cell protein extracts of the type strains of Phaeobacter and Leisingera as well as those of the neighboring genera Litorimicrobium, Nautella, Oceanibulbus, Sediminimonas, Salinihabitans, Seohaeicola, Roseobacter, and Ruegeria were analyzed by MALDI-TOF MS (Maier and Kostrzewa, 2007) using a Microflex L20 mass spectrometer (Bruker Daltonics) equipped with a N2 laser. Sample preparation for MALDI-TOF MS protein analysis was carried out according to the ethanol/formic acid extraction protocol recommended by Bruker Daltonics as described in detail by Tóth et al. (2008). The MALDI-TOF mass spectra were analyzed with the BioTyper software (version 3.1, Bruker Daltonics).
Results
The comprehensive 16S rRNA gene alignment for the Rhodobacteraceae contained 245 organisms and 1314 characters. The resulting ML and MP trees had a log likelihood of -32912.16 and a number of steps of 6329, respectively, and are shown in Supplementary File 1 together with the bootstrapping values. As expected, the 16S rRNA gene analysis overall suffered from poor resolution (average branch support 45.3% under ML, 39.7% under MP). The Leisingera and Phaeobacter species were distributed over several clusters, hence none of the two genera formed a monophyletic group. With the exception of the strongly supported (99/98%) sister-group relationship between P. arcticus and P. leonis, the moderately supported (86/92%) group comprising L. methylohalidivorans and L. aquimarina and the weakly supported (69/<60%) group containing P. aquaemixtae, P. caeruleus, and P. daeponensis, the internal edges of these clades were unsupported. Other species that might potentially form the sister group of any of the Phaeobacter and Leisingera clades included Pelagicola litoralis and P. antarcticum. In contrast, Nautella italica, S. saemankumensis, and L. taeanense were placed in more isolated positions, whereas S. qiaohouensis and Salinihabitans flavidus formed a moderately supported (89/62%) clade that comprised the sister group of the cluster containing Ruegeria. So the evidence that any of these genera were intermixed with either Leisingera or Phaeobacter was negligible, but to assume a close relationship of other genera with Leisingera or Phaeobacter would be even more speculative. For this reason, the forthcoming analyses were restricted to the genera Leisingera, Pelagicola, Phaeobacter, and Puniceibacterium. Additionally, only Litorimicrobium, Nautella, Ruegeria, Salinihabitans, Sediminimonas, and Seohaeicola were included to serve as a close outgroup and Roseobacter and Oceanibulbus for rooting the tree, yielding 14 organisms in the phylogenomic and 28 organisms in an additional 16S rRNA analysis.
The core-gene amino-acid supermatrix comprised 1550 genes and 502,216 characters, whereas the “full” supermatrix contained 4582 genes and 1,390,199 characters before, 2527 genes and 767,593 characters after cleaning with MARE. The “Ciccarelli” data matrix contained 7811 characters, whereas the “Wu-Eisen” matrix comprised 7905 characters. For all five matrices, the selected model was PROTGAMMALGF [the LG model of amino acid evolution (Le and Gascuel, 2008) in conjunction with gamma-distributed substitution rates (Yang, 1993) and empirical amino acid frequencies]. The resulting trees had log likelihoods of (i) -5132414.78, (ii) -12814043.60, (iii) -7798812.71, (iv) -57494.18 and (v) -53469.14, respectively. The core-gene, MARE-filtered supermatrix as well as “Wu-Eisen” trees were topologically identical. This topology is shown in Figure 1 together with ML and MP bootstrap support values from all five supermatrix analyses if larger than 60%. The tree inferred from the unfiltered supermatrix and “Ciccarelli” matrix showed a distinct grouping of P. arcticus DSM 23566T, i.e., as sister group of the clade comprising P. inhibens T5T and P. gallaeciensis CIP 105210T. The best MP trees found had lengths of (i) 714,607, (ii) 1,749,584, (iii) 1,091,094, (iv) 6225, (v) 5499 steps, respectively, (not counting counting uninformative characters) and were topologically identical to the ML core-gene and MARE-filtered supermatrix trees. The “Ciccarelli” tree showed yet another grouping of P. arcticus DSM 23566T, i.e., as sister group of the clade comprising L. aquimarina DSM 24565T, L. methylohalidivorans MB2T, P. caeruleus 13T, P. daeponensis TF-218T, P. inhibens T5T, and P. gallaeciensis CIP 105210T. Support was maximum (100%) for all branches under ML and MP but the previously described deviating ones (Figure 1). The trees agreed regarding a maximally supported monophyletic group comprising P. daeponensis, P. caeruleus, L. methylohalidivorans as well as L. aquimarina, regarding another clade with maximum support containing P. gallaeciensis and P. inhibens, and regarding the placement of Ruegeria spp. as sister group of Leisingera and Phaeobacter to the exclusion of L. nanhaiensis. Removal of the outgroup and subsequent phylogenetic inference yielded trees with the same topology that would have been obtained by pruning the outgroup from the tree depicted in Figure 1 (data not shown), indicating that the position of L. nanhaiensis is not due to long branch attraction.
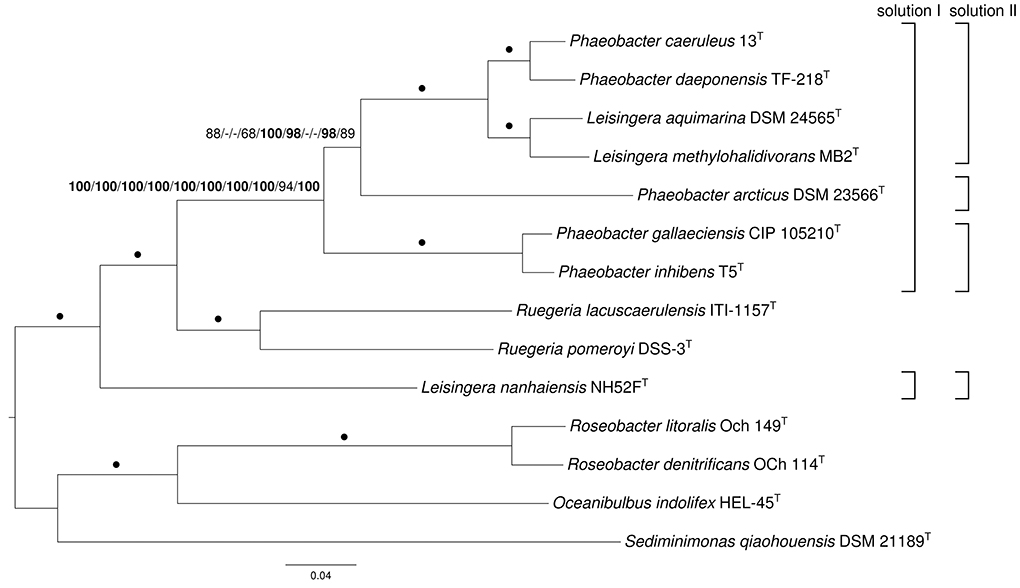
Figure 1. Phylogenetic tree inferred from the core-gene matrix under the maximum likelihood (ML) criterion and rooted with Oceanibulbus, Roseobacter and Sediminimonas. The branches are scaled in terms of the expected number of substitutions per site. Numbers above the branches (from left to right) are bootstrapping support values (if larger than 60%) from (i) ML core-genes; (ii) maximum-parsimony (MP) core-genes; (iii) ML unfiltered supermatrix; (iv) MP unfiltered supermatrix; (v) ML MARE-filtered supermatrix; (vi) MP MARE-filtered supermatrix; (vii) ML “Ciccarelli” matrix; (viii) MP “Ciccarelli” matrix; (ix) ML “Wu-Eisen” matrix; (x) MP “Wu-Eisen” matrix analysis. Values larger than 95% are shown in bold; dots indicate branches with maximum support under all settings. On the right side two potential new taxonomic arrangements into genera are shown that are in agreement with the tree.
