 Hyun Ju Kim1,2†
Hyun Ju Kim1,2† Haeyoung Jeong1,3†
Haeyoung Jeong1,3† Seungwoo Hwang3
Seungwoo Hwang3 Moo-Seung Lee2
Moo-Seung Lee2 Yong-Jik Lee4
Yong-Jik Lee4 Dong-Woo Lee4*
Dong-Woo Lee4* Sang Jun Lee1,2*
Sang Jun Lee1,2*- 1Biosystems and Bioengineering Program, University of Science and Technology (UST), Daejeon, South Korea
- 2Infection and Immunity Research Center, Korea Research Institute of Bioscience & Biotechnology (KRIBB), Daejeon, South Korea
- 3Korean Bioinformation Center, Korea Research Institute of Bioscience & Biotechnology (KRIBB), Daejeon, South Korea
- 4School of Applied Biosciences, Kyungpook National University, Daegu, South Korea
Microbial adaptations often occur via genomic mutations under adverse environmental conditions. This study used Escherichia coli ΔadhE cells as a model system to investigate adaptation to anaerobic conditions, which we then compared with the adaptive mechanisms of two closely related E. coli strains, K-12 and B. In contrast to K-12 ΔadhE cells, the E. coli B ΔadhE cells exhibited significantly delayed adaptive growth under anaerobic conditions. Adaptation by the K-12 and B strains mainly employed anaerobic lactate fermentation to restore cellular growth. Several mutations were identified in the pta or pflB genes of adapted K-12 cells, but mostly in the pta gene of the B strains. However, the types of mutation in the adapted K-12 and B strains were similar. Cellular viability was affected directly by severe redox imbalance in B ΔadhE cells, which also impaired their ability to adapt to anaerobic conditions. This study demonstrates that closely related microorganisms may undergo different adaptations under the same set of adverse conditions, which might be associated with the specific metabolic characteristics of each strain. This study provides new insights into short-term microbial adaptation to stressful conditions, which may reflect dynamic microbial population changes in nature.
Introduction
Stress has been defined as “any deviation from optimal growth conditions that results in a reduced growth rate” (Storz and Hengge-Aronis, 2000). Sources of bacterial stress include irradiation, heat shock, osmotic pressure, pH changes, starvation, oxygen radicals, aerobic to anaerobic transition, and oxygen deprivation (Patschkowski et al., 2000; Moat et al., 2002b). In addition, imbalances in intracellular metabolites, such as sugar phosphate accumulation (Lee et al., 2014), nucleotide depletion (Lee et al., 2009), and elevated NADPH levels (Auriol et al., 2011) due to the absence of relevant metabolic enzymes, can cause retarded cellular growth, which are also forms of stress.
Microorganisms have evolved a variety of alternative and/or bypass pathways to maintain their metabolic functionality in response to different environmental conditions. Under unfavorable growth conditions, microbes can sustain their viability by activating various biochemical pathways to maintain homeostasis (or the cellular growth rate), where their metabolism may be regulated by two-component systems (Lynch and Lin, 1996; Bekker et al., 2006), sigma factors (Battesti et al., 2011), regulator proteolysis (Mettert and Kiley, 2005; Mika and Hengge, 2005), small RNAs (Durand and Storz, 2010), alarmones (Mechold et al., 2013), and other mechanisms. To cope with severe environmental stress, adaptation can emerge via the acquisition of random mutations, which may be positively selected in a population if the mutations generate a beneficial phenotype (Rando and Verstrepen, 2007).
Recent studies show that mutations beneficial for optimal cell growth can be identified after long-term evolution and adaptation (Barrick et al., 2009; Conrad et al., 2010). Specific mutations such as rpoC mutations are found repeatedly after adaptation in minimal growth medium (Conrad et al., 2010), whereas diverse beneficial mutations can be detected in microbial populations that evolve at high temperature (Tenaillon et al., 2012). Efforts have been made to study the evolutionary pathways utilized under stress conditions (such as antibiotic resistance) using continuous culture (Toprak et al., 2012). Time course experiments may also be helpful for elucidating how stress affects microbial populations and when compensatory mutants arise during adaptation to adverse conditions.
Under anaerobic conditions, Escherichia coli convert glucose into formate, acetate, ethanol, lactate, and succinate (Clark, 1989; Moat et al., 2002a). The mixed acid fermentation is required to recycle redox cofactors, which can occur via several reactions catalyzed by alcohol or acid dehydrogenases in cells (Figure 1). Alteration or impairment of the redox balance caused by a lack of appropriate electron acceptors, carriers, or redox-related enzymes can result in stress in microbial cells (Gonzalez-Siso et al., 2009). In the absence of alcohol dehydrogenase (encoded by adhE), which plays a key role in the maintenance of redox balance via NADH oxidation (or NAD+ recycling) under anaerobic conditions, cells experience severe redox-stress during the transition from aerobic to anaerobic conditions (Gupta and Clark, 1989; Galinina et al., 2012). E. coli mutant cells that lack the adhE gene are unable to grow anaerobically, whereas additional pta (phosphotransacetylase) mutations enable adhE mutants to grow via lactate fermentation (Gupta and Clark, 1989) (Figure 1).
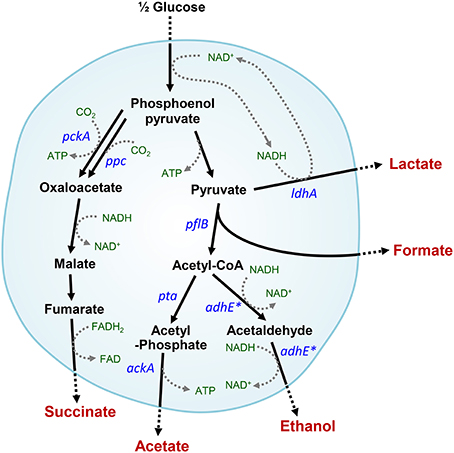
Figure 1. Intracellular anaerobic NAD+ recycling in mixed acid fermentation by lactate dehydrogenase in ΔadhE mutant cells. Metabolic pathways affected by the adhE mutation are indicated by asterisks.
In our previous genome-wide analysis of the redox reactions that are important for anaerobic growth (Kim et al., 2013), we serendipitously found that E. coli K-12 ΔadhE smoothly grew via lactate fermentation in liquid culture under anaerobic conditions. By contrast, BL21(DE3) ΔadhE cells exhibited highly delayed anaerobic growth. When fermentation broths of K-12 ΔadhE and BL21(DE3) ΔadhE strains were respectively taken to regrow in the same fresh broth, growth rates of K-12 ΔadhE and BL21(DE3) ΔadhE strains were accelerated without any lag periods and comparable to that exhibited by the wild-type strain. The present study examined the different adaptations (genomic mutations and metabolic characteristics) of two closely related E. coli strains (K-12 and B) grown under the same stress conditions. This study provides insights into how microbial populations can change rapidly and adapt to adverse environmental conditions via genomic mutations, which might be a common feature of microbial outbreaks and population changes in nature.
Materials and Methods
Bacterial Strains
E. coli strains are listed in Table 1. The E. coli B (REL606) strain was kindly provided by Richard E. Lenski at Michigan State University. The E. coli B strains BL21 and BL21(DE3) were purchased from Promega (Madison, WI) and Invitrogen (Carlsbad, CA), respectively. The E. coli K-12 strains, MG1655, BW25113, and W3110 were kindly provided by Sankar Adhya at the NIH, the Coli Genetic Stock Center (CGSC) at Yale University, and Korean Collection for Type Cultures (KCTC) at KRIBB, respectively.
