 Kelsey Roe
Kelsey Roe Sébastien Gibot2
Sébastien Gibot2 Saguna Verma
Saguna Verma- 1Department of Tropical Medicine, Medical Microbiology and Pharmacology, John A. Burns School of Medicine, University of Hawaii at Manoa, Honolulu, HI, USA
- 2Service de Réanimation Médicale, University Hospital of Nancy, Nancy, France
The triggering receptor expressed on myeloid cells (TREM) family of protein receptors is rapidly emerging as a critical regulator of a diverse array of cellular functions, including amplification of inflammation. Although the ligand(s) for TREM have not yet been fully identified, circumstantial evidence indicates that danger- and pathogen-associated molecular patterns (DAMPs and PAMPs) can induce cytokine production via TREM-1 activation. The discovery of novel functions of TREM, such as regulation of T-cell proliferation and activation of antigen-presenting cells, suggests a larger role of TREM proteins in modulation of host immune responses to microbial pathogens, such as bacteria and fungi. However, the significance of TREM signaling in innate immunity to virus infections and the underlying mechanisms remain largely unclear. The nature and intensity of innate immune responses, specifically production of type I interferon and inflammatory cytokines is a crucial event in dictating recovery vs. adverse outcomes from virus infections. In this review, we highlight the emerging roles of TREM-1, including synergy with classical pathogen recognition receptors. Based on the literature using viral PAMPs and other infectious disease models, we further discuss how TREM-1 may influence host-virus interactions and viral pathogenesis. A deeper conceptual understanding of the mechanisms associated with pathogenic and/or protective functions of TREM-1 in antiviral immunity is essential to develop novel therapeutic strategies for the control of virus infection by modulating innate immune signaling.
Introduction
The robust induction of innate immunity is the first line of defense against virus infections and depends on the ability of the host immune cells to detect invading pathogens and alert adaptive immune cells. Viral pathogens and replicative intermediates present pathogen-associated molecular patterns (PAMPs), such as viral double-stranded (ds) RNA, uncapped single-stranded (ss) RNA and viral proteins, which are detected by pattern recognition receptors (PRRs) expressed on immune cells. Typically, binding of virus PAMPs to PRRs namely Toll-like receptors (TLRs), RIG-I-like receptors (RLRs) and NOD-like receptors (NLRs) cause activation of downstream signaling resulting in the production of antiviral type I interferon (IFN) and pro-inflammatory cytokines (Kumar et al., 2011). A timely production of IFN-α/β and pro-inflammatory cytokines is critical for the clearance of the virus during early phase of the infection and fine-tuning the innate-adaptive interface for long-lasting protection. The activation of these PRRs is very tightly regulated at multiple steps to prevent tissue damage due to uncontrolled production of cytokines. Thus, overall virus disease outcome depends on the magnitude and the nature of innate immune events triggered following virus entry. Understanding the host factors that participate in positive or negative regulation of innate immunity to virus infections is currently the focus of intense research to seek out cues for designing therapeutics to modulate inflammation at the molecular level. Several novel innate immune molecules, including members of triggering receptors expressed on myeloid cells (TREM) family of receptors, have been recently characterized to play an important role in modulating the intensity of innate immune responses.
Since the discovery of TREM in 2000, they have been described as critical immunomodulators in several inflammatory disorders of both infectious and non-infectious etiology (Ford and McVicar, 2009). TREM-1 is the most well-characterized member of the TREM family and plays an important role in the amplification of inflammation, crosstalk with other PRR pathways and activation of antigen-presenting cells (APC). A substantial amount of data supports the link between TREM-1 signaling and several diseases, such as polymicrobial septic shock and inflammatory bowel disease, and in animal models of pneumonia and asthma (Ford and McVicar, 2009; Klesney-Tait et al., 2012). Although the data on the functions of TREM-1 in virus infections is limited, in vitro studies using viral PAMPs suggest the ability of viruses to modulate TREM signaling. In this review, we first discuss the current understanding of the immunoregulatory functions of the TREM family, in particular TREM-1, and then highlight potential roles of TREM-1 in antiviral immunity.
TREM Family of Receptors
Members of the TREM family (designated TREM-1 to TREM-4) belong to the immunoglobulin variable (IgV) domain receptor superfamily of proteins (Bouchon et al., 2000). They are cell surface activating receptors with a transmembrane region containing charged lysine residues and a short cytoplasmic tail lacking signaling motifs (Bouchon et al., 2000). Signaling through these receptors is facilitated by the adaptor protein DNAX-activating protein of 12 kDa (DAP12) (Bouchon et al., 2000). TREM-1 was designated as CD354 by the Ninth Workshop on Human Leukocyte Differentiation Antigens in 2011 (Matesanz-Isabel et al., 2011).
The human TREM family members, which share low sequence homology with each other, are located on chromosome 6p21.1 (Radaev et al., 2003; Kelker et al., 2004b) and cluster with the related NKp44 receptor (Allcock et al., 2003). Several related receptors, such as the TREM-like transcripts (TLT) 1-5 also map to this region of the human genome and share the V-domain of the Ig superfamily but unlike TREM, they also contain an immunoreceptor tyrosine inhibitory motif in their cytoplasmic tail (Allcock et al., 2003). In the laboratory mouse, the TREM genes map to chromosome 17C3 (Chung et al., 2002; Watarai et al., 2008; Paradowska-Gorycka and Jurkowska, 2013). TREM-3, which is not found in humans and shares 43% sequence similarity with TREM-1, is located directly next to TREM-1 in the mouse genome, and is predicted to be functionally similar to TREM-1 (Chung et al., 2002; Radaev et al., 2003). pDC-TREM (also called TREM-4 and not expressed in human cells) is present on mature mouse plasmacytoid dendritic cells (pDCs) and share ~20% amino acid homology with TREM-1 and TREM-2 (Watarai et al., 2008). Two independent groups have solved the structure of the extracellular IgV domain of TREM-1. Although, the results have been conflicting, both groups have validated that TREM-1 belongs to the Ig superfamily (Radaev et al., 2003; Kelker et al., 2004a,b).
Cellular Localization
TREM-1 and TREM-2 were first identified on lipopolysaccharide (LPS)-stimulated monocytes and neutrophils (Bouchon et al., 2000), following which the initial search for TREMs was mostly limited to immune cells. The types of human immune cells expressing high levels of TREM-1 includes monocytes, neutrophils, granulocytes, DCs and natural killer (NK) cells, and low level expression on T cells and all subsets of B cells (Allcock et al., 2003; Matesanz-Isabel et al., 2011; Rigo et al., 2011). However, more recent studies have characterized the expression of TREM family members in several human and mouse non-immune cells and tissues. Studies describing the presence of TREMs in different cell types and species are summarized in Table 1. The presence of TREM-1 is now reported in varying human and mouse non-immune cells such as epithelial cells and fibroblasts, and in tissues such as lymph nodes, spinal cord, lung, heart, and placenta (Gingras et al., 2002; Chen et al., 2008; Wu et al., 2012; Zangi et al., 2012). TREM-2 has the widest range of expression so far and includes myeloid cells, osteoclasts and microglia, and tissues such as kidney, liver, heart, brain, and lung (Schmid et al., 2002; Paloneva et al., 2003). As seen in Table 1, the literature on the expression patterns of other TREM and TLT family members is limited and mostly reports presence on mouse immune cells. However, this expanding list of cell types and species is likely to continue growing as further research on TREMs is undertaken.