The ortholog-content matrix contained 13,676 characters, and the resulting best trees had a log likelihood of -77923.13 and a length of 19,909 steps, respectively. The gene-content matrix comprised 9844 characters and yielded best trees with a log likelihood of -54954.97 and a parsimony score of 13,580, respectively. Both ML trees were topologically identical and are shown in Figure 2 with bootstrap support values for ML and MP if larger than 60%. The MP gene-content tree showed the monophyly of Ruegeria, whereas P. arcticus DSM 23566T was grouped as in the supermatrix tree (see Figure 1). Like the supermatrix trees, a maximally supported clade comprising P. daeponensis, P. caeruleus, L. methylohalidivorans as well as L. aquimarina and another one containing P. gallaeciensis and P. inhibens were revealed. In addition to Ruegeria spp., in the gene- and ortholog-content trees Oceanibulbus and Sediminimonas were indicated as more closely related than L. nanhaiensis to the remaining Leisingera and Phaeobacter species. None of the branches in conflict with the supermatrix trees were particularly well supported.
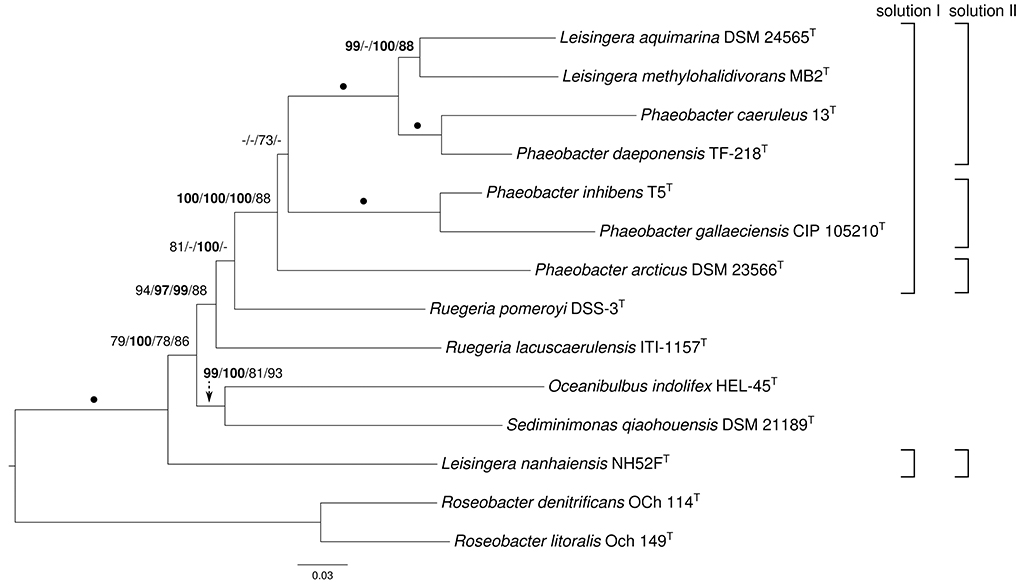
Figure 2. Phylogeny inferred from the ortholog-content matrix under the maximum likelihood (ML) criterion and rooted with Roseobacter. The branches are scaled in terms of the expected number of substitutions per site. Numbers above the branches (from left to right) are bootstrapping support values (if larger than 60%) from (i) ML ortholog-content matrix; (ii) maximum-parsimony (MP) ortholog-content matrix; (iii) ML gene-content matrix; (iv) MP gene-content matrix analysis. Values larger than 95% are shown in bold; dots indicate branches with maximum support under all settings. On the right side two potential new taxonomic arrangements into genera are shown that are in agreement with the tree.
The rRNA gene matrix of selected organisms contained 1503 characters and yielded a highest likelihood of -6252.76 and a minimal length of 790 steps in unconstrained search, -6292.42 and 807 steps in constrained search. The constrained trees were neither significantly worse in the MP-based Kishino–Hasegawa test as implemented in PAUP* (α = 0.01) nor in the ML-based Shimodaira–Hasegawa test as implemented in RAxML (α = 0.01), indicating no conflict between the genomic data and 16S rRNA gene. One of the best constrained MP trees is shown in Figure 3 together with ML and MP bootstrap support values from both constrained and unconstrained analyses. Without the constraint, the 16S rRNA gene yielded little overall support, but it maximally supported the sister-group relationship of P. leonis with P. arcticus. L. nanhaiensis was placed apart from the other Leisingera species, but without support, not even in the constrained analysis. Importantly, none of the species not present in the phylogenomic analysis were additionally placed in the clade containing the other Leisingera species together with P. caeruleus and P. daeponensis, even though this was not enforced by the constraint.
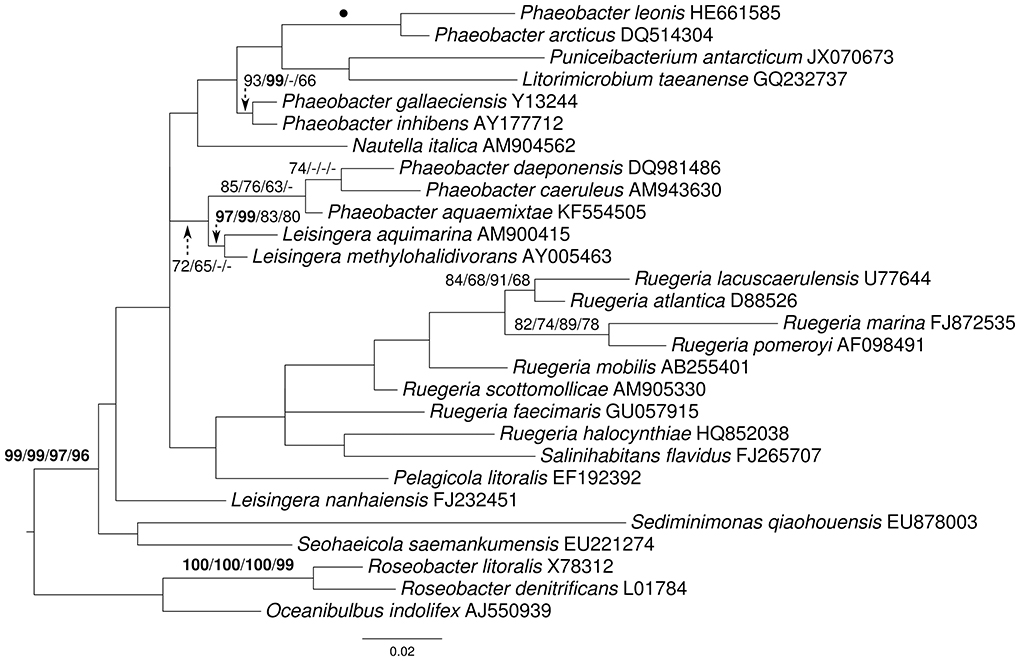
Figure 3. Phylogeny inferred from the 16S rRNA gene matrix under the maximum likelihood (ML) criterion and the maximally supported branches of the topology depicted in Figure 1 as backbone constraint. Rooting was done with Oceanibulbus and Roseobacter. The branches are scaled in terms of the expected number of substitutions per site. Numbers above the branches (from left to right) are bootstrapping support values (if larger than 60%) from (i) constrained ML, (ii) constrained maximum-parsimony (MP), (iii) unconstrained ML, and (iv) unconstrained MP analysis. Values larger than 95% are shown in bold; dots indicate branches with maximum support under all settings.
Again, the topology (Figure 3) differed to some degree from those inferred from the other data matrices, but it also showed a weakly (72/65%) supported monophyletic group containing P. daeponensis, P. caeruleus, P. aquaemixtae, L. methylohalidivorans, and L. aquimarina as well as another clade containing P. gallaeciensis and P. inhibens (93/99% support). Further, as in the previous trees, L. nanhaiensis was shown to branch first, before Ruegeria spp. Thus, none of the branches in conflict with the previous trees obtained particularly high bootstrap supported. The 16S rRNA gene sequences also clearly separated L. nanhaiensis from the other Leisingera species but yielded few support otherwise.
The dendrogram from the MALDI-TOF MS analysis is shown in Figure 4. It confirms the following groupings of taxonomic interest in the current study: P. arcticus as sister group of P. leonis, both set apart from the type species P. gallaeciensis and other Phaeobacter species; P. gallaeciensis as sister group of P. inhibens; a group comprising P. caeruleus, P. daeponensis, L. aquimarina and the type species L. methylohalidivorans; and L. nanhaiensis set apart from the other Leisingera species and also from L. taeanense. Rather, L. nanhaiensis is found as sister taxon of the group P. inhibens and P. gallaeciensis.