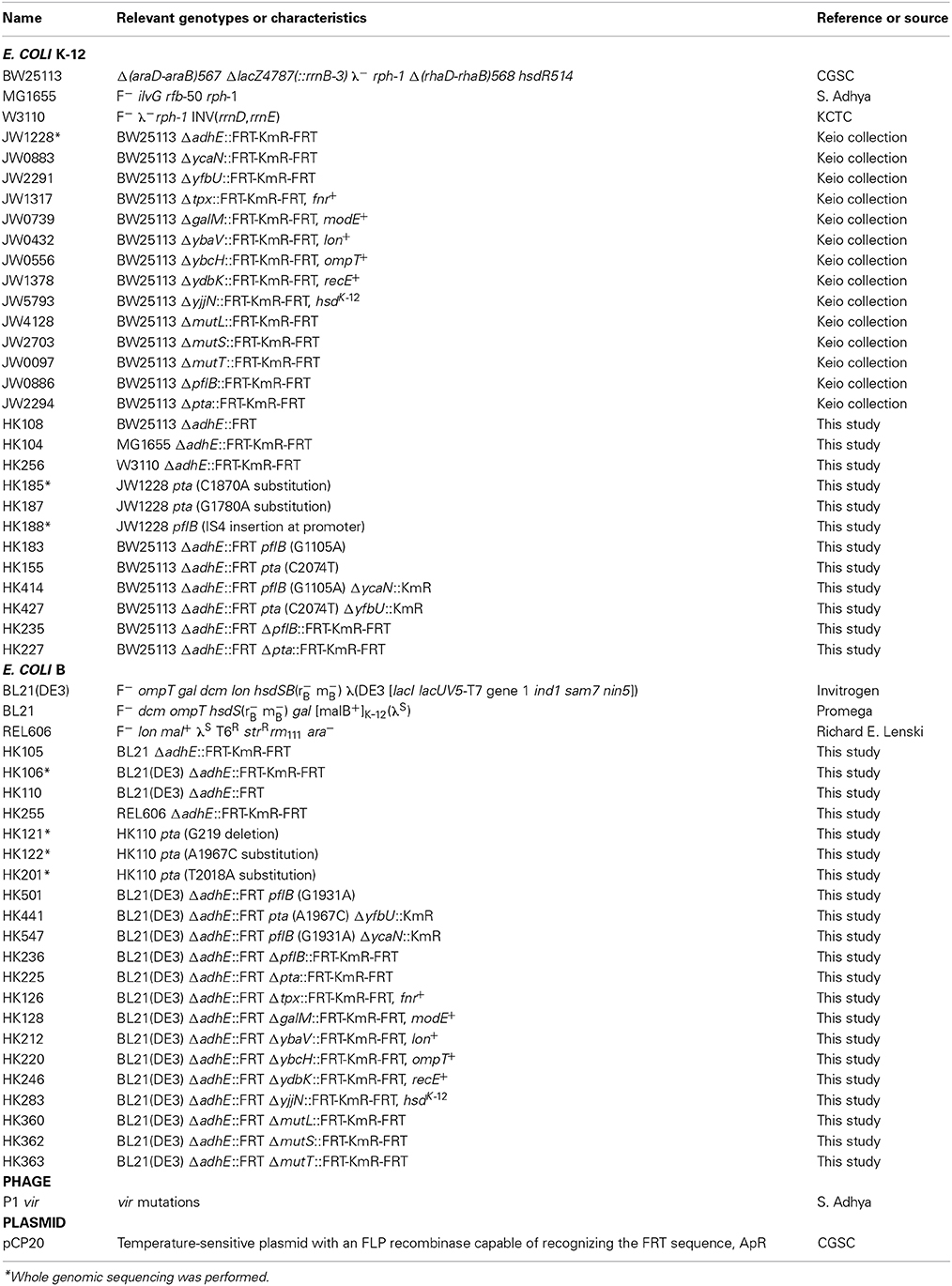
Table 1. Bacterial strains, phages, and plasmids used in this study.
Individual gene knockout mutants of E. coli in the Keio collection (Baba et al., 2006) were purchased from Open Biosystems (Lafayette, CO). The open reading frames in the individual genes were replaced by kanamycin markers. The mutations were transferred to other strains by standard P1 transduction (Miller, 1992). To transfer the ΔadhE mutation, P1 vir phage lysates of kanamycin-resistant strain BW25113 ΔadhE (JW1228) in the Keio collection were used to transduce the MG1655, W3110, BL21(DE3), REL606, and BL21 strains. If needed, the plasmid pCP20 (Cherepanov and Wackernagel, 1995) was transformed into E. coli strains to delete the kanamycin resistance gene from their chromosomes by FLP recombinase at 30°C. Subsequently, plasmid pCP20 with a temperature-sensitive replication origin was cured at 42°C, resulting in the kanamycin-sensitive strains.
To introduce point mutations of the pta and pflB genes into E. coli K-12 ΔadhE strains, ΔycaN::KmR and ΔyfbU::KmR cassettes obtained from Keio collection were electroporated into mutant cells to tag pflB(G1105A) and pta(C2074T) mutations, respectively, harboring pKD46 plasmids after lambda recombinase was fully induced by L-arabinose. Subsequently, P1 lysates of the resulting cells were used to transduce kanamycin marker-tagged pflB(G1105A) and pta(C2074T) mutations into kanamycin-sensitive BW25113 ΔadhE backgrounds. In B strain, the same above strategy was employed to individually introduce pta(A1967C) and pflB(G1931A) mutations into kanamycin-sensitive BL21(DE3) ΔadhE (Table 1).
To transfer several authentic genes of K-12 strain to B strain background, we used Pl lysates of JW1317, JW0739, JW0432, JW0556, JW1378, and JW5793 to transduce fnr+, modE+, lon+, ompT+, recE+, and hsdK-12 genes (tagged by adjacent kanamycin resistance genes) into BL21(DE3) ΔadhE strain to make HK126, HK128, HK212, HK220, HK246, and HK283 strains. To introduce deletion mutations of ΔmutL, ΔmutS, and ΔmutT genes of Keio collection into BL21(DE3) ΔadhE strain, we carried out P1 transduction to make HK360, HK362, and HK363 strains. Other strains were genetically constructed using P1 phage transduction when needed, and also verified by PCR or DNA sequencing (Table 1).
Anaerobic Culture
LB broth and yeast extract were purchased from Becton Dickinson (Sparks, MD). D-glucose, sodium bicarbonate, sodium phosphate monobasic monohydrate, potassium phosphate dibasic, and sodium sulfide nonahydrate were purchased from Sigma-Aldrich (St. Louis, MO). Bacterial seed cultures were grown in 5 mL LB broth at 37°C with shaking at 180 rpm. One milliliter of seed culture was inoculated into a 125 mL serum vial containing 100 mL of fermentation medium, as described previously (Kim et al., 2013). If needed, D-gluconate (final 50 mM), D-fructose, and D-mannitol were respectively added to the fermentation medium as a major carbon source instead of D-glucose. The headspace in serum vials was filled with N2 gas and Na2S·9H2O (final 1 mM) was added to yield strictly anaerobic conditions. Bacterial cells were cultured anaerobically at 37°C with shaking at 180 rpm. Cell growth was monitored by measuring optical density at 600 nm using an Ultrospec 8000 spectrophotometer (GE Healthcare, Uppsala, Sweden). The cell cultures were diluted 1:10 using the same media to measure the optical density accurately.
Metabolite Analysis
The concentrations of metabolites including D-glucose and lactate in the culture were determined by high-performance liquid chromatography (RID-10A RI monitor, Shimadzu, Japan) using an Aminex HPX-87H column (300 × 7.8 mm, Hercules, BioRad) as described previously (Lee et al., 2005). After centrifugation of the cell culture broth, the supernatant was passed through a 0.2 μm syringe filter. The column was isocratically eluted at 47°C with a flow rate of 0.5 mL min−1 using 0.01 N H2SO4. The intracellular concentrations of NAD+ and NADH were measured using a NAD+/NADH quantification kit (BioVision Inc., Milpitas, CA). Cells were harvested by centrifugation at 10,000 × g for 10 min and resuspended in cold phosphate buffered saline solution with an OD600 of 1.0. Preparation of NADH extraction buffers and all subsequent steps were performed according to the manufacturer's protocols.
