 Alejandra Alvarado1,2
Alejandra Alvarado1,2 Lilia E. Montañez-Hernández1
Lilia E. Montañez-Hernández1 Sandra L. Palacio-Molina1
Sandra L. Palacio-Molina1 Ricardo Oropeza-Navarro3
Ricardo Oropeza-Navarro3 Miriam P. Luévanos-Escareño1
Miriam P. Luévanos-Escareño1 Nagamani Balagurusamy1*
Nagamani Balagurusamy1*
- 1Laboratorio de Biorremediación, Escuela de Ciencias Biológicas, Universidad Autónoma de Coahuila, Torreón, México
- 2Department of Ecophysiology, Max Planck Institute for Terrestrial Microbiology, Marburg, Germany
- 3Instituto de Biotecnología – Universidad Nacional Autónoma de México, Cuernavaca, México
Anaerobic digestion (AD) is a biological process where different trophic groups of microorganisms break down biodegradable organic materials in the absence of oxygen. A wide range of AD technologies is being used to convert livestock manure, municipal and industrial wastewaters, and solid organic wastes into biogas. AD gains importance not only because of its relevance in waste treatment but also because of the recovery of carbon in the form of methane, which is a renewable energy and is used to generate electricity and heat. Despite the advances on the engineering and design of new bioreactors for AD, the microbiology component always poses challenges. Microbiology of AD processes is complicated as the efficiency of the process depends on the interactions of various trophic groups involved. Due to the complex interdependence of microbial activities for the functionality of the anaerobic bioreactors, the genetic expression of mcrA, which encodes a key enzyme in methane formation, is proposed as a parameter to monitor the process performance in real time. This review evaluates the current knowledge on microbial groups, their interactions, and their relationship to the performance of anaerobic biodigesters with a focus on using mcrA gene expression as a tool to monitor the process.
Introduction
Anaerobic digestion (AD) is a series of unique processes that involve the reduction and oxidation of organic molecules by the complex metabolic interactions between several microbial groups. AD is widely used worldwide for the treatment of organic wastes, such as animal manures, municipal, and industrial wastewaters, and solid organic wastes such as sludge, crop, and food wastes. Carbon present in the biomass is recovered in the form of methane; a renewable form of energy and the effluent can be used as organic fertilizer as it is rich in nutrients (Cho et al., 2013).
For years, several studies have focused on optimization of the design of biodigesters (Ince, 1998; Raynal et al., 1998; Bouallagui et al., 2005; Martí-Herrero, 2011), or treatment conditions (Kim et al., 2002, 2003) and the characterization and preparation of adequate waste mixtures to obtain proper C:N ratio (Hills, 1979; Yen and Brune, 2007). However, the performance and efficiency of AD depends greatly on the interactions between different active microbial groups (Talbot et al., 2008; Ali Shah et al., 2014). Therefore, characterization of the microbial community structure and the comprehension of the metabolic networks are critical to improve digestion efficiency (Shin et al., 2010). Several molecular biological analytical tools, including polymerase chain reaction (PCR) and its many variants, denaturing gradient gel electrophoresis (DGGE), fluorescence in situ hybridization (FISH), restriction fragment length polymorphism (RFLP) among others, have been applied in the study of the microbial communities in AD (Montero et al., 2009; Shin et al., 2010; Nelson et al., 2011; Supaphol et al., 2011).
Usually AD is conceptually divided into three or four stages, hydrolysis and/or fermentation, acetogenesis and methanogenesis. During the first stage insoluble particles of cellulose or hemicellulose contained in the substrates are hydrolyzed and converted into simple and soluble products, which are catabolized by fermentative bacteria into alcohol and fatty acids. Subsequent steps involve the oxidation of such alcohols and fatty acids with carbon chain longer than C2 to acetate by syntrophic bacteria and their activity depends on the removal of hydrogen either by CO2 or sulfate reduction. Finally, during methanogenesis, acetate and other methyl-containing C1 compounds are reduced to methane by aceticlastic and methylotrophic methanogens and CO2 is reduced by H2-oxidizing methanogens. Methanogens belong to the domain Archaea and are characterized by their substrate specificity, slow growth rate, and susceptibility to environmental changes, but their growth and activity is vital for the efficient functioning of AD process (Balagurusamy and Ramasamy, 1999).
Methanogenesis requieres reduction of the methyl group of methyl coenzyme M to CH4 by the enzyme methyl coenzyme M reductase (MCR), involving a nickel-containing factor F430 (Pramanik and Kim, 2013). All known genomes of the methanogenic archaea encode at least one copy of the mcrBDCGA operon, which is composed of two alpha (mcrA), beta (mcrB), and gamma (mcrG) subunits (Luo et al., 2002). Moreover, all known methanogens express MCR, which catalyzes the last step in the methanogenesis (Ferry, 1999). Therefore, the presence of this enzyme is a reliable diagnostic indicator of methanogenesis in diverse environments (Reeve et al., 1997; Luton et al., 2002; Steinberg and Regan, 2009; Palacio-Molina et al., 2013). Currently various groups are involved in developing strategies to combine the analysis of differential gene expression of mcr alpha subunit and the traditional approaches to monitor the performance of biodigesters on real time basis. The present paper discusses the currently available knowledge on this new strategy for management of AD process.
Anaerobic Digestion: the Players
Although many of the microorganisms involved in the process are still to be identified or cultured, at least 11 groups have been reported to interact with each other in a series of specific reactions in anaerobic ecosystems (Figure 1).
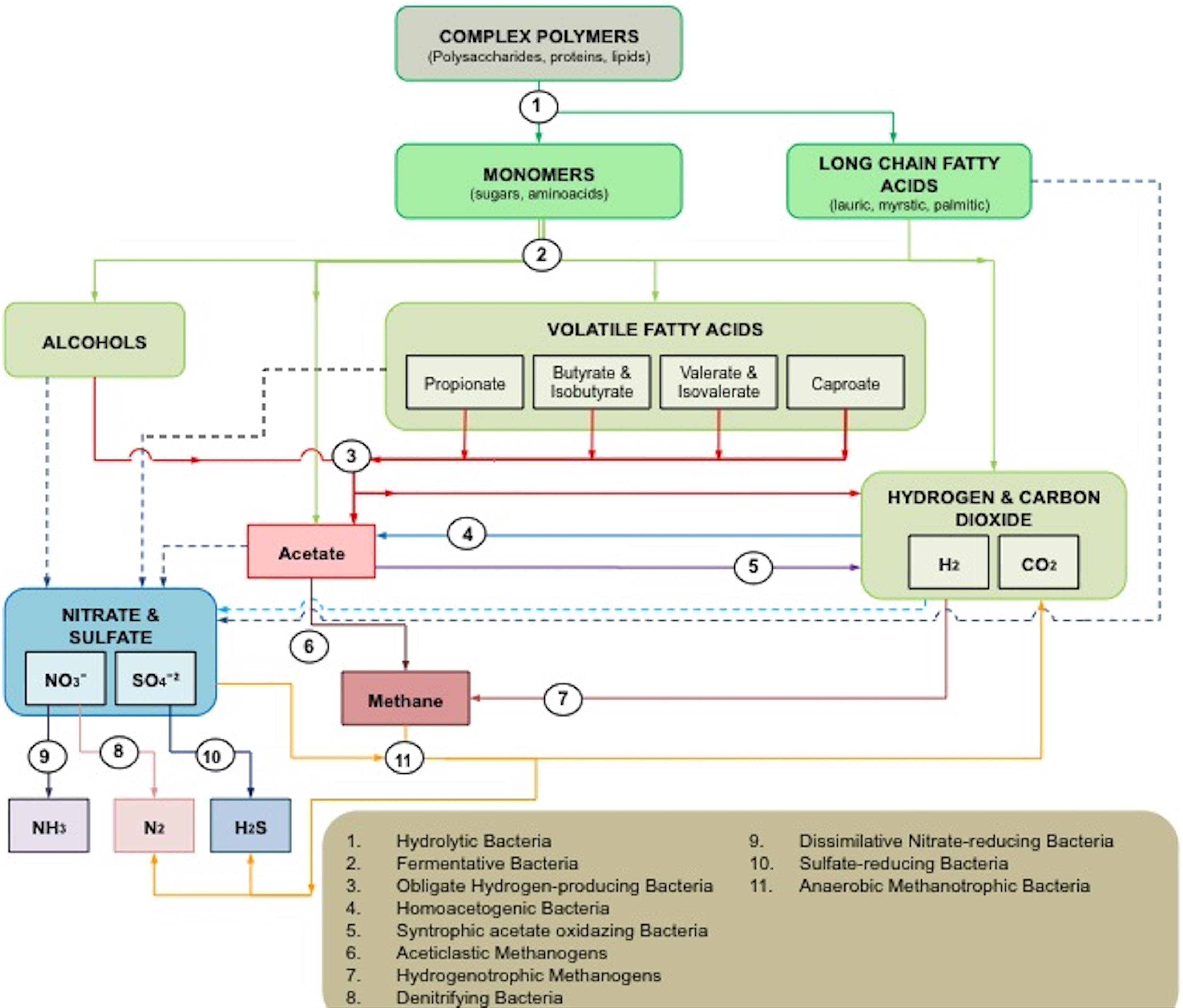
FIGURE 1. Microbial interactions in anaerobic ecosystems. Solid lines represent the usual pathways in anaerobic digestion. Dash lines indicate alternative pathways in which the final electron acceptors are either sulfates or nitrates.
Hydrolytic bacteria are extremely diverse in anaerobic biodigesters, reflecting their enormous metabolic flexibility. One of the most important polysaccharides in biodigesters is cellulose, the main substrate of anaerobic cellulolytic bacteria. Cellulolytic anaerobes possess cellulosome, a multienzymatic complex, which degrades cellulose by binding to the substrate. In general, hydrolysis of polysaccharides is a slow process under anaerobic conditions. The rate and efficiency of cellulose hydrolysis in a biodigester is intrinsically related with the abundance of particulate-bound hydrolytic bacteria. In fact, the performance of a bioreactor may vary depending on the hydrolytic species that form the microbial community (Ren et al., 2007).
In one study, the microbial community was examined by DGGE and dot-blot hybridization during the start-up of two acidophilic systems at mesophilic (35°C) and thermophilic (55°C) conditions (Liu et al., 2002b). The reactors were fed dairy wastewater and inoculated with granular sludge. In both systems, mesophilic and thermophilic Bacteria predominated during hydrolysis, specifically the phylum Firmicutes. Actually, it is well known that in cellulolytic environments, Clostridium predominates (Balagurusamy, 2007). In addition, Acetivibrio, Bacteroides, Selenomonas, and Ruminococcus are some of the most common hydrolytic bacteria in the anaerobes bioreactors (Balagurusamy and Ramasamy, 1999).