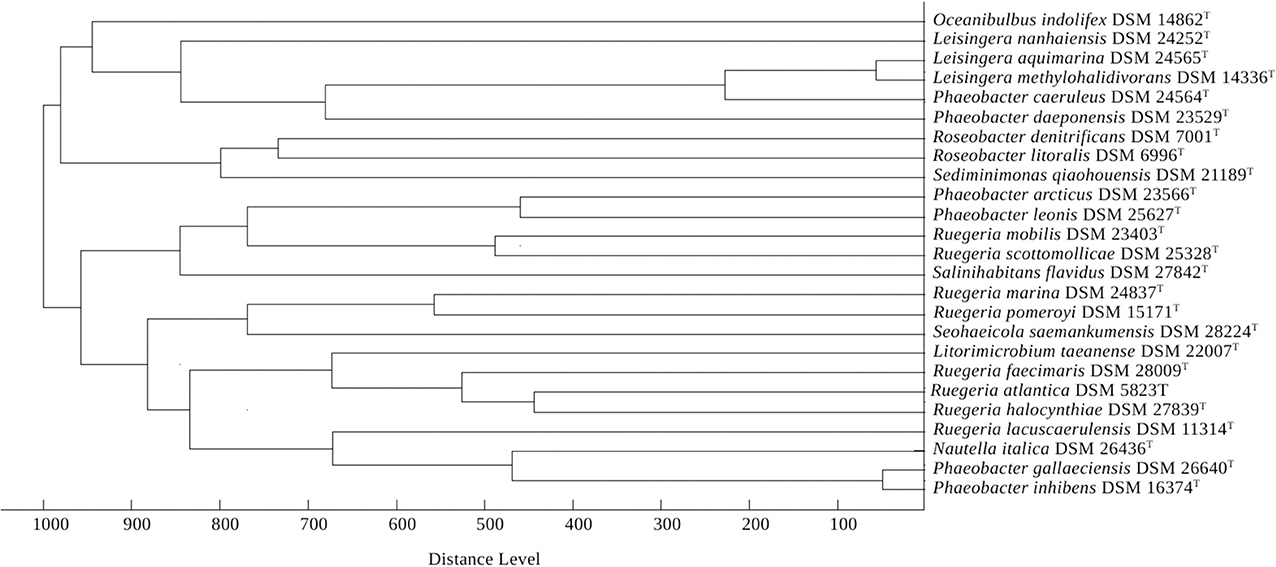
Figure 4. Score-oriented dendrogram generated by the BioTyper software (version 3.1, Bruker Daltonics) showing the similarity of MALDI-TOF mass spectra of cell extracts of type strains of selected species within the genera Leisingera, Phaeobacter, Litorimicrobium, Nautella, Oceanibulbus, Roseobacter, Salinihabitans, Seohaeicola, Sediminimonas, and Ruegeria.
An overview of the taxonomically relevant phenotypic characters collected from the literature and the G+C content values inferred from the genome sequences is given in Table 1. They are discussed in detail below in the light of the phylogenetic analyses. Supplementary File 2 contains the complete set of phenotypic characters analyzed in the course of this study.
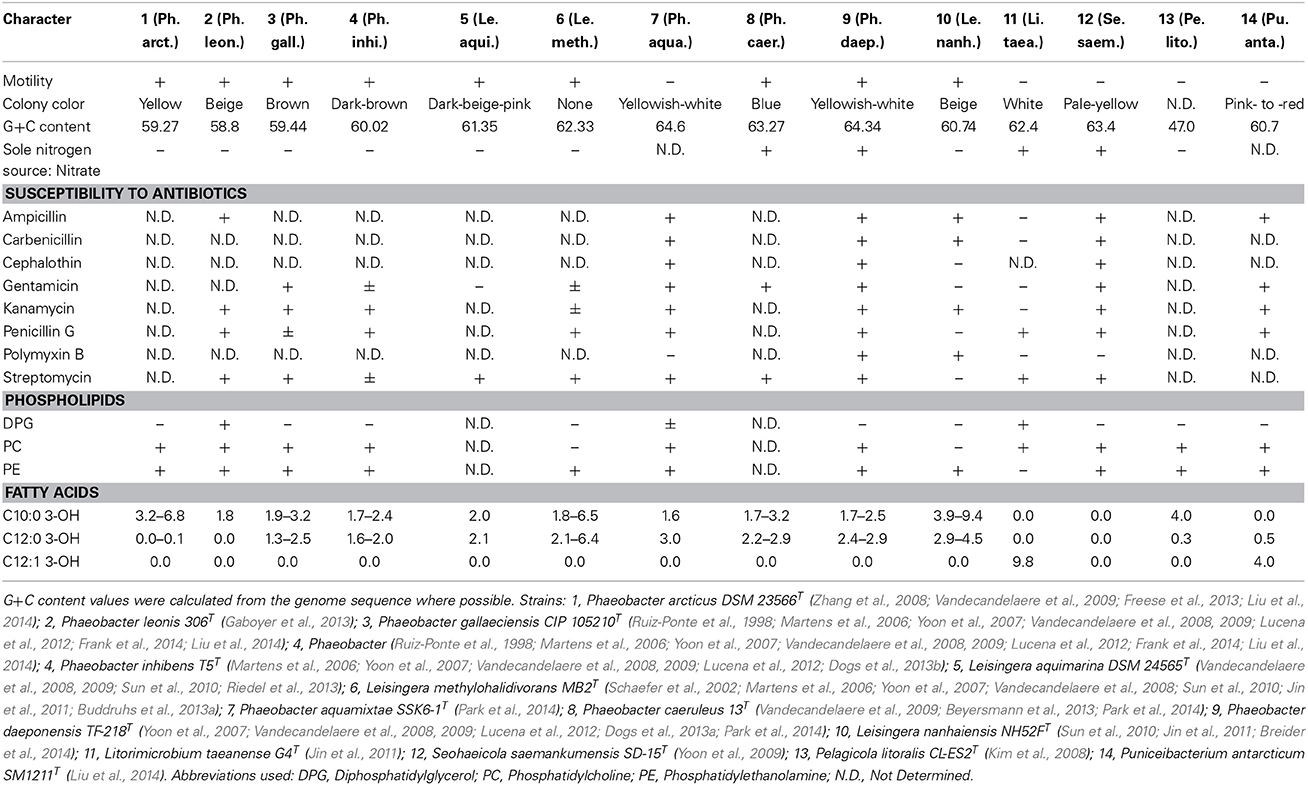
Table 1. Major phenotypic and genomic properties that differentiate Leisingera spp., Phaeobacter spp., Pseudophaeobacter spp., and Sedimentitalea nanhaiensis from each other and from probably closely related genera.
Discussion
The conducted phylogenomic analyses aimed at generating several distinct genome-scale data matrices for assessing whether, and in which respects, their distinct analyses corroborated each other, instead of generating a single data matrix and subsequent phylogenetic inference whose sensitivity to issues such as gene selection remained essentially unknown (Klenk and Göker, 2010). The conducted analyses indeed corroborated each other regarding the genealogy of the organisms investigated, as no well-supported conflict between the resulting topologies was found. The sole exception might be the single branch in Figure 1 that was not maximally supported by all analyses, but for taxonomic classifications inferred from phylogenomic analyses a remaining set of only ambiguously supported branches would not matter anyway, as not all subtrees could be assigned a taxon name in a Linnean system because of its limited number of taxonomic ranks (Wiley and Lieberman, 2011).
Importantly, the paired-site tests indicate that there is no significant conflict of the topologies from phylogenomic analyses with the 16S rRNA gene data either. The conflicts between the current classification are thus due to the insufficient resolution of the 16S rRNA gene in this group (Figure 3). This problem can hardly be avoided in current microbial taxonomy as long as it relies only on 16S rRNA gene data for phylogenies, simply because newly described species must be placed in some genus. Whereas one might be tempted to presume that stability in taxonomic classification might come from using a standardized set of characters (such as the 16S rRNA gene, or any other standardized set of genes), this is not actually the case. As in previous studies (Spring et al., 2010; Anderson et al., 2011; Göker et al., 2011; Abt et al., 2012, 2013; Frank et al., 2014; Stackebrandt et al., 2014; Verbarg et al., 2014) and listed in Supplementary File 3, the analyses conducted here confirmed that more characters (up to entire genomes) yield better resolved phylogenies (Figure 1) and thus better substantiated classifications. This is particularly striking for the two 31-gene data sets (Ciccarelli et al., 2006; Wu and Eisen, 2008) which, even though the gene sets are partially overlapping, yield distinct phylogenies (Figure 1). These observations strongly argue for a “total evidence” approach (Kluge, 1989; Lienau and DeSalle, 2009). Of course the trees should also be correct, but it is possible to detect artifacts such as long-branch attraction (Bergsten, 2006), and there is no indication for those in the current analyses. In the following we discuss the phylogenomic outcomes in the light of the phenotypic data known for Leisingera, Phaeobacter and their probable close relatives and assess several possible taxonomic rearrangements.