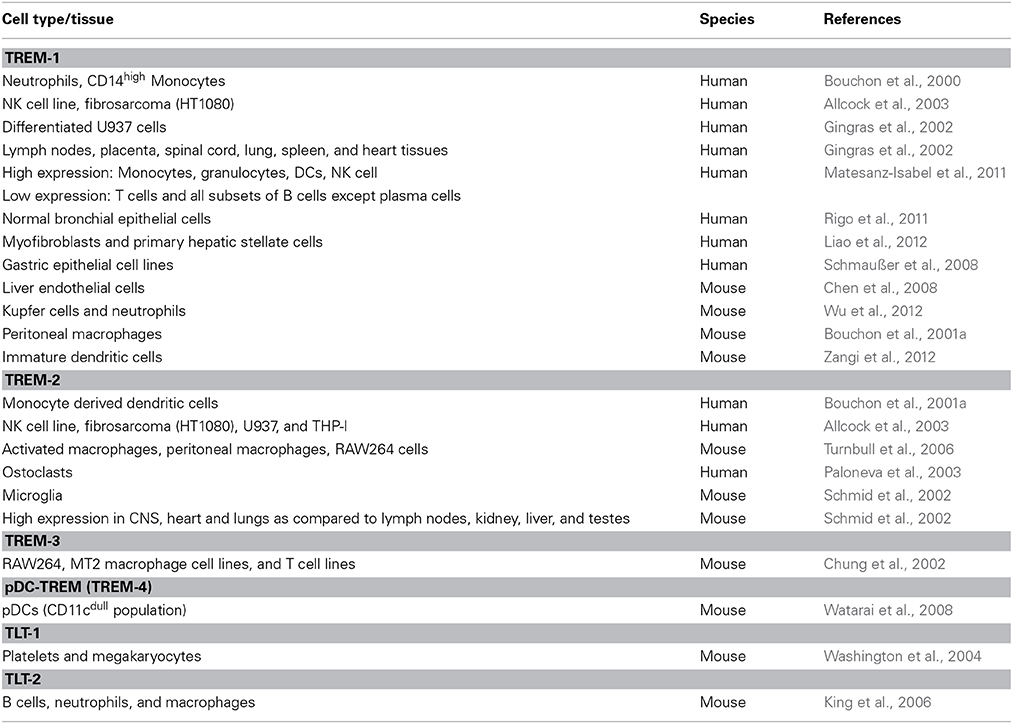
Table 1. Cellular expression profiles of the TREM family proteins.
Soluble TREM as Marker of Disease Severity
Although TREM family members are typically membrane-bound receptors, multiple members are now reported to exist in a soluble form in the clinical samples of patients from several inflammatory conditions. So far, soluble forms of TREM-1, TREM-2, and TLT-1 have been identified (Klesney-Tait et al., 2006). However, it is possible that other members of the family might also be recognized in this form. The soluble form of TREM-1, a 27 kDa glycosylated peptide, is most likely produced by the cleavage of the extracellular ectodomain of the membrane-bound form by matrix metalloproteinases (Gómez-Piña et al., 2007). Using in vitro time-course analysis, Gómez-Piña and colleagues demonstrated that increased levels of sTREM-1 correlated with decreased cell surface TREM-1 expression after 6 h of LPS stimulation of CD14+ monocytes (Gómez-Piña et al., 2007). It remains probable, however, that sTREM-1 may be produced through alternate pathways, such as alternate spicing (Gingras et al., 2002).
In clinical conditions, sTREM was first identified in the plasma of patients with sepsis (Gibot et al., 2004c) and in bronchial lavage specimens of pneumonia patients (Gibot et al., 2004a). Elevated levels of sTREM-1 have now been found in multiple infectious and chronic inflammatory diseases such as pneumonia, pleural effusion, intra-abdominal infections, inflammatory bowel disorders, inflammatory rheumatoid disorders, and lung cancer as reviewed previously (Barraud and Gibot, 2011). In addition to the serum, two studies reported increased sTREM-1 levels in cerebral spinal fluid (CSF) of bacterial meningitis patients implying that it may be a marker of differentiating bacterial vs. non-bacterial meningitis (Determann et al., 2006; Bishara et al., 2007). Although the sample size of non-bacterial cases was very small as compared to bacterial meningitis cases, nonetheless, these studies emphasize that TREM-1 signaling may be modulated during CNS infections. Initial interpretations of these data led to the proposal of developing sTREM-1 as a biomarker for the diagnosis of acute inflammatory diseases, such as septic shock. However, this observation was not supported by subsequent studies and is now considered as a possible indicator of increased severity of the disease (Determann et al., 2006; Bucova et al., 2012). At present, the function of sTREM is unknown, but based on the data on other soluble forms of membrane receptors such as ICAM-1 and VCAM-1 (Page and Liles, 2013), it is possible that sTREM-1 and sTREM-2 may negatively regulate TREM receptor signaling via neutralization of the respective ligands.
TREM Ligands
Identification of the ligands for any receptor is a crucial step in establishing a link between a signaling receptor and disease pathogenesis. The search for TREM family ligands has been elusive, although multiple putative ligands have been proposed. Haselmayer and co-workers identified a membrane-bound ligand for TREM-1 on the surface of platelets, which upon interacting with TREM-1, amplified LPS-induced neutrophil activation (Haselmayer et al., 2007). Evidence for the presence of a soluble ligand for TREM-1 comes from a study showing that exposure of human monocytes to the serum of septic patients and LPS increased production of tumor necrosis factor-α (TNF-α), which was blocked by TREM-1/Fc fusion peptide (Wong-Baeza et al., 2006). Moreover, a few DAMPs and PAMP are described as possible TREM-1 ligands, indicating that TREM-1 could act as a PRR in a similar capacity as TLRs. Mohamadzadeh and colleagues used a viral replicon system to demonstrate that the surface glycoprotein, but not the nuclear protein, of Marburg virus was able to bind to a TREM-1/Fc fusion peptide (Mohamadzadeh et al., 2006). In the list of potential DAMPs, two independent studies have proposed high-mobility group box 1 protein (HMGB1) or heat shock protein 70 (HSP70) as possible ligands for TREM-1 (El Mezayen et al., 2007; Wu et al., 2012). HMGB1 and HSP70 present in the necrotic cell lysates of myeloid cells were responsible for significant induction of the proinflammatory cytokine expression, which was reduced by blocking TREM-1, thus confirming the role of TREM-1 in cytokine expression cascade via responding to these endogenous DAMPs. Collectively, these data suggest that TREM-1 may not have a single ligand, as most activating receptors do, but might recognize multiple epitopes and bind to a range of viral ligands.
TREM-1 Signaling and Inflammation
Activation of TREM-1 signaling is initiated upon binding of the ligand to the receptor, which triggers the association and phosphorylation of immunoreceptor tyrosine-based activation motif of the adaptor protein DAP12. The signaling pathways triggered downstream to DAP12 phosphorylation are very specific to the TREM family member. In vitro studies in relevant cell types such as neutrophils and macrophages demonstrate that phosphorylation of DAP12 by Src family kinases results in the recruitment and activation of non-receptor tyrosine kinase Syk. The Syk, in turn, activates the downstream signaling molecules including PI3K, PLCγ, ERK1/2, and MAP kinases that regulate NF-κB activation and expression of inflammatory genes in a cell-specific manner (Bouchon et al., 2000; Tessarz and Cerwenka, 2008; Ford and McVicar, 2009). Although the exact pathway remains unclear, TREM-1 signaling via phosphorylation of Syk also regulates calcium influx, which further activates MAPK/ERK pathway (Arts et al., 2013). In neutrophils, TREM-1 also regulates neutrophil degranulation and production of reactive oxygen species in addition to cytokines and chemokines (Bouchon et al., 2001a; Radsak et al., 2004; Haselmayer et al., 2007) On the other hand, activation of DAP12 by TREM-2 is shown to promote anti-inflammatory response in a cell-specific manner (Paradowska-Gorycka and Jurkowska, 2013). In contrast to TREM-1, TREM-2 signaling does not involve NF-κB translocation and is shown to induce DAP12-dependent calcium influx followed by activation of ERK and PI3K (Bouchon et al., 2001b). Although the overlap and cross-talk between TREM-1 and TREM-2 signaling pathway is not yet clear, several studies demonstrating downregulation of cytokines such as TNF-α by TREM-2 (Takahashi et al., 2005) emphasize opposing physiological roles of these two receptors.
Amplification of inflammation is the best-characterized function attributed to TREM-1. Because of the absence of a well-characterized ligand, an agonist antibody to TREM-1 is routinely used to over activate TREM-1 signaling. Bouchon and colleagues first showed that the key functional outcome of the artificial over-activation of TREM-1 receptor in monocytes was the increased production of cytokines, such as TNF-α, monocyte chemoattractant protein 1 (MCP-1) and interleukin-1β (IL-1β) following LPS treatment (Bouchon et al., 2000). Following this observation, several studies demonstrated the ability of TREM-1 to amplify inflammation during septic shock. Pharmacological inhibition of TREM-1 by the use of synthetic peptides or fusion protein repeatedly prevented hyper-responsiveness and death during various experimental septic shock models: endotoxemia in mice or monkeys (Bouchon et al., 2001a; Derive et al., 2014), bacterial pneumonia in rats (Gibot et al., 2006), polymicrobial peritonitis in rodents and pigs (Gibot et al., 2004b; Derive et al., 2013). By contrast, genetic invalidation of TREM-1 leads to contrasting results: while some report a decreased bacterial clearance and survival during Pneumococcal pneumonia and Klebsiella pneumonia liver abscesses in mice (Hommes et al., 2014; Lin et al., 2014), the opposite has been described in the setting of Leishmania major infection (Weber et al., 2014). Further, using siRNA silencing of TREM-1 in the mouse, Gibot and colleagues demonstrated the importance of the balanced activation of TREM-1 signaling during sepsis. This study showed that partial silencing of TREM-1 was protective during peritonitis, while complete silencing was lethal to septic mice (Gibot et al., 2007). This effect of TREM-1-dependent enhancement of inflammatory response is also observed in non-infectious disease models including hemorrhagic shock and pancreatitis (acute inflammation) and chronic inflammatory bowel diseases and inflammatory arthritis (Barraud and Gibot, 2011). TREM-1-deficient mice displayed significantly attenuated disease that was associated with reduced inflammatory infiltrates and diminished expression of pro-inflammatory cytokines, thus representing an attractive target for treatment of chronic inflammatory disorders (Weber et al., 2014). Such data are significant in suggesting that TREM-1 is not simply an inflammatory amplifier, but also plays a regulatory role in influencing the disease outcome.
