 Camila N. Signori1,2,3‡
Camila N. Signori1,2,3‡ François Thomas3†‡
François Thomas3†‡ Alex Enrich-Prast1,2,4*
Alex Enrich-Prast1,2,4* Ricardo C. G. Pollery2
Ricardo C. G. Pollery2 Stefan M. Sievert3*
Stefan M. Sievert3*- 1Instituto de Microbiologia Professor Paulo de Góes, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
- 2Laboratório de Biogeoquímica, Instituto de Biologia, Universidade Federal do Rio de Janeiro, Centro de Ciências da Saúde, Rio de Janeiro, Brazil
- 3Biology Department, Woods Hole Oceanographic Institution, Woods Hole, MA, USA
- 4Department of Environmental Change, Linköping University, Linköping, Sweden
The Southern Ocean is currently subject to intense investigations, mainly related to its importance for global biogeochemical cycles and its alarming rate of warming in response to climate change. Microbes play an essential role in the functioning of this ecosystem and are the main drivers of the biogeochemical cycling of elements. Yet, the diversity and abundance of microorganisms in this system remain poorly studied, in particular with regards to changes along environmental gradients. Here, we used amplicon sequencing of 16S rRNA gene tags using primers covering both Bacteria and Archaea to assess the composition and diversity of the microbial communities from four sampling depths (surface, the maximum and minimum of the oxygen concentration, and near the seafloor) at 10 oceanographic stations located in Bransfield Strait [northwest of the Antarctic Peninsula (AP)] and near the sea ice edge (north of the AP). Samples collected near the seafloor and at the oxygen minimum exhibited a higher diversity than those from the surface and oxygen maximum for both bacterial and archaeal communities. The main taxonomic groups identified below 100 m were Thaumarchaeota, Euryarchaeota and Proteobacteria (Gamma-, Delta-, Beta-, and Alphaproteobacteria), whereas in the mixed layer above 100 m Bacteroidetes and Proteobacteria (mainly Alpha- and Gammaproteobacteria) were found to be dominant. A combination of environmental factors seems to influence the microbial community composition. Our results help to understand how the dynamic seascape of the Southern Ocean shapes the microbial community composition and set a baseline for upcoming studies to evaluate the response of this ecosystem to future changes.
Introduction
The Southern Ocean is currently subject to intense investigations, mainly related to its importance for global biogeochemical cycles and its alarming rate of warming in response to climate change (e.g., Smetacek and Nicol, 2005; Schofield et al., 2010; Luria et al., 2014). This area of the world's ocean accounts for a third of the global ocean CO2 uptake, and thus plays a major role in the Earth's carbon cycle. The Southern Ocean is a large and heterogeneous biogeochemical system, comprising both highly productive coastal and ice-edge waters that contrast with open ocean waters where primary production is to a large extent limited by the availability of the micronutrient iron. In particular, the Western Antarctic Peninsula (WAP) is considered as one of the main areas experiencing rapid regional warming (Anisimov et al., 2007), and is the only one with a maritime climate—making it an ideal place to monitor, understand and predict the impacts of climate change on marine ecosystems (Schofield et al., 2010).
Marine microorganisms play a critical role in ecosystem functioning and are the main drivers of the biogeochemical cycling of elements. They dominate in abundance, and diversity, and are responsible for the majority of the metabolic activity of the ocean (Azam and Malfatti, 2007). The processing of organic matter by Bacteria and Archaea is considered a major carbon-flow pathway, and its variability can change the overall pattern of carbon flux (Azam, 1998). Yet, the diversity, activity, and abundance of microorganisms in the Southern Ocean remains poorly studied, in particular with regards to changes along environmental gradients (e.g., Wilkins et al., 2013a,b; Luria et al., 2014). The need of studying the microbial diversity in conjunction with physical and chemical observations (considering both temporal and spatial scales) to understand the role of the microbes in oceanic ecosystems has been previously noted (Karl, 2007). Yet, it is only through recent advances in sequencing technology that it has become feasible to perform these kinds of studies at the required scale and resolution (DeLong and Karl, 2005; Sogin et al., 2006; Frias-Lopez et al., 2008; Yooseph et al., 2010). Some more recent studies have shown that oceanographic factors such as the physical-chemical and biological properties of the water masses (Agogué et al., 2011), advection of cells by water currents (Wilkins et al., 2013a), and seasonal evolution of the upper water column (Luria et al., 2014) have a role in shaping the microbial community structure in the Southern Ocean.
The Southern Ocean is characterized by a complex interplay of environmental variables that may influence the composition of the microbial communities, and at present, studies are still at the stage of establishing reference values for assessing the temporal and spatial dynamics of the microbial communities in the water column (e.g., Wilkins et al., 2013b; Luria et al., 2014). In a recent study, Luria et al. (2014) studied the microbial communities at 4 stations and 2 depths (10 and 100 m) on the continental shelf of the WAP, providing a reference point for future investigations. In the present study, we assessed the composition and structure of bacterial and archaeal communities at 10 stations in a region to the north of that studied by Luria et al. (2014), collecting samples from 4 different depths (from surface to seafloor) influenced by contrasted water masses. Combined with the measurement of various environmental parameters, our study contributes to an increased understanding of the factors that drive the diversity of microbes in this dynamic and rapidly changing region of the Southern Ocean, which is paramount to predict responses of this important biome to future changes.
Materials and Methods
The Study Area
The Bransfield Strait (BS) is influenced by different water masses. In this area, cold and salty waters enter from the Weddell Sea (called Transitional Zonal Water with Weddell Sea influence, TWW), while water from the Bellingshausen Sea forms an overlaying well-stratified relatively warm and fresh surface layer (called Transitional Zonal Water with Bellingshausen influence, TBW) (Tokarczyk, 1987; Garcia et al., 1994; Sangrà et al., 2011). BS is also an area with a high biological productivity, all the way from phytoplankton and zooplankton to whales (Zhou et al., 2002; Dalla Rosa et al., 2008). The dynamic hydrographic structure is also reflected in the chlorophyll distribution, as seen by Figueiras et al. (1998), with concentrations lower than 0.5 mg.m−3 in surface Weddell Sea waters and higher than 1 mg.m−3 in surface waters with Bellingshausen Sea influence.