In all phylogenetic analyses conducted, L. nanhaiensis was unambiguously distinct from the remaining species of the genus, which had a closer relationship to both Phaeobacter and Ruegeria. In contrast to the other Leisingera species, L. nanhaiensis shows a lower DNA G+C content and no susceptibility to the antibiotic streptomycin (Table 1). In the original 16S rRNA gene analysis conducted by Sun et al. (2010), L. nanhaiensis was placed on a long branch as sister group of the other Leisingera species, but with low bootstrap support. The inference method used was neighbor joining based on a Kimura-2-parameter evolutionary model, which might be too simplistic for these data (for instance, it does not distinguish between rapidly and slowly evolving alignment positions) (Felsenstein, 2004). Another reason for the differences between the outcomes of the 16S rRNA gene analyses might be distinct taxon sampling. The 16S rRNA gene tree topologies inferred in the studies on the Leisingera and Phaeobacter genomes varied considerably depending on the included species from other genera (Beyersmann et al., 2013; Buddruhs et al., 2013a; Dogs et al., 2013a,b; Freese et al., 2013; Riedel et al., 2013; Breider et al., 2014). This is as expected, since the 16S rRNA gene trees for the group hardly contain branches with relevant support. Quite in contrast, the phylogenomic analyses yielded high support, and there is no reason to assume that any of these branches could be easily affected by taxon sampling. As mentioned above, more characters yield better resolved phylogenies and thus more reliable taxonomic classifications; the present study is no exception from this rule. Thus, there is no evidence for the sister-group relationship of L. nanhaiensis to the type species L. methylohalidivorans and the other Leisingera species and evidence from more than a million characters against it (Figure 1).
A potential solution is to include L. nanhaiensis in another already established genus of Rhodobacteraceae. But given the already evident taxonomic problems in the Roseobacter group that have been caused by a low resolution of the 16S rRNA gene, the mere fact that no sister-group relationship between L. nanhaiensis and another genus has any statistical support argues against this proposal. Including L. nanhaiensis in an already existing genus would avoid introducing a novel genus, but would be too risky, because future phylogenomic studies might easily demonstrate such a group to be non-monophyletic. Characters other than the 16S rRNA gene neither support such a merging. For instance, the MALDI-TOF MS analysis is in conflict with a sister-group-relationship of L. nanhaiensis and L. taeanense (Figure 4); the analysis would rather suggest an affiliation to P. gallaeciensis and P. inhibens, but this would be in strong conflict with the phylogenomic analyses (Figures 1, 2). Chemotaxonomically, L. nanhaiensis can be distinguished from L. taeanense G4T by the presence of phosphatidylethanolamine and C10:0 3-OH (about 4–9%), C12:0 3-OH (about 3–5%) in only the former and the presence of (about 10%) C12:1 3-OH, phosphatidylcholine and diphosphatidylglycerol in only the latter (Table 1). Moreover, they can be distinguished by their motility, anaerobic growth with nitrate and the susceptibility to ampicillin, carbenicillin, kanamycin, penicillin G, polymyxin B and streptomycin (Table 1). Similarly, L. nanhaiensis can be distinguished from S. saemankumensis regarding anaerobic growth with nitrate, motility, fatty acids and polar lipids and susceptibility/resistance to antibiotics (Table 1). The fatty acid composition of L. nanhaiensis is quite similar to the one of P. litoralis but the two can be distinguished by the >10% higher G+C content and the absence of phosphatidylcholine in the former (Table 1). Differences between L. nanhaiensis and Puniceibacter antarcticum are the motility of the former, the presence of (about 4%) C12:1 3-OH in the latter, and the colony color (Table 1). Other characteristics which distinguish L. nanhaiensis from other species are shown in Table 1. The MALDI-TOF MS analysis shows L. nanhaiensis as sister taxon of the group P. inhibens and P. gallaeciensis. However, no other evidence (16S rRNA, Figure 4; phenotypic characteristics, Table 1) for including L. nanhaiensis in Phaeobacter was found.
The differences mentioned above might partially be regarded as few, but one should not overlook that given the frequently ambiguous phenotypic characters in the entire Roseobacter group, it is unlikely that, on average, more phenotypic differences would appear if L. nanhaiensis would be compared to any other genus within the group. This is supported by the suggested “mix-and-match” genome arrangement (Moran et al., 2007) found within the Roseobacter group, which could make the genomes of organisms of this group very flexible. Accordingly the physiology of the organisms is not necessarily reflected by their phylogeny, and it has been found that trophic strategies correlate better than phylogeny (Newton et al., 2010).
The third potential alternative for creating monophyletic taxa, i.e., to merge even more genera (such as all ingroup genera in Figure 3), can be rejected for two reasons. First, it would be less taxonomically conservative as it involved more name changes. Second, given the large number of unsupported branches in the 16S rRNA analysis (Figure 3), merging all these genera would be phylogenetically even more uncertain than the inclusion of L. nanhaiensis into another genus. We thus propose to reclassify L. nanhaiensis as Sedimentitalea nanhaiensis gen. nov., comb. nov. The description of the genus Leisingera is emended accordingly.
The discrepancies between phylogeny and classification that remain after the removal of L. nanhaiensis from the genus Leisingera (Figures 1–3) could be solved either by merging Leisingera and Phaeobacter (solution I) or by assigning P. arcticus and P. leonis to a new genus and reclassifying P. aquaemixtae, P. caeruleus, and P. daeponensis as members of the genus Leisingera. As Leisingera has priority over Phaeobacter, solution I would involve one change at the genus level (removal of the genus Phaeobacter) and seven changes at the species level (assignment of the seven Phaeobacter species to Leisingera). In contrast, solution II would involve one change at the genus level (introduction of one new genus) and five changes at the species level (assignment of five Phaeobacter species to another genus). Solution II thus involves fewer overall taxonomic changes. Another disadvantage of solution I is that the clade comprising only Phaeobacter and Leisingera except for L. nanhaiensis is unsupported in the 16S rRNA gene analysis (Figure 3). That is, a merging of Leisingera (except L. nanhaiensis) and Phaeobacter would bear the risk of creating a group that turns out as non-monophyletic once more genomes from the other organisms now included only in the 16S rRNA tree (Figure 3) become available for a phylogenomic analysis. The MALDI-TOF MS analysis (Figure 4) is also in conflict with the merging of the two genera. Furthermore, solution I would lead to a combination of organisms with very different physiological and chemotaxonomic features within one genus. The number of common features for the different species would be reduced and the genus description could only comprise very general features, also found for species of other genera within the Rhodobacteracae and thus not suitable for a discrimination. Indeed, as shown in the following, the genus Phaeobacter as currently circumscribed is already phenotypically very heterogeneous.