Genome Analysis
The genomic DNAs of E. coli strains were purified using the Wizard Genomic DNA purification kit purchased from Promega (Madison, WI). Genome sequences from the parental strains and their derivatives were obtained from an Illumina HiSeq 2000 platform. The 101-cycle paired end reads, with 2.46-3.88 Gb range (30,901,022 reads on average), were produced from 500 bp genomic libraries and processed by the CASAVA 1.9 pipeline. Pretreatment of the reads, reference mapping, and variant detection were carried out using CLC Genomics Workbench version 4.5. Reads shorter than 50 nt were filtered out after quality trimming using a modified Mott algorithm (quality cutoff 0.01), which allowed one or less ambiguous base (N) per read. For reference mapping, the genome sequences of E. coli K-12 subst. MG1655 (NC_000913.2) and BL21(DE3) (CP001509.3) were used for K-12 and B lineage, respectively. The default mapping parameters were applied (similarity = 0.8, length fraction = 0.5). Variants in SNP and DIP (deletions, insertions, and polymorphisms) called by the CLC Genomics Workbench were validated to identify mutations that had occurred only in the descendants, but not in the parental strains [BW25113 ΔadhE and BL21(DE3) ΔadhE]. Nucleotide discrepancies in the adhE region owing to P1 transduction, which was used for the disruption of the adhE gene, were excluded and later confirmed by comparing de novo-assembled contigs. The pta and pflB genes including promoter regions were amplified by PCR using chromosomal DNAs of E. coli parental and derivative strains as templates, and mutations in the corresponding genes were identified by Sanger DNA sequencing.
Viability Test
Both K-12 ΔadhE and B ΔadhE cells were grown anaerobically in the fermentation medium. All cell cultures (1 ml) were collected from the medium during anaerobic fermentation as mentioned above, and samples diluted with PBS solution were then spread on LB agar plates. After 24 h of aerobic incubation at 37°C, the number of culturable cells was counted for by multiplying the number of colonies on LB agar plates with the corresponding dilution fold. Viability values were obtained by calculating the log base 10 of numbers of culturable cells per 1 ml. For mutant frequency test, ΔadhE mutant cells were aerobically grown in LB broth for 12 h at 37°C, diluted with PBS solution, spread on LB agar and incubated aerobically for 24 h at 37°C. The same volumes of ΔadhE mutant cells aerobically grown in LB for 12 h at 37°C were spread on the fermentation medium agar and anaerobically incubated using AnaeroGen™ anaerobic pouch system (Oxoid, Hampshire, UK) for 72 h at 37°C.
Inoculum Dilution Test
Colonies of K-12 ΔadhE and B ΔadhE strains, and adapted K-12 ΔadhE and B strains ΔadhE were singly isolated and inoculated for starter cultures in LB broth. Cell cultures (1 ml) were harvested, resuspended in sterile water (1 ml), serially diluted (1:10), and inoculated in the fermentation medium. Cell growth was monitored by measuring optical densities at 600 nm of culture broths sampled every 3 h.
Results
Comparative Anaerobic Fermentation by ΔadhE Mutant Strains of E. coli K-12 and B
Under anaerobic conditions, wild-type E. coli K-12 cells (BW25113) completely consumed glucose in 8 h via mixed acid fermentation pathways (Figure 1), which produced succinate, lactate, acetate, ethanol, and formate from D-glucose (Table 2). The BW25113 ΔadhE mutant cells (JW1228) mainly produced lactate under anaerobic conditions (Figure 2A). The complete consumption of 50 mM glucose resulted in the production of lactate (>80 mM) by the mutant within 24 h. To verify the effect of the single ΔadhE gene mutation on lactic acid fermentation, the ΔadhE mutation was introduced into other E. coli K-12 (MG1655, and W3110) and B strains [REL606, BL21(DE3), and BL21] via P1 transduction, and anaerobic fermentation was induced. In the E. coli MG1655 ΔadhE (HK104) and W3110 ΔadhE (HK256) strains, the anaerobic lactate fermentations were completed within 24 h, which was similar to the BW25113 ΔadhE (JW1228) strain (Figures 2B,C).
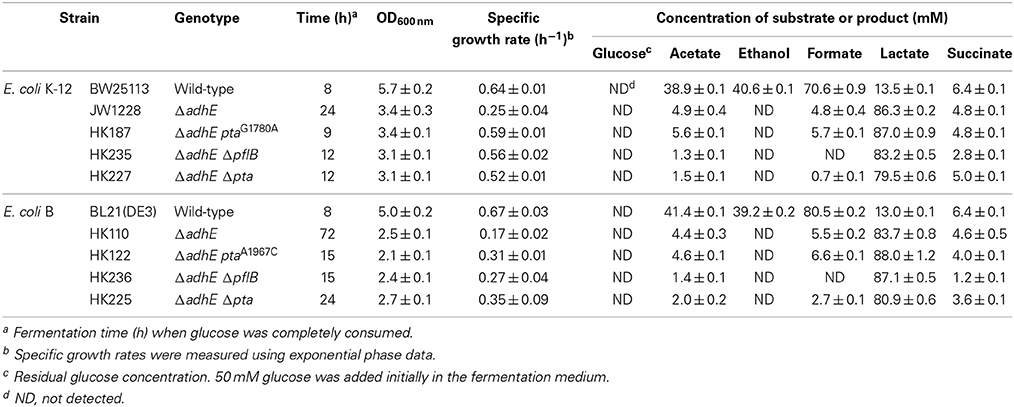
Table 2. Anaerobic fermentation profiles of wild-type cells and ΔadhE derivatives of E. coli K-12 and B strains.
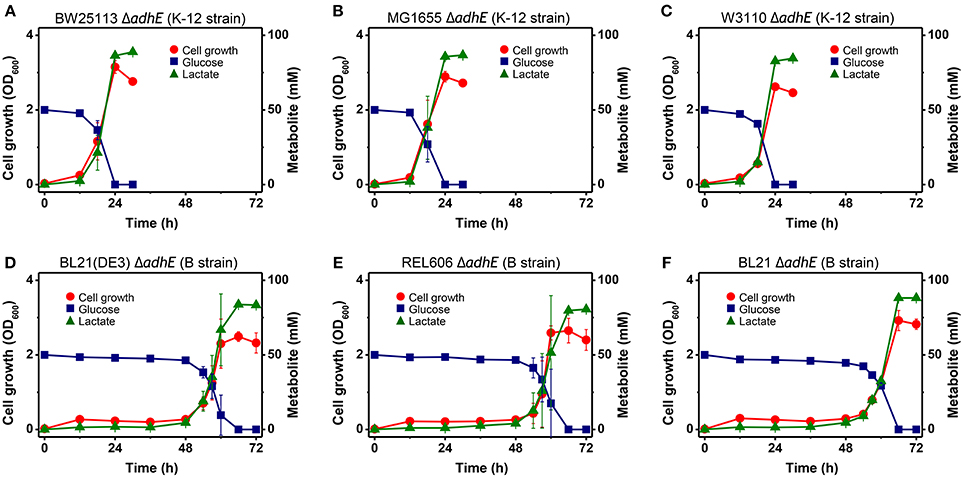
Figure 2. Anaerobic lactic acid fermentation profiles of various E. coli K-12 (A–C) and B (D–F) strains with ΔadhE mutations. (A) BW25113 ΔadhE (JW1228), (B) MG1655 ΔadhE (HK104), (C) W3110 ΔadhE (HK256), (D) BL21(DE3) ΔadhE (HK110), (E) REL606 ΔadhE (HK255), and (F) BL21 ΔadhE (HK105).
Unexpectedly, the ΔadhE mutants in the B strain background (BL21(DE3), REL606, and BL21) showed long lag periods (approximately 48 h after inoculation), which were followed by exponential growth, rapid glucose consumption, and lactate production (Figures 2D–F). Genotypic and phenotypic verification of the cells was performed by 16S rRNA gene sequencing and single colony isolation on MacConkey agar containing D-galactose or D-lactose before and after anaerobic fermentation to exclude the possibility of additional microbial contamination. The results indicate that the K-12 and B strains lacking alcohol dehydrogenase performed the same lactate fermentation for anaerobic cellular growth. However, the growth speed of E. coli ΔadhE cells differed in a strain-dependent manner under anaerobic conditions.