Sampling Strategy
The research cruise was conducted by the Brazilian Navy vessel Npo. Almirante Maximiano (H44) during the austral summer, February to March 2013. Ten oceanographic stations were selected (PB49, PB53, PB57, PB59, PB62, PB66, PB72, PB76A, PO01.01, PO01.21B—see Figure 1) from the Bransfield Strait (located in the northwest of the Antarctic Peninsula) to the open ocean and near the sea ice edge (north of the AP), to be able to evaluate changes in the microbial community along an approximately 500 km long transect. Seawater and physical data (temperature, salinity and dissolved oxygen) were collected using a combined Sea-Bird CTD/Carrousel 911 system equipped with 24 5-l Niskin bottles. Sampling depths (surface, maximum and minimum of dissolved oxygen and near the bottom) were selected according to the CTD profiles. For microbial diversity approximately 1.8 l of seawater were filtered onto Sterivex filters (Millipore) with a pore size of 0.2 μm using a peristaltic pump. Filters were immediately frozen at −80°C. Due to competing water demands on the cruise, the data are from single, unreplicated samples. In general, within-sample replication for water samples in this area yields to low variability in the community composition (Luria et al., 2014).
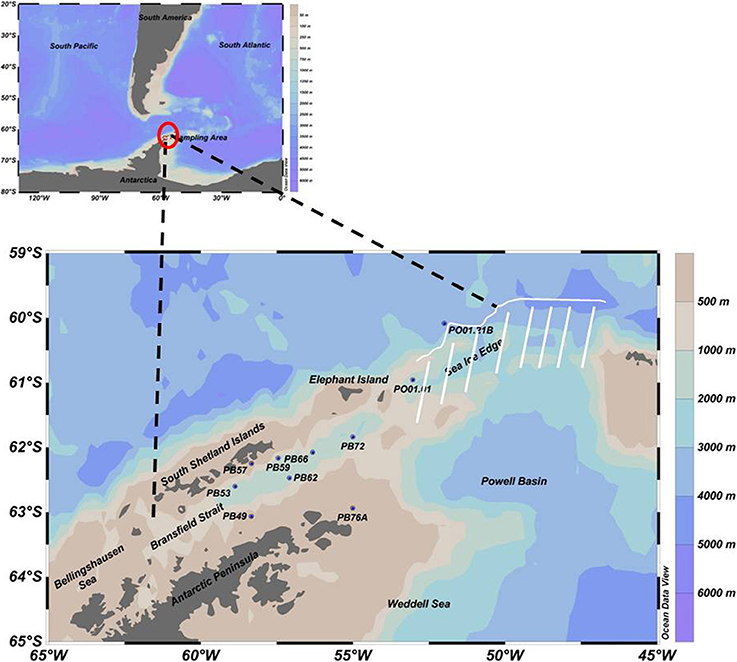
Figure 1. Sampling map with 10 selected oceanographic stations in the Southern Ocean, from the Bransfield Strait (northwest of the AP) to the open ocean near the sea ice edge (north of the AP), represented by the white line. Due to ice, navigation was impossible in the region indicated by the white stripes.
Environmental Factors
Concentrations of inorganic nutrients (nitrate, nitrite, ammonia, silicate, and phosphate) were determined using a flow injection autoanalyzer (FIAstar 5000, Foss Tecator, Denmark). Samples for chlorophyll-a were filtered onto Whatman GF/F filters under vacuum pressure and analyzed using a Shimadzu High Performance Liquid Chromatography (HPLC).
DNA Extraction and Sequencing
For DNA extraction, a protocol previously developed by Boström et al. (2004) and Manganelli et al. (2009) for low biomass samples was slightly modified by replacing t-RNA with linear acrylamide as co-precipitant. The DNA concentration in each sample was quantified by a fluorescent PicoGreen assay (Invitrogen), using a thermocycler (MX3005P, Stratagene). The composition of the microbial community was analyzed using a 16S rRNA gene based approach. Amplicons for sequencing were generated using the universal primers S-D-Bact-0785-a-S-18 and S-*-Univ-1392-a-A-15, recently identified as a suitable primer pair for Bacteria and Archaea after a thorough evaluation (Klindworth et al., 2013). This primer pair amplifies a 608 bp product and targets V5-V8 hypervariable regions of bacterial and archaeal 16S rRNA, with overall coverages of 86.5% and 67.0%, respectively (SILVA TestPrime 1.0, http://www.arb-silva.de/search/testprime/, tested in October 2014, allowing for 1 mismatch with no mismatches in the last 4 bases of the 3′end). A single-step 28 cycles PCR using HotStarTaq Plus Master Mix Kit (Qiagen, Valencia, CA) was used on 5 ng of DNA under the following conditions: 94°C for 3 min, followed by 28 cycles of 94°C for 30 s; 53°C for 40 s and 72°C for 1 min; after which a final elongation step at 72°C for 5 min was performed. Following PCR, all barcoded amplicon products (see Supplement Table 1 for details) from different samples were quantified using Qubit (Life Technologies), mixed in equal concentrations and purified using Agencourt Ampure beads (Agencourt Bioscience Corporation, MA, USA). After these steps, samples were sequenced using FLX titanium instruments and reagents (Roche 454), following the manufacturer's guidelines. The PCR and sequencing were carried out at Molecular Research Lab (Shallowater, Texas, USA). All sequence data have been deposited in the National Center for Biotechnology Information Sequence Read Archives (SRA) under BioProject ID PRJNA269510.