P. gallaeciensis and P. inhibens produce a brown, diffusible pigment and show a strong inhibitory activity against bacteria, based on production of the antibiotic tropodithietic acid (TDA) (Martens et al., 2006). This is in contrast to what was described for P. aquaemixtae, P. caeruleus and P. daeponensis (see Yoon et al., 2007; Vandecandelaere et al., 2009; Park et al., 2014 and Table 1). The colony color of these species was described as yellowish-white for P. aquaemixtae and P. daeponensis but blue for P. caeruleus, although P. daeponensis also forms blue colonies when grown on YTSS medium (Dogs et al., 2013a). P. arcticus and P. leonis show a yellow or beige colony color in contrast to the brown or dark brown color described for P. gallaeciensis and P. inhibens (Table 1). A yellow-brown extracellular pigment is correlated with the production of TDA in members of the Roseobacter group (Geng et al., 2008; Berger et al., 2011). Thus, it can be assumed that P. aquaemixtae, P. arcticus, P. caeruleus, P. daeponensis, and P. leonis do not produce TDA, which is confirmed by the absence of genes involved in TDA production in the known genomes of these organisms. Other phenotypical differences which distinguish P. gallaeciensis and P. inhibens on the one hand from P. aquaemixtae, P. caeruleus, P. daeponensis, L. aquimarina, and L. methylohalidivorans on the other hand are the utilization of D-mannose, D-maltose, D-cellobiose, D-galactose, D-xylose and the tolerance to ampicillin (Supplementary File 2). The incapability to utilize D-mannose and D-maltose was previously included in the description of the genus Leisingera (Vandecandelaere et al., 2009). It is more difficult to separate P. arcticus and P. leonis from Leisingera phenotypically, but the two are the sole species in the genera Phaeobacter and Leisingera which hardly or not at all form the fatty acid C12:0 3-OH. Moreover, P. arcticus and P. leonis are phylogenetically (Figures 1–3), regarding the MALDI-TOF MS data (Figure 4) and the G+C content (Table 1) obviously distinct from Leisingera.
P. aquaemixtae, P. caeruleus, and P. daeponensis also show a higher G+C content (63–64%) than P. gallaeciensis (59.4%) and P. inhibens (60%). Besides the phylogenetic analyses, showing that P. aquaemixtae, P. caeruleus, and P. daeponensis cluster together with L. methylohalidivorans (the type species of Leisingera) and L. aquimarina, the phenotypic and genomic characteristics of these three Phaeobacter species are also more similar to those of L. aquimarina with its dark beige-pink color and G+C content of 61.3%, and of L. methylohalidivorans, which is non-pigmented and has a G+C content of 62.3% (Table 1). P. gallaeciensis CIP 105210T was tested negatively for genes coding for tmm or gammaglutamylmethylamide synthetase (gmaS). These genes were used as functional markers for the utilization of methylated amines as alternative nitrogen source (Chen, 2012). L. aquimarina and L. methylohalidivorans were both tested positively for the two genes and are able to use monomethylamine (MMA) and trimethylamine (TMA) as sole nitrogen source (Chen, 2012). The genomes of P. caeruleus and P. daeponensis also possess tmm and gmaS sequences, in contrast to the strains of P. inhibens (i.e., T5T, 2.10, DSM 17395) and P. gallaeciensis (CIP105210T) (Chen, 2012; this study).
The conventional DDH experiments conducted by Vandecandelaere et al. (2009) resulted in the highest similarity of P. caeruleus with L. methylohalidivorans (55 ± 1%). A significantly lower similarity (40 ± 5%) was found with P. gallaeciensis, indicating a closer affiliation to the genus Leisingera. A comparison with DDH similarities calculated in silico for genome-sequenced strains using GGDC 2.0 supports the results of the phylogenomic analyses. Comparatively high similarity (36.2 ± 2.57) was observed between P. gallaeciensis CIP 105210T and P. inhibens T5T, compared to values between 20.6 ± 2.46 and 22.6 ± 2.46 for the similarities to the other species of the genera Phaeobacter and Leisingera (Dogs et al., 2013b). The similarities between the strains L. aquimarina DSM 24565T, L. methylohalidivorans MB2T, P. caeruleus 13T, and P. daeponensis TF-218T ranged from 27.9 ± 2.43 to 40.3 ± 2.51, compared to lower values of 19.2 ± 2.28 to 21.5 ± 2.34 for the similarities to the other strains (Beyersmann et al., 2013; Buddruhs et al., 2013a; Dogs et al., 2013a; Riedel et al., 2013). The DDH result for L. aquimarina DSM 24565T and L. methylohalidivorans MB2T was 32.4 ± 2.46 (Riedel et al., 2013), the result for P. caeruleus 13T and P. daeponensis TF-218T was 40.3 ± 2.51 (Beyersmann et al., 2013), in accordance with the branching within the trees (Figures 1–3). The in silico DDH analysis of P. arcticus DSM 23566T showed only low similarity values (20.4 ± 2.32 to 22.9 ± 2.37) when compared to the other Phaeobacter and Leisingera strains (Freese et al., 2013), indicating a lower degree of relatedness.
Based on these polyphasic results we propose to reclassify P. aquaemixtae as Leisingera aquaemixtae comb. nov., P. caeruleus as Leisingera caerulea comb. nov., P. daeponensis as Leisingera daeponensis comb. nov., P. arcticus as type species of the new genus Pseudophaeobacter as Pseudophaeobacter arcticus gen. nov., comb. nov., and P. leonis as Pseudophaeobacter leonis comb. nov. The descriptions of the genera Phaeobacter and Leisingera are emended accordingly.
The proposed reclassifications lead to a homogenous Leisingera-Phaeobacter cluster, consisting of monophyletic genera for Phaeobacter (including only P. gallaeciensis and P. inhibens) and Leisingera (including L. aquaemixtae, L. aquimarina, L. caerulea, L. daeponensis and L. methylohalidivorans) with Pseudophaeobacter (including P. arcticus and P. leonis) and Sedimentitalea (including S. nanhaiensis) as separate lineages. Reduction of the genus Phaeobacter to the species P. gallaeciensis and P. inhibens returns to the original genus description given by Martens et al. (2006) and allows a much better discrimination of the genus Phaeobacter against the closely related genera. Equivalently, the changes suggested in solution II allow for a better discrimination of the genus Leisingera and the newly proposed genera Pseudophaeobacter and Sedimentitalea. Classification of new species would subsequently be based on much clearer taxonomic definitions. Given the low resolution of the 16S rRNA gene within Rhodobacteraceae, more precisely defined genera will also reduce the future risk of creating non-monophyletic groups.
Description of Sedimentitalea, gen. nov.
Sedimentitalea (Se.di.men.ti.ta'le.a. L. n. sedimentum, sediment; L. fem. n. talea, a rod; N.L. Sedimentitalea, fem. a rod isolated from sediment).
Gram negative, oxidase and catalase positive, motile rod-shaped bacteria, 0.6–0.8 μm wide and 1.6–3.0 μm long. Sodium ions are essential for growth. The major polar lipids are phosphatidylglycerol, phosphatidylethanolamine, an unidentified phospholipid, an unidentified lipid and an aminolipid. The fatty acid composition (>1%) is C18:1 ω 7c, an unknown fatty acid (equivalent chain length of 11.799), C16:0 2-OH, C10:0 3-OH, C16:0, 11-methyl C18:1 ω 7c and C12:0 3-OH. The G+C content is about 60–61%.
On the basis of 16S rRNA gene sequence and particularly phylogenomic analysis, the genus represents a separate branch within the family Rhodobacteraceae of the class Alphaproteobacteria. The type species (and currently sole species) of the genus is S. nanhaiensis.
Description of Sedimentitalea Nanhaiensis, comb. nov.
S. nanhaiensis (nan.hai.en'sis. N.L. fem. adj. nanhaiensis referring to Nanhai, the Chinese name for the South China Sea, from where the type strain was isolated).
Basonym: Leisingera nanhaiensis (Sun et al., 2010).
The description is the same as for L. nanhaiensis (Sun et al., 2010) as emended by Breider et al. (2014). The type strain is NH52FT (LMG 24841T, DSM 24252T).
Description of Pseudophaeobacter, gen. nov.
Pseudophaeobacter (Pseu.do.phae.o.bac'ter. Gr. adj. pseudes false; N.L. masc. n. Phaeobacter, a bacterial genus; N.L. masc. n. Pseudophaeobacter false Phaeobacter).
Gram-negative, aerobic, oxidase and catalase positive, rod-shaped bacteria. The cells are 1.0–2.6 μm long and 0.3–0.5 μm wide. Sodium ions are essential for growth. Ubiquinone-10 is the principal isoprenoid quinone. The main polar lipids present are phosphatidylethanolamine, phosphatidylglycerol, phosphatidylcholine and an unidentified aminolipid. The predominant fatty acids (>1%) are C18:1 cis7ω, C18:1 cis7ω methyl, an unknown fatty acid (equivalent chain length of 11.799), C16:0 (hexadecanoic acid), C10:0 3-OH. The G+C content of the currently two species of this genus is 58.8–59.2%.