It is now established that for the culmination of balanced and effective innate immunity, it is crucial to have crosstalk between multiple innate immune signaling pathways. Many recent studies support a model of synergy between TREM-1 and other PRRs, although the precise mechanisms are yet unclear. Available data indicates that the synergy between TLRs and TREMs might be at two levels. First, is the ability of TLR ligands to increase the mRNA expression of TREM-1, and second, the amplification of TLR-induced inflammatory response by TREM-1. Exposure of immune cells to several PAMPs, such as LPS (a TLR-4 ligand) and lipoteichoic acid (a TLR-2 ligand), and microbial pathogens such as Pseudomonas aeruginosa have shown to increase TREM-1 mRNA (Bleharski et al., 2003; Zeng et al., 2007; Zheng et al., 2010). In a TLR-2 dependent manner, soluble fungal antigens were shown to up-regulate the expression of TREM-1 transcripts in macrophages (Buckland et al., 2011). Similarly, expression of TREM-1 mRNA following activation of macrophages by LPS was dependent on the TLR-4/NF-κ B pathway (Zeng et al., 2007). The reverse was not true, TREM-1 signaling had no effect on TLR-4 expression. The mechanisms by which TREM-1 amplifies TLR-initiated inflammation are still being investigated. However, available data suggest that the synergy is at the level of NF-κB and IRAK-1 and appears to be cell-specific (Arts et al., 2013). Activation of TREM-1 using agonistic monoclonal antibody in combination with the ligands for TLR-4 was shown to synergistically amplify the production of proinflammatory cytokines in monocytes (Bouchon et al., 2000). More recent studies by Ornatowska and colleagues used pathway-specific microarray analysis to show that TREM-1 silencing did not alter expression of TLR-4, but reduced the expression of adaptor protein Myd88 and cytokines such as IL-1b and IL-10, thus emphasizing that regulating expression of downstream signaling molecules may be one of the mechanisms of TREM-1/TLR cross talk (Ornatowska et al., 2007). Likewise, Hu and group further support the role of MyD88 protein as the point of cross talk between TREM-1 and TLR-4 signaling in the infection of corneal epithelial cells with fungi Aspergillus fumigatus (Hu et al., 2014). Additionally, artificial activation of TREM-1 is also shown to down-regulate expression of Tollip and ST2, negative regulators of TLR-2 and TLR-4 pathways (Lagler et al., 2009; Wu et al., 2011) These studies collectively emphasize the fact that cross talk between TLRs and TREM-1 is at multiple levels.
Most of the TREM research so far has focused on non-viral infections and autoimmune diseases. However, characterization of several additional roles of TREM-1 such as modulation of T-cell proliferation and APC activation clearly argues for its crucial immunomodulatory role in virus infections. Below we present essential elements of antiviral immune responses and then discuss how TREM-1 signaling fits into these immune events based on the current literature on TREMs in innate-adaptive interface.
Antiviral Immunity
Immune responses to virus infections are as diverse and complex as the viruses that induce them. However, there are specific events shared by many viruses, which are important determinants of virus clearance vs. immunopathology. An important feature of an efficient innate response to the entry of both DNA viruses, such as herpes simplex virus and cytomegalovirus, and RNA viruses, including Influenza A virus, West Nile virus (WNV) and chikungunya virus (CHIKV), is the rapid detection by PRRs, notably endosomal TLRs (TLR-3, TLR-7/8 and TLR-9), RLRs (RIG-I and MDA5) and the NLRs (NLRP3 and NOD2) (Lund et al., 2004; Wang et al., 2004; Varani et al., 2007; Zhang et al., 2007; Daffis et al., 2008; Kumar et al., 2013). The production of inflammatory cytokines is one of the hallmarks of PRR activation in innate immune cells including dendritic cells (DCs), and is required for the recruitment and activation of inflammatory cells such as macrophages, NK cells and neutrophils to the site of infection (Saitoh et al., 2012; Jenne et al., 2013). The profile of cytokines produced by innate immune cells dictates the adaptive immune response and the virus disease outcome. Unlike bacterial infections, another major innate immune response to virus infection is the production of type I IFN. The paracrine and autocrine secretion of IFN renders cells “antiviral” by inducing several interferon-stimulated genes (ISGs). These ISGs confer an antiviral state by blocking virus replication at different levels such as early-stage virus infection, inhibition of post-transcriptional modification and virus maturation, activation of macrophages and DC and stimulation of NK cells to kill virus-infected cells.
As an immediate effect of innate immune activation, effector cells such as NK cells and CD8+ T cells are recruited at the site of infection. These cells act to kill virus-infected cells and macrophages clear the resulting debris. Further, depending on the cytokines induced by the APCs, different types of T helper cell responses are induced. Recruited CD4+ T cells progress toward a TH1 phenotype in most viral infections, eventually leading to the induction of several components of adaptive immunity. However, viruses such as human immunodeficiency virus type 1 (HIV-1), HSV and hepatitis C virus (HCV) also drive TH17 and T regulatory cells (TREG) cell expansion (Rouse and Sehrawat, 2010). Influenza virus is another example where TH17 cell responses mediate recruitment of neutrophils, which then become responsible for the associated lung pathology (Bermejo-Martin et al., 2009). Humoral immunity provided by specific neutralizing antibodies is also an essential component of the adaptive immune response to virus infection that inhibits virus attachment, internalization and protection against subsequent infection. At the later stages of infection, resolution of inflammation and the return to homeostasis is mediated by anti-inflammatory components, classically TREG and the cytokines IL-10 and TGF-β, to prevent tissue damage after virus clearance. Viruses, such as HSV, HCV, and HIV use the strategy of increased TREG functions to facilitate persistent infection (Veiga-Parga et al., 2013). Thus, induction of an effective and balanced innate immune response is an important determinant of virus disease outcome and is fine-tuned by multiple immune components. Understanding the specific mechanisms associated with the fine control of innate immune signaling pathways has been greatly enhanced because of the identification of several novel host molecules involved with either blocking or facilitating the synergy between important immune signaling pathways. TREMs family of proteins represents this class of novel innate immune molecules, which can influence the innate immune responses to viruses. The potential roles of TREM-1 in the different arms of viral immunity are discussed in the sections below.
Activation of TREM-1 Signaling by Viruses
In 2006, Mohamadzadeh and colleagues first reported activation of TREM-1 signaling in filovirus-infected neutrophils. Although Marburg and Ebola viruses do not replicate in primary human neutrophils, they increased TREM-1 expression following internalization, which correlated with phosphorylation of DAP12 and ERK1/2. Additionally, this study also suggested that the surface glycoprotein (GP) of filoviruses may act as a ligand for TREM-1 (Mohamadzadeh et al., 2006). In the clinical scenario, increase in the neutrophils has been reported during the human Ebola disease (Martini, 1973; Fisher-Hoch et al., 1985). Further, in vitro studies have demonstrated that the soluble variant of Ebola GP can interact with neutrophils (Yang et al., 1998). Therefore, it is possible that the interaction between TREM-1 on neutrophils with the GP protein during infection contributes to the “cytokine storm” associated with lethal filovirus disease (Wauquier et al., 2010). Similarly, exposure of PBMC to the gp41 protein of HIV-1 has been shown to up-regulate the mRNA expression of TREM-1 (Denner et al., 2013). Another recent study by Suthar and co-workers used transcriptional profiling and pathway modeling to show that TREM-1 signaling was enriched in the liver following infection with WNV in mice (Suthar et al., 2013). Although this study implies that TREM-1 signaling might be one of the pathways responsible for restricting tissue tropism, the precise role of TREM-1 in WNV pathogenesis has not been explored.