Accelerated Anaerobic Growth by Adapted ΔadhE Mutant Strains
During the characterization of the anaerobic lactate fermentation reaction in K-12 ΔadhE and B ΔadhE strains, we observed that several anaerobic cultures showed unusually rapid growth. For example, the variant strains HK187 and HK122 were obtained as pure isolates from fermentation broths of BW25113 ΔadhE (JW1228) and BL21(DE3) ΔadhE (HK110), respectively, and then cultured anaerobically in serum bottles. The progeny strain (HK187) completed lactate fermentation within 12 h, whereas anaerobic fermentation by the parental JW1228 cells required 24 h (Figure 3A). Moreover, the adapted progeny cells (HK122) derived from BL21(DE3) ΔadhE did not exhibit a long lag period and they completed lactate fermentation within 15 h (Figure 3B), indicating that anaerobic growth by the K-12 and B mutant strains was modified after they underwent anaerobic fermentation. We randomly isolated 37 and 45 additional adapted strains from separate anaerobic cultures of JW1228 and HK110, respectively. Most of the adapted K-12 and B variants produced lactate as a major fermentation product within 12 and 24 h, respectively, which was faster compared to their parental strains (Table 3).
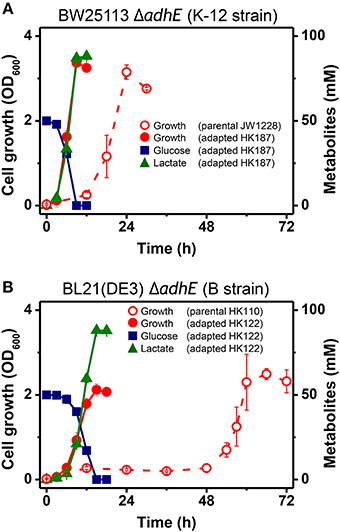
Figure 3. Rapid anaerobic cellular growth of anaerobically-adapted E. coli K-12 ΔadhE mutants (A) and B ΔadhE mutants (B).
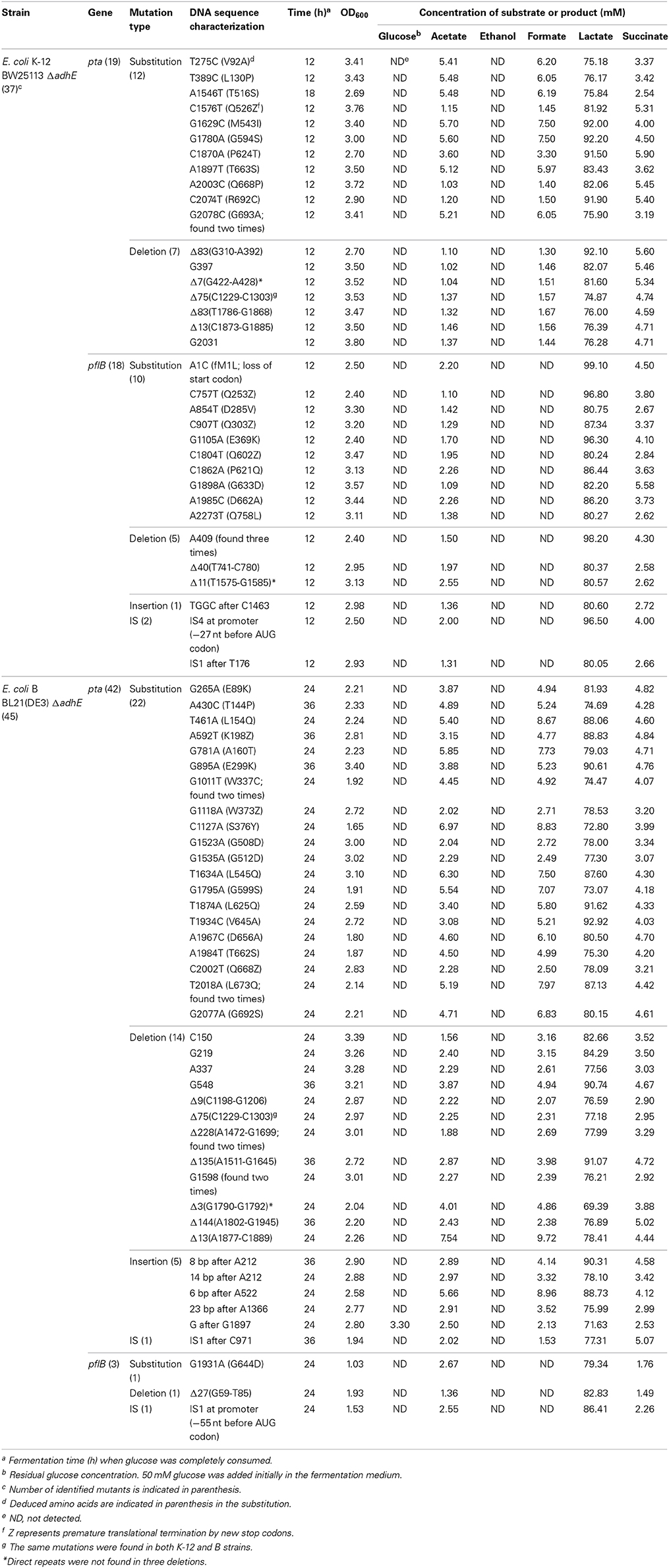
Table 3. Mutation analysis and fermentation profiles of anaerobically-adapted E. coli K-12 ΔadhE and B ΔadhE cells.
Genetic Analysis of Adapted ΔadhE Mutant Cells
To investigate the genetic mechanism that facilitated anaerobic adaptation, seven parental and adapted cells of the K-12 and B strains were analyzed by whole genome sequencing (Table 4). Next generation sequencing showed that, compared with the parental K-12 ΔadhE (JW1228) and B ΔadhE (HK106) strains, the adapted progeny strains (HK185, HK188, HK121, HK122, and HK201) contained only single mutations in either the pta or pflB gene; however, there were no other mutations in their genomes (Table 4). Therefore, we further analyzed the nucleotide sequences of the pta and pflB genes of the adapted progeny strains by Sanger sequencing.
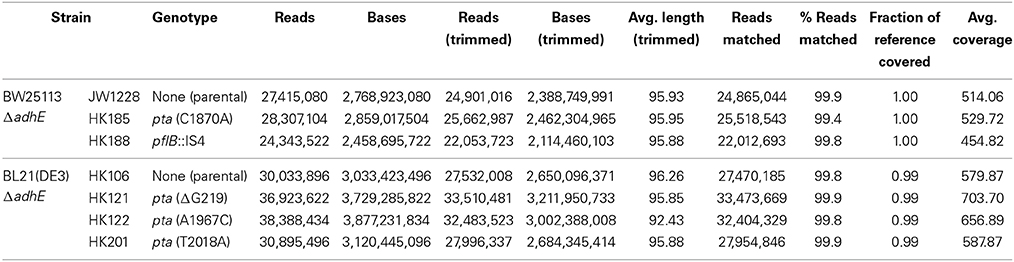
Table 4. Genomic analysis of E. coli ΔadhE parental and adapted progeny strains.
The results of the mutational analysis of the adapted K-12 and B strains are presented in Table 3. Of the 37 adapted K-12 adhE mutant cells, 19 harbored mutations in the pta gene and 18 harbored mutations in the pflB gene, including point mutations, deletions, and insertion sequence (IS) elements that caused missense, nonsense, and frame shift mutations. By contrast, the B ΔadhE strains exhibited biased mutations in the pta gene, where most of the adapted strains (42/45 mutant strains) harbored pta mutations, with the exception of three pflB mutations (Figure 4A). These results indicate that despite the close relationship between the E. coli K-12 and B strains (>99% nucleotide identity) (Jeong et al., 2009), compensatory mutations occurred in preferred target genes under the same anaerobic conditions. Comparative genome studies of K-12 and B strains led us to consider the individual introduction of authentic fnr, modE, lon, ompT, recE, and hsd genes of the K-12 strain into the BL21(DE3) ΔadhE mutant, which would remove its long lag phase (~48 h). As a result, the lag phases (~36 h) of fnr+ and lon+ cells of BL21(DE3) ΔadhE backgrounds were not as short as those (~12 h) of the K-12 ΔadhE cells during anaerobic adaptation, and those of other strains (modE+, ompT+, recE+, and hsdK−12) of BL21(DE3) ΔadhE cells were not affected (Figure 5).