16S rRNA Gene Reads Processing and Statistical Analyses
The 16S rRNA gene sequences were denoised with Acacia using the default parameters (Bragg et al., 2012) and further processed in the QIIME 1.7.0 pipeline (Caporaso et al., 2010). Reads were filtered for length (400 ≤ length ≤ 1000), quality score (mean, ≥25), number of ambiguous bases (=0), and length of homopolymer runs (<6). Sequences were clustered at 97% similarity using usearch (Edgar, 2010) including de novo and reference-based chimera checking. Taxonomy was assigned to each Operational Taxonomic Unit (OTU) using RDP classifier (Wang et al., 2007) with the Greengenes 13_5 database (McDonald et al., 2012). Sequences assigned to chloroplasts and mitochondria were removed from the dataset for further analysis. Multiple rarefactions at 594 reads per sample were conducted for alpha- and beta-diversity analyses. Differences in alpha-diversity estimates between groups of samples were tested using Student t-test in R (version 3.0.1). The weighted normalized Unifrac distance (Lozupone and Knight, 2005) was used for UPGMA (Unweighted Pair Group Method with Arithmetic Mean) cluster analysis with 100 resamplings and adonis statistical test with 999 permutations. The program iTOL (Interactive Tree Of Life, version 2.1, (available at http://itol.embl.de/index.shtml) was used for dendrogram visualization (Letunic and Bork, 2007, 2011). In addition, we plotted the weighted normalized UniFrac distances of all surface samples, which were collected at the same water depth (5 m), against geographical distance. Geographical distance is the actual distance between two stations, and it was calculated from latitude and longitude, assuming a spherical Earth, by using the following formula: d = acos(sin φ 1 · sin φ 2 + cos φ 1 · cos φ 2 · cos Δ λ) · R, where φ 1 is latitude of station 1, φ 2 is latitude of station 2, Δ λ is the difference in longitude, and R is the radius of the Earth. Non-metric multidimensional scaling (nMDS) was performed in R (version 3.0.1) on a Bray-Curtis dissimilarity matrix, with fitting of the environmental parameters using the envfit function from the vegan package (Oksanen et al., 2013). Differences in the relative abundances of taxa between groups were evaluated with a Welch's t-test as implemented in the STAMP program (Parks and Beiko, 2010), with FDR correction of the p-values to account for multiple testing, and filtering of the results based on effect sizes (difference between proportions >1 or ratio of proportions >4).
Results
Sampling Site Characteristics: Environmental Factors
The surface waters in Bransfield Strait were characterized by elevated temperatures and higher concentrations of oxygen, nitrite, ammonia and phytoplankton biomass (chlorophyll-a), when compared to the deep waters, which exhibited higher salinity as well as higher concentrations of nitrate, phosphate and silicate (Table 1). The highest chlorophyll-a value (5.75 mg.m−3) was found at the oceanographic station closest to the continent (PB57, at the Admiralty Bay entrance—South Shetland Islands). We found the highest concentration of silicate (66.36 μM, station PO01.21B) in the deepest sample (3012 m) close to the sea ice edge north of the AP (Table 1). When all the 30 samples were plotted according to in situ temperature, salinity, and dissolved oxygen, three groups of samples could clearly be separated, indicating that they belong to different water masses (Figure 2). The first group was influenced by the Antarctic Surface Water (AASW), with temperatures lower than 1°C, salinities between 33.0 and 34.5 psu, and a water depth greater than 200 m (Gordon and Huber, 1995). The second group of samples was affected by the Modified Deep Warm Water (mWDW), transitional waters with temperatures from −0.7 to −1.7°C and salinities between 34.4 and 34.6 psu (Robertson et al., 2002; Duarte, 2006). The third group was influenced by the Circumpolar Deep Water (CDW), with temperatures of 0.2–1.7°C and salinities of 34.5–34.8 psu (Santoso et al., 2006).
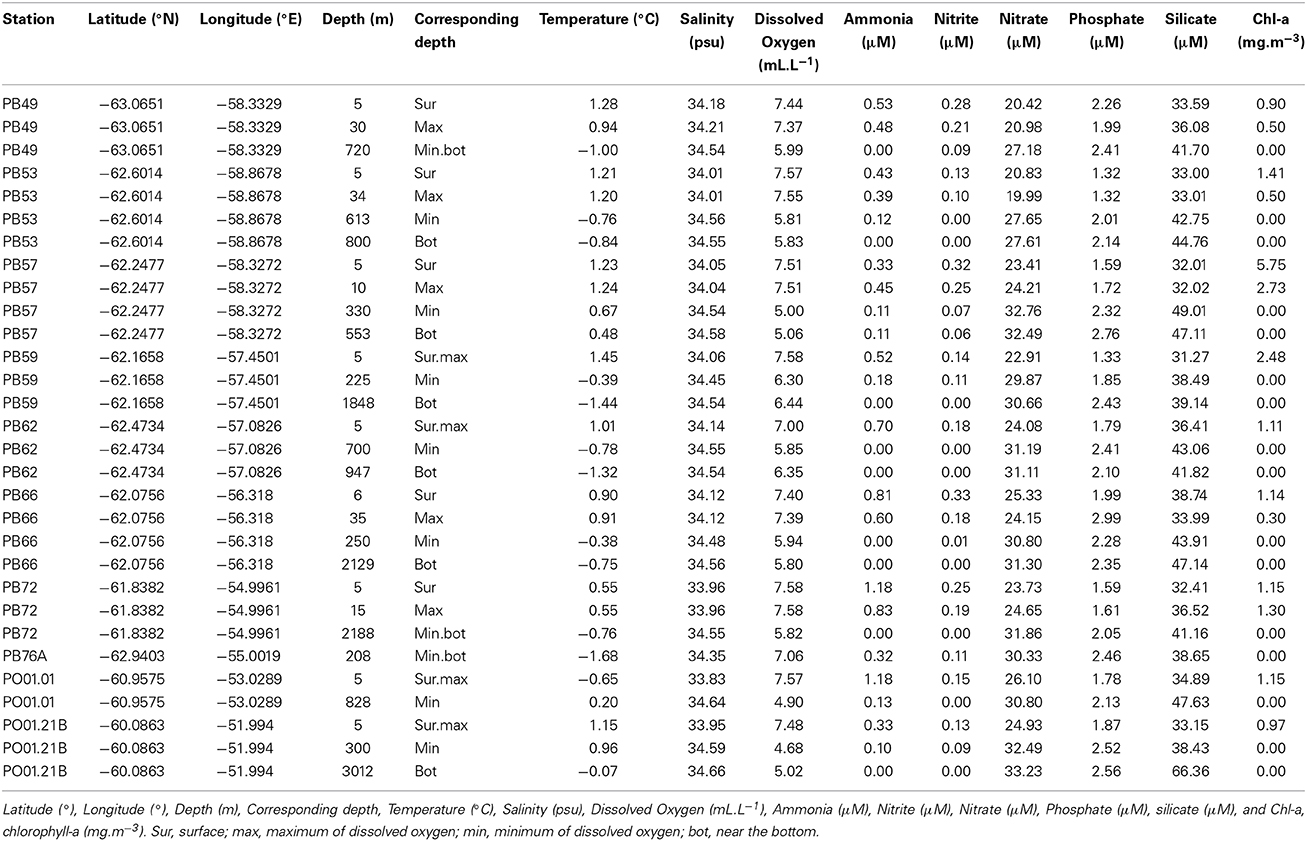
Table 1. Environmental parameters measured for each sample in the Western Antarctic Peninsula (WAP).