In the rumen, the most similar natural environment to biodigesters, Ruminococcus albus and R. flavefaciens are the predominant gram-positive, fiber-degrading bacteria, while Fibrobacter succinogenes is the most abundant Gram-negative (Wanapat and Cherdthong, 2009). Commonly, hydrolytic bacteria adhere to the substrate particles along with some anaerobic fungi, which are also present in biodigesters. However, the growth of anaerobic hydrolytic fungi is slower than that of bacteria, which explains their limited presence in the community structure studies. Neocallimastix sp., is one the most studied anaerobic fungi in rumen, which also include Orpimomyces, Anaeromyces, Piromyces, and Caecomyces (Lynd et al., 2002; Gallert and Winter, 2005). As hydrolysis of complex compounds is catalyzed by a defined group of specific enzymes such as cellulases, proteases and lipases, this step is known as one of the most catalytically active and one that could greatly benefit from new monitoring strategies involving the analysis of gene expression profiles of key enzymes (Amani et al., 2010).
In general, hydrolysis of recalcitrant materials, such as lignin, cellulose, or hemicelluloses is a relatively slow process, and hence is often a limiting-step, normally overcome in thermophilic treatments, especially when solids contents are kept below 7%. In comparison, hydrolysis of proteins and lipids is faster (Ortega-Charleston, 2008). Proteins are generally hydrolyzed to amino acids by proteases. Microorganisms that are responsible of this reaction include species of the genera Bacteroides, Butyrivibrio, Clostridium, Fusobacterium, Selenomonas, and Streptococcus (Amani et al., 2010).
Monomeric compounds generated after hydrolysis are taken by fermentative bacteria and transformed to alcohols, volatile fatty acids (VFAs), CO2, or H2. In biodigesters, alcohols and VFAs are further transformed into the substrates for methanogenesis, namely, acetate, formate, H2, and CO2. In the presence of electron acceptors such as sulfate and nitrate, the intermediates for methanogenesis are diverted to anaerobic respiration.
Representatives of domain Bacteria are largely responsible for fermentation reactions. Among the fermentative microorganisms in the rumen are several species of Clostridium and R. albus. Meanwhile in the biodigesters fed with cow manure, members of Clostridium, Eubacterium, and Bacteroides are the abundant ones (Sivakumaran et al., 1991; Delbes et al., 2000). Streptococcus sp., Lactobacillus sp. and Propionibacterium are also fermentative microorganisms commonly found in the biodigesters, producing lactate or lactate and ethanol plus CO2 and H2 (Insam et al., 2010). From the phylogenetic point of view, acidogenic bacteria are widely diverse. Most of the microorganisms of this group found in biodigersters include members of the genera Clostridium, Eubacterium, and Ruminococcus (Drake et al., 2013).
The products generated by fermentation are typically VFAs, which decrease the pH and are the most common cause of failure in anaerobic systems (Chen et al., 2008; Wang et al., 2009). This acidification is a consequence of the imbalance between fermentative and syntrophic bacteria, especially obligatory hydrogen acetogenic bacteria. Although the simple monitoring of the profile of VFAs can help to prevent failures, differences between microbial populations are already very large when a decrease in the pH has been perceived (Amani et al., 2010). Hence, knowledge on the activities of microbial communities might help to anticipate this common failure even before it arises. This is probably the most important reason that microbial diversity alone is not helpful in the monitoring of anaerobic biodigesters.
Further, inhibition thresholds of VFAs vary greatly, as they depend on multiple factors, such as temperature, characteristics of feeding, source of inoculum, type of system, organic load, their state of ionization, among many others (Chen et al., 2008). But in general, acetic acid requires higher concentrations; about 2.4 g L-1, than the other acids to be inhibitory, and it is also a major substrate for methane production. In contrast, concentrations of propionic acid below of 900 mg L-1 are a sign of good performance (Wang et al., 2009).
During AD syntrophic acetogenic bacteria oxidize VFAs greater than C3 into hydrogen, acetate, and CO2 in association with methanogens or sulfate reducing bacteria. The oxidation of VFAs by syntrophic acetogenic bacteria is not a thermodynamically favorable process (ΔG ≥ 0). These reactions are favorable only under low partial pressure of hydrogen, which is achieved either by methanogenesis or sulfate reduction (Schink, 1997). Therefore, hydrogen metabolism is crucial in AD since at high partial pressures of hydrogen ( > 10 Pa o 10-4 atm) will result in accumulation of VFAs and will result in acidification of biodigesters. It has been reported that hydrogen partial pressure in biodigesters should not exceed 10-6 atm for the efficient oxidation of VFAs (Schink, 1997; Sieber et al., 2012).
Methane formation is the final step in AD, and it is also the most sensitive to imbalance. As a matter of fact, the amount of viable methanogens is probably the most effective indicator of a stable and effective system. Methanogenic communities are not as diverse as the others within the digesters, and they possess a specialized metabolism, characteristics that make them more likely to be inhibited. Among the seven orders of known methanogens, three are found with more frequency in the biodgesters: Methanobacteriales, Methanomicrobiales, and Methanosarcinales (Demirel and Scherer, 2008). Members of the order Methanococcales are rarely found in biodigesters; however, there is a report about the finding of these microorganisms in granular sludge treating brewery wastewater. (Liu et al., 2002a) The fifth order, Methanopyrales, includes only one hyperthemophilic species, which is unlikely to be found into anaerobic biodigesters (Bapteste et al., 2005). The recently recognized sixth order of Methanocellales contains only one genus, Methanocella, a hydrogenotrophic methanogen that was first isolated from a propionate-degrading culture obtained from rice paddy soils (Sakai et al., 2008). Meanwhile, the newest proposed order, Methanoplasmatales, was derived from samples of hindguts of termites and wood-feeding cockroaches. Methanoplasmatales includes members, which were previously believed to be distantly related to a different lineage in the phylum Euryarchaeota (Paul et al., 2012).
Members of Methanobacteriales, Methanococcales, and Methanomicrobiales utilize CO2 as electron acceptor. Hydrogen is commonly used as electron donor in this case, but some species also use formate and/or alcohols such as ethanol or isopropanol. With the exception of Methanosphaera (from Methanobacteriales), members of these orders cannot use acetate or one-single carbon compounds (Bonin and Boone, 2006).
Methanosarcinales are the most diverse in terms of metabolism. Acetate, hydrogen, formate, ethanol, isopropanol, and methylated compounds can be metabolized by members from this order (Kendall and Boone, 2006). Methanosaetaceae is the only family within Methanosarcinales that includes strictly aceticlastic anaerobes (Smith and Ingram-Smith, 2007).
In natural environments, such as swamps or rumen, populations of hydrogenotrophic methanogens are predominant, while in biodigesters; usually there are more aceticlastic methanogens (Ferry, 2010). This difference appears to be related to the amount of substrates and the presence of relatively high levels of various inhibitory compounds in the biodigesters, such as ammonia, H2S, and VFAs.
In general, the methanogenic pathway itself has captured the curiosity of many for decades. The pioneer work of several scientists has allowed us to know the biochemically distinctive features of methanogens. However, our understanding of how methanogenesis is coupled to energy conservation has been slower to develop (Leigh et al., 2011).
Methanogenesis and Methanogens
Methanogenesis usually occurs in a variety of natural anaerobic environments such as marine and freshwater sediments, rice paddies, landfill, animal digestive tracts, and hydrothermal vents. However, it has been demonstrated that these microorganisms are also able to grow in aerated places like deserts soils (Aśchenbach et al., 2013). Annually, approximately 600 million metric tons of methane is produced. Due to its potential greenhouse effect, which is 21 times higher that of CO2, methane emission into the atmosphere is an important concern (EPA, 2014). In this sense, AD represents an economical and effective alternative for reducing the emission of methane from organic wastes since it recovers methane as an energy source.
Methanogens can obtain energy for growth by converting a limited number of substrates to methane under anaerobic conditions. In thermodynamic terms, methanogenesis will only occur when other electron acceptors such as oxygen, nitrate, and sulfate are absent, as methanogens require a low redox potential, around -300 mv for growth and activity. Given that CO2 is the only electron acceptor that does not owe its abundance to photosynthesis, methanogenesis was a favored metabolism early on earth (Kasting and Siefert, 2002).
For methanogens, methane is actually a waste product. The heterodisulfide CoM-S-S-CoB formed as an intermediate in the pathway is of vital importance for the cell since its reduction is coupled to energy conservation, making the heterodisulfide the terminal electron acceptor in the respiratory chain of methanogens (Hedderich and Whitman, 2013). Methanogens use 2-mercaptoethanesulfonate (coenzyme M or CoM) as the terminal methyl carrier in methanogenesis and have four enzymes for CoM biosynthesis. Coenzyme B-Coenzyme M heterodisulfide reductase (Hdr), required for the final reaction of methanogenesis, is divided into two types, cytoplasmic HdrABC in most methanogens and membrane-bound HdrED in Methanosarcina species (Kaster et al., 2011).
Currently, only two types of methanogenic pathways are known, (1) methanogenesis from H2/CO2 or formate, (2) from acetate and methyl group containing C1 compounds. The conversion of methyl group to methane is common in both pathways as shown in Figure 2 (Ferry, 2011).