However, indirect evidence supports the ability of viruses to induce TREMs. Bleharski and colleagues first showed that poly (I:C), a ligand for TLR-3, can induce the transcription of TREM-1 in primary monocytes (Bleharski et al., 2003). Later, studies by Begum and co-workers could not validate the increase of TREM-1 mRNA following stimulation of monocytes with poly (I:C), however the reason for this discrepancy might be the different time points of analysis, 6 h after stimulation, as compared to 24 h time point used in the previous study (Begum et al., 2004). Watarai and colleagues demonstrated that TREM-4 mRNA expression increased in mouse pDCs following TLR-7 or -9 activation and led to DAP12-phosphorylation, activation of ERK1/2 signaling and ultimately IFN-α secretion (Watarai et al., 2008). Although such studies are limited, they strongly suggest that diverse RNA and DNA viruses that produce poly (I:C) and CpG DNA may be capable of inducing TREMs.
Similarly, our understanding of whether viruses can induce production of sTREM-1 is unclear and so far comes from only one clinical study. Ruiz in-Pacheco and colleagues recently demonstrated increased levels of sTREM-1 in the serum of dengue virus (DENV)-infected patients during the early stages of infection (first 5 days) as compared to healthy individuals (Ruiz-Pacheco et al., 2014). At this point, one can only speculate the role of sTREM in pathogenesis of DENV, an important global human pathogen, however, this is an important finding and provides direct evidence of modulation of TREM-1 in response to virus infection. Elevated levels of sTREMs could signify either a virus-induced compensatory mechanism to counteract inflammatory process, or a host-induced mechanism to control tissue damage by attenuating downstream inflammatory signals. Therefore, such clinical studies will be highly relevant to identify the potential of sTREM-1 as a marker of disease severity in acute virus infections as with influenza virus and CHIKV.
Impact of TREM-1 on Virus-Associated Inflammation
The function of TREM-1 in modulating virus-associated inflammation appears to be supported more by in vitro studies using viral PAMPs, than actual virus infections (Figure 1). Activation of TREM-1 by TLR-9 ligand CpG DNA enhanced TNF-α production in mouse bone marrow-derived dendritic cells (BMDCs) (Hara et al., 2007). Similarly, Netea and colleagues noted increased production of TNF-α in TREM-1-activated human PBMC following stimulation with poly (I:C) and CpG (Netea et al., 2006). This was also the first study to indicate that TREM-1 synergizes with NLR pathways. NLR ligands amplified the production of TNF-α, IL-1β, and IL-6 when TREM-1 signaling was activated (Netea et al., 2006). Similarly, TLR-TREM synergistic activation of neutrophils is not restricted to TLR-2 and TLR-4 but also occurs with TLR-7 and TLR-8, which are common TLRs responding to virus PAMPs (Radsak et al., 2004). Filoviruses are the only viruses, where the function of TREM-1 in regulating production of pro-inflammatory cytokines such as TNF-α and IL-1β is documented (Mohamadzadeh et al., 2006). The impact of TREM-1 in regulating inflammation in other virus diseases awaits discovery. Comprehensive analysis of virus infections in vitro as well as in TREM-1 deficient mice will be required before we fully understand the cooperation between TREM-1 and other viral PRRs in the context of the virus-associated inflammation.
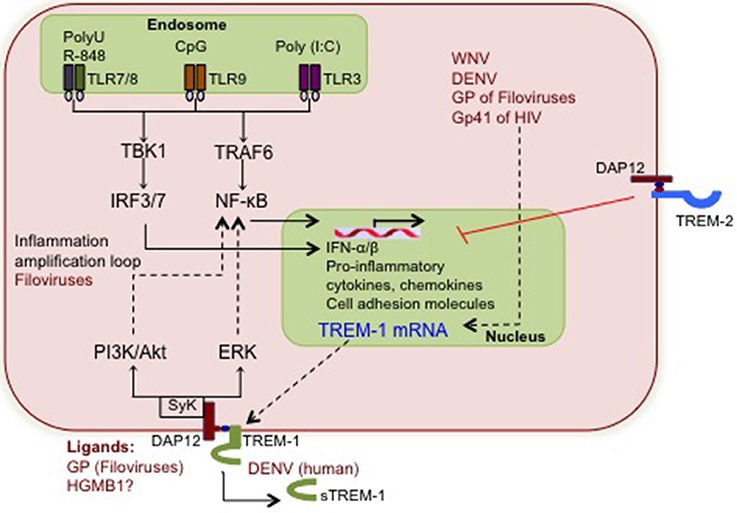
Figure 1. The putative interactions between viruses and TREM-1 signaling. During early stages of infection, viral nucleic acids and some proteins are detected by TLR-3, TLR-7/8 and TLR-9, which induce the mRNA production of pro-inflammatory cytokines, chemokines, and cell surface receptors including TREM-1. Viruses shown to increase TREM-1 mRNA and/or soluble TREM-1 levels are depicted in red, although the associated pathways are not clear. Increased TREM-1 receptor responds to yet uncharacterized viral or putative host ligands and activates signaling via DAP12 and Syk tyrosine kinase. Downstream PI3K and ERK signaling further activate NF-κ B and synergizes with the TLR cascade to amplify inflammation. The potential interactions between virus-induced TREM-1 and other cellular pathways such as type I IFN and apoptosis are not yet defined.
Modulation of Type I IFN by TREM-1
The ability of the innate immune cells to rapidly produce type I IFN is one of the major determinants of the virus disease outcome. However, its role in other non-viral disease models, including bacterial infections, shock, and autoimmunity is less well-defined and complicated. With the emphasis of TREM-1 studies mainly in bacterial infection models, the role of TREM-1 in the regulation of type I IFNs has not been investigated so far. However, other TREM members have been shown to positively or negatively regulate type I IFN responses. In plasmacytoid DCs, stimulation of TREM-4 resulted in increased IFN-α secretion, and was dependent on the phosphorylation of DAP12 and PI3K and ERK1/2 pathways (Watarai et al., 2008). Conversely, mouse BMDCs deficient in TREM-2 had increased transcriptional levels of type I IFN following TLR-9 activation (Ito and Hamerman, 2012). Therefore, the fact that TREM-1 can synergize with TLR-3 and TLR-7 and signal through the ERK1/2 pathway leads us to speculate that TREM-1 might play a role in positively regulating type I IFN levels.
Role of TREM-1 in APC Activation, Migration, and T-Cell Priming
The availability of recently developed TREM-1-deficient mice have led to studies describing several novel functions of TREM family members in addition to amplifying inflammation. Wu and colleagues developed a TREM-1 knockout (KO) mouse and demonstrated that TREM-1 was essential for the activation of Kupffer cells in the diethylnitrosamine model of hepatocellular carcinoma and contributed to chronic liver damage. TREM-1 KO macrophages were not as responsive as WT to signals from necrotic hepatocytes and exhibited attenuated APC responses including reduced production of IL-1β, IL-6, TNF-α, and CCL2 (Wu et al., 2012). In line with this, the role of TREM-1 in leukocyte recruitment is clearly demonstrated by Klesney-Tait and co-workers who used a double knockout TREM-1/3 mice for Pseudomons aeruginosa infection (Klesney-Tait et al., 2012). TREM-1/3 deficiency increased mortality of infected mice, which correlated with higher local and systemic cytokine production. However, although TREM-1/3-deficient neutrophils had intact bacterial killing and chemotaxis properties, histologic examination of TREM-1/3-deficient lungs revealed decreased neutrophil infiltration of the airways (Klesney-Tait et al., 2012).
Another novel function attributed to TREM-1 recently is its ability to influence the differentiation of primary monocytes into immature DCs. TREM-1 governs the upregulation of the surface expression of CD86 and MHC class II, rendering them more efficient at eliciting T-cell proliferative activity (Bleharski et al., 2003). Likewise, it has been demonstrated that signaling through TREM-1 on hypoxic iDCs up-regulates T cell co-stimulatory molecules, including CD83, CD86, and HLA-DR. Co-culture of these hypoxic iDCs with T-cells resulted in increased the cell proliferation and IFN-γ and IL-17 production (Pierobon et al., 2013). However, alternatively, Ito and Hamerman used TREM-2 deficient BMDCs to demonstrate that TREM-2 inhibited TLR-induced DC maturation and antigen presentation to T cells (Ito and Hamerman, 2012) thus supporting the dynamic opposite roles of TREM-1 and TREM-2 at the level of antigen presentation as well.