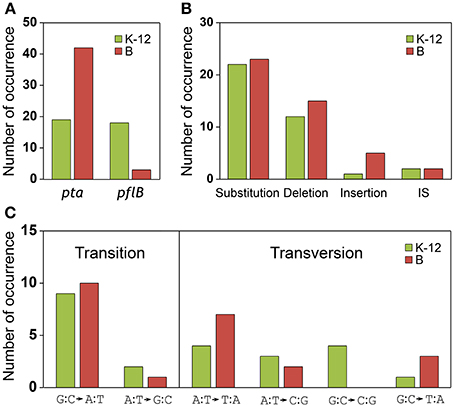
Figure 4. Analysis of compensatory mutations for anaerobic adaptation in ΔadhE mutants of K-12 and B strains. Thirty seven K-12 mutants (derived from BW25113 ΔadhE) and 45 B mutants [derived from BL21(DE3) ΔadhE] were analyzed by DNA sequencing. (A) Mutation occurrence in pta and pflB genes, (B) types of mutations, and (C) types of substitutions were compared in respect of K-12 and B strains.
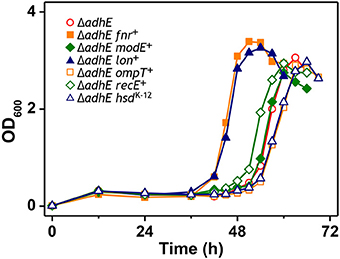
Figure 5. Anaerobic growth of BL21(DE3) ΔadhE cells supplemented with authentic fnr, modE, lon, ompT, recE, and hsd genes of the K-12 strain.
To confirm the effects of pta and pflB mutations on the growth phenotypes in the ΔadhE background, we introduced point mutations back into the pta and pflB genes in the K-12 ΔadhE and B ΔadhE strains, respectively, and monitored the growth of the mutants relative to that of their parental strains (Figure 6). We found that K-12 ΔadhE cells harboring pta(C2074T) or pflB(G1105A) mutations grew faster than the parental strain and completed fermentation within 12 h. In addition, there were no significant differences in the growth phenotypes of the pta(C2074T) and pflB(G1105A) mutants in the ΔadhE background under anaerobic conditions (Figure 6A). In the B strain background, ΔadhE pta(A1967C) and ΔadhE pflB(G1931A) cells lacked the long lag periods observed in the ΔadhE parental cells and they completed lactate fermentation within 12 and 24 h, respectively (Figure 6B). The ΔadhE pta(A1967C) mutants consumed glucose in 12 h and reached a higher final cell density than the ΔadhE pflB(G1931A) mutants, which consumed glucose in 24 h. These data showed that the pta or pflB mutations appeared to result in shorter lag times during the adaptation of ΔadhE cells (both K-12 and B strains) to anaerobic conditions.
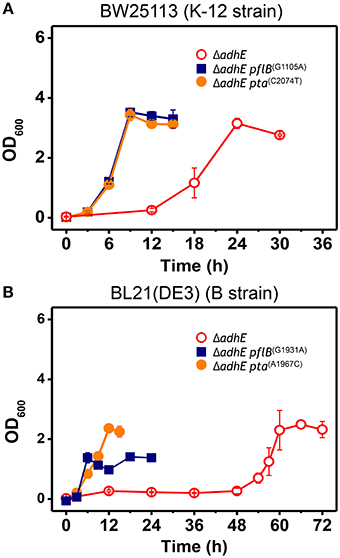
Figure 6. Accelerated growth of ΔadhE mutants of E. coli K-12 and B strains by introduction of pta or pflB point mutations. (A) BW25113 ΔadhE cells carrying the pflB(G1105A) or pta(C2074T) mutation in the genome instead of the authentic pflB or pta gene, and (B) BL21(DE3) ΔadhE cells carrying pflB(G1931A) or pta(A1967C).
Occurrence of Various Types of Mutations During Anaerobic Adaptation
We compared the types of mutations that occurred in the adapted K-12 and B strains. In adapted cells, base substitutions were most frequent, which cause nonsense or missense mutations (Figure 4B). Deletions were also frequent, but insertions and IS elements were rarely observed in the adapted strains. Among the base substitutions, G:C to A:T transitions and A:T to T:A transversions were observed frequently in both the K-12 and B strains (Figure 4C). We also found repeat sequences in the insertions and deletions in the pta and pflB genes (Figure 7). Insertions of tandem repeat sequences (4–23 bp) were confirmed in the K-12 and B strains. Direct repeat-mediated deletions (9–228 bp) were observed frequently in both strains. We found five deletions with imperfect direct repeats (even with 2 bp mismatches out of 10 bp repeats) in the adapted K-12 and B cells.

Figure 7. Repeated sequence mediated insertions and deletions in adapted K-12 and B mutants. (A) Insertional mutations were generated by precise tandem repeats, and (B) deletions between perfect or imperfect direct repeats were observed. Mismatched bases in direct repeats were indicated by asterisks.
We observed that the mutant frequency (3.0 ± 0.9 × 10−6) in BL21(DE3) ΔadhE cells was slightly lower than that (1.2 ± 0.3 × 10−5) in BW25113 ΔadhE cells. To test whether mutator gene knockouts affect the long lag period of BL21(DE3) ΔadhE cells, we introduced three individual mutations of mutator genes (ΔmutL, ΔmutS, and ΔmutT) into BL21(DE3) ΔadhE cells and cultured the cells under anaerobic conditions. As a result, the adaptation periods of the B strains were not shortened as those of the K-12 strains (Figure 8).
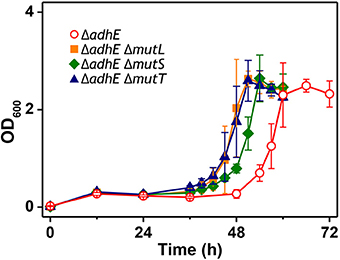
Figure 8. Effect of mutator gene knockouts on anaerobic adaptation of BL21(DE3) ΔadhE cells.
Redox Cofactor Recycling for Anaerobic Growth
The phenotypic characteristics of the K-12 ΔadhE and B ΔadhE cells were investigated by growth on several carbon sources under different reducing conditions (Figure 9). Assuming that E. coli cells fully metabolize D-gluconate, D-glucose (or D-fructose), and D-mannitol to pyruvate under anaerobic conditions, the moles of NADH generated would be 1, 2, and 3 (per six carbons), respectively. K-12 and B cells harboring ΔadhE mutations did not grow in the presence of reduced D-mannitol, even within 120 h. However, the K-12 ΔadhE and B ΔadhE cells grew well in a medium that contained oxidized D-gluconate and there was no lag period under anaerobic conditions. These results indicate that ΔadhE mutants might experience severe problems with NAD+ recycling during anaerobic metabolism. Therefore, we next determined the intracellular levels of NAD+/NADH in the K-12 and B strains.
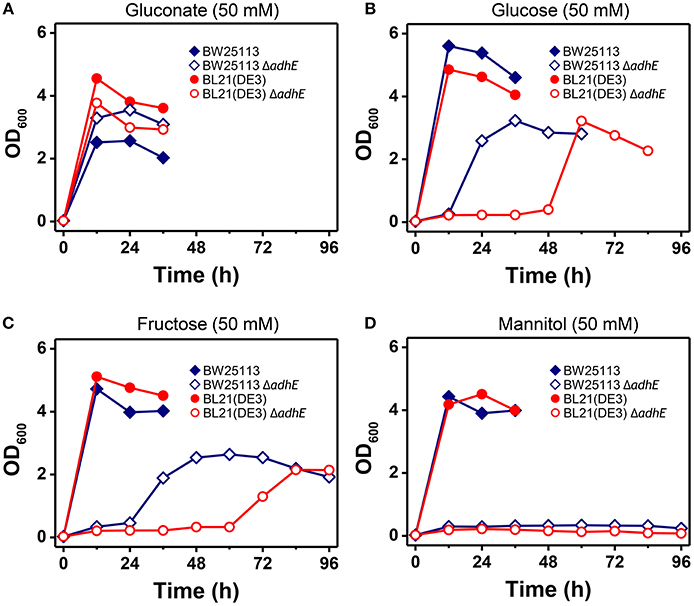
Figure 9. Anaerobic growth of E. coli K-12 ΔadhE and B ΔadhE cells when various carbon sources with different oxidized states were used. (A) D-Gluconate, (B) D-glucose, (C) D-fructose, and (D) D-mannitol were respectively added to the fermentation medium as a major carbon source.