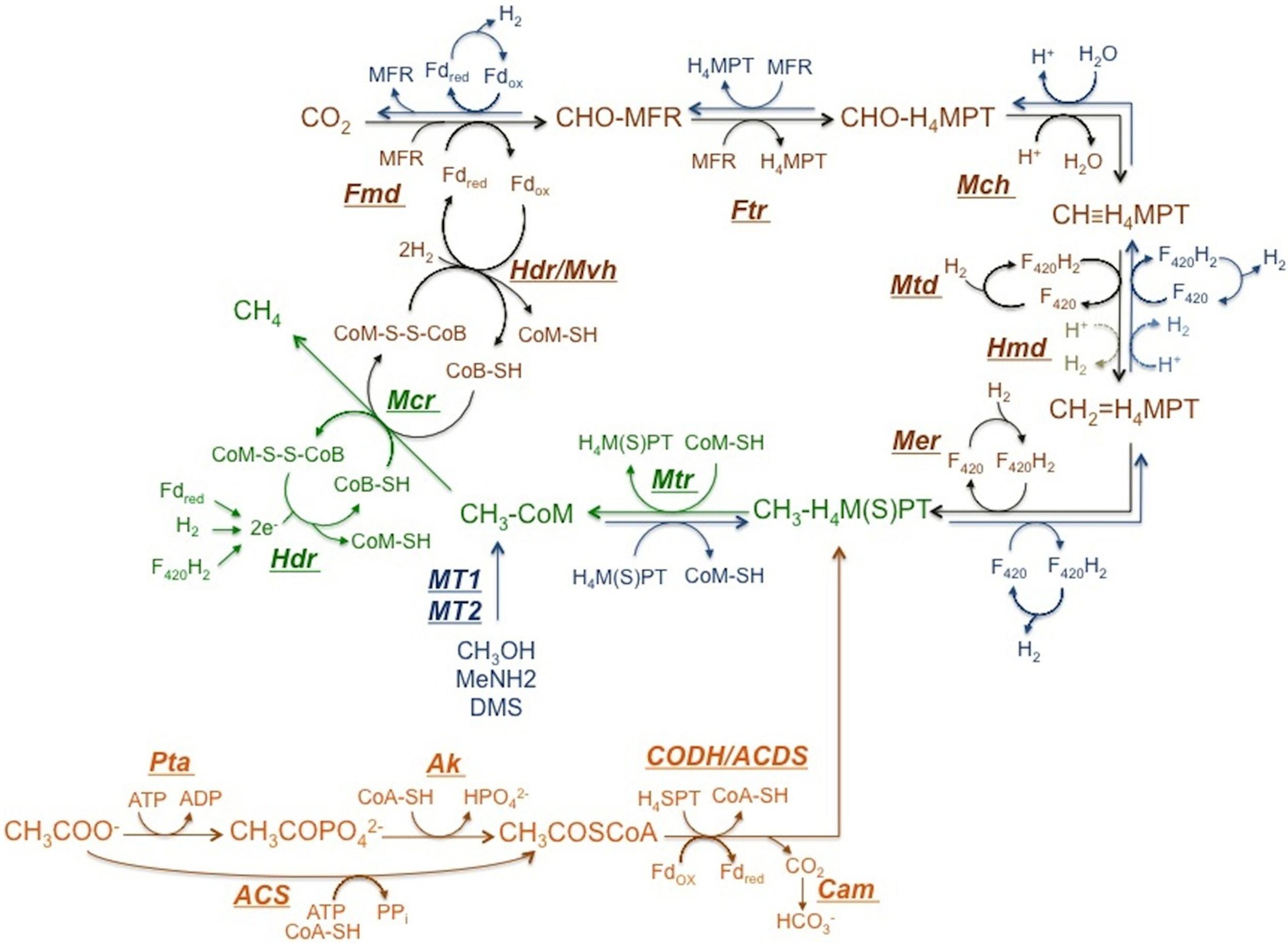
FIGURE 2. Methanogenic Pathways. Reactions in brown correspond to hydrogenotrophic pathway and reactions in orange belong to the aceticlastic pathway. Reactions in blue represent the pathway employed when methyl group containing C1 compounds are the substrate. Reactions in green are common in all pathways. ATP, adenosine triphosphate; H4SPT, tetrahydrosarcinapterin; H4MPT, tetrahydromethanopterin, Fd, ferredoxin; CoA-SH, coenzyme A; CoM-SH, coenzyme M; CoB-SH, coenzyme B; MFR, methanofuran; F420, coenzyme F420; Fmd, CHO-MFR dehydrogenase; Ftr, CHO-MFR:H4M(S)PT formyltransferase; Mch, CH≡H4M(S)PT cyclohydrolase; Mtd, CH2 = H4M(S)PT dehydrogenase dependent of F420; Hmd, CH2 = H4M(S)PT dehydrogenase independent of F420; Mer, CH2 = H4M(S)PT reductase; Ak, acetate kinase; Pta, phosphotransacetylase; ACS, AMP-forming acetyl-CoA synthetase; CODH/ACDS, CO dehydrogenase/acetyl-CoA synthase; Cam, carbonic anhydrase; Mtr, CH3-H4M(S)PT:CoM methyltransferase; Mcr, methyl coenzyme M reductase; Hdr, heterodisulfide reductase; Hdr/Mvh, heterodisulfide reductase/ cytoplasmic F420 -non-reducing hydrogenase complex.
In order to establish an accurate classification of methanogens, phylogenetic analysis have been made with the purpose of organize these microorganisms according to their evolutionary history (Fox et al., 1977; Bapteste et al., 2005). Initially, Bapteste et al. (2005) divided the five orders of methanogens known until that date into two major groups which they named Classes. Class I included the orders Methanobacteriales, Methanococcales and Methanopyrales and Class II comprised Methanomicrobiales and Methanosarcinales. However, it was acknowledged that Methanomicrobiales shared more traits with Class I members than with Methanosarcinales. Therefore, in 2009, an updated view for methanogens differentiation was presented. This new classification divided methanogens into three classes according to seven core methanogenesis enzymes and cofactor biosynthesis (Anderson et al., 2009). In this arrangement, Methanomicrobiales and Methanosarcinales, orders that used to be grouped in the same class, were separated into Class II and Class III, respectively, due to several unique protein signatures observed. Nevertheless, with the discovery of two novel orders, Methanocellales and Methanoplasmatales, the classification of methanogens must be updated.
On the other hand, methanogens also can be divided into two categories based on the presence or lack of cytochromes (Thauer et al., 2008). All members of Methanosarcinales possess cytochromes and methanophenazine while members of the remaining orders lack of both of them. Additionally, Methanosarcinales have the ability to grow on acetate, methanol, and H2/CO2 with a higher growth yield.
It is well documented that methanogenic communities in biodigesters are susceptible to environmental changes, especially low pH and temperature (Demirel and Scherer, 2008). However, methanogenesis in natural ecosystems is known to proceed in cold and acidic conditions that are inhospitable for biodigesters (Steinberg and Regan, 2008; Aśchenbach et al., 2013). This difference between biodigesters and natural ecosystems could be attributed to differences in the composition of the methanogenic communities (Liu and Whitman, 2008; Steinberg and Regan, 2008; Liu, 2010). Therefore, a better knowledge of these differences might lend insights into community-based strategies to increase digester stability with reduced chemical and energy inputs necessary to maintain narrow operating conditions (Steinberg and Regan, 2008).
Community studies of methanogenic population most frequently involve culture-independent techniques and molecular analysis has taken a major role in recent years. Recently biochemical markers using archaeol (2,3-diphytanyl-O-sn-glycerol) also have been developed (McCartney et al., 2013a). In the case of molecular analysis, various methanogen specific primers targeting 16S rRNA gene have been developed (Castro et al., 2004; Yu et al., 2005; Zhou et al., 2011). To eliminate potential problems with non-specific amplification, some researchers have developed primers for the gene sequence of the alpha subunit of the MCR, mcrA (Springer et al., 1995; Hales et al., 1996; Luton et al., 2002; Denman et al., 2007; Steinberg and Regan, 2008). Phylogenetic inference with mcrA sequence is similar to that obtained with 16S rRNA, suggesting non-lateral gene transfer. Due to the fact that methanogens may be examined exclusively from other bacteria present in the biodigesters, mcrA has been increasingly used for phylogenetic analysis coupled with or independently of 16S rRNA studies. Primers and methods targeting both genes for monitoring of methanogens have been also reviewed (Narihiro and Sekiguchi, 2011).
Comparisons Between mcrA and 16S rRNA
Methyl coenzyme M reductase is the unique enzyme that catalyzes the reduction of CH3-CoM to CH4 and is highly conserved in all methanogens. Two izoenzymes of MCR designated MCR I and MCR II are known and their respective operons are shown in Figure 3. The operon encoding MCR I, mcrBDCGA, prevails in all known methanogens while MCR II operon, mrtBDGA, is only found in some members from the orders Methanobacteriales and the Methanococcales (Garcia et al., 2000; Luton et al., 2002). Kinetic parameters are different for both isoenzymes and expression of either MCR I or MCR II seems to be dependent on hydrogen concentrations (Reeve et al., 1997). MCR II in Methanothermobacter thermoautotrophicus (formely known as Methanobacterium thermoautotrophicum) was shown to be expressed only in the earlier stages of batch cultures, then it was replaced with MCR I in late growth and stationary phases where hydrogen concentrations were lower (Pihl et al., 1994; Nölling et al., 1995).
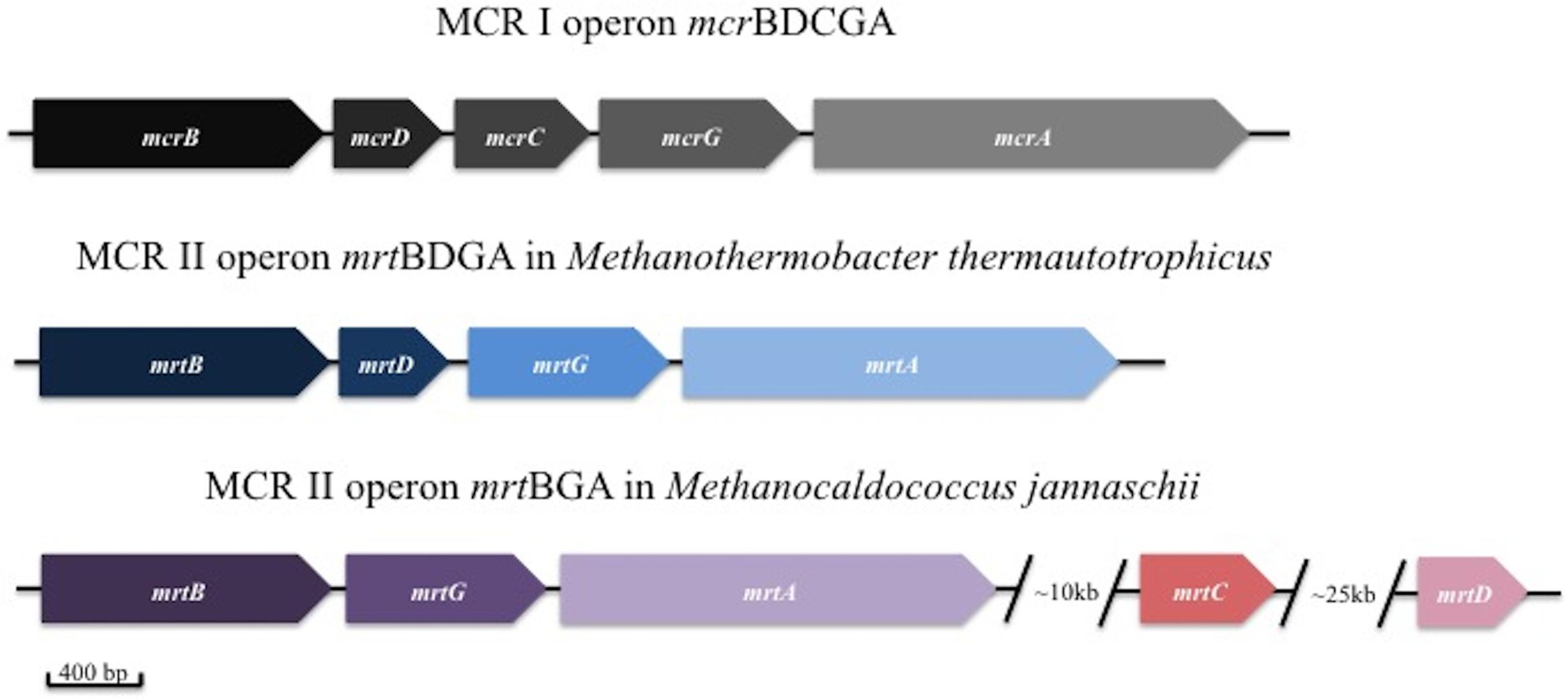
FIGURE 3. Methyl coenzyme M reductase I (MCR I) and MCR II operon. Structure of MCR I operon rarely varies in all methanogen species while MCR II operon is different between species containing these genes (modified from Hallam et al., 2003).