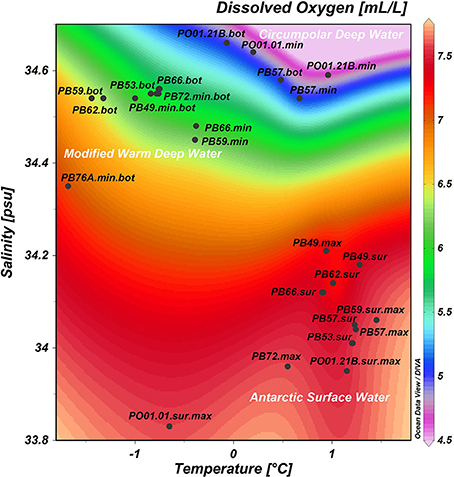
Figure 2. Samples distribution according to in situ measurements of temperature (°C), salinity (psu) and dissolved oxygen (mL.L−1). In white: water masses sampled in the Southern Ocean. Sur, surface; max, maximum of dissolved oxygen; min, minimum of dissolved oxygen; bot, near the bottom.
Overall Diversity
After quality checking and data filtering, a total of 146,287 sequences were obtained from 30 water samples, with an average of 4876 reads per sample (range 594–10,574 reads per sample). Clustering of these reads at 97% similarity threshold resulted in 1,506 OTUs: 172 for Archaea (11.4%) and 1,328 for Bacteria (88.2%). Considering all sequences retrieved in the present study (Figure 3), Gammaproteobacteria accounted for the largest fraction (35.1% of the total)—mainly represented by the orders Oceanospirillales (17.4%), Vibrionales (9.1%, dominated by the genus Pseudoalteromonas) and Alteromonadales (7.1%). Bacteroidetes was the second most represented class (19.7%), with representatives mostly from the order Flavobacteriales (19.2%, dominated by the psychrophilic and proteorhodopsin-containing Polaribacter). In addition, we also identified Alphaproteobacteria (7.6%, mainly represented by the order Rhodobacterales and to a lesser extent SAR11), Deltaproteobacteria (3.8%, SAR324), Planctomycetes (2.1%), SAR 406 (1.5%), and other taxa with contributions lower than 1%. Archaea were mainly represented by Thaumarchaeota (Nitrosopumilus) with 14.2% and Euryarchaeota (class Thermoplasmata, Marine Groups II and III) with 13% of the total number of sequences.
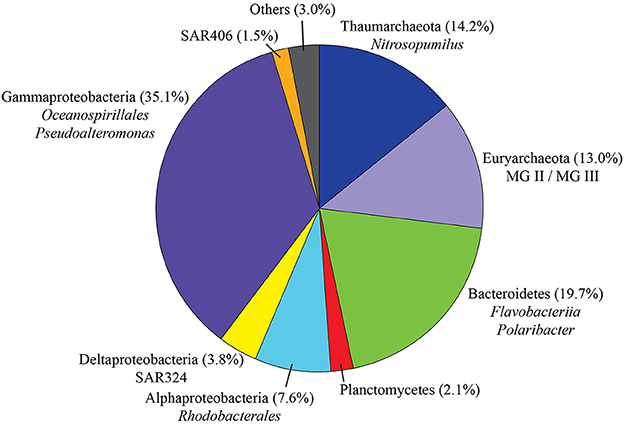
Figure 3. Taxonomic distribution of all sequences retrieved during this study (n = 146,287). “Others” comprises taxa accounting for less than 1% of the total.
Alpha Diversity
After rarefaction to even sequencing depth, the number of observed OTUs per sample ranged from 63 to 167 (Table 2). Richness and diversity were significantly greater in samples originating from deep waters (oxygen minimum and seafloor) than in samples collected at shallower depth (surface and oxygen maximum). Considering all the indices, the lowest values of diversity were found at the following oceanographic stations: PB53.Sur, PB57.Sur, PB59.Sur.Max, PB72.Sur, PO01.01.Sur.Max, and PO.21B.Sur.Max, which were influenced by the Antarctic Surface Water (AASW), i.e., characterized by higher temperatures and dissolved oxygen and lower salinities. The highest values were found at PB49.Min.Bot, PB53.Bot, and PB62.Bot, influenced by the modified Warm Deep Water, with relatively lower temperatures and oxygen and higher salinities (Figure 2).