The intracellular NAD+/NADH ratio is a biomarker of metabolic activity (Heikal, 2010). Thus, we measured the viability and redox cofactor ratio [NAD+/NADH] of ΔadhE mutant cells in the K-12 and B strains to elucidate how cells adapt to anaerobic stress (Figure 10). Under anaerobic conditions, the BW25113 ΔadhE cells maintained their intracellular redox balance and viability until 6 h after inoculation into a fermentation medium that contained D-glucose. The viability and redox balance were affected slightly between 6 and 12 h, but recovered after 12 h (Figure 10A). The cellular viability and redox balance of the BL21(DE3) ΔadhE cells were markedly affected between 6 and 36 h, but subsequently recovered as cellular growth increased (based on the optical density) (Figure 10B).
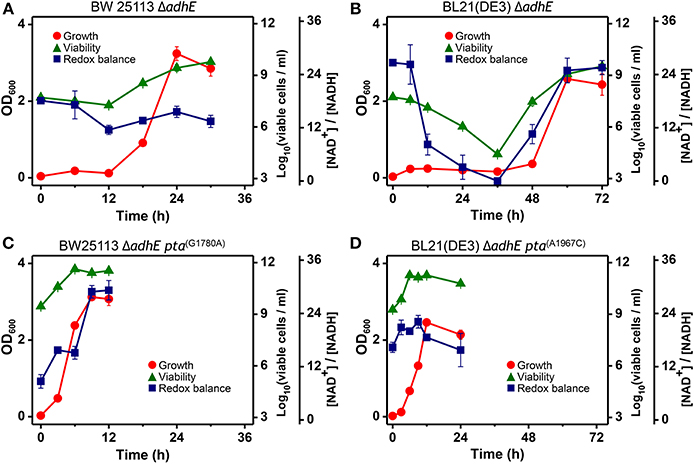
Figure 10. Analysis of intracellular redox balance and viability of anaerobically grown cells of BW25113 ΔadhE (A), BL21(DE3) ΔadhE (B), adapted BW25113 ΔadhE pta(G1780A) (C), and adapted BL21(DE3) ΔadhE pta(A1967C) (D).
In contrast to the optical density, the redox ratio [NAD+/NADH] facilitates real-time monitoring of the metabolic state of cells exposed to stress. The NAD+/NADH ratios indicated that ΔadhE mutant cells underwent severe anaerobic stress, whereas the adapted mutant cells survived. The aerobic-to-anaerobic transition ability and the viability of K-12 cells were superior to those of B cells under anaerobic stress conditions. We analyzed the NAD+/NADH ratios of the adapted mutants of both K-12 and B strains, BW25113 ΔadhE pta(G1780A) and BL21(DE3) ΔadhE pta(A1967C) (Figures 10C,D). The viable cell numbers and the NAD+/NADH ratios did not decrease when the adapted mutant cells were grown under anaerobic conditions. These results indicate that the adapted mutants could recycle NAD+ efficiently under anaerobic conditions.
We next performed seed dilution experiments to determine the effect of the inoculated cell numbers on adaptation under anaerobic conditions. The results showed that the K-12 ΔadhE cells grew and completed lactate fermentation when <10 cells were inoculated into anaerobic culture vials, indicating that these cells could maintain their viability during anaerobic growth (Figure 11A). In the B strain background, smaller inocula of ΔadhE cells (107 and 108 cells) grew faster than large inocula (109 cells); this was observed repeatedly and indicated that compensatory mutations occurred randomly during the anaerobic growth of B ΔadhE cells, regardless of the inoculum size (Figure 11C). Furthermore, the B ΔadhE cells could not adapt and grow within 72 h when <105 cells were inoculated probably because the B ΔadhE cells failed to generate a population large enough to acquire spontaneous pta or pflB mutations. However, the growth speed of the adapted K-12 and B cells appeared to be proportional to the number of inoculated cells (Figures 11B,D).
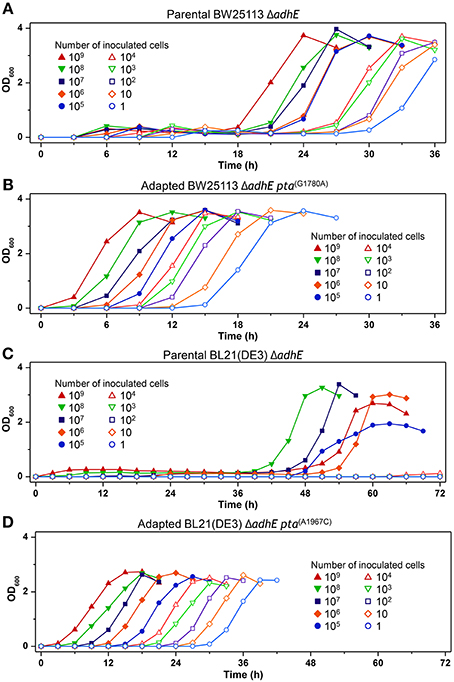
Figure 11. Inoculum dilution effects on anaerobic growth profiles of parental and adapted cells of BW25113 ΔadhE (A), BL21(DE3) ΔadhE (B), adapted BW25113 ΔadhE pta(G1780A) (C), and adapted BL21(DE3) ΔadhE pta(A1967C) (D).
Discussion
Routine microbial experiments and even extraordinary achievements in bacterial genetics have begun with the colonies formed on agar plates under certain selectable conditions. When broth cultures start with those colonies, we might assume or wish that the genotypes of inoculated cells are not changed in short period of time (~days). Here we found that the quick adaptive growth of ΔadhE cells is accompanied by genomic mutations during anaerobic liquid culture, and also that closely related K-12 and B strains followed different trajectories in adapting to harsh environments.
The genome sequences of K-12 and B cells are very similar (>99% identity) (Jeong et al., 2009), but the main differences between these two strains are the genomic locations of their IS elements, which indicates the important roles of these elements in genomic plasticity and divergence (Schneider et al., 2002). Recently, Yoon et al. determined the differences between E. coli B and K-12 strains by performing a comprehensive genome-wide analysis of metabolic networks to identify the genetic bases of their phenotypes via in silico complementation testing (Yoon et al., 2012). This analysis showed that E. coli B has a greater capacity for amino acid biosynthesis, fewer proteases, lacks flagella, and possesses an additional type II secretion system, whereas E. coli K-12 shows higher expression of heat shock genes and is less susceptible to certain stress conditions. In the present study, the K-12 ΔadhE strains adapted rapidly to anaerobic stress and grew faster than the B ΔadhE strains (Figures 3A,B).
A recent study showed that B strains lack certain anaerobic respiration-related proteins and enzymes, such as H2-oxidizing hydrogenases, formate dehydrogenase, nitrate reductase, and the molybdenum-responsive transcriptional regulator (ModE) (Pinske et al., 2011). Given the results of previous comparative genome studies of K-12 and B strains (Jeong et al., 2009; Studier et al., 2009), we anticipated that the individual introduction of authentic fnr, modE, lon, ompT, recE, and hsd genes of the K-12 strain into the BL21(DE3) ΔadhE mutant would abolish its long lag phase. However, we failed to remove the delayed long lag phase of B mutant strains under anaerobic conditions (Figure 5), suggesting that several genes might play concerted roles in the specific anaerobic metabolism processes of K-12 and B strains.
This characterization of adaptation by closely related strains raises intriguing but fundamental questions. First, what is the mechanism responsible for the different compensatory mutations in K-12 and B mutants?
The types of mutations found in pta and pflB genes were similar, regardless of the strain (Figures 4B,C). Substitutions such as G:C to A:T transitions and A:T to T:A transversions occurred frequently in both strains during adaptation to redox-stress conditions. The insertions were generated by tandem repeat sequences (Figure 7A) and the deletions were mediated by direct repeats (Figure 7B), which might have been caused by slipped-strand mis-pairing during DNA replication and/or homologous recombination; both are regarded as general molecular mechanisms that facilitate bacterial evolution (Albertini et al., 1982; Levinson and Gutman, 1987).