On the basis of 16S rRNA gene sequence and particularly phylogenomic analysis, the genus represents a separate branch within the family Rhodobacteraceae of the class Alphaproteobacteria. The type species is P. arcticus.
Description of Pseudophaeobacter Arcticus, comb. nov.
P. arcticus (arc'ti.cus. L. masc. adj. arcticus northern, arctic, referring to the site from where the type strain was isolated).
Basonym: Phaeobacter arcticus (Zhang et al., 2008).
The description of the species is the same as given for P. arcticus by Zhang et al. (2008). The type strain is 20188T (JCM 14644T, DSM 23566T).
Description of Pseudophaeobacter Leonis, comb. nov.
(le.o'nis., L. gen n. leonis, of a lion, named after sinus Leonis, the Medieval Latin name of the Gulf of Lion, in reference to the origin of the type strain).
Basonym: Phaeobacter leonis (Gaboyer et al., 2013).
The description of the species is the same as given for P. leonis by Gaboyer et al. (2013). The type strain is 306T (DSM 25627T, CIP 110369T).
Emended Description of the Genus Leisingera (Schaefer et al., 2002)
Leisingera (Lei.sin'ge.ra. N.L. fem. n. Leisingera in honor of Thomas Leisinger, on the occasion of his retirement and for his contributions to our understanding of the biochemistry of bacterial methyl halide metabolism).
The description given by Schaefer et al. (2002) and emended by Martens et al. (2006) and Vandecandelaere et al. (2008) is no longer appropriate due to the addition of further species previously classified in Phaeobacter. The description is thus as given by Vandecandelaere et al. (2008) with the following modifications.
Colony color can vary from non-pigmented to yellowish-white to dark beige-pink and blue, depending on the medium used. The G+C content ranges from 61.3 to 64.6%. Does not degrade casein or hydrolyse aesculin. Positive for leucine aramylase activity, but no activity is detected for lipase (C14), cystine arylamidase, trypsin, α-chymotrypsin, α-galactosidase, β-glucoronidase, α-glucosidase, N-acetyl-β-glucosaminidase, α-mannosidase, and α-fucosidase. Do not assimilate D-maltose or D-mannose. Susceptible to streptomycin. Able to utilize methylated amines as alternative nitrogen source. The type species is L. methylohalidivorans. Its type strain is MB2T (ATCC BAA-92T, DSM 14336T).
Description of Leisingera Aquaemixtae, comb. nov.
L. aquaemixtae (a.quae.mi'xtae. L. fem. n. aqua water; L. fem. part. adj. mixta mixed; N.L. fem. gen. n. aquaemixtae of mixed waters).
Basonym: Phaeobacter aquaemixtae (Park et al., 2014).
The description is the same as that given for P. aquaemixtae (Park et al., 2014). The type strain is SSK6-1T (KTCC 32538T, CECT 8399T).
Description of Leisingera Caerulea, comb. nov.
L. caerulea (cae.ru'le.a. L. fem. adj. caerulea dark blue colored, referring to the colony color of the isolates).
Basonym: Phaeobacter caeruleus (Vandecandelaere et al., 2009).
The description is the same as that for P. caeruleus (Vandecandelaere et al., 2009). The type strain is LMG 24369T (CCUG 55859T, DSM 24564T).
Description of Leisingera Daeponensis, comb. nov.
L. daeponensis (dae.po.nen'sis. N.L. fem. adj. daeponensis of Daepo, Korea, where the type strain was isolated).
Basonym: Phaeobacter daeponensis (Yoon et al., 2007).
The description is the same as that for P. daeponensis (Yoon et al., 2007) as emended by Vandecandelaere et al. (2008) and Dogs et al. (2013a). The type strain is TF-218T (KCTC 12794T, DSM 23529T).
Emended Description of the Genus Phaeobacter (Martens et al., 2006)
The emended description given by Yoon et al. (2007) is no longer appropriate due to the reclassification of P. daeponensis as L. daeponensis. The description is thus as given by Martens et al. (2006) with the following modifications.
Phaeobacter colonies are brownish to dark brown and a diffusible brownish pigment is produced. Produce TDA. Nitrate is not reduced. Facultatively anaerobic by reduction of nitrite. The G+C content is in the range 59.4–60.0%. The type species is P. gallaeciensis. Its type strain is BS107T = CIP 105210T = DSM 26640T.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the German Research Foundation (DFG) Transregio-SFB 51, which is gratefully acknowledged.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2014.00416/abstract
References
Abt, B., Göker, M., Scheuner, C., Han, C., Lu, M., Misra, M., et al. (2013). Genome sequence of the thermophilic fresh-water bacterium Spirochaeta caldaria type strain (H1T), reclassification of Spirochaeta caldaria, Spirochaeta stenostrepta, and Spirochaeta zuelzerae in the genus Treponema as Treponema caldaria comb. nov., Treponema stenostrepta comb. nov., and Treponema zuelzerae comb. nov., and emendation of the genus Treponema. Stand. Genomic Sci. 8, 88–105. doi: 10.4056/sigs.3096473
Abt, B., Han, C., Scheuner, C., Lu, M., Lapidus, A., Nolan, M., et al. (2012). Complete genome sequence of the termite hindgut bacterium Spirochaeta coccoides type strain (SPN1T), reclassification in the genus Sphaerochaeta as Sphaerochaeta coccoides comb. nov. and emendations of the family Spirochaetaceae and the genus Sphaerochaeta. Stand. Genomic Sci. 6, 194–209. doi: 10.4056/sigs.2796069
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Anderson, I. J., Scheuner, C., Göker, M., Mavromatis, K., Hooper, S. D., Porat, I., et al. (2011). Novel insights into the diversity of catabolic metabolism from ten haloarchaeal genomes. PLoS ONE 6:e20237. doi: 10.1371/journal.pone.0020237
Arahal, D. R., Macian, M. C., Garay, E., and Pujalte, M. J. (2005). Thalassobius mediterraneus gen. nov., sp. nov., and reclassification of Ruegeria gelatinovorans as Thalassobius gelatinovorus comb. nov. Int. J. Syst. Evol. Microbiol. 55, 2371–2376. doi: 10.1099/ijs.0.63842-0
Auch, A. F., Klenk, H.-P., and Göker, M. (2010a). Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand. Genomic Sci. 2, 142–148. doi: 10.4056/sigs.541628
Auch, A. F., Von Jan, M., Klenk, H.-P., and Göker, M. (2010b). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Berger, M., Neumann, A., Schulz, S., Simon, M., and Brinkhoff, T. (2011). Tropodithietic acid production in Phaeobacter gallaeciensis is regulated by N-acyl homoserine lactone-mediated quorum sensing. J. Bacteriol. 193, 6576–6585. doi: 10.1128/JB.05818-11
Bergsten, J. (2006). A review of long-branch attraction. Cladistics 21, 163–193. doi: 10.1111/j.1096-0031.2005.00059.x
Beyersmann, P. G., Chertkov, O., Petersen, J., Fiebig, A., Chen, A., Pati, A., et al. (2013). Genome sequence of Phaeobacter caeruleus type strain (DSM 24564T), a surface-associated member of the marine Roseobacter clade. Stand. Genomic Sci. 8, 403–419. doi: 10.4056/sigs.3927626
Breider, S., Teshima, H., Petersen, J., Chertkov, O., Dalingault, H., Chen, A., et al. (2014). Genome sequence of Leisingera nanhaiensis strain DSM 24252T isolated from marine sediment. Stand. Genomic Sci. 9, 687–703. doi: 10.4056/sigs.3828824
Brinkhoff, T., Giebel, H. A., and Simon, M. (2008). Diversity, ecology, and genomics of the Roseobacter clade: a short overview. Arch. Microbiol. 189, 531–539. doi: 10.1007/s00203-008-0353-y