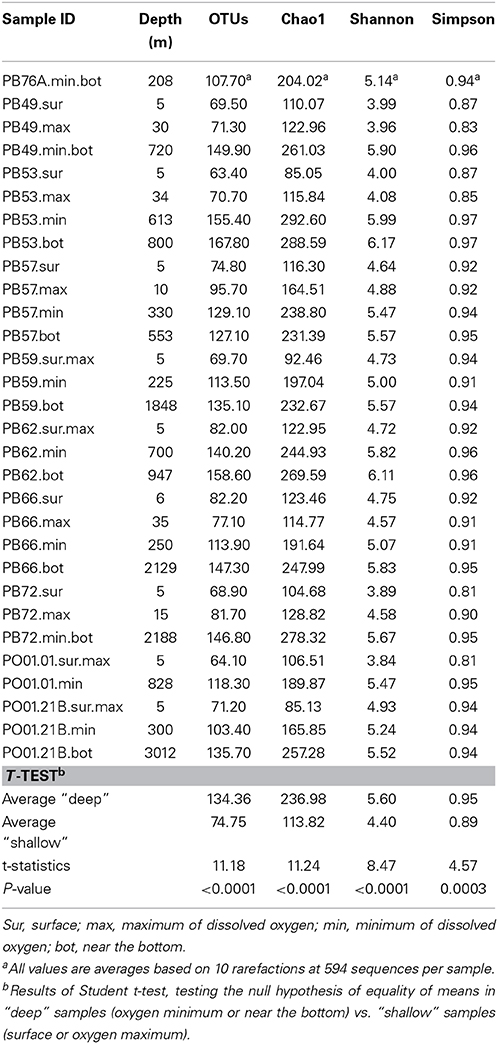
Table 2. Values of alpha-diversity estimates.
Microbial Community Structure and Environmental Parameters
Beta-diversity analysis using UPGMA clustering revealed a clear separation of samples originating either from the surface or oxygen maximum from samples originating either from near the seafloor or the oxygen minimum (Figure 4), which was statistically supported by adonis analysis (F = 28.76, r2 = 0.51, P < 0.001). This was further corroborated by the non-metric multidimensional scaling (nMDS) analyses, which also revealed a clear distinction between surface and deeper samples (Figure 5). nMDS further revealed that all the environmental parameters were significantly (p < 0.05) correlated with microbial community composition, although the highest correlations were observed for salinity (r2 = 0.93), dissolved oxygen (r2 = 0.83), and nitrate (r2 = 0.79). The surface samples were dominated by Gammaproteobacteria (relative abundance between 19.3 and 76.0%, Figure 4), Bacteroidetes (18.3–66.1%), and Alphaproteobacteria (3.4–27.0%). Nearly 50% of the sequences from deeper samples belonged to archaeal OTUs, with relative abundance ranging from 14.6 to 38.6% for Thaumarchaeota and from 11.5 to 38.5% for Euryarchaeota. Deltaproteobacteria and Planctomycetes were also mainly found in deep waters. As in surface waters, the class Gammaproteobacteria was also found to be abundant in the deep waters, with percentages varying from 12.0 to 40.1%. For surface samples (all collected at 5 m water depth), weighted normalized Unifrac distance between pairs of samples was plotted against geographical distance (Supplementary Figure 1). In general, Unifrac distance was positively correlated to geographical distance, except for 5 outlier pairs of samples (in red in Supplementary Figure 1A). These five outlier pairs were between PO01.21B Sur.Max and PB62 Sur.Max, PB59 Sur.Max, PB57 Surface, PB49 Surface, and PB53 Surface. When they were removed from the analysis, there was a strong correlation between Unifrac distance and geographical distance (Supplementary Figure 1B).
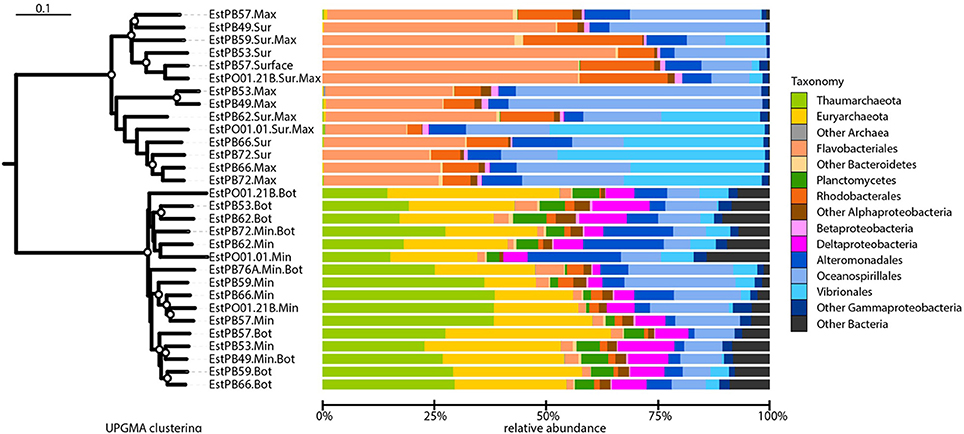
Figure 4. Beta-diversity analysis and taxonomic composition of Southern Ocean samples collected from near the seafloor (Bot), at the minimum or maximum oxygen depth (Min or Max, respectively), or at the surface (Sur). Samples IDs are as shown in Table 2. Left: unweighted pair group method with arithmetic mean (UPGMA) dendrogram based on weighted-normalized unifrac distances. Bootstrap values higher than 50% based on 100 resamplings are shown as white circles on the branches. The bar represents 10% dissimilarity. Right: relative abundances of archaeal and bacterial taxa. Some major taxa are shown at the order level for better taxonomic resolution.
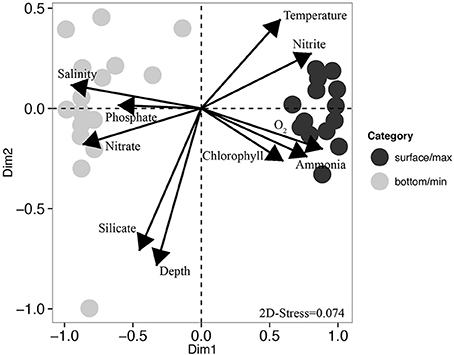
Figure 5. Non-metric multidimensional scaling (nMDS) ordination. Dark circles represent samples collected either at the surface or at the oxygen maximum, and gray circles represent samples collected either at the oxygen minimum or close to the seafloor. Each arrow represents one environmental gradient significantly correlated to the ordination (envfit, p < 0.05). The arrow points to the direction of the most rapid change in the environment (direction of the gradient) and its length is proportional to the correlation between ordination and environmental variable (strength of the gradient).