Earlier, Rouvière and Wolfe (1987) showed that phylogenic relationships obtained with different subunits of MCR corresponded at genus level of methanogens with those derived with 16S rRNA gene. After the genes encoding for MCR subunits were available, the mcrA gene, which encodes the α-subunit of MCR I was selected as a phylogenetic tool for the analysis of members of the family Methanosarcinaceae (Springer et al., 1995). The relationships calculated with the sequences of mcrA and 16S rRNA showed high similarity. Subsequent studies have also confirmed that similar phylogenetic relationships can be obtained by use of 16S rRNA and mcrA in methanogens (Luton et al., 2002; Bapteste et al., 2005). Both strategies showed higher concurrence within the same environmental sample. However, it was also reported that 16S rRNA library of the biodigester showed less diversity than the library of mcrA gene (Springer et al., 1995; Steinberg and Regan, 2008).
Since then, mcrA gene has been established as a molecular marker for methanogenic archaea (Lueders et al., 2001) and several studies have identified the presence of mcrA with methanogenic activity. In 2003, environmental mcrA sequences were reported for the first time from a eutrophic lake (Earl et al., 2003) as well as in salt marsh sediments (Castro et al., 2004). Subsequent studies of vertebrate guts also revealed the presence of mcrA genes in the cow rumen (Denman et al., 2007); feces of pigs, chickens and horses (Ufnar et al., 2007a); the guts of humans (Scanlan et al., 2008; Mihajlovski et al., 2010), and the foregut of wallabies (Evans et al., 2009). A comparison between 16S rRNA and mcrA clone frequencies in samples of insect guts showed their strong accordance (Paul et al., 2012). As a result of this comparison arose the differentiation of a separate linage into a new order of methanogens, the Methanoplasmatales. It can be observed that use of mcrA gene is a potential tool in the analysis of methanogen diversity in samples from different and varied sources (Ellis et al., 2012; Lwin and Matsui, 2014).
One of the advantages of mcrA gene is that only one or two copies of mcrA have been found in sequenced methagenogens genomes, making it a more precise tool for estimating the number of these archaeas in the biodigesters than the 16S rRNA gene, which can have up to four copies per genome (Lee et al., 2009). Also, a strong correlation between mcrA copy number and methane production has been reported in H2/CO2- enriched cultures (Morris et al., 2014). Moreover, transcription of mcrA has been used to demonstrate that methanogens are metabolically active (Juottonen et al., 2008), as it is well known that these microorganisms are capable of dormancy when conditions are not optimal (Speece, 1983). Thus, identifying active members of the methanogenic population can provide a real insight into the digester performance. Likewise, because transcription is more closely related to activity, determination of mcrA transcript number promises to be a better indicator of good performance rather than the only mcrA copy number (Morris, 2011). This was proven by studies in paddy field soils, where it was discovered that under different environmental conditions, abundance of mcrA transcrips changed while mcrA gene copy number remained almost the same, suggesting that only certain members of the methanogenic community were metabolically active and responsible for methane emissions (Watanabe et al., 2009; Ma et al., 2012).
However, it was reported that quantification of gene transcript abundance in peat soils was not a reliable method since the presence of inactive and dormant cells overestimated the final values (Freitag and Prosser, 2009). Besides, MCR activity is strongly temperature dependent (Goenrich et al., 2005), and it is still unknown if post-translational modifications affect the expression of the enzyme (Kahnt et al., 2007). Therefore, the analysis of mcrA transcripts solely may not be efficient as a tool for monitoring biodigesters. Moreover, it should be taken into consideration that sequences of isoenzyme mrtA can also be part of the targets of the mcrA primers. Hence, overestimation of transcripts is also possible (Nettmann et al., 2008), if members of Methanobacteriales and Methanococcales are present in the samples, since mrtA has been observed only in both of these orders (Luton et al., 2002).
Linking the mcrA Gene Transcripts to the Dynamics of the Methanogenic Community in Anaerobic Biodigesters
Some of the problems in using and comparing methods of mcrA expression studies are the choice of primers (McCartney et al., 2013b) and the differences in the PCR conditions (Steinberg and Regan, 2008). A comparison between the methods and the outcomes in each experiment requires a much more detailed analysis and for those interested on other molecular methods for environmental monitoring of methanogens, Narihiro and Sekiguchi’s (2011) review is suggested. This review discusses only the relationship between the mcrA gene expression and methanogenic activity, thrusting forward our knowledge on the importance of mcrA as a tool to monitor the functioning of the biodigester. The use of mcrA in different studies related to anaerobic biodigesters and natural anaerobic environments is summarized in Table 1.
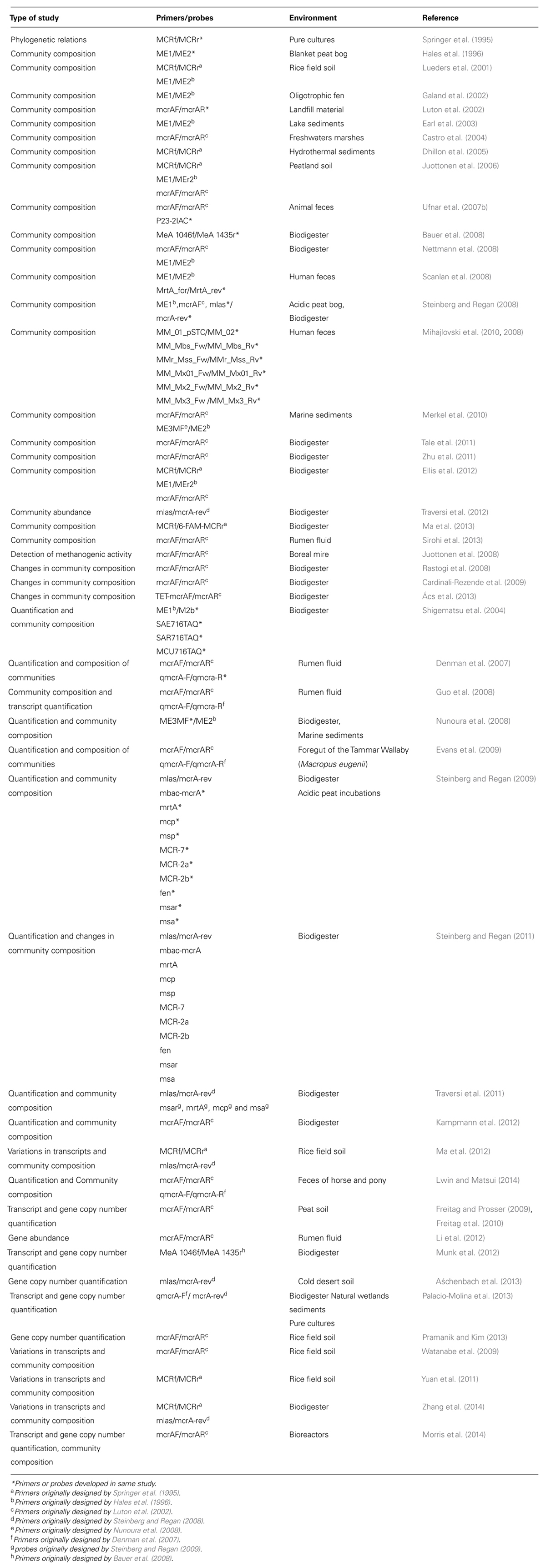
TABLE 1. Application of mcrA gene in studies related to biodigester and other natural anaerobic environments.
Correlation Between Operational and Environmental Conditions of Biodigesters and mcrA
Usually, in balanced anaerobic reactors it is reported that the majority of the methanogens are aceticlastic. The effect of dilution rate and their relation to methanogenic pathways using 13C-labeled acetate and phylogenetic analysis of mcrA gene transcripts showed that transcrips of Methanosarcina species were the most abundant at high dilutions and that aceticlastic pathway was the major pathway for cleavage of acetate and methane production at those dilutions (Shigematsu et al., 2004). However, at low dilution rates, transcripts of Methanoculleus were the most abundant ones and the pathway shifted towards syntrophic acetate oxidation where hydrogenotrophic pathway was the major source for methane production. Traversi et al. (2011) reported a positive correlation between the biogas production and the presence of Methanosarcina and Methanosaeta were found in biodigesters even when most of the mcrA genes corresponded to members of Methanomicrobiales. It was proposed that the abundance of Methanosarcina was a better indicator to understand the efficiency AD process. However, other investigators have had a different experience, and the use of Methanosarcina species alone is not sufficient to monitor the efficiency of the biodigesters. For example, in bioreactors recovered from organic overload by addition of propionate-degrading microorganisms, mcrA gene copies obtained from samples of these bioreactors were associated with Methanospirillum hungatei and Methanobacterium beijingense, both hydrogenotrophic methanogens (Tale et al., 2011). The study does not report the presence of Methanosarcina species, and methanogenic activity is attributed to M. hungatei and M. beijingense.
Recently, analysis of mcrA-based libraries showed that methanogenic populations shifted substantially with modifications in substrate composition (Ács et al., 2013). Microbial community analysis of a large scale mesophilic biodigester with swine manure as substrate showed that 123 clones of mcrA library were assigned to 28 OTUs, of which Methanobrevibacter spp. (an hydrogenotrphic methanogen) was the most abundant (Zhu et al., 2011). Similarly, the predominance of hydrogenotrophic phyla (60–90%) over aceticlastic ones in six large-scale biodigesters fed with different industrial wastes has been reported (Regueiro et al., 2012). A higher predominance of hydrogenotrophic methanogens was found in a continuous anaerobic biodigester treated a mixture of fruit and meal leftovers (Cardinali-Rezende et al., 2009). Similarly, a higher proportion of OTUs clustered within the order of Methanomicrobiales for both mcrA and 16S rRNA libraries (79–88%) in an agricultural biogas plant fed with cattle manure and maize silage under mesophilic conditions (39°C) was reported (Nettmann et al., 2008). Likewise, it was stated that H2/CO2 was the main substrate for methanogenesis in acidic peat (Castro et al., 2004). It was also observed that casein addition modified the population of fermenting bacteria, as well as the available hydrogen and the methanogenic community. After 5 weeks, Methanoculleus marisnigri increased almost twice when casein was added, and with addition of pig blood, Methanomicrobiaceae increased its abundance by 10 times. Clones related to Methanocorpusculum parvum, Methanomassiliicoccus luminyensis, and Methanoculleus bourgensis were more abundant after casein addition and decreased with pig blood. M. luminyensis is a methanogen that produces methane from H2 and methanol (Dridi et al., 2012), whereas M. marisnigri, M. parvum, and M. bourgensis as members of the Methanomicrobiales are strictly hydrogenotrophic methanogens. Similarly in another study with casein, starch and cream as substrates showed that copy numbers of mcrA were higher in casein fed biodigesters than the other two substrates (Kampmann et al., 2012). In the starch-fed reactor, the predominant methanogenic populations were Methanoculleus bourgensis and Methanobrevibacter millerae. These methanogens utilize H2 and CO2 for their metabolism. Similarly, the dominance of Methanobrevibacter and Methanospirillum together with uncharacterized methanogens was reported in biodigesters fed with swine manure (Zhu et al., 2011). Hydrogenotrophic methanogens, specifically of the genus Methanoculleus and of the order Methanomicrobiales were reported to be predominant in pulp mill wastewater treating biodigesters (Yang et al., 2013).