APC activation is essential during virus infection for the stimulation of TH1 responses and subsequent development of neutralizing antibodies vital for viral clearance. In addition to APC activation, TREM signaling also can skew T-cell responses in a TH1 or TH17 direction. Pierbon and colleagues, for instance showed that TREM-1 signaling in hypoxic iDCs was responsible for TH1/TH17 priming (Pierobon et al., 2013), while two independent studies showed a reduction in systemic TH1 responses following inhibition of TREM-1. In a model of Pseudomonas aeruginosa-induced keratitis, inhibition of TREM-1 reduced IFN-γ responses (TH1 phenotype), while increasing TH2 cytokines including IL-4 and -5 (Wu et al., 2011). Similar results were obtained in a model of cardiac allografts, in which TREM-1 inhibition led to increased allograft survival by dampening the differentiation and proliferation of IFN-γ secreting CD4+ T cells (Schiechl et al., 2013). In addition, TREM-1 signaling can be influenced by a TH1 environment; treatment of primary human NK cells with the classic TH1 cytokines IL-12 and -18 led to the activation of TREM-1 signaling events, as measured by microarray analysis (Grangeiro de Carvalho et al., 2011). Currently, there is no evidence supporting the hypothesis that TREM-1 may modulate innate-adaptive immune interface in virus infections. However, because viral ligands can activate TREM-1 signaling and cytokines production, there is high likely hood that cytokines governed by TREM-1 may influence activation of APCs, which in turn may impact protective T-cell functions and viral clearance.
Return to Homeostasis
Anti-inflammatory reactions are important in any immune reaction to return the host to homeostasis. In virus infections, one of the factors that determine the balance between virus clearance and tissue damage is the timely induction of anti-inflammatory molecules. Available data indicate that TGF-β and IL-10 treatment of primary monocytes synergistically down-regulates cell surface expression of TREM-1 (Schenk et al., 2005), but whether TREM-1 signaling can influence production of TGF-β and IL-10 has not been clearly defined. Nonetheless, this study strongly supports the notion that TREM-1 is the target of anti-inflammatory cytokines and that attenuating TREM-1- associated information might be one of the events in the immune homeostasis process. In microglia, TREM-2 participates in the process of tissue debris clearance and resolution of latent inflammatory reactions in Nasu-Hakola disease, a recessively inherited chronic neurodegenerative disease (Colonna, 2003). Similar anti-inflammatory and protective functions of TREM-2 have been proposed for other acute neuroinflammatory diseases, such as multiple sclerosis (Piccio et al., 2007) and Alzheimer's disease (Guerreiro et al., 2013; Jonsson et al., 2013), implying that alterations in the immune homeostasis may be mediated by TREMs in infections with neurotropic viruses, such as HIV, WNV or tick-borne encephalitis virus (TBEV).
Conclusions and Future Perspectives
The immune functions of the TREM family are broad and diverse (Table 2) and may contribute to antiviral immunity. One of the pertinent questions is, can TREM-1 signaling respond to infection with globally important human viruses? Of particular interest would be the viruses associated with acute inflammation as the major cause of pathology such as orthomyxoviruses (influenza virus), filoviruses (Ebola and Marburg viruses), flaviviruses (DENV, WNV, and TBEV) and alphaviruses (CHIKV). More importantly, it is essential to characterize the role of TREM-1-dependent responses in disease outcome, i.e., protective vs. pathogenic. For example, it is likely that TREM-1 signaling may promote inflammation and cause substantial tissue damage thereby contributing to disease pathogenesis of acute infections as with influenza virus or CHIKV. In this regard, Weber and colleagues recently demonstrated that TREM-1 deficient mice were protected from severe influenza disease without affecting virus clearance, although this study did not look into the innate immune markers governed by TREM-1 (Weber et al., 2014). On the other hand, in virus infections with WNV, TBEV or Japanese encephalitis virus, activation of TREM-1 might be protective and enhance the robustness of innate immune responses in the periphery thereby facilitating efficient virus clearance and reduced neuroinvasion. This notion is supported by our unpublished data demonstrating increased mortality and morbidity in WNV-infected TREM-1/3-deficient mice. Our unpublished data further demonstrates that WNV can induce TREM-1 mRNA in a cell-type specific manner.
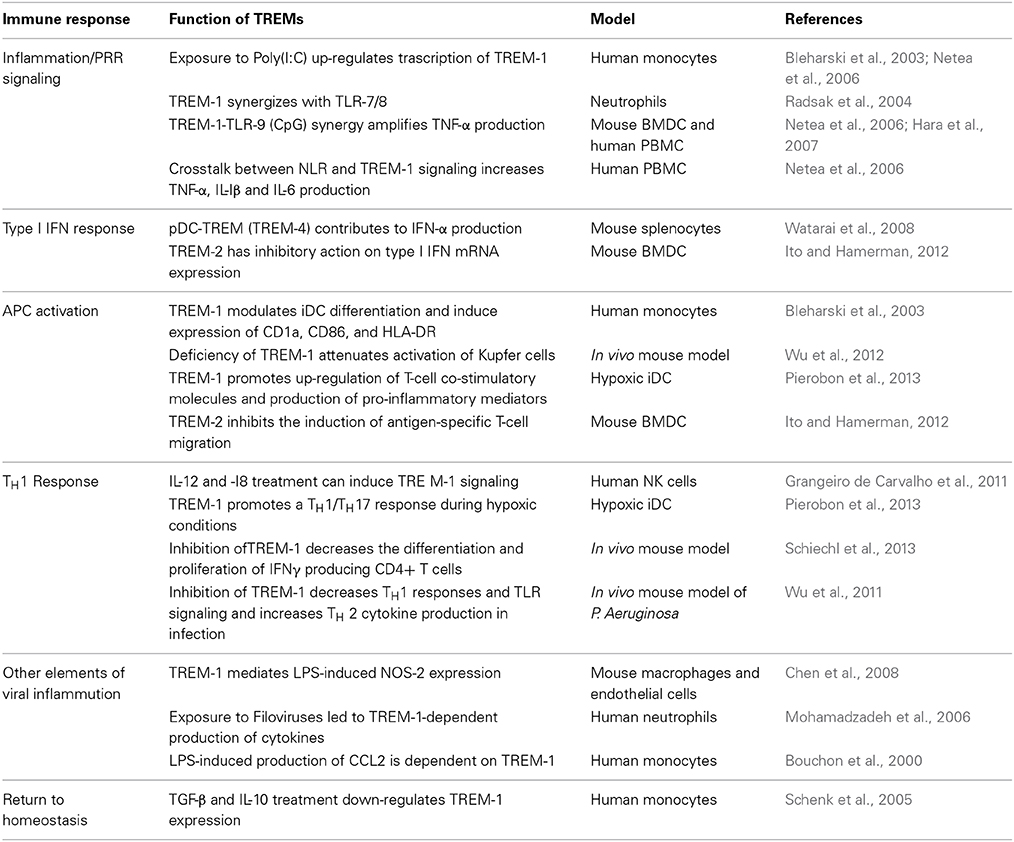
Table 2. The role of the TREM family receptors in immune responses relevant to antiviral immunity.
The question of how TREM-1 might influence disease outcome differently in acute vs. chronic infections with HIV or HCV also remains to be explained. Identification of inflammatory cytokines and chemokines regulated by TREM-1 will provide valuable insights into the role of TREM-1 in APC activation, T-cell responses and anti-viral immunity. Further, given that the type I IFN system is a powerful line of defense, characterization of the function of TREM-1 in modulating IFN levels and development of effective neutralizing antibody responses will further enhance our understanding of the complex network of antiviral immunity and fine-tuning of adaptive immunity.
In conclusion, this review describes the possible roles TREM-1 might play during virus infections. Future research using newly developed TREM-1 knockout mice and clinical samples from infected patients should focus on assigning protective or pathogenic functions for different viruses, examining the underlying cell- and tissue-type specific mechanisms of action, and identification of potential ligands responsible for TREM-1 receptor activation. There is such a large impact of the innate immune responses on the outcome of various viral diseases, therefore knowledge of the interactions between TREM-1 and viral PRRs (TLRs, NLRs, and RLRs) may lead to a better understanding of the pathophysiology of viral diseases. Further advancement in this field is crucial before TREM-1 can be proposed as an immunotherapeutic target to ultimately promote virus clearance with minimum tissue damage.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by institutional funds and grants from the Centers of Biomedical Research Excellence (P20GM103516), National Institute of General Medical Sciences, and Research Centers in Minority Institutions Program (G12MD007601, U54MD008149), National Institute on Minority Health and Health Disparities, National Institutes of Health.