To determine which taxa were most contributing to the observed difference between shallow (surface or maximum oxygen) and deep samples (seafloor or minimum oxygen), we tested for difference in the relative abundances between the two groups, at the phylum, family and OTU taxonomic resolution (Figure 6). At the phylum and family levels (Figures 6A,B), deep samples were significantly enriched in Euryarchaeota (Marine Group II and III), Crenarchaeota (Cenarchaeacea), Planctomycetes (Pirellulaceae), SAR406 (group AT714017), and Chloroflexi. Shallow samples showed higher contributions of Bacteroidetes (Flavobacteriaceae and Cryomorphaceae), Rhodobacteraceae and Oceanospirillaceae (Figures 6A,B). In total, 19 OTUs showed significant variations between deep and shallow samples (Figure 6C). Nine of them were more abundant in deep waters than in shallow waters, including 8 archaeal OTUs (related to Nitrosopumilus, MG II and MG III) and one deltaproteobacterial OTU (related to SAR324). The remaining 10 OTUs were significantly more abundant in shallow samples, including 6 flavobacterial OTUs (2 related to Polaribacter), 3 gammaproteobacterial OTUs (related to Oceanospirillales and Alteromonadales) and one alphaproteobacterial OTU (Octadecabacter).
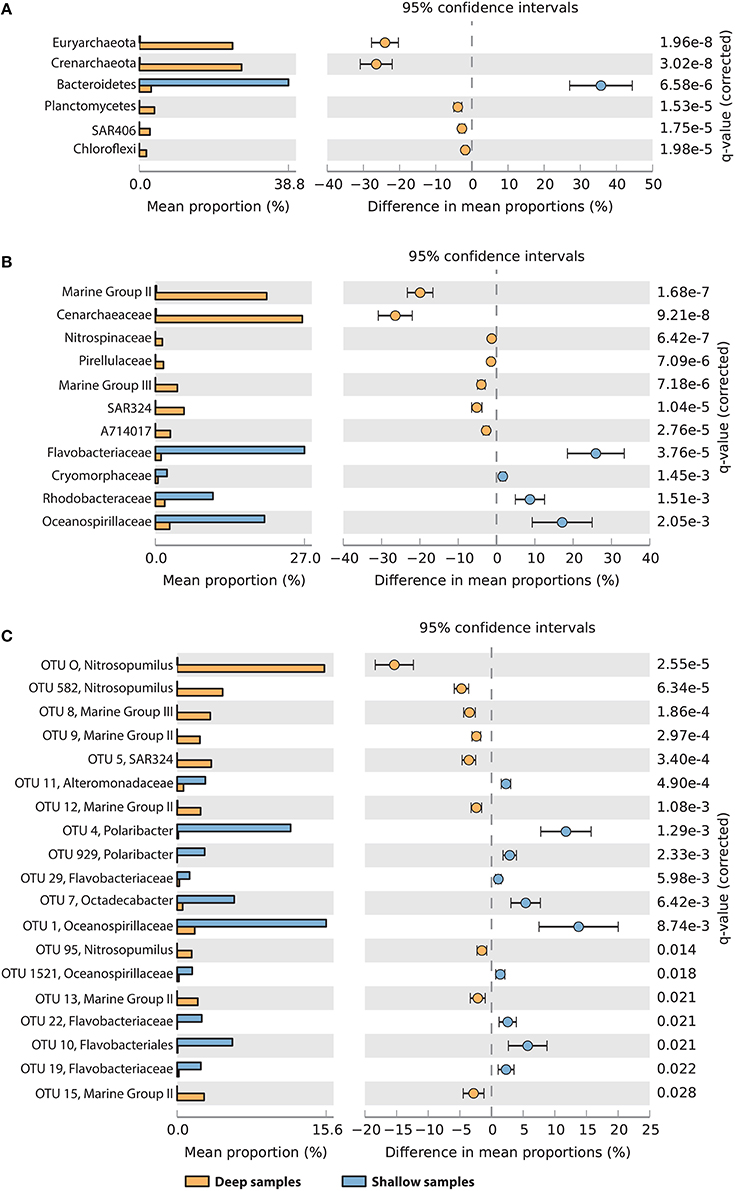
Figure 6. Taxa showing significantly different relative abundances between “deep” samples (yellow, oxygen minimum, or seafloor) and “shallow” samples (blue, oxygen maximum or surface), at the phylum (A), family (B), and OTU (C) levels. For OTUs, the best taxonomic assignment is reported. Data were compared using Welch's t-test with FDR correction of p-values. Differences were considered significant if q < 0.05 and filtered for effect sizes (difference between proportions >1 or ratio between proportions >4).
Discussion
Surface vs. Deep-Sea Microbial Community Diversity and Structure
The present work showed a clear distinction of the microbial community structure according to depth and characteristics of the water masses. The clear differences in microbial community structure based on depth were consistent with previous studies of strong vertical stratification in community composition (DeLong et al., 2006; Brown et al., 2009; Luria et al., 2014). Results from nMDS analyses showed that the differences in microbial communities could be explained by a combination of environmental factors, with salinity, dissolved oxygen, and nitrate—all factors that are influenced by water column vertical stratification—having the strongest influence. A similar trend was observed by Luria et al. (2014), who found that salinity and silicate were the factors best explaining the differences in bacterial community composition, whereas for the eukaryotic community composition it was temperature and oxygen. However, while some of the factors determined in our study might directly influence the community composition, for example the oxygen concentration, we cannot exclude the possibility that they might co-vary with other parameters that we did not measure, like the organic matter composition.