All these studies contradict the previously established ratio of acetate and H2/CO2 on methane production in biodgesters, 70 and 30%, respectively (Ahring, 2003). Although it is reported that stirred tank reactor conditions affected the conglomeration and structure of Methanosarcina and Methanosaeta and thereby decreased the aceticlastic activity (Kampmann et al., 2012), the predominance of hydrogenotrophic methanogens suggested that there was an increase in hydrogen concentration in these biodigesters. It is possible that the type of substrate as well as the presence and activity of syntrophic bacteria resulted in additional hydrogen and promoted hydrogenotrophic methanogenic activity.
Steinberg and Regan (2011) studied the effects of different organic loading rates on the diversity of methanogenic community in two lab-scale semi-batch reactors, one inoculated with acidic sediments and the other with anaerobic sludge. mcrA copies affiliated with Methanoregula boonei and Methanoregula formicica were present in the reactor with acidic sediments. M. boonei has been reported to show growth in acidic pH of 5 (Bräuer et al., 2006), while M. formicica grows at near neutral pH in syntrophic relationship with VFA oxidizers. However, mcrA gene copies belonging to either Methanosarcina species or members of the family Methanobacteriaceae were the dominants ones in the two biodigesters after increase in the organic loading rate (Steinberg and Regan, 2011). Likewise, an increase in Methanosarcina and Methanosaeta species was related to recovery of biodigesters performance after overloading conditions (Fernandez et al., 2000; McMahon et al., 2004; Scully et al., 2005; Hori et al., 2006). Methanosarcina species have been reported to grow better under high loading rates, with high acetate turnover and Methanosaeta species are favored in habitats with low acetate turn over (Rastogi et al., 2008). Methanosarcina has been defined as “the robust methanogen,” because its proven ability to tolerate the four most common causes of stress in biodigesters, viz., temperature variations, organic loading rates, concentration of ammonium, and other salts (De Vrieze et al., 2012). Moreover, Methanosarcina has also been observed in high acidic environments, including natural wetlands (Cadillo-Quiroz et al., 2008), which suggest that this type of aceticlastic, acid-resistant methanogens might represent a good choice as inoculum for waste treatment at higher organic loads and to overcome adverse acidic pH conditions.
Palacio-Molina et al. (2013) reported that the mcrA gene transcription and methanogenic activity correlated to the predominant methanogenic community in one of the wetlands studied. They further reported that this correlation could not be found in samples obtained from another wetland and biodigesters. A study on the methanogenic population of a biogas plant treating cattle manure at different seasons found that mcrA clones related to the genus Methanocorpusculum were able to grow at temperatures as low as 1–5°C, and were highly abundant in both summer (36°C) and winter (25°C; Rastogi et al., 2008). In contrast, clones related to Methanosaeta concilii were present only during summer.
Zhang et al. (2014) studied the response of methanogens to different concentrations of ammonia using mcrA transcripts. While T-RFLP analysis showed that members of Methanoseataceae were the dominant ones in all samples, the abundance of transcripts displayed variations according to the ammonia concentrations. In the case of transcripts, Methanobacteriales recorded higher number at high concentrations of ammonia. Transcripts of Methanosarcinaceae increased during the last stages of the experiments and this coincided with the decrease in concentration of free ammonia. In another study, addition of tea saponins recorded only 8% decrease in methane production by rumen microorganism, but decreased mcrA gene transcription by 76% (Guo et al., 2008). This reduction was attributed to a 79% decrease on protozoa population. It is well known that methanogens are associated with ciliates protozoa of the genera Entodinium, Polyplastron, Epidinium, and Ophryoscolex (Hook et al., 2010).
Even though mcrA gene is mainly employed to determine the presence and community composition of methanogens, transcriptional analysis of this gene can give us a major insight to the dynamics and performance of anaerobic digesters. Inspite of observed variations, mcrA gene could become an important tool for the monitoring of presence and activity in methanogens in different environments in combination with other unique biochemical properties of methanogens.
Conclusion
It is a common perception and widely accepted that aceticlastic methanogens contribute nearly 70% of methane produced in animal wastes fed biodigesters. Most of the time, data on methanogenic community analysis support this idea. However, analysis of mcrA gene expression has broadened our knowledge on the composition and activity of methanogenic communities in biodigesters and in other anaerobic environments. It is clear that hydrogenotrophic methanogens are widely distributed, active and under some operational conditions even dominate over aceticlasic methanogens. Hence, making assumptions based only on the presence and abundance of certain methanogens groups is not a valid parameter to monitor the state of biodigesters. It can be concluded that gene expression of mcrA can be a potential tool in determining the active members of the methanogenic community since it gives a better insight on the metabolic dynamics within biodigesters, However, the use of mcrA gene expression alone or in combination with other parameters such as fluorescence as biomarkers to monitor the state of biodigester on real time basis needs further research on determining the exact of relationship between the transcripts, fluorescence, and methanogenic activity.
Author Contributions
Alejandra Alvarado, Lilia E. Montañez-Hernández, and Nagamani Balagurusamy equally contributed on the structure and manuscript of the review. All authors equally contributed on analysis, revision, and approved the final version of the manuscript. All authors agree to accountable for the accuracy and integrity of the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
Authors thank Dr. W. B. Whitman, Professor of Microbiology, The University of Georgia, Athens, GA, USA for his valuable revision and comments.
References
Ács, N., Kovács, E., Wirth, R., Bagi, Z., Strang, O., Herbel, Z.,et al. (2013). Changes in the Archaea microbial community when the biogas fermenters are fed with protein-rich substrates. Bioresour. Technol. 131, 121–127. doi: 10.1016/j.biortech.2012.12.134
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ahring, B. (2003). “Perspectives for anaerobic digestion,” in Biomethanation I, eds B. Ahring, I. Angelidaki, E. C. Macario, H. N. Gavala, J. Hofman-Bang, A. J. L. Macario et al. (Berlin: Springer), 1–30.
Ali Shah, F., Mahmood, Q., Maroof Shah, M., Pervez, A., and Ahmad Asad, S. (2014). Microbial ecology of anaerobic digesters: the key players of anaerobiosis. ScientificWorldJournal 2014, 1–21. doi: 10.1155/2014/183752
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Amani, T., Nosrati, M., and Sreekrishnan, T. R. (2010). Anaerobic digestion from the viewpoint of microbiological, chemical, and operational aspects – a review. Environ. Rev. 18, 255–278. doi: 10.1139/A10-011
Anderson, I., Ulrich, L. E., Lupa, B., Susanti, D., Porat, I., and Hooper, S. D.et al. (2009). Genomic characterization of methanomicrobiales reveals three classes of methanogens. PLoS ONE 4:e5797. doi: 10.1371/journal.pone.0005797
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aschenbach, K., Conrad, R., Řeháková, K., Doležal, J., Janatková, K., and Angel, R. (2013). Methanogens at the top of the world: occurrence and potential activity of methanogens in newly deglaciated soils in high-altitude cold deserts in the Western Himalayas. Front. Microbiol. 4:359. doi: 10.3389/fmicb.2013.00359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Balagurusamy, N. (2007). A preliminary study on molecular characterization of the eubacteria in a thermophilic, poultry waste fed anaerobic digester. Rev. Mex. Ing. Quím. 6, 237–242.
Balagurusamy, N., and Ramasamy, K. (1999). Biogas technology: an Indian perspective. Curr. Sci. 77, 44–55.
Bapteste,É., Brochier, C., and Boucher, Y. (2005). Higher-level classification of the archaea: evolution of methanogenesis and methanogens. Archaea 1, 353–363. doi: 10.1155/2005/859728
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bauer, C., Korthals, M., Gronauer, A., and Lebuhn, M. (2008). Methanogens in biogas production from renewable resources – a novel molecular population analysis approach. Water Sci. Technol. 58, 1433. doi: 10.2166/wst.2008.514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bonin, A. S., and Boone, D. R. (2006). “The order Methanobacteriales,” in The Prokaryotes, eds M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (New York: Springer), 231–243.