Buchan, A., Gonzalez, J. M., and Moran, M. A. (2005). Overview of the marine Roseobacter lineage. Appl. Environ. Microb. 71, 5665–5677. doi: 10.1128/AEM.71.10.5665-5677.2005
Buddruhs, N., Chertkov, O., Petersen, J., Fiebig, A., Chen, A., Pati, A., et al. (2013a). Complete genome sequence of the marine methyl-halide oxidizing Leisingera methylohalidivorans type strain (DSM 14336T), a member of the Roseobacter clade. Stand. Genomic Sci. 9, 128–141. doi: 10.4056/sigs.4297965
Buddruhs, N., Pradella, S., Göker, M., Päuker, O., Michael, V., Pukall, R., et al. (2013b). Molecular and phenotypic analyses reveal the non-identity of the Phaeobacter gallaeciensis type strain deposits CIP 105210T and DSM 17395. Int. J. Syst. Evol. Microbiol. 63, 4340–4349. doi: 10.1099/ijs.0.053900-0
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chen, Y. (2012). Comparative genomics of methylated amine utilization by marine Roseobacter clade bacteria and development of functional gene markers (tmm, gmaS). Environ. Microbiol. 14, 2308–2322. doi: 10.1111/j.1462-2920.2012.02765.x
Ciccarelli, F. D., Doerks, T., von Mering, C., Creevey, C. J., Snel, B., and Bork, P. (2006). Toward automatic reconstruction of a highly resolved tree of life. Science 311, 1283–1287. doi: 10.1126/science.1123061
Dogs, M., Teshima, H., Petersen, J., Chertkov, O., Dalingault, H., Chen, A., et al. (2013a). Genome sequence of Phaeobacter daeponensis type strain (DSM 23529T), a facultatively anaerobic bacterium isolated from marine sediment, and emendation of Phaeobacter daeponensis. Stand. Genomic Sci. 9, 142–159. doi: 10.4056/sigs.4287962
Dogs, M., Voget, S., Teshima, H., Petersen, J., Fiebig, A., Davenport, K., et al. (2013b). Genome sequence of Phaeobacter inhibens type strain (T5T), a secondary-metabolite producing member of the marine Roseobacter clade, and emendation of the species Phaeobacter inhibens. Stand. Genomic Sci. 9, 334–350. doi: 10.4056/sigs.4448212
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Enright, A. J., Van Dongen, S. M., and Ouzounis, C. A. (2002). An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584. doi: 10.1093/nar/30.7.1575
Felsenstein, J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376. doi: 10.1007/BF01734359
Fitch, W. M. (1977). Toward defining the course of evolution: minimum change on a specified tree topology. Syst. Zool. 20, 406–416. doi: 10.2307/2412116
Frank, O., Pradella, S., Rohde, M., Scheuner, C., Klenk, H.-P., Göker, M., et al. (2014). Complete genome sequence of the Phaeobacter gallaeciensis type strain CIP 105210T (= DSM 26640T = BS107T). Stand. Genomic Sci. 9. doi: 10.4056/sigs.5179110
Freese, H., Dalingault, H., Petersen, J., Pradella, S., Davenport, K., Teshima, H., et al. (2013). Genome sequence of the phage-gene rich marine Phaeobacter arcticus type strain DSM 23566T. Stand. Genomic Sci. 8, 450–464. doi: 10.4056/sigs.383362
Gaboyer, F., Tindall, B. J., Ciobanu, M.-C., Duthoit, F., Le Romancer, M., and Alain, K. (2013). Phaeobacter leonis sp. nov., a novel alphaproteobacterium from Mediterranean Sea sediments. Int. J. Syst. Evol. Microbiol. 63, 3301–3306. doi: 10.1099/ijs.0.046128-0
Geng, H., Bruhn, J. B., Nielsen, K. F., Gram, L., and Belas, R. (2008). Genetic dissection of tropodithietic acid biosynthesis in marine roseobacters. Appl. Environ. Microb. 74, 1535–1545. doi: 10.1128/AEM.02339-07
Göker, M., and Klenk, H.-P. (2013). Phylogeny-driven target selection for genome-sequencing (and other) projects. Stand. Genomic Sci. 8, 360–374. doi: 10.4056/sigs.3446951
Göker, M., Scheuner, C., Klenk, H.-P., Stielow, J. B., and Menzel, W. (2011). Codivergence of mycoviruses with their hosts. PLoS ONE 6:e22252. doi: 10.1371/journal.pone.0022252
Goloboff, P. A. (2003). Parsimony, likelihood, and simplicity. Cladistics 19, 91–103. doi: 10.1111/j.1096-0031.2003.tb00297.x
Jin, H.-M., Lee, H.-J., Kim, J.-M., Park, M.-S., Lee, K., and Jeon, C. E. (2011). Litorimicrobium taeanense gen. nov., sp. nov., isolated from a sandy beach. Int. J. Syst. Evol. Microbiol. 61, 1392–1396. doi: 10.1099/ijs.0.025007-0
Kim, Y.-G., Hwang, C. Y., and Cho, B. C. (2008). Pelagicola litoralis gen. nov., sp. nov., isolated from coastal water in Korea. Int. J. Syst. Evol. Microbiol. 58, 2102–2106. doi: 10.1099/ijs.0.65820-0
Klenk, H.-P., and Göker, M. (2010). En route to a genome-based taxonomy of Archaea and Bacteria? Syst. Appl. Microbiol. 33, 175–182. doi: 10.1016/j.syapm.2010.03.003
Kluge, A. G. (1989). A concern for evidence and a phylogenetic hypothesis of relationships among Epicrates (Boidae, Serpentes). Syst. Zool. 38, 7–25. doi: 10.2307/2992432
Le, S. Q., and Gascuel, O. (2008). An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320. doi: 10.1093/molbev/msn067
Lee, C., Grasso, C., and Sharlow, M. F. (2002). Multiple sequence alignment using partial order graphs. Bioinformatics 18, 452–464. doi: 10.1093/bioinformatics/18.3.452
Li, L., Stoeckert, C. J. Jr., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Lienau, E. K., and DeSalle, R. (2009). Evidence, content and corroboration and the tree of life. Acta Biotheor. 57, 187–199. doi: 10.1007/s10441-008-9066-5
Liu, C., Zhang, X.-Y., Su, H.-N., Zhou, M.-Y., Chen, B., Li, H., et al. (2014). Puniceibacterium antarcticum gen. nov., sp. nov., isolated from Antarctic seawater. Int. J. Syst. Evol. Microbiol. 64, 1566–1572. doi: 10.1099/ijs.0.057695-0
Lucena, T., Pujalte, M. J., Ruvira, M. A., Garay, E., Macián, M. C., and Arahal, D. R. (2012). Tropicibacter multivorans sp. nov., an aerobic alphaproteobacterium isolated from surface seawater. Int. J. Syst. Evol. Microbiol. 62, 844–848. doi: 10.1099/ijs.0.030973-0
Maier, T., and Kostrzewa, M. (2007). Fast and reliable MALDI-TOF MS-based microorganism identification. Chem. Today 25, 68–71. doi: 10.1038/nmeth870
Martens, T., Heidorn, T., Pukall, R., Simon, M., Tindall, B. J., and Brinkhoff, T. (2006). Reclassification of Roseobacter gallaeciensis Ruiz-Ponte et al., 1998 as Phaeobacter gallaeciensis gen. nov., comb. nov., description of Phaeobacter inhibens sp. nov., reclassification of Ruegeria algicola (Lafay et al., 1995) Uchino et al., 1999 as Marinovum algicola gen. nov., comb. nov., and emended description of the genera Roseobacter, Ruegeria and Leisingera. Int. J. Syst. Evol. Microbiol. 56, 1293–1304. doi: 10.1099/ijs.0.63724-0
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 14:60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., Klenk, H.-P., and Göker, M. (2014). Taxonomic use of the G+C content and DNA:DNA hybridization in the genomic age. Int. J. Syst. Evol. Microbiol. 64, 352–356. doi: 10.1099/ijs.0.056994-0
Meusemann, K., Von Reumont, B. M., Simon, S., Roeding, F., Strauss, S., Kück, P., et al. (2010). A phylogenomic approach to resolve the arthropod tree of life. Mol. Biol. Evol. 27, 2451–2464. doi: 10.1093/molbev/msq130
Moran, M. A., Belas, R., Schell, M. A., Gonzalez, J. M., Sun, F., Sun, S., et al. (2007). Ecological genomics of marine roseobacters. Appl. Environ. Microb. 73, 4559–4569. doi: 10.1128/AEM.02580-06