Communities from above 100 m depth were dominated by Gammaproteobacteria (mainly from the orders Oceanospirillales, Vibrionales and Alteromonadales), Bacteroidetes (mainly Polaribacter-related) and Alphaproteobacteria, whereas Archaea and Gammaproteobacteria prevailed in deeper waters, confirming results of previous studies (Ghiglione and Murray, 2012; Grzymski et al., 2012; Jamieson et al., 2012; Williams et al., 2012; Wilkins et al., 2013b; Luria et al., 2014). The identified microbes in the surface waters correspond to a predominantly heterotrophic summer community subsisting on organic matter produced by photoautotrophs, a model previously derived for a coastal site off Palmer Station using “omic” approaches (Grzymski et al., 2012; Williams et al., 2012). In contrast, the deep-water communities correspond to the winter surface community, which is characterized by a higher proportion of potentially chemolithoautotrophic bacteria and archaea (Grzymski et al., 2012; Williams et al., 2012). In fact, the pronounced differences in composition when comparing the austral summer and winter microbial communities and the similarities between the temporal (seasonal) and the spatial (depth) variation has been noted early on by Murray et al. (1998), and been confirmed in a number of subsequent studies (Murray and Grzymski, 2007; Manganelli et al., 2009; Ghiglione and Murray, 2012; Grzymski et al., 2012; Williams et al., 2012). A similar pattern was recently reported by Luria et al. (2014), who sampled at the surface and at 100 m just below the euphotic zone at 4 stations on the continental shelf of the WAP. Luria et al. (2014) speculate that the reason the winter surface community resembles the deeper communities is due to the fact that the summer surface community develops in the cold, salty Winter Water as it warms and freshens in spring and summer. However, in contrast to the deep communities identified in the present study, the summer communities at 100 m reported by Luria et al. (2014) still contain a signal of the community in the winter surface layer, which becomes isolated at about 100 m depth by the summer warming, for example in the form of a larger contribution by Alphaproteobacteria and Bacteroidetes, indicating that the base of the photic zone represents a transition between the summer surface community and the deep-water communities. The fact that the communities in the deep-ocean are similar to the winter surface community further suggests that the conditions in the water column below the photic zone are quite homogenous, and it appears that deep-water communities are carried to the surface, where they can thrive in the winter and get outcompeted by the summer community (Murray et al., 1998; Luria et al., 2014).
Surface Community
Communities found in surface and maximum oxygen samples were dominated by heterotrophic bacteria, likely consuming the phytoplankton-derived organic matter. This includes members of the Oceanospirillales and Alteromonadales within Gammaproteobacteria, which were significantly more abundant in shallow waters than in deep samples. Gammaproteobacteria are known to play an important ecological role in the degradation of organic carbon by producing extracellular hydrolytic enzymes (such as amylases, proteases, lipases, and DNAses) (Dang et al., 2009), although some Oceanospirillales have also been shown to possess genes for carbon fixation (Calvin cycle), and thus have the potential to grow as chemoautotrophs (Swan et al., 2011). The greater relative abundance of Bacteroidetes at the surface compared to deeper waters also likely reflects their potential association with phytoplankton, since many cultivated species of this phylum have specialized in the degradation of high molecular weight compounds derived from primary producers (Wilkins et al., 2013b). The most representative genus belonging to the Bacteroidetes was found to be Polaribacter—a gas-vacuolated, proteorhodopsin-containing Flavobacteria that is prevalent in Antarctica and Arctic seawater (Wilkins et al., 2013b). Grzymski et al. (2012) reported sequences affiliated with the genus Polaribacter only in summer (comprising 11% of the 16S rRNA gene reads), and speculate that they are seeded from phytoplankton blooms following the melting of sea ice. The class Alphaproteobacteria was mainly represented by the order Rhodobacterales, known as a common and dominant primary surface colonizing group (Dang et al., 2008). We also identified sequences belonging to the SAR11 clade, which has been identified as a major player of microbial communities in the surface ocean, including the Southern Ocean (Brown et al., 2012).
Interestingly, we found that the surface communities became more dissimilar from each other with geographical distance, indicating that different regions harbor distinct communities. This is in contrast to results reported by Luria et al. (2014), who did not find a relationship with geographical distance. Luria et al. (2014) argue that similar conditions observed in summer lead to similar surface communities, whereas differences in communities might be expected at times when conditions change, for example in response to transitions between seasons (spring-summer or summer-fall). We sampled in late summer at the transition to fall, which would fit with this scenario. As an exception to the trend with geographical distance, we found that the surface community of PO01.21B, which was the station furthest away from shore, was more similar to samples obtained closer to shore than distance would suggest. One possible explanation could be that PO01.21B was located 30 m away from the sea ice edge, leading to the development of communities that are more similar to communities in coastal waters, which appear to be dominated by Flavobacteria. Also, these stations are all characterized by elevated temperature and chlorophyll-a concentrations, suggesting that increased activity due to higher temperatures and the availability of organic matter might be driving this difference.