References
Allcock, R. J. N., Barrow, A. D., Forbes, S., Beck, S., and Trowsdale, J. (2003). The human TREM gene cluster at 6p21.1 encodes both activating and inhibitory single IgV domain receptors and includes NKp44. Eur. J. Immunol. 33, 567–577. doi: 10.1002/immu.200310033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arts, R. J. W., Joosten, L. A. B., van der Meer, J. W. M., and Netea, M. G. (2013). TREM-1: intracellular signaling pathways and interaction with pattern recognition receptors. J. Leukoc. Biol. 93, 209–215. doi: 10.1189/jlb.0312145
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barraud, D., and Gibot, S. (2011). Triggering receptor expressed on myeloid cell 1. Crit. Care Clin. 27, 265–279. doi: 10.1016/j.ccc.2010.12.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Begum, N. A., Ishii, K., Kurita-Taniguchi, M., Tanabe, M., Kobayashi, M., Moriwaki, Y., et al. (2004). Mycobacterium bovis BCG cell wall-specific differentially expressed genes identified by differential display and cDNA subtraction in human macrophages. Infect. Immun. 72, 937–948. doi: 10.1128/IAI.72.2.937-948.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bermejo-Martin, J. F., Ortiz de Lejarazu, R., Pumarola, T., Rello, J., Almansa, R., Ramírez, P., et al. (2009). Th1 and Th17 hypercytokinemia as early host response signature in severe pandemic influenza. Crit. Care Lond. Engl. 13, R201. doi: 10.1186/cc8208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bishara, J., Hadari, N., Shalita-Chesner, M., Samra, Z., Ofir, O., Paul, M., et al. (2007). Soluble triggering receptor expressed on myeloid cells-1 for distinguishing bacterial from aseptic meningitis in adults. Eur. J. Clin. Microbiol. Infect. Dis. 26, 647–650. doi: 10.1007/s10096-007-0343-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bleharski, J. R., Kiessler, V., Buonsanti, C., Sieling, P. A., Stenger, S., Colonna, M., et al. (2003). A role for triggering receptor expressed on myeloid cells-1 in host defense during the early-induced and adaptive phases of the immune response. J. Immunol. 170, 3812–3818. doi: 10.4049/jimmunol.170.7.3812
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bouchon, A., Dietrich, J., and Colonna, M. (2000). Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J. Immunol. 164, 4991–4995. doi: 10.4049/jimmunol.164.10.4991
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bouchon, A., Facchetti, F., Weigand, M. A., and Colonna, M. (2001a). TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature 410, 1103–1107. doi: 10.1038/35074114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bouchon, A., Hernández-Munain, C., Cella, M., and Colonna, M. (2001b). A Dap12-mediated pathway regulates expression of Cc chemokine receptor 7 and maturation of human dendritic cells. J. Exp. Med. 194, 1111–1122. doi: 10.1084/jem.194.8.1111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buckland, K. F., Ramaprakash, H., Murray, L. A., Carpenter, K. J., Choi, E. S., Kunkel, S. L., et al. (2011). Triggering receptor expressed on myeloid cells-1 (TREM-1) modulates immune responses to Aspergillus fumigatus during fungal asthma in mice. Immunol. Invest. 40, 692–722. doi: 10.3109/08820139.2011.578270
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bucova, M., Suchankova, M., Dzurilla, M., Vrlik, M., Novosadova, H., Tedlova, E., et al. (2012). Inflammatory marker sTREM-1 reflects the clinical stage and respiratory tract obstruction in allergic asthma bronchiale patients and correlates with number of neutrophils. Mediators Inflamm. 2012, 1–8. doi: 10.1155/2012/628754
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, L. C., Laskin, J. D., Gordon, M. K., and Laskin, D. L. (2008). Regulation of TREM expression in hepatic macrophages and endothelial cells during acute endotoxemia. Exp. Mol. Pathol. 84, 145–155. doi: 10.1016/j.yexmp.2007.11.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chung, D.-H., Seaman, W. E., and Daws, M. R. (2002). Characterization of TREM-3, an activating receptor on mouse macrophages: definition of a family of single Ig domain receptors on mouse chromosome 17. Eur. J. Immunol. 32, 59–66. doi: 10.1002/1521-4141(200201)32:1<59::AID-IMMU59>3.0.CO;2-U
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Colonna, M. (2003). TREMs in the immune system and beyond. Nat. Rev. Immunol. 3, 445–453. doi: 10.1038/nri1106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Daffis, S., Samuel, M. A., Suthar, M. S., Gale, M., and Diamond, M. S. (2008). Toll-like receptor 3 has a protective role against West Nile virus infection. J. Virol. 82, 10349–10358. doi: 10.1128/JVI.00935-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Denner, J., Eschricht, M., Lauck, M., Semaan, M., Schlaermann, P., Ryu, H., et al. (2013). Modulation of cytokine release and gene expression by the immunosuppressive domain of gp41 of HIV-1. PLoS ONE 8:e55199. doi: 10.1371/journal.pone.0055199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Derive, M., Boufenzer, A., Bouazza, Y., Groubatch, F., Alauzet, C., Barraud, D., et al. (2013). Effects of a TREM-like transcript 1-derived peptide during hypodynamic septic shock in pigs. [Miscellaneous Article]. Shock 39, 176–182. doi: 10.1097/SHK.0b013e31827bcdfb
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Derive, M., Boufenzer, A., and Gibot, S. M. D. (2014). Attenuation of responses to endotoxin by the triggering receptor expressed on myeloid cells-1 inhibitor LR12 in nonhuman primate. [Miscellaneous Article]. Anesthesiology 120, 935–942. doi: 10.1097/ALN.0000000000000078
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Determann, R. M., Weisfelt, M., de Gans, J., van der Ende, A., Schultz, M. J., and van de Beek, D. (2006). Soluble triggering receptor expressed on myeloid cells 1: a biomarker for bacterial meningitis. Intensive Care Med. 32, 1243–1247. doi: 10.1007/s00134-006-0240-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
El Mezayen, R., El Gazzar, M., Seeds, M. C., McCall, C. E., Dreskin, S. C., and Nicolls, M. R. (2007). Endogenous signals released from necrotic cells augment inflammatory responses to bacterial endotoxin. Immunol. Lett. 111, 36–44. doi: 10.1016/j.imlet.2007.04.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fisher-Hoch, S. P., Platt, G. S., Neild, G. H., Southee, T., Baskerville, A., Raymond, R. T., et al. (1985). Pathophysiology of shock and hemorrhage in a fulminating viral infection (Ebola). J. Infect. Dis. 152, 887–894. doi: 10.1093/infdis/152.5.887
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ford, J. W., and McVicar, D. W. (2009). TREM and TREM-like receptors in inflammation and disease. Curr. Opin. Immunol. 21, 38–46. doi: 10.1016/j.coi.2009.01.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gibot, S., Alauzet, C., Massin, F., Sennoune, N., Faure, G. C., Béné, M.-C., et al. (2006). Modulation of the triggering receptor expressed on myeloid cells–1 pathway during pneumonia in rats. J. Infect. Dis. 194, 975–983. doi: 10.1086/506950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gibot, S., Cravoisy, A., Levy, B., Bene, M.-C., Faure, G., and Bollaert, P.-E. (2004a). Soluble triggering receptor expressed on myeloid cells and the diagnosis of pneumonia. N. Engl. J. Med. 350, 451–458. doi: 10.1056/NEJMoa031544
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gibot, S., Kolopp-Sarda, M.-N., Béné, M.-C., Bollaert, P.-E., Lozniewski, A., Mory, F., et al. (2004b). A soluble form of the triggering receptor expressed on myeloid cells-1 modulates the inflammatory response in murine sepsis. J. Exp. Med. 200, 1419–1426. doi: 10.1084/jem.20040708
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gibot, S., Kolopp-sarda, M., René, M. C., Cravoisy, A., Levy, B., Faure, G. C., et al. (2004c). Plasma level of a triggering receptor expressed on myeloid cells-1: its diagnostic accuracy in patients with suspected sepsis. Ann. Intern. Med. 141, 9–15. doi: 10.7326/0003-4819-141-1-200407060-00009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gibot, S., Massin, F., Marcou, M., Taylor, V., Stidwill, R., Wilson, P., et al. (2007). TREM-1 promotes survival during septic shock in mice. Eur. J. Immunol. 37, 456–466. doi: 10.1002/eji.200636387