The compensatory mutations that occurred in the pta genes K-12 and B strains were found mostly in the C-terminal catalytic domain of the deduced amino acid sequences (Campos-Bermudez et al., 2010) (Table 3). According to the crystal structure of Pta (phosphotransacetylase) from Methanosarcina thermophile (Iyer et al., 2004), the pta gene mutation in the adapted HK155 strain corresponded to Arg692, which is a proposed putative CoA binding site. Therefore, we assume that pta gene mutations might reduce or block further metabolic flux to acetyl phosphate, which can act as a phosphoryl and acetyl donor during cellular signal transduction (Klein et al., 2007; Weinert et al., 2013).
According to the fermentation profiles of adapted mutants (Table 3), it seems that two distinct sets of pta mutants exist compared to the metabolite profiles of parental ΔadhE cells (Table 2): one with strong reduction of both acetate and formate production such as BW25113 ΔadhE cells with C1576T, A2003C, C2074T, or seven individual deletion mutations in pta gene, and the other with no significant impact on both acetate and formate production such as the reconstructed HK187and HK122 strains or the adapted BW25113ΔadhE cells with the T275C mutation in pta gene. Thus, it is likely that some pta mutations may restore anaerobic growth while not affecting the formation of acetate and formate, indicating some Pta enzymes in those adapted cells may not be fully inactivated. Moreover, this dichotomy within pta mutants is more obvious in the K-12 progeny than in the B derivatives.
In case of adapted mutants via pflB gene mutations, all pflB mutants could not produce formate, one of reaction products of the PflB enzyme, and exhibited reduced acetate formation (Table 3) as observed for ΔadhE ΔpflB cells (Table 2), arguing for the inactivation of PflB enzymes in those adapted mutants. It is not clear why compensatory pflB gene mutations occurred frequently in K-12 ΔadhE strains but rarely in B ΔadhE strains. As shown in Figure 6, the growth of the ΔadhE pflB(G1931A) mutants was poorer than that of the ΔadhE pta(A1967C) mutants in the BL21 (DE3) background, which might explain why we detected fewer pflB mutations among the total range of compensatory mutations.
The second question is why did these strains adapt differently in stress conditions?
As mentioned above, we tested whether adaptation could be accelerated by mutator gene knockouts (ΔmutL, ΔmutS, and ΔmutT). It is reported that mutT mutations enhance the point mutation rate by 150-fold (Wielgoss et al., 2013); however, these mutations could not shorten the delayed lag phase of the B strains compared to the growth of K-12 strains (Figure 8). These results demonstrate that the mutation rate is not the main explanation for the delayed adaptation of B strains compared with K-12 strains under anaerobic conditions.
The redox cofactor balance [NAD+/NADH] in the K-12 ΔadhE strains was affected slightly under anaerobic conditions, whereas that of the B ΔadhE cells was affected markedly (Figure 10). The recovery of the redox balance [NAD+/NADH] in adapted K-12 ΔadhE cells was even higher than that in the parental K-12 ΔadhE cells (Figure 10C). This indicates that alternative redox balance metabolic pathways might be available in K-12 ΔadhE cells. Figure 10 shows that the redox balance was significantly associated with the viability of stressed cells. Thus, it is probable that the culturable cell number of B ΔadhE strains is critical for the generation of adapted cells harboring compensatory mutations under severely stressful conditions, which may explain why the B ΔadhE strains exhibited delayed adaptive growth compared with the K-12 ΔadhE strains. This hypothesis was also strongly supported by the inoculum dilution experiments, which showed that the adaptation speed depended on the number of cells inoculated (Figure 11). We assume that severely stressed B ΔadhE cells lost their viability and could not restore cellular growth via adaptations. Under the same anaerobic conditions, K-12 cells may adapt rapidly by altering their metabolic fluxes via alternative and/or bypass reactions to restore their redox balance, because [NAD+/NADH] was not severely affected by the absence of AdhE (Figure 10). The present study clearly demonstrated that the requirements for short and long lag periods during microbial adaptation depended mainly on the fraction of metabolically active cells present when bacteria were exposed to adverse conditions.
Conclusion
Our study identified an unusual short-term microbial adaptation to stressful conditions, which was accompanied by genomic mutations that have been observed frequently during other examples of molecular evolution. We found that even closely related strains possess distinct metabolic characteristics that allow them to adapt in different ways to the same environmental conditions. In biotechnological aspects, engineered bacterial strains may rapidly evolve to cope with adverse metabolic fluxes, which might have been overlooked during the development and fermentation of industrial strains. These events may occur without our knowledge in microbial experiments in the laboratory as well as microbial outbreaks in nature.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2012042114; NRF-2011-0019745), the Next-Generation BioGreen21 Program (SSAC-PJ009524), Rural Development Administration, and KRIBB Research Innovative and Initiative Programs.
References
Albertini, A. M., Hofer, M., Calos, M. P., and Miller, J. H. (1982). On the formation of spontaneous deletions: the importance of short sequence homologies in the generation of large deletions. Cell 29, 319–328. doi: 10.1016/0092-8674(82)90148-9
Auriol, C., Bestel-Corre, G., Claude, J. B., Soucaille, P., and Meynial-Salles, I. (2011). Stress-induced evolution of Escherichia coli points to original concepts in respiratory cofactor selectivity. Proc. Natl. Acad. Sci. U.S.A. 108, 1278–1283. doi: 10.1073/pnas.1010431108
Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., et al. (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008. doi: 10.1038/msb4100050
Barrick, J. E., Yu, D. S., Yoon, S. H., Jeong, H., Oh, T. K., Schneider, D., et al. (2009). Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 461, 1243–1247. doi: 10.1038/nature08480
Battesti, A., Majdalani, N., and Gottesman, S. (2011). The RpoS-mediated general stress response in Escherichia coli. Annu. Rev. Microbiol. 65, 189–213. doi: 10.1146/annurev-micro-090110-102946
Bekker, M., Teixeira De Mattos, M. J., and Hellingwerf, K. J. (2006). The role of two-component regulation systems in the physiology of the bacterial cell. Sci. Prog. 89, 213–242. doi: 10.3184/003685006783238308
Campos-Bermudez, V. A., Bologna, F. P., Andreo, C. S., and Drincovich, M. F. (2010). Functional dissection of Escherichia coli phosphotransacetylase structural domains and analysis of key compounds involved in activity regulation. FEBS J. 277, 1957–1966. doi: 10.1111/j.1742-4658.2010.07617.x
Cherepanov, P. P., and Wackernagel, W. (1995). Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9–14. doi: 10.1016/0378-1119(95)00193-A
Clark, D. P. (1989). The fermentation pathways of Escherichia coli. FEMS Microbiol. Rev. 5, 223–234.
Conrad, T. M., Frazier, M., Joyce, A. R., Cho, B. K., Knight, E. M., Lewis, N. E., et al. (2010). RNA polymerase mutants found through adaptive evolution reprogram Escherichia coli for optimal growth in minimal media. Proc. Natl. Acad. Sci. U.S.A. 107, 20500–20505. doi: 10.1073/pnas.0911253107
Durand, S., and Storz, G. (2010). Reprogramming of anaerobic metabolism by the FnrS small RNA. Mol. Microbiol. 75, 1215–1231. doi: 10.1111/j.1365-2958.2010.07044.x
Galinina, N., Lasa, Z., Strazdina, I., Rutkis, R., and Kalnenieks, U. (2012). Effect of ADH II deficiency on the intracellular redox homeostasis in Zymomonas mobilis. Sci. World J. 2012:742610. doi: 10.1100/2012/742610
Gonzalez-Siso, M. I., Garcia-Leiro, A., Tarrio, N., and Cerdan, M. E. (2009). Sugar metabolism, redox balance and oxidative stress response in the respiratory yeast Kluyveromyces lactis. Microb. Cell Fact. 8:46. doi: 10.1186/1475-2859-8-46
Gupta, S., and Clark, D. P. (1989). Escherichia coli derivatives lacking both alcohol dehydrogenase and phosphotransacetylase grow anaerobically by lactate fermentation. J. Bacteriol. 171, 3650–3655.