Bouallagui, H., Touhami, Y., Ben Cheikh, R., and Hamdi, M. (2005). Bioreactor performance in anaerobic digestion of fruit and vegetable wastes. Process Biochem. 40, 989–995. doi: 10.1016/j.procbio.2004.03.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bräuer, S. L., Cadillo-Quiroz, H., Yashiro, E., Yavitt, J. B., and Zinder, S. H. (2006). Isolation of a novel acidiphilic methanogen from an acidic peat bog. Nature 442, 192–194. doi: 10.1038/nature04810
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cadillo-Quiroz, H., Yashiro, E., Yavitt, J. B., and Zinder, S. H. (2008). Characterization of the archaeal community in a minerotrophic fen and terminal restriction fragment length polymorphism-directed isolation of a novel hydrogenotrophic methanogen. Appl. Environ. Microbiol. 74, 2059–2068. doi: 10.1128/AEM.02222-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cardinali-Rezende, J., Debarry, R. B., Colturato, L. F. D. B., Carneiro, E. V., Chartone-Souza, E., and Nascimento, A. M. A. (2009). Molecular identification and dynamics of microbial communities in reactor treating organic household waste. Appl. Microbiol. Biotechnol. 84, 777–789. doi: 10.1007/s00253-009-2071-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Castro, H., Ogram, A., and Reddy, K. R. (2004). Phylogenetic characterization of methanogenic assemblages in eutrophic and oligotrophic areas of the florida everglades. Appl. Environ. Microbiol. 70, 6559–6568. doi: 10.1128/AEM.70.11.6559-6568.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, Y., Cheng, J. J., and Creamer, K. S. (2008). Inhibition of anaerobic digestion process: a review. Bioresour. Technol. 99, 4044–4064. doi: 10.1016/j.biortech.2007.01.057
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cho, S.-K., Im, W.-T., Kim, D.-H., Kim, M.-H., Shin, H.-S., and Oh, S.-E. (2013). Dry anaerobic digestion of food waste under mesophilic conditions: performance and methanogenic community analysis. Bioresour. Technol. 131, 210–217. doi: 10.1016/j.biortech.2012.12.100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Vrieze, J., Hennebel, T., Boon, N., and Verstraete, W. (2012). Methanosarcina: the rediscovered methanogen for heavy duty biomethanation. Bioresour. Technol. 112, 1–9. doi: 10.1016/j.biortech.2012.02.079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Delbes, C., Moletta, R., and Godon, J.-J. (2000). Monitoring of activity dynamics of an anaerobic digester bacterial community using 16S rRNA polymerase chain reaction–single-strand conformation polymorphism analysis. Environ. Microbiol. 2, 506–515. doi: 10.1046/j.1462-2920.2000.00132.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Demirel, B., and Scherer, P. (2008). The roles of acetotrophic and hydrogenotrophic methanogens during anaerobic conversion of biomass to methane: a review. Rev. Environ. Sci. Biotechnol. 7, 173–190. doi: 10.1007/s11157-008-9131-1
Denman, S. E., Tomkins, N. W., and McSweeney, C. S. (2007). Quantitation and diversity analysis of ruminal methanogenic populations in response to the antimethanogenic compound bromochloromethane: monitoring of rumen methanogenic Archaea. FEMS Microbiol. Ecol. 62, 313–322. doi: 10.1111/j.1574-6941.2007.00394.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dhillon, A., Lever, M., Lloyd, K. G., Albert, D. B., Sogin, M. L., and Teske, A. (2005). Methanogen diversity evidenced by molecular characterization of methyl coenzyme M reductase A (mcrA) genes in hydrothermal sediments of the Guaymas basin. Appl. Environ. Microbiol. 71, 4592–4601. doi: 10.1128/AEM.71.8.4592-4601.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Drake, H. L., Küsel, K., and Matthies, C. (2013). “Acetogenic prokaryotes,” in The Prokaryotes, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin: Springer), 3–60. doi: 10.1007/978-3-642-30141-4_61
Dridi, B., Fardeau, M.-L., Ollivier, B., Raoult, D., and Drancourt, M. (2012). Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int. J. Syst. Evol. Microbiol. 62, 1902–1907. doi: 10.1099/ijs.0.033712-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Earl, J., Hall, G., Pickup, R. W., Ritchie, D. A., and Edwards, C. (2003). Analysis of methanogen diversity in a hypereutrophic lake using PCR-RFLP analysis of mcr sequences. Microb. Ecol. 46, 270–278. doi: 10.1007/s00248-003-2003-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ellis, J. T., Tramp, C., Sims, R. C., and Miller, C. D. (2012). Characterization of a methanogenic community within an algal fed anaerobic digester. ISRN Microbiol. 2012, 1–12. doi: 10.5402/2012/753892
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
EPA. (2014). Inventory of U.S. Greenhouse Gas Emissions and Sinks: 1990 – 2012. Washington, DC: U.S. Environmental Protection Agency.
Evans, P. N., Hinds, L. A., Sly, L. I., McSweeney, C. S., Morrison, M., and Wright, A.-D. G. (2009). Community composition and density of methanogens in the foregut of the tammar wallaby (Macropus eugenii). Appl. Environ. Microbiol. 75, 2598–2602. doi: 10.1128/AEM.02436-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fernandez, A. S., Hashsham, S. A., Dollhopf, S. L., Raskin, L., Glagoleva, O., Dazzo, F. B.,et al. (2000). Flexible ommunity structure correlates with stable community function in methanogenic bioreactor communities perturbed by glucose. Appl. Environ. Microbiol. 66, 4058–4067. doi: 10.1128/AEM.66.9.4058-4067.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferry, J. G. (1999). Enzymology of one-carbon metabolism in methanogenic pathways. FEMS Microbiol. Rev. 23, 13–38. doi: 10.1111/j.1574-6976.1999.tb00390.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferry, J. G. (2010). The chemical biology of methanogenesis. Planet. Space Sci. 58, 1775–1783. doi: 10.1016/j.pss.2010.08.014
Ferry, J. G. (2011). Fundamentals of methanogenic pathways that are key to the biomethanation of complex biomass. Curr. Opin. Biotechnol 22, 351–357. doi: 10.1016/j.copbio.2011.04.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fox, G. E., Magrum, L. J., Balch, W. E., Wolfe, R. S., and Woese, C. R. (1977). Classification of methanogenic bacteria by 16S ribosomal RNA characterization. Proc. Natl. Acad. Sci. U.S.A. 47, 4537–4541. doi: 10.1073/pnas.74.10.4537
Freitag, T. E., and Prosser, J. I. (2009). Correlation of methane production and functional gene transcriptional activity in a peat soil. Appl. Environ. Microbiol. 75, 6679–6687. doi: 10.1128/AEM.01021-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Freitag, T. E., Toet, S., Ineson, P., and Prosser, J. I. (2010). Links between methane flux and transcriptional activities of methanogens and methane oxidizers in a blanket peat bog: methane flux and transcriptional activities in a peat bog. FEMS Microbiol. Ecol. 73, 157–165. doi: 10.1111/j.1574-6941.2010.00871.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Galand, P. E., Saarnio, S., Fritze, H., and Yrjälä, K. (2002). Depth related diversity of methanogen archaea in finnish oligotrophic fen. FEMS Microbiol. Ecol. 42, 441–449. doi: 10.1111/j.1574-6941.2002.tb01033.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gallert, C., and Winter, J. (2005). “Bacterial metabolism in wastewater treatment systems,” in Environmental Biotechnology: Concepts and Applications, eds H.-J. Jördening and J. Winter (Weinheim: Wiley-VCH), 1–48.
Garcia, J.-L., Patel, B. K., and Ollivier, B. (2000). Taxonomic, phylogenetic, and ecological diversity of methanogenic archaea. Anaerobe 6, 205–226. doi: 10.1006/anae.2000.0345
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goenrich, M., Duin, E. C., Mahlert, F., and Thauer, R. K. (2005). Temperature dependence of methyl-coenzyme M reductase activity and of the formation of the methyl-coenzyme M reductase red2 state induced by coenzyme B. JBIC J. Biol. Inorg. Chem. 10, 333–342. doi: 10.1007/s00775-005-0636-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guo, Y. Q., Liu, J.-X., Lu, Y., Zhu, W. Y., Denman, S. E., and McSweeney, C. S. (2008). Effect of tea saponin on methanogenesis, microbial community structure and expression of mcrA gene, in cultures of rumen micro-organisms. Lett. Appl. Microbiol. 47, 421–426. doi: 10.1111/j.1472-765X.2008.02459.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hales, B. A., Edwards, C., Ritchie, D. A., Hall, G., Pickup, R. W., and Saunders, J. R. (1996). Isolation and identification of methanogen-specific DNA from blanket bog peat by PCR amplification and sequence analysis. Appl. Environ. Microbiol. 62, 668–675.
Hallam, S. J., Girguis, P. R., Preston, C. M., Richardson, P. M., and DeLong, E. F. (2003). Identification of methyl coenzyme M reductase a (mcrA) genes associated with methane-oxidizing archaea. Appl. Environ. Microbiol. 69, 5483–5491. doi: 10.1128/AEM.69.9.5483-5491.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hedderich, R., and Whitman, W. B. (2013). “Physiology and biochemistry of the methane-producing archaea,” in The Prokaryotes, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Springer Berlin Heidelberg), 635–662.
Hills, D. J. (1979). Effects of carbon: nitrogen ratio on anaerobic digestion of dairy manure. Agric. wastes 1, 267–278. doi: 10.1016/0141-4607(79)90011-8
Hook, S. E., Wright, A.-D. G., and McBride, B. W. (2010). Methanogens: methane producers of the rumen and mitigation strategies. Archaea. 2010, 1–11. doi: 10.1155/2010/945785
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hori, T., Haruta, S., Ueno, Y., Ishii, M., and Igarashi, Y. (2006). Dynamic transition of a methanogenic population in response to the concentration of volatile fatty acids in a thermophilic anaerobic digester. Appl. Environ. Microbiol. 72, 1623–1630. doi: 10.1128/AEM.72.2.1623-1630.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ince, O. (1998). Performance of a two-phase anaerobic digestion system when treating dairy wastewater. Water Res. 32, 2707–2713. doi: 10.1016/S0043-1354(98)00036-0
Insam, H., Franke-Whittle, I., and Goberna, M. (2010). “Microbes in aerobic and anaerobic waste treatment,” in Microbes at Work, eds H. Insam, I. Franke-Whittle, and M. Goberna (Berlin: Springer), 1–34.
Juottonen, H., Galand, P. E., and Yrjälä, K. (2006). Detection of methanogenic archaea in peat: comparison of PCR primers targeting the mcrA gene. Res. Microbiol. 157, 914–921. doi: 10.1016/j.resmic.2006.08.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Juottonen, H., Tuittila, E.-S., Juutinen, S., Fritze, H., and Yrjälä, K. (2008). Seasonality of rDNA-and rRNA-derived archaeal communities and methanogenic potential in a boreal mire. ISME J. 2, 1157–1168. doi: 10.1038/ismej.2008.66
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kahnt, J., Buchenau, B., Mahlert, F., Krüger, M., Shima, S., and Thauer, R. K. (2007). Post-translational modifications in the active site region of methyl-coenzyme M reductase from methanogenic and methanotrophic archaea: methanogenic archaea methyl-coenzyme M reductase. FEBS J. 274, 4913–4921. doi: 10.1111/j.1742-4658.2007.06016.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kampmann, K., Ratering, S., Baumann, R., Schmidt, M., Zerr, W., and Schenell, S. (2012). Hydrogenotrophic methanogens dominate in biogas reactors fed with defined substrates. Syst. Appl. Microbiol. 35, 404–413. doi: 10.1016/j.syapm.2012.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaster, A.-K., Moll, J., Parey, K., and Thauer, R. K. (2011). Coupling of ferredoxin and heterodisulfide reduction via electron bifurcation in hydrogenotrophic methanogenic archaea. Proc. Natl. Acad. Sci. U.S.A. 108, 2981–2986. doi: 10.1073/pnas.1016761108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kasting, J. F., and Siefert, J. L. (2002). Life and the evolution of earth’s atmosphere. Science 296, 1066–1068. doi: 10.1126/science.1071184
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kendall, M., and Boone, D. (2006). “The order Methanosarcinales,” in The Prokaryotes, eds M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (New York: Springer), 244–256.