Munoz, R., Yarza, P., Ludwig, W., Euzéby, J., Amann, R., Schleifer, K. H., et al. (2011). Release LTPs104 of the All-Species Living Tree. Syst. Appl. Microbiol. 34, 169–170. doi: 10.1016/j.syapm.2011.03.001
Newton, R. J., Griffin, L. E., Bowles, M. M., Meile, C., Gifford, S., Givens, C. E., et al. (2010). Genome characteristics of a generalist marine bacterial lineage. ISME J. 4, 784–798. doi: 10.1038/ismej.2009.150
Park, S., Park, D.-S., Bae, K. S., and Yoon, J.-H. (2014). Phaeobacter aquaemixtae sp. nov., isolated from the junction between the ocean and a freshwater spring. Int. J. Syst. Evol. Microbiol. 64, 1378–1383. doi: 10.1099/ijs.0.057646-0
Pattengale, N. D., Alipour, M., Bininda-Emonds, O. R. P., Moret, B. M. E., and Stamatakis, A. (2010). How many bootstrap replicates are necessary? J. Comput. Biol. 17, 337–354. doi: 10.1089/cmb.2009.0179
Riedel, T., Teshima, H., Petersen, J., Fiebig, A., Davenport, K., Daligault, H., et al. (2013). Genome sequence of the Leisingera aquimarina type strain (DSM 24565T), a member of the marine Roseobacter clade rich in extrachromosomal elements. Stand. Genomic Sci. 8, 389–402. doi: 10.4056/sigs.3858183
Ruiz-Ponte, C., Cilia, V., Lambert, C., and Nicolas, J. L. (1998). Roseobacter gallaeciensis sp. nov., a new marine bacterium isolated from rearings and collectors of the scallop Pecten maximus. Int. J. Syst. Bacteriol. 48, 537–542. doi: 10.1099/00207713-48-2-537
Schaefer, J. K., Goodwin, K. D., McDonald, I. R., Murrell, J. C., and Oremland, R. S. (2002). Leisingera methylohalidivorans gen. nov., sp. nov., a marine methylotroph that grows on methyl bromide. Int. J. Syst. Evol. Microbiol. 52, 851–859. doi: 10.1099/ijs.0.01960-0
Siddall, M. E., and Whiting, M. F. (1999). Long-branch abstractions. Cladistics 15, 9–24. doi: 10.1111/j.1096-0031.1999.tb00391.x
Spring, S., Scheuner, C., Lapidus, A., Lucas, S., Glavina Del Rio, T., Tice, H., et al. (2010). The genome sequence of Methanohalophilus mahii SLPT reveals differences in the energy metabolism among members of the Methanosarcinaceae inhabiting freshwater and saline environments. Archaea 2010:690737. doi: 10.1155/2010/690737
Stackebrandt, E., Scheuner, C., Göker, M., and Schumann, P. (2014). “Family Intrasporangiaceae,” in The Prokaryotes—Actinobacteria, 4th Edn., eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin: Springer), in press.
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Stamatakis, A., Hoover, P., and Rougemont, J. (2008). A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771. doi: 10.1080/10635150802429642
Sun, F., Wang, B., Liu, X., Lai, Q., Du, Y., Li, G., et al. (2010). Leisingera nanhaiensis sp. nov., isolated from marine sediment. Int. J. Syst. Evol. Microbiol. 60, 275–280. doi: 10.1099/ijs.0.010439-0
Swofford, D. L. (2002). PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4.0 b10. Sunderland, MA: Sinauer Association.
Thompson, J. D., Thierry, J.-C. C., and Poch, O. (2003). RASCAL: rapid scanning and correction of multiple sequence alignments. Bioinformatics 19, 1155–1161. doi: 10.1093/bioinformatics/btg133
Tóth, E. M., Schumann, P., Borsodi, A. K., Kéki, Z., Kovács, A. L., and Márialigeti, K. (2008). Wohlfahrtiimonas chitiniclastica gen. nov., sp. nov., a new gammaproteobacterium isolated from Wohlfahrtia magnifica (Diptera: Sarcophagidae). Int. J. Syst. Evol. Microbiol. 58, 976–981. doi: 10.1099/ijs.0.65324-0
Uchino, Y., Hirata, A., Yokota, A., and Sugiyama, J. (1998). Reclassification of marine Agrobacterium species: proposals of Stappia stellulata gen. nov., comb. nov., Stappia aggregata sp. nov., nom. rev., Ruegeria atlantica gen. nov., comb. nov., Ruegeria gelatinovora comb. nov., Ruegeria algicola comb. nov., and Ahrensia kieliense gen. nov., sp. nov., nom. rev. J. Gen. Appl. Microbiol. 44, 201–210. doi: 10.2323/jgam.44.201
Vandecandelaere, I., Segaert, E., Mollica, A., Faimali, M., and Vandamme, P. (2008). Leisingera aquimarina sp. nov., isolated from a marine electroactive biofilm, and emended descriptions of Leisingera methylohalidivorans Schaefer et al., 2002, Phaeobacter daeponensis Yoon et al., 2007 and Phaeobacter inhibens Martens et al., 2006. Int. J. Syst. Evol. Microbiol. 58, 2788–2793. doi: 10.1099/ijs.0.65844-0
Vandecandelaere, I., Segaert, E., Mollica, A., Faimali, M., and Vandamme, P. (2009). Phaeobacter caeruleus sp. nov., a blue-coloured, colony-forming bacterium isolated from a marine electroactive biofilm. Int. J. Syst. Evol. Microbiol. 59, 1209–1214. doi: 10.1099/ijs.0.002642-0
Verbarg, S., Göker, M., Scheuner, S., Schumann, P., and Stackebrandt, E. (2014). “The families Erysipelotrichaceae emend., Coprobacillaceae fam nov., and Turicibacteraceae fam. nov.,” in The Prokaryotes, 4th Edn., eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin: Springer), in press.
Wiley, E. O., and Lieberman, B. S. (2011). Phylogenetics. Theory and Practice of Phylogenetic Systematics. Hoboken, NJ: Wiley-Blackwell. doi: 10.1002/9781118017883
Wu, M., and Eisen, J. A. (2008). A simple, fast, and accurate method of phylogenomic inference. Genome Biol. 9:R151. doi: 10.1186/gb-2008-9-10-r151
Yang, Z. (1993). Maximum-likelihood estimation of phylogeny from DNA sequences when substitution rates differ over sites. Mol. Biol. Evol. 10, 1396–1401.
Yoon, J.-H., Kang, S.-J., Lee, S.-Y., and Oh, T.-K. (2007). Phaeobacter daeponensis sp. nov., isolated from a tidal flat of the Yellow Sea in Korea. Int. J. Syst. Evol. Microbiol. 57, 856–861. doi: 10.1099/ijs.0.64779-0
Yoon, J.-H., Kang, S.-J., Lee, S.-Y., Oh, K.-H., and Oh, T.-K. (2009). Seohaeicola saemankumensis gen. nov., sp. nov., isolated from a tidal flat. Int. J. Syst. Evol. Microbiol. 59, 2675–2679. doi: 10.1099/ijs.0.011312-0
Yurkov, V. V., and Beatty, J. T. (1998). Aerobic anoxygenic phototrophic bacteria. Microbiol. Mol. Biol. Rev. 62, 695–724.
Keywords: marine microbiology, Roseobacter group, phylogenomics, supermatrix, gene content, genus boundaries
Citation: Breider S, Scheuner C, Schumann P, Fiebig A, Petersen J, Pradella S, Klenk H-P, Brinkhoff T and Göker M (2014) Genome-scale data suggest reclassifications in the Leisingera-Phaeobacter cluster including proposals for Sedimentitalea gen. nov. and Pseudophaeobacter gen. nov.. Front. Microbiol. 5:416. doi: 10.3389/fmicb.2014.00416
Received: 22 May 2014; Accepted: 22 July 2014;
Published online: 11 August 2014.
Edited by:
Martin G. Klotz, University of North Carolina at Charlotte, USAReviewed by:
Martin G. Klotz, University of North Carolina at Charlotte, USAAharon Oren, The Hebrew University of Jerusalem, Israel
Copyright © 2014 Breider, Scheuner, Schumann, Fiebig, Petersen, Pradella, Klenk, Brinkhoff and Göker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Göker, Department of Microorganisms, Leibniz Institute DSMZ - German Collection of Microorganisms and Cell Cultures, Inhoffenstraße 7 B, 38124 Braunschweig, Germany e-mail:bWFya3VzLmdvZWtlckBkc216LmRl
†These authors have contributed equally to this work.