Heikal, A. A. (2010). Intracellular coenzymes as natural biomarkers for metabolic activities and mitochondrial anomalies. Biomark Med. 4, 241–263. doi: 10.2217/bmm.10.1
Iyer, P. P., Lawrence, S. H., Luther, K. B., Rajashankar, K. R., Yennawar, H. P., Ferry, J. G., et al. (2004). Crystal structure of phosphotransacetylase from the methanogenic archaeon Methanosarcina thermophila. Structure 12, 559–567. doi: 10.1016/j.str.2004.03.007
Jeong, H., Barbe, V., Lee, C. H., Vallenet, D., Yu, D. S., Choi, S. H., et al. (2009). Genome sequences of Escherichia coli B strains REL606 and BL21(DE3). J. Mol. Biol. 394, 644–652. doi: 10.1016/j.jmb.2009.09.052
Kim, H. J., Hou, B. K., Lee, S. G., Kim, J. S., Lee, D. W., and Lee, S. J. (2013). Genome-wide analysis of redox reactions reveals metabolic engineering targets for d-lactate overproduction in Escherichia coli. Metab. Eng. 18C, 44–52. doi: 10.1016/j.ymben.2013.03.004
Klein, A. H., Shulla, A., Reimann, S. A., Keating, D. H., and Wolfe, A. J. (2007). The intracellular concentration of acetyl phosphate in Escherichia coli is sufficient for direct phosphorylation of two-component response regulators. J. Bacteriol. 189, 5574–5581. doi: 10.1128/JB.00564-07
Lee, S. J., Lee, D. Y., Kim, T. Y., Kim, B. H., Lee, J., and Lee, S. Y. (2005). Metabolic engineering of Escherichia coli for enhanced production of succinic acid, based on genome comparison and in silico gene knockout simulation. Appl. Environ. Microbiol. 71, 7880–7887. doi: 10.1128/AEM.71.12.7880-7887.2005
Lee, S. J., Trostel, A., and Adhya, S. (2014). Metabolite changes signal genetic regulatory mechanisms for robust cell behavior. MBio 5, e00972–e00913. doi: 10.1128/mBio.00972-13
Lee, S. J., Trostel, A., Le, P., Harinarayanan, R., Fitzgerald, P. C., and Adhya, S. (2009). Cellular stress created by intermediary metabolite imbalances. Proc. Natl. Acad. Sci. U.S.A. 106, 19515–19520. doi: 10.1073/pnas.0910586106
Levinson, G., and Gutman, G. A. (1987). Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol. Biol. Evol. 4, 203–221.
Lynch, A. S., and Lin, E. C. (1996). Transcriptional control mediated by the ArcA two-component response regulator protein of Escherichia coli: characterization of DNA binding at target promoters. J. Bacteriol. 178, 6238–6249.
Mechold, U., Potrykus, K., Murphy, H., Murakami, K. S., and Cashel, M. (2013). Differential regulation by ppGpp versus pppGpp in Escherichia coli. Nucleic Acids Res. 41, 6175–6189. doi: 10.1093/nar/gkt302
Mettert, E. L., and Kiley, P. J. (2005). ClpXP-dependent proteolysis of FNR upon loss of its O2-sensing [4Fe-4S] cluster. J. Mol. Biol. 354, 220–232. doi: 10.1016/j.jmb.2005.09.066
Mika, F., and Hengge, R. (2005). A two-component phosphotransfer network involving ArcB, ArcA, and RssB coordinates synthesis and proteolysis of sigmaS (RpoS) in E. coli. Genes Dev. 19, 2770–2781. doi: 10.1101/gad.353705
Miller, J. H. (1992). A Short Course in Bacterial Genetics. A. Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
Moat, A. G., Foster, J. W., and Spector, M. P. (2002a). “Chapter 11. Fermentation pathways,” in Microbial Physiology, 4th Edn., eds A. G. Moat, J. W. Foster, and M. P. Spector (New York, NY: Wiley-Liss, Inc.), 412–433. doi: 10.1002/0471223867.ch11
Moat, A. G., Foster, J. W., and Spector, M. P. (2002b). “Chapter 18. Microbial stress responses,” in Microbial Physiology, 4th Edn., eds A. G. Moat, J. W. Foster, and M. P. Spector (New York, NY: Wiley-Liss, Inc.), 582–611. doi: 10.1002/0471223867.ch18
Patschkowski, T., Bates, D. M., and Kiley, P. J. (2000). “Chapter 5. Meachanism for sensing and responding to oxygen deprivation,” in Bacterial Stress Responses, eds G. Storz and R. Hengge-Aronis (Washington, DC: ASM Press), 61–78.
Pinske, C., Bonn, M., Kruger, S., Lindenstrauss, U., and Sawers, R. G. (2011). Metabolic deficiences revealed in the biotechnologically important model bacterium Escherichia coli BL21(DE3). PLoS ONE 6:e22830. doi: 10.1371/journal.pone.0022830
Rando, O. J., and Verstrepen, K. J. (2007). Timescales of genetic and epigenetic inheritance. Cell 128, 655–668. doi: 10.1016/j.cell.2007.01.023
Schneider, D., Duperchy, E., Depeyrot, J., Coursange, E., Lenski, R., and Blot, M. (2002). Genomic comparisons among Escherichia coli strains B, K-12, and O157:H7 using IS elements as molecular markers. BMC Microbiol. 2:18. doi: 10.1186/1471-2180-2-18
Storz, G., and Hengge-Aronis, R. (2000). “Preface,” in Bacterial Stress Responses, eds G. Storz and R. Hengge-Aronis (Washington, DC: ASM Press), xiii–xiv.
Studier, F. W., Daegelen, P., Lenski, R. E., Maslov, S., and Kim, J. F. (2009). Understanding the differences between genome sequences of Escherichia coli B strains REL606 and BL21(DE3) and comparison of the E. coli B and K-12 genomes. J. Mol. Biol. 394, 653–680. doi: 10.1016/j.jmb.2009.09.021
Tenaillon, O., Rodriguez-Verdugo, A., Gaut, R. L., Mcdonald, P., Bennett, A. F., Long, A. D., et al. (2012). The molecular diversity of adaptive convergence. Science 335, 457–461. doi: 10.1126/science.1212986
Toprak, E., Veres, A., Michel, J. B., Chait, R., Hartl, D. L., and Kishony, R. (2012). Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat. Genet. 44, 101–105. doi: 10.1038/ng.1034
Weinert, B. T., Iesmantavicius, V., Wagner, S. A., Scholz, C., Gummesson, B., Beli, P., et al. (2013). Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol. Cell 51, 265–272. doi: 10.1016/j.molcel.2013.06.003
Wielgoss, S., Barrick, J. E., Tenaillon, O., Wiser, M. J., Dittmar, W. J., Cruveiller, S., et al. (2013). Mutation rate dynamics in a bacterial population reflect tension between adaptation and genetic load. Proc. Natl. Acad. Sci. U.S.A. 110, 222–227. doi: 10.1073/pnas.1219574110
Keywords: anaerobic condition, alcohol dehydrogenase, adaptation, genomic mutation, pta gene, pflB gene, redox balance
Citation: Kim HJ, Jeong H, Hwang S, Lee M-S, Lee Y-J, Lee D-W and Lee SJ (2014) Short-term differential adaptation to anaerobic stress via genomic mutations by Escherichia coli strains K-12 and B lacking alcohol dehydrogenase. Front. Microbiol. 5:476. doi: 10.3389/fmicb.2014.00476
Received: 26 May 2014; Accepted: 25 August 2014;
Published online: 09 September 2014.
Edited by:
Mickael Desvaux, Institut National de la Recherche Agronomique, FranceReviewed by:
Thomas Hindré, Laboratory Adaptation and Pathogenesis of Microorganisms, UMR 5163, University J. Fourier, CNRS, FranceIsabelle Meynial-Salles, Institut National des Sciences Appliquées, France
Christophe Nguyen-The, Institut National de la Recherche Agronomique, France
Copyright © 2014 Kim, Jeong, Hwang, Lee, Lee, Lee and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong-Woo Lee, School of Applied Biosciences, Kyungpook National University, Daehakro 80, Daegu 702-701, South Korea e-mail:bGVlaGljYW1Aa251LmFjLmty;
Sang Jun Lee, Infection and Immunity Research Center, Korea Research Institute Bioscience & Biotechnology (KRIBB), 125 Gwahak-ro, Yuseong-gu, Daejeon 305-806, South Korea e-mail:bGVlc2pAa3JpYmIucmUua3I=
†These authors have contributed equally to this work.