Kim, J., Park, C., Kim, T. H., Lee, M., Kim, S., Kim, S. W.,et al. (2003). Effects of various pretreatments for enhanced anaerobic digestion with waste activated sludge. J. Biosci. Bioeng. 95, 271–275. doi: 10.1016/S1389-1723(03)80028-2
Kim, M., Ahn, Y. H., and Speece, R. E. (2002). Comparative process stability and efficiency of anaerobic digestion; mesophilic vs. thermophilic. Water Res. 36, 4369–4385. doi: 10.1016/S0043-1354(02)00147-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, C., Kim, J., Hwang, K., O’Flaherty, V., and Hwang, S. (2009). Quantitative analysis of methanogenic community dynamics in three anaerobic batch digesters treating different wastewaters. Water Res. 43, 157–165. doi: 10.1016/j.watres.2008.09.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leigh, J. A., Albers, S.-V., Atomi, H., and Allers, T. (2011). Model organisms for genetics in the domain Archaea: methanogens, halophiles, Thermococcales and Sulfolobales. FEMS Microbiol. Rev. 35, 577–608. doi: 10.1111/j.1574-6976.2011.00265.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, L., Schoenhals, K. E., Brady, P. A., Estill, C. T., Perumbakkam, S., and Craig, A. M. (2012). Flaxseed supplementation decreases methanogenic gene abundance in the rumen of dairy cows. Animal 6, 1784–1787. doi: 10.1017/S175173111200078X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, W.-T., Chan, O.-C., and Fang, H. H. (2002a). Characterization of microbial community in granular sludge treating brewery wastewater. Water Res. 36, 1767–1775. doi: 10.1016/S0043-1354(01)00377-3
Liu, W.-T., Chan, O.-C., and Fang, H. H. (2002b). Microbial community dynamics during start-up of acidogenic anaerobic reactors. Water Res. 36, 3203–3210. doi: 10.1016/S0043-1354(02)00022-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, Y. (2010). “Taxonomy of methanogens,” in Handbook of Hydrocarbon and Lipid Microbiology, ed. K. N. Timmis (Berlin: Springer-Verlag), 550–558.
Liu, Y., and Whitman, W. B. (2008). Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann. N. Y. Acad. Sci. 1125, 171–189. doi: 10.1196/annals.1419.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lueders, T., Chin, K.-J., Conrad, R., and Friedrich, M. (2001). Molecular analyses of methyl-coenzyme M reductase a-subunit (mcrA) genes in rice field soil and enrichment cultures reveal the methanogenic phenotype of a novel archaeal lineage. Environ. Microbiol. 3, 194–204. doi: 10.1046/j.1462-2920.2001.00179.x
Luo, H.-W., Zhang, H., Suzuki, T., Hattori, S., and Kamagata, Y. (2002). Differential expression of methanogenesis genes of Methanothermobacter thermoautotrophicus (formerly Methanobacterium thermoautotrophicum) in pure culture and in cocultures with fatty acid-oxidizing syntrophs. Appl. Environ. Microbiol. 68, 1173–1179. doi: 10.1128/AEM.68.3.1173-1179.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luton, P. E., Wayne, J. M., Sharp, R. J., and Riley, P. W. (2002). The mcrA gene as an alternative to 16S rRNA in the phylogenetic analysis of methanogen populations in landfill. Microbiology 148, 3521–3530.
Lwin, K.-O., and Matsui, H. (2014). Comparative analysis of the methanogen diversity in horse and pony by using mcrA gene and archaeal 16S rRNA gene clone libraries. Archaea 2014, 1–10. doi: 10.1155/2014/483574
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lynd, L. R., Weimer, P. J., van Zyl, W. H., and Pretorius, I. S. (2002). Microbial cellulose utilization: fundamentals and biotechnology. Microbiol. Mol. Biol. Rev. 66, 506–577. doi: 10.1128/MMBR.66.3.506-577.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ma, J., Zhao, B., Frear, C., Zhao, Q., Yu, L., Li, X.,et al. (2013). Methanosarcina domination in anaerobic sequencing batch reactor at short hydraulic retention time. Bioresour. Technol. 137, 41–50. doi: 10.1016/j.biortech.2013.03.101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ma, K., Conrad, R., and Lu, Y. (2012). Responses of methanogen mcrA genes and their transcripts to an alternate dry/wet cycle of paddy field soil. Appl. Environ. Microbiol. 78, 445–454. doi: 10.1128/AEM.06934-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martí-Herrero, J. (2011). Reduced hydraulic retention times in low-cost tubular digesters: two issues. Biomass Bioenergy 35, 4481–4484. doi: 10.1016/j.biombioe.2011.07.020
McCartney, C. A., Bull, I. D., and Dewhurst, R. J. (2013a). Chemical markers for rumen methanogens and methanogenesis. Animal 7, 409–417. doi: 10.1017/S1751731113000694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McCartney, C. A., Bull, I. D., Waters, S. M., and Dewhurst, R. J. (2013b). Technical note: comparison of biomarker and molecular biological methods for estimating methanogen abundance. J. Anim. Sci. 91, 5724–5728. doi: 10.2527/jas.2013-6513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McMahon, K. D., Zheng, D., Stams, A. J. M., Mackie, R. I., and Raskin, L. (2004). Microbial population dynamics during start-up and overload conditions of anaerobic digesters treating municipal solid waste and sewage sludge. Biotechnol. Bioeng. 87, 823–834. doi: 10.1002/bit.20192
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Merkel, A. Y., Chernykh, N. A., Kanapatskii, T. A., and Pimenov, N. V. (2010). Detection of methanotrophic archaea in pockmark sediments (Gdansk Deep, Baltic Sea) by sequence analysis of the gene encoding the α subunit of methyl-coenzyme M reductase. Microbiology 79, 849–852. doi: 10.1134/S0026261710060196
Mihajlovski, A., Alric, M., and Brugère, J.-F. (2008). A putative new order of methanogenic Archaea inhabiting the human gut, as revealed by molecular analyses of the mcrA gene. Res. Microbiol. 159, 516–521. doi: 10.1016/j.resmic.2008.06.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mihajlovski, A., Doré, J., Levenez, F., Alric, M., and Brugère, J.-F. (2010). Molecular evaluation of the human gut methanogenic archaeal microbiota reveals an age-associated increase of the diversity. Environ. Microbiol. Rep. 2, 272–280. doi: 10.1111/j.1758-2229.2009.00116.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Montero, B., García-Morales, J. L., Sales, D., and Solera, R. (2009). Analysis of methanogenic activity in a thermophilic-dry anaerobic reactor: use of fluorescent in situ hybridization. Waste Manag. 29, 1144–1151. doi: 10.1016/j.wasman.2008.08.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Morris, R. (2011). Relating Methanogen Community Structure to Function in Anaerobic Wastewater Digesters. Degree of Doctor of Philosophy, Marquette University, Milwaukee, WI.
Morris, R., Schauer-Gimenez, A., Bhattad, U., Kearney, C., Struble, C. A., Zitomer, D.,et al. (2014). Methyl coenzyme M reductase (mcrA) gene abundance correlates with activity measurements of methanogenic H2/CO2 -enriched anaerobic biomass: mcrA genes and transcripts. Microbial Biotechnol. 7, 77–84. doi: 10.1111/1751-7915.12094
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Munk, B., Bauer, C., Gronauer, A., and Lebuhn, M. (2012). A metabolic quotient for methanogenic Archaea. Water Sci. Technol. 66, 2311–2317. doi: 10.2166/wst.2012.436
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Narihiro, T., and Sekiguchi, Y. (2011). Oligonucleotide primers, probes and molecular methods for the environmental monitoring of methanogenic archaea: probes for methanogens. Microbial Biotechnol. 4, 585–602. doi: 10.1111/j.1751-7915.2010.00239.x
Nelson, M. C., Morrison, M., and Yu, Z. (2011). A meta-analysis of the microbial diversity observed in anaerobic digesters. Bioresour. Technol. 102, 3730–3739. doi: 10.1016/j.biortech.2010.11.119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nettmann, E., Bergmann, I., Mundt, K., Linke, B., and Klocke, M. (2008). Archaea diversity within a commercial biogas plant utilizing herbal biomass determined by 16S rDNA and mcrA analysis. J. Appl. Microbiol. 105, 1835–1850. doi: 10.1111/j.1365-2672.2008.03949.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nölling, J., Pihl, T. D., Vriesema, A., and Reeve, J. N. (1995). Organization and growth phase-dependent transcription of methane genes in two regions of the Methanobacterium thermoautotrophicum genome. J. Bacteriol. 9, 2460–2468.
Nunoura, T., Oida, H., Miyazaki, J., Miyashita, A., Imachi, H., and Takai, K. (2008). Quantification of mcrA by fluorescent PCR in methanogenic and methanotrophic microbial communities. FEMS Microbiol. Ecol. 64, 240–247. doi: 10.1111/j.1574-6941.2008.00451.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ortega-Charleston, L. (2008). Hyperthermophilic Anaerobic Digestion of Food Waste. Ph.D. thesis, McGill University, Montreal.
Palacio-Molina, S. L., Oropeza-Navarro, R., and Balagurusamy, N. (2013). Quantitative analysis of mcrA trascripts and its correlation with methanogenic activity in two natural wetlands. Oral Presented at the II Congreso de Bioquímica y Biología Molecular de Bacterias, Cuatrocienegas, Coahuila.
Paul, K., Nonoh, J. O., Mikulski, L., and Brune, A. (2012). “Methanoplasmatales,” thermoplasmatales-related Archaea in termite guts and other environments, are the seventh order of methanogens. Appl. Environ. Microbiol. 78, 8245–8253. doi: 10.1128/AEM.02193-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pihl, T. D., Sharma, S., and Reeve, J. N. (1994). Growth phase-dependent transcription of the genes that encode the two methyl coenzyme M reductase isoenzymes and N5-methyltetrahydromethanopterin: coenzyme M methyltransferase in Methanobacterium thermoautotrophicum. J. Bacteriol. 176, 6384–6391.
Pramanik, P., and Kim, P. J. (2013). Effect of limited nickel availability on methane emission from EDTA treated soils: coenzyme M an alternative biomarker for methanogens. Chemosphere 90, 873–876. doi: 10.1016/j.chemosphere.2012.07.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rastogi, G., Ranade, D. R., Yeole, T. Y., Patole, M. S., and Shouche, Y. S. (2008). Investigation of methanogen population structure in biogas reactor by molecular characterization of methyl-coenzyme M reductase A (mcrA) genes. Bioresour. Technol. 99, 5317–5326. doi: 10.1016/j.biortech.2007.11.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Raynal, J., Delgenes, J. P., and Moletta, R. (1998). Two-phase anaerobic digestion of solid wastes by a multiple liquefaction reactors process. Bioresour. Technol. 65, 97–103. doi: 10.1016/S0960-8524(98)00009-1
Reeve, J. N., Nölling, J., Morgan, R. M., and Smith, D. R. (1997). Methanogenesis: genes, genomes, and who’s on first? J. Bacteriol. 179, 5975–5986.