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gingras, M.-C., Lapillonne, H., and Margolin, J. F. (2002). TREM-1, MDL-1, and DAP12 expression is associated with a mature stage of myeloid development. Mol. Immunol. 38, 817–824. doi: 10.1016/S0161-5890(02)00004-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gómez-Piña, V., Soares-Schanoski, A., Rodríguez-Rojas, A., Del Fresno, C., García, F., Vallejo-Cremades, M. T., et al. (2007). Metalloproteinases shed TREM-1 ectodomain from lipopolysaccharide-stimulated human monocytes. J. Immunol. 179, 4065–4073. doi: 10.4049/jimmunol.179.6.4065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grangeiro de Carvalho, E., Bonin, M., Kremsner, P. G., and Kun, J. F. J. (2011). Plasmodium falciparum-infected erythrocytes and IL-12/IL-18 induce diverse transcriptomes in human NK Cells: IFN-α/β pathway versus TREM signaling. PLoS ONE 6:e24963. doi: 10.1371/journal.pone.0024963
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer's disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hara, H., Ishihara, C., Takeuchi, A., Imanishi, T., Xue, L., Morris, S. W., et al. (2007). The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat. Immunol. 8, 619–629. doi: 10.1038/ni1466
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haselmayer, P., Grosse-Hovest, L., von Landenberg, P., Schild, H., and Radsak, M. P. (2007). TREM-1 ligand expression on platelets enhances neutrophil activation. Blood 110, 1029–1035. doi: 10.1182/blood-2007-01-069195
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hommes, T. J., Hoogendijk, A. J., Dessing, M. C., van't Veer, C., Florquin, S., Colonna, M., et al. (2014). Triggering receptor expressed on myeloid cells-1 (TREM-1) improves host defence in pneumococcal pneumonia. J. Pathol. 233, 357–367. doi: 10.1002/path.4361
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hu, L., Du, Z., Zhao, G., Jiang, N., Lin, J., Wang, Q., et al. (2014). Role of TREM-1 in response to Aspergillus fumigatus infection in corneal epithelial cells. Int. Immunopharmacol. 23, 288–293. doi: 10.1016/j.intimp.2014.09.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ito, H., and Hamerman, J. A. (2012). TREM-2, triggering receptor expressed on myeloid cell-2, negatively regulates TLR responses in dendritic cells. Eur. J. Immunol. 42, 176–185. doi: 10.1002/eji.201141679
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jenne, C. N., Wong, C. H. Y., Zemp, F. J., McDonald, B., Rahman, M. M., Forsyth, P. A., et al. (2013). Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe 13, 169–180. doi: 10.1016/j.chom.2013.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer's disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kelker, M. S., Debler, E. W., and Wilson, I. A. (2004a). Crystal structure of mouse triggering receptor expressed on myeloid cells 1 (TREM-1) at 1.76 Å. J. Mol. Biol. 344, 1175–1181. doi: 10.1016/j.jmb.2004.10.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kelker, M. S., Foss, T. R., Peti, W., Teyton, L., Kelly, J. W., Wüthrich, K., et al. (2004b). Crystal structure of human triggering receptor expressed on myeloid cells 1 (TREM-1) at 1.47 Å. J. Mol. Biol. 342, 1237–1248. doi: 10.1016/j.jmb.2004.07.089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
King, R. G., Herrin, B. R., and Justement, L. B. (2006). Trem-like transcript 2 is expressed on cells of the myeloid/granuloid and B lymphoid lineage and is up-regulated in response to inflammation. J. Immunol. 176, 6012–6021. doi: 10.4049/jimmunol.176.10.6012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klesney-Tait, J., Keck, K., Li, X., Gilfillan, S., Otero, K., Baruah, S., et al. (2012). Transepithelial migration of neutrophils into the lung requires TREM-1. J. Clin. Invest. 123, 138–149. doi: 10.1172/JCI64181
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klesney-Tait, J., Turnbull, I. R., and Colonna, M. (2006). The TREM receptor family and signal integration. Nat. Immunol. 7, 1266–1273. doi: 10.1038/ni1411
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kumar, H., Kawai, T., and Akira, S. (2011). Pathogen recognition by the innate immune system. Int. Rev. Immunol. 30, 16–34. doi: 10.3109/08830185.2010.529976
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kumar, M., Roe, K., Orillo, B., Muruve, D. A., Nerurkar, V. R., Gale, M., et al. (2013). Inflammasome adaptor protein apoptosis-associated speck-like protein containing CARD (ASC) is critical for the immune response and survival in West Nile virus encephalitis. J. Virol. 87, 3655–3667. doi: 10.1128/JVI.02667-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lagler, H., Sharif, O., Haslinger, I., Matt, U., Stich, K., Furtner, T., et al. (2009). TREM-1 activation alters the dynamics of pulmonary IRAK-M expression in vivo and improves host defense during pneumococcal pneumonia. J. Immunol. 183, 2027–2036. doi: 10.4049/jimmunol.0803862
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liao, R., Sun, T.-W., Yi, Y., Wu, H., Li, Y.-W., Wang, J.-X., et al. (2012). Expression of TREM-1 in hepatic stellate cells and prognostic value in hepatitis B-related hepatocellular carcinoma. Cancer Sci. 103, 984–992. doi: 10.1111/j.1349-7006.2012.02273.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, Y.-T., Tseng, K.-Y., Yeh, Y.-C., Yang, F.-C., Fung, C.-P., and Chen, N.-J. (2014). TREM-1 promotes survival during klebsiella pneumoniae liver abscess in mice. Infect. Immun. 82, 1335–1342. doi: 10.1128/IAI.01347-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lund, J. M., Alexopoulou, L., Sato, A., Karow, M., Adams, N. C., Gale, N. W., et al. (2004). Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. U.S.A. 101, 5598–5603. doi: 10.1073/pnas.0400937101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martini, G. A. (1973). Marburg virus disease. Postgrad. Med. J. 49, 542–546. doi: 10.1136/pgmj.49.574.542
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matesanz-Isabel, J., Sintes, J., Llinàs, L., de Salort, J., Lázaro, A., and Engel, P. (2011). New B-cell CD molecules. Immunol. Lett. 134, 104–112. doi: 10.1016/j.imlet.2010.09.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mohamadzadeh, M., Coberley, S. S., Olinger, G. G., Kalina, W. V., Ruthel, G., Fuller, C. L., et al. (2006). Activation of triggering receptor expressed on myeloid cells-1 on human neutrophils by marburg and ebola viruses. J. Virol. 80, 7235–7244. doi: 10.1128/JVI.00543-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Netea, M. G., Azam, T., Ferwerda, G., Girardin, S. E., Kim, S.-H., and Dinarello, C. A. (2006). Triggering receptor expressed on myeloid cells-1 (TREM-1) amplifies the signals induced by the NACHT-LRR (NLR) pattern recognition receptors. J. Leukoc. Biol. 80, 1454–1461. doi: 10.1189/jlb.1205758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ornatowska, M., Azim, A. C., Wang, X., Christman, J. W., Xiao, L., Joo, M., et al. (2007). Functional genomics of silencing TREM-1 on TLR4 signaling in macrophages. AJP Lung Cell. Mol. Physiol. 293, L1377–L1384. doi: 10.1152/ajplung.00140.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Page, A. V., and Liles, W. C. (2013). Biomarkers of endothelial activation/dysfunction in infectious diseases. Virulence 4, 507–516. doi: 10.4161/viru.24530
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paloneva, J., Mandelin, J., Kiialainen, A., Böhling, T., Prudlo, J., Hakola, P., et al. (2003). DAP12/TREM2 deficiency results in impaired osteoclast differentiation and osteoporotic features. J. Exp. Med. 198, 669–675. doi: 10.1084/jem.20030027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paradowska-Gorycka, A., and Jurkowska, M. (2013). Structure, expression pattern and biological activity of molecular complex TREM-2/DAP12. Hum. Immunol. 74, 730–737. doi: 10.1016/j.humimm.2013.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Piccio, L., Buonsanti, C., Mariani, M., Cella, M., Gilfillan, S., Cross, A. H., et al. (2007). Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur. J. Immunol. 37, 1290–1301. doi: 10.1002/eji.200636837
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pierobon, D., Bosco, M. C., Blengio, F., Raggi, F., Eva, A., Filippi, M., et al. (2013). Chronic hypoxia reprograms human immature dendritic cells by inducing a proinflammatory phenotype and TREM-1 expression. Eur. J. Immunol. 43, 949–966. doi: 10.1002/eji.201242709
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Radaev, S., Kattah, M., Rostro, B., Colonna, M., and Sun, P. D. (2003). Crystal structure of the human myeloid cell activating receptor TREM-1. Structure 11, 1527–1535. doi: 10.1016/j.str.2003.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Radsak, M. P., Salih, H. R., Rammensee, H.-G., and Schild, H. (2004). Triggering receptor expressed on myeloid cells-1 in neutrophil inflammatory responses: differential regulation of activation and survival. J. Immunol. 172, 4956–4963. doi: 10.4049/jimmunol.172.8.4956