Regueiro, L., Veiga, P., Figueroa, M., Alonso-Gutierrez, J., Stams, A. J. M., Lema, J. M.,et al. (2012). Relationship between microbial activity and microbial community structure in six full-scale anaerobic digesters. Microbiol. Res. 167, 581–589. doi: 10.1016/j.micres.2012.06.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ren, Z., Ward, T. E., Logan, B. E., and Regan, J. M. (2007). Characterization of the cellulolytic and hydrogen-producing activities of six mesophilic Clostridium species: clostridial hydrogen from cellulose. J. Appl. Microbiol. 103, 2258–2266. doi: 10.1111/j.1365-2672.2007.03477.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rouvière, P. E., and Wolfe, R. S. (1987). Use of subunits of the methylreductase protein for taxonomy of methanogenic bacteria. Arch. Microbiol. 148, 253–259. doi: 10.1007/BF00456701
Sakai, S., Imachi, H., Hanada, S., Ohashi, A., Harada, H., and Kamagata, Y. (2008). Methanocella paludicola gen. nov., sp. nov., a methane-producing archaeon, the first isolate of the lineage “Rice Cluster I”, and proposal of the new archaeal order Methanocellales ord. nov. Int. J. Syst. Evol. Microbiol. 58, 929–936. doi: 10.1099/ijs.0.65571-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scanlan, P. D., Shanahan, F., and Marchesi, J. R. (2008). Human methanogen diversity and incidence in healthy and diseased colonic groups using mcrA gene analysis. BMC Microbiol. 8:79. doi: 10.1186/1471-2180-8-79
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schink, B. (1997). Energetics of syntrophic cooperation in methanogenic degradation. Microbiol. Mol. Biol. Rev. 61, 262–280.
Scully, C., Collins, G., and O’Flaherty, V. (2005). Assessment of anaerobic wastewater treatment failure using terminal restriction fragment length polymorphism analysis. J. Appl. Microbiol. 99, 1463–1471. doi: 10.1111/j.1365-2672.2005.02743.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shigematsu, T., Tang, Y., Kobayashi, T., Kawaguchi, H., Morimura, S., and Kida, K. (2004). Effect of dilution rate on metabolic pathway shift between aceticlastic and nonaceticlastic methanogenesis in chemostat cultivation. Appl. Environ. Microbiol. 70, 4048–4052. doi: 10.1128/AEM.70.7.4048-4052.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shin, S. G., Han, G., Lim, J., Lee, C., and Hwang, S. (2010). A comprehensive microbial insight into two-stage anaerobic digestion of food waste-recycling wastewater. Water Res. 44, 4838–4849. doi: 10.1016/j.watres.2010.07.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sieber, J. R., McInerney, M. J., and Gunsalus, R. P. (2012). Genomic insights into syntrophy: the paradigm for anaerobic metabolic cooperation. Annu. Rev. Microbiol. 66, 429–452. doi: 10.1146/annurev-micro-090110-102844
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sirohi, S. K., Chaudhary, P. P., Singh, N., Singh, D., and Puniya, A. K. (2013). The 16S rRNA and mcrA gene based comparative diversity of methanogens in cattle fed on high fibre based diet. Gene 523, 161–166. doi: 10.1016/j.gene.2013.04.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sivakumaran, S., Balagurusamy, N., and Ramasamy, K. (1991). “Cellulase complex in biogas slurry,” in Biological Nitrogen Fixation and Biogas Technology, eds S. Kannaiyan, K. Ramasamy, K. Ilamuguru, and K. Kumar (Coimbatore: Tamil Nadu Agricultural University), 101–110.
Smith, K. S., and Ingram-Smith, C. (2007). Methanosaeta, the forgotten methanogen? Trends Microbiol. 15, 150–155. doi: 10.1016/j.tim.2007.02.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Speece, R. E. (1983). Anaerobic biotechnology for industrial wastewater treatment. Environ. Sci. Technol. 17, 416A–427A. doi: 10.1021/es00115a725
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Springer, E., Sachs, M. S., Woese, C. R., and Boone, D. R. (1995). Partial gene sequences for the a subunit of methyl-coenzyme M reductase (mcrI) as a phylogenetic tool for the family Methanosarcinaceae. Int. J. Syst. Bacteriol. 45, 554–559. doi: 10.1099/00207713-45-3-554
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steinberg, L. M., and Regan, J. M. (2008). Phylogenetic comparison of the methanogenic communities from an acidic, oligotrophic fen and an anaerobic digester treating municipal wastewater sludge. Appl. Environ. Microbiol. 74, 6663–6671. doi: 10.1128/AEM.00553-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steinberg, L. M., and Regan, J. M. (2009). mcrA-Targeted real-time quantitative PCR method to examine methanogen communities. Appl. Environ. Microbiol. 75, 4435–4442. doi: 10.1128/AEM.02858-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steinberg, L. M., and Regan, J. M. (2011). Response of lab-scale methanogenic reactors inoculated from different sources to organic loading rate shocks. Bioresour. Technol. 102, 8790–8798. doi: 10.1016/j.biortech.2011.07.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Supaphol, S., Jenkins, S. N., Intomo, P., Waite, I. S., and O’Donnell, A. G. (2011). Microbial community dynamics in mesophilic anaerobic co-digestion of mixed waste. Bioresour. Technol. 102, 4021–4027. doi: 10.1016/j.biortech.2010.11.124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Talbot, G., Topp, E., Palin, M. F., and Massé, D. I. (2008). Evaluation of molecular methods used for establishing the interactions and functions of microorganisms in anaerobic bioreactors. Water Res. 42, 513–537. doi: 10.1016/j.watres.2007.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tale, V. P., Maki, J. S., Struble, C. A., and Zitomer, D. H. (2011). Methanogen community structure-activity relationship and bioaugmentation of overloaded anaerobic digesters. Water Res. 45, 5249–5256. doi: 10.1016/j.watres.2011.07.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thauer, R. K., Kaster, A.-K., Seedorf, H., Buckel, W., and Hedderich, R. (2008). Methanogenic archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6, 579–591. doi: 10.1038/nrmicro1931
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Traversi, D., Villa, S., Acri, M., Pietrangeli, B., Degan, R., and Gilli, G. (2011). The role of different methanogen groups evaluated by Real-Time qPCR as high-efficiency bioindicators of wet anaerobic co-digestion of organic waste. AMB Express 1, 1–7.
Traversi, D., Villa, S., Lorenzi, E., Degan, R., and Gilli, G. (2012). Application of a real-time qPCR method to measure the methanogen concentration during anaerobic digestion as an indicator of biogas production capacity. J. Environ. Manage. 111, 173–177. doi: 10.1016/j.jenvman.2012.07.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ufnar, J. A., Wang, S. Y., Ufnar, D. F., and Ellender, R. D. (2007a). Methanobrevibacter ruminantium as an indicator of domesticated-ruminant fecal pollution in surface waters. Appl. Environ. Microbiol. 73, 7118–7121. doi: 10.1128/AEM.00911-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ufnar, J. A., Ufnar, D. F., Wang, S. Y., and Ellender, R. D. (2007b). Development of a swine-specific fecal pollution marker based on host differences in methanogen mcrA Genes. Appl. Environ. Microbiol. 73, 5209–5217. doi: 10.1128/AEM.00319-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wanapat, M., and Cherdthong, A. (2009). Use of real-time PCR technique in studying rumen cellulolytic bacteria population as affected by level of roughage in swamp buffalo. Curr. Microbiol. 58, 294–299. doi: 10.1007/s00284-008-9322-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, Y., Zhang, Y., Wang, J., and Meng, L. (2009). Effects of volatile fatty acid concentrations on methane yield and methanogenic bacteria. Biomass Bioenergy 33, 848–853. doi: 10.1016/j.biombioe.2009.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watanabe, T., Kimura, M., and Asakawa, S. (2009). Distinct members of a stable methanogenic archaeal community transcribe mcrA genes under flooded and drained conditions in Japanese paddy field soil. Soil Biol. Biochem. 41, 276–285. doi: 10.1016/j.soilbio.2008.10.025
Yang, C., Wang, W., Du, M., Li, C., Ma, C., and Xu, P. (2013). Pulp mill wastewater sediment reveals novel methanogenic and cellulolytic populations. Water Res. 47, 683–692. doi: 10.1016/j.watres.2012.10.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yen, H. W., and Brune, D. E. (2007). Anaerobic co-digestion of algal sludge and waste paper to produce methane. Bioresour. Technol. 98, 130–134.
Yu, Y., Lee, C., Kim, J., and Hwang, S. (2005). Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol. Bioeng. 89, 670–679. doi: 10.1002/bit.20347
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yuan, Y., Conrad, R., and Lu, Y. (2011). Transcriptional response of methanogen mcrA genes to oxygen exposure of rice field soil: transcriptional response of mcrA genes to aeration stress. Environ. Microbiol. Rep. 3, 320–328. doi: 10.1111/j.1758-2229.2010.00228.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, C., Yuan, Q., and Lu, Y. (2014). Inhibitory effects of ammonia on methanogen mcrA transcripts in anaerobic digester sludge. FEMS Microbiol. Ecol. 87, 368–377. doi: 10.1111/1574-6941.12229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, M., McAllister, T. A., and Guan, L. L. (2011). Molecular identification of rumen methanogens: technologies, advances and prospects. Animal Feed Science Technol. 166–167, 76–86. doi: 10.1016/j.anifeedsci.2011.04.005
Zhu, C., Zhang, J., Tang, Y., Zhengkai, X., and Song, R. (2011). Diversity of methanogenic archaea in a biogas reactor fed with swine feces as the mono-substrate by mcrA analysis. Microbiol. Res. 166, 27–35. doi: 10.1016/j.micres.2010.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: anaerobic digestion, microbial interactions, methanogens, mcrA gene expression, monitoring biodigesters
Citation: Alvarado A, Montañez-Hernández LE, Palacio-Molina SL, Oropeza-Navarro R, Luévanos-Escareño MP and Balagurusamy N (2014) Microbial trophic interactions and mcrA gene expression in monitoring of anaerobic digesters. Front. Microbiol. 5:597. doi: 10.3389/fmicb.2014.00597
Received: 30 August 2014; Accepted: 22 October 2014;
Published online: 12 November 2014.
Edited by:
Weiwen Zhang, Tianjin University, ChinaReviewed by:
Jeffrey Scott McLean, University of Washington, USAYingying Wang, Nankai University, China
Daniel Puyol, University of Queensland, Australia
Copyright © 2014 Alvarado, Montañez-Hernández, Palacio-Molina, Oropeza-Navarro, Luévanos-Escareño and Balagurusamy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nagamani Balagurusamy, Laboratorio de Biorremediación, Escuela de Ciencias Biológicas, Universidad Autónoma de Coahuila, Carretera Torreón-Matamoros Km. 7.5, Torreón, Coahuila 27000, México e-mail:Ym5hZ2FtYW5pQHVhZGVjLmVkdS5teA==