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rigo, I., McMahon, L., Dhawan, P., Christakos, S., Yim, S., Ryan, L. K., et al. (2011). Induction of triggering receptor expressed on myeloid cells (TREM-1) in airway epithelial cells by 1,25(OH)2 vitamin D3. Innate Immun. 18, 250–257. doi: 10.1177/1753425911399796
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rouse, B. T., and Sehrawat, S. (2010). Immunity and immunopathology to viruses: what decides the outcome? Nat. Rev. Immunol. 10, 514–526. doi: 10.1038/nri2802
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ruiz-Pacheco, J. A., Vivanco-Cid, H., Izaguirre-Hernández, I. Y., Estrada-García, I., Arriaga-Pizano, L., Chacón-Salinas, R., et al. (2014). TREM-1 modulation during early stages of dengue virus infection. Immunol. Lett. 158, 183–188. doi: 10.1016/j.imlet.2014.01.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saitoh, T., Komano, J., Saitoh, Y., Misawa, T., Takahama, M., Kozaki, T., et al. (2012). Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 12, 109–116. doi: 10.1016/j.chom.2012.05.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schenk, M., Bouchon, A., Birrer, S., Colonna, M., and Mueller, C. (2005). Macrophages expressing triggering receptor expressed on myeloid cells-1 are underrepresented in the human intestine. J. Immunol. 174, 517–524. doi: 10.4049/jimmunol.174.1.517
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schiechl, G., Brunner, S. M., Kesselring, R., Martin, M., Ruemmele, P., Mack, M., et al. (2013). Inhibition of innate co-receptor TREM-1 signaling reduces CD4+ T cell activation and prolongs cardiac allograft survival. Am. J. Transplant. 13, 1168–1180. doi: 10.1111/ajt.12186
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schmaußer, B., Endrich, S., Beier, D., Moran, A. P., Burek, C. J., Rosenwald, A., et al. (2008). Triggering receptor expressed on myeloid cells-1 (TREM-1) expression on gastric epithelium: implication for a role of TREM-1 in Helicobacter pylori infection. Clin. Exp. Immunol. 152, 88–94. doi: 10.1111/j.1365-2249.2008.03608.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schmid, C. D., Sautkulis, L. N., Danielson, P. E., Cooper, J., Hasel, K. W., Hilbush, B. S., et al. (2002). Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J. Neurochem. 83, 1309–1320. doi: 10.1046/j.1471-4159.2002.01243.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suthar, M. S., Brassil, M. M., Blahnik, G., McMillan, A., Ramos, H. J., Proll, S. C., et al. (2013). A systems biology approach reveals that tissue tropism to West Nile virus is regulated by antiviral genes and innate immune cellular processes. PLoS Pathog 9:e1003168. doi: 10.1371/journal.ppat.1003168
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takahashi, K., Rochford, C. D. P., and Neumann, H. (2005). Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 201, 647–657. doi: 10.1084/jem.20041611
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tessarz, A. S., and Cerwenka, A. (2008). The TREM-1/DAP12 pathway. Immunol. Lett. 116, 111–116. doi: 10.1016/j.imlet.2007.11.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Turnbull, I. R., Gilfillan, S., Cella, M., Aoshi, T., Miller, M., Piccio, L., et al. (2006). Cutting edge: TREM-2 attenuates macrophage activation. J. Immunol. 177, 3520–3524. doi: 10.4049/jimmunol.177.6.3520
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Varani, S., Cederarv, M., Feld, S., Tammik, C., Frascaroli, G., Landini, M. P., et al. (2007). Human cytomegalovirus differentially controls B cell and T cell responses through effects on plasmacytoid dendritic cells. J. Immunol. 179, 7767–7776. doi: 10.4049/jimmunol.179.11.7767
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Veiga-Parga, T., Sehrawat, S., and Rouse, B. T. (2013). Role of regulatory T cells during virus infection. Immunol. Rev. 255, 182–196. doi: 10.1111/imr.12085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, T., Town, T., Alexopoulou, L., Anderson, J. F., Fikrig, E., and Flavell, R. A. (2004). Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 10, 1366–1373. doi: 10.1038/nm1140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Washington, A. V., Schubert, R. L., Quigley, L., Disipio, T., Feltz, R., Cho, E. H., et al. (2004). A TREM family member, TLT-1, is found exclusively in the α-granules of megakaryocytes and platelets. Blood 104, 1042–1047. doi: 10.1182/blood-2004-01-0315
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watarai, H., Sekine, E., Inoue, S., Nakagawa, R., Kaisho, T., and Taniguchi, M. (2008). PDC-TREM, a plasmacytoid dendritic cell-specific receptor, is responsible for augmented production of type I interferon. Proc. Natl. Acad. Sci. U.S.A. 105, 2993–2998. doi: 10.1073/pnas.0710351105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wauquier, N., Becquart, P., Padilla, C., Baize, S., and Leroy, E. M. (2010). Human fatal zaire ebola virus infection is associated with an aberrant innate immunity and with massive lymphocyte apoptosis. PLoS Negl Trop Dis 4:e837. doi: 10.1371/journal.pntd.0000837
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weber, B., Schuster, S., Zysset, D., Rihs, S., Dickgreber, N., Schürch, C., et al. (2014). TREM-1 deficiency can attenuate disease severity without affecting pathogen clearance. PLoS Pathog 10:e1003900. doi: 10.1371/journal.ppat.1003900
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wong-Baeza, I., González-Roldán, N., Ferat-Osorio, E., Esquivel-Callejas, N., Aduna-Vicente, R., Arriaga-Pizano, L., et al. (2006). Triggering receptor expressed on myeloid cells (TREM-1) is regulated post-transcriptionally and its ligand is present in the sera of some septic patients. Clin. Exp. Immunol. 145, 448–455. doi: 10.1111/j.1365-2249.2006.03158.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, J., Li, J., Salcedo, R., Mivechi, N. F., Trinchieri, G., and Horuzsko, A. (2012). The proinflammatory myeloid cell receptor TREM-1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res. 72, 3977–3986. doi: 10.1158/0008-5472.CAN-12-0938
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, M., Peng, A., Sun, M., Deng, Q., Hazlett, L. D., Yuan, J., et al. (2011). TREM-1 Amplifies corneal inflammation after Pseudomonas aeruginosa infection by modulating toll-like receptor signaling and Th1/Th2-Type immune responses. Infect. Immun. 79, 2709–2716. doi: 10.1128/IAI.00144-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, Z., Delgado, R., Xu, L., Todd, R. F., Nabel, E. G., Sanchez, A., et al. (1998). Distinct cellular interactions of secreted and transmembrane Ebola virus glycoproteins. Science 279, 1034–1037. doi: 10.1126/science.279.5353.1034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zangi, L., Klionsky, Y. Z., Yarimi, L., Bachar-Lustig, E., Eidelstein, Y., Shezen, E., et al. (2012). Deletion of cognate CD8 T cells by immature dendritic cells: a novel role for perforin, granzyme A, TREM-1, and TLR7. Blood 120, 1647–1657. doi: 10.1182/blood-2012-02-410803
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zeng, H., Ornatowska, M., Joo, M. S., and Sadikot, R. T. (2007). TREM-1 expression in macrophages is regulated at transcriptional level by NF-κ B and PU.1. Eur. J. Immunol. 37, 2300–2308. doi: 10.1002/eji.200737270
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, S.-Y., Jouanguy, E., Ugolini, S., Smahi, A., Elain, G., Romero, P., et al. (2007). TLR3 deficiency in patients with herpes simplex encephalitis. Science 317, 1522–1527. doi: 10.1126/science.1139522
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zheng, H., Heiderscheidt, C. A., Joo, M., Gao, X., Knezevic, N., Mehta, D., et al. (2010). MYD88-dependent and -independent activation of TREM-1 via specific TLR ligands. Eur. J. Immunol. 40, 162–171. doi: 10.1002/eji.200839156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: TREM-1, innate immune response, virus pathogenesis, inflammation, antiviral immunity
Citation: Roe K, Gibot S and Verma S (2014) Triggering receptor expressed on myeloid cells-1 (TREM-1): a new player in antiviral immunity? Front. Microbiol. 5:627. doi: 10.3389/fmicb.2014.00627
Received: 09 September 2014; Accepted: 03 November 2014;
Published online: 26 November 2014.
Edited by:
Mei-Ru Chen, National Taiwan University, TaiwanReviewed by:
Dahlene N. Fusco, Massachusetts General Hospital, USALi-Chung Hsu, National Taiwan University, Taiwan
Copyright © 2014 Roe, Gibot and Verma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saguna Verma, Department of Tropical Medicine, Medical Microbiology and Pharmacology, John A. Burns School of Medicine, University of Hawaii at Manoa, 651 Ilalo Street, Honolulu, HI 96813, USA e-mail:c2FndW5hQGhhd2FpaS5lZHU=