Deep Community
The deep-water masses we sampled (Circumpolar Deep Water, Antarctic Intermediate Water, and Antarctic Bottom Water) were dominated by Thaumarchaeota (Nitrosopumilus), Euryarchaeota (MGII and III), and Gammaproteobacteria, largely in line with the few studies that have analyzed the microbial communities of deep waters of the Southern Ocean (López-García et al., 2001; Manganelli et al., 2009). Thaumarchaeota may be responsible for nitrification in the deep ocean (Könneke et al., 2005; Wuchter et al., 2006; Nakagawa et al., 2007; Kalanetra et al., 2009), whereas Euryarchaeota belonging to the MGII clade have been shown to be motile, proteorhodopsin-containing photoheterotrophs that have the potential to degrade proteins and lipids (Iverson et al., 2012). In contrast to deep waters, we detected almost no 16S rRNA genes from Archaea above 100 m depth, in line with previous studies sampling surfaces communities in austral summer (Murray et al., 1998; Murray and Grzymski, 2007; Manganelli et al., 2009; Ghiglione and Murray, 2012; Grzymski et al., 2012; Williams et al., 2012). The ammonia-oxidizing Thaumarchaeota have been shown to be very sensitive to photoinhibition (Merbt et al., 2012), which could explain their decline during periods of extended illumination, for example during the summer in Southern Ocean waters when light levels are highest (Murray et al., 1998; Grzymski et al., 2012). In line with this hypothesis, metaproteomic analysis found that Thaumarchaeota/Nitrosopumilus maritimus represented 30% of all microbial proteins in winter coastal Antarctic Peninsula samples, whereas no proteins were detected in the summer (Williams et al., 2012). On the other hand, the fact that we did not find any euryarchaeotal sequences in the surface samples is somewhat surprising, considering that some have been shown to contain proteorhodopsin, and thus could grow as photoheterotrophs (Iverson et al., 2012). Indeed, in contrast to Thaumarchaeota, Euryarchaeota have been frequently identified in surface waters in other oceanic regions, including the summer surface community in the Southern Drake Passage (Manganelli et al., 2009). This discrepancy might in part be explained by the partial coverage of the primers we used toward Euryarchaeota (61.9%). However, their complete absence in surface samples in our dataset suggests that further work is required to elucidate the factors that govern the distribution of Euryarchaeota in the Southern Ocean. Although less prevalent than Archaea and Gammaproteobacteria, Deltaproteobacteria (dominated by the SAR324 clade) and Planctomycetes were almost exclusively found in deep waters. Deltaproteobacterial sequences belonging to SAR324 have previously been found in deeper layers and also as part of the Antarctic surface winter community (López-García et al., 2001; Grzymski et al., 2012; Luria et al., 2014), where they are likely to contribute to chemosynthetic production due to their versatile metabolism (Swan et al., 2011; Sheik et al., 2014). We further detected sequences belonging to the SAR406 clade, which has been frequently identified in the deep-ocean (Rappé et al., 2000), including Antarctic deep waters (López-García et al., 2001). Their ecological role remains to be elucidated.
Conclusions
We found a clear separation between surface and deeper samples of Bransfield Strait and near the ice edge north of the AP, considering the microbial composition, richness, and diversity. Communities originating from the transitional waters (from the modified Warm Deep Water) showed the highest diversity, whereas communities influenced by surface waters (Antarctic Surface Water) exhibited the lowest. A combination of environmental factors drives the microbial community structure in Bransfield Strait, especially salinity, dissolved oxygen, and nitrate which showed the highest correlation indices with the ordination. Among the groups identified in this study, Thaumarchaeota (Nitrosopumilus-related phylotypes), Gammaproteobacteria (Oceanospirillales), and Deltaproteobacteria (SAR 324) are likely to contribute to chemosynthetic production in deep waters of the Southern Ocean, and also at the surface in winter, when light is a limiting factor. On the other hand, groups like Bacteroidetes, Gammaproteobacteria (Vibrionales, Alteromodales), Euryarchaeota (MGII) and Alphaproteobacteria (Rhodobacterales) are likely to have a heterotrophic metabolism and contribute to organic matter degradation. Our results represent a baseline study of the microbial community in this critical area of the Southern Ocean, and also extend the recent survey of Luria et al. (2014) further to the north. These data will help to understand how the dynamic seascape of the Southern Ocean shapes the microbial community composition and provide an invaluable reference point to evaluate how the system might respond to future changes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the captain Newton Pinto Homem (XXXI Operantar) and the crew of the Npo. Almirante Maximiano (H41) for their assistance in collecting samples. We also thank the scientific team from the Projects ProOasis, Polar Canion, Interbiota and INCT-Mar-COI for sharing their knowledge and the environmental data, as well as Sievert's lab team (Jesse, Lara, and Emelia) for their help. We thank both reviewers for their comments and suggestions that improved the paper. This work was supported by the Brazilian National Counsel of Technological and Scientific Development (Polar Canion CNPq 556848/2009-8, ProOasis CNPq 565040/2010-3, Interbiota CNPq 407889/2013-2 and INCT-MAR-COI). Alex Enrich-Prast received a CNPq Productivity fellowship. Camila N. Signori was supported by a WHOI Mary Sears Visitor Award (for the microbial community analyses) and by the Brazilian Federal Agency for Support and Evaluation of Graduate Education (CAPES) for the “Doctorate Sandwich” scholarship (n. 18835/12-0).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2014.00647/abstract
References
Agogué, H., Lamy, D., Neal, P. R., Sogin, M. L., and Herndl, G. J. (2011). Water mass-specificity of bacterial communities in the North Atlantic revealed by massively parallel sequencing. Mol. Ecol. 20, 258–274. doi: 10.1111/j.1365-294X.2010.04932.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Anisimov, O. A., Vaughan, D. G., Callaghan, T. V., Furgal, C., Marchant, H., Prowse, T. D., et al. (2007). “Polar regions (Arctic and Antarctic),” in Climate Change 2007: Impacts, Adaptation and Vulnerability. Contribution of Working Group II to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, eds M. L. Parry, O. F. Canziani, J. P. Palutikof, P. J. van der Linden, and C. E. Hanson (Cambridge: Cambridge University Press), 653–685.
Azam, F. (1998). Microbial control of oceanic carbon flux: the plot thickens. Science 280, 694–696. doi: 10.1126/science.280.5364.694
Azam, F., and Malfatti, F. (2007). Microbial structuring of marine ecosystems. Nat. Rev. Microbiol. 5, 782–791. doi: 10.1038/nrmicro1747
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boström, K. H., Simu, K., Hagström, A., and Riemann, L. (2004). Optimization of DNA extraction for quantitative marine bacterioplankton community analysis. Limnol. Oceanogr. Methods 2, 365–373. doi: 10.4319/lom.2004.2.365
Bragg, L., Stone, G., Imelfort, M., Hugenholtz, P., and Tyson, G. W. (2012). Fast, accurate error-correction of amplicon pyrosequences using Acacia. Nat. Methods 9, 425–426. doi: 10.1038/nmeth.1990
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brown, M. V., Lauro, F. M., DeMaere, M. Z., Muir, L., Wilkins, D., Thomas, T., et al. (2012). Global biogeography of SAR11 marine bacteria. Mol. Syst. Biol. 8, 595. doi: 10.1038/msb.2012.28
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brown, M. V., Philip, G. K., Bunge, J. A., Smith, M. C., Bissett, A., Lauro, F. M., et al. (2009). Microbial community structure in the North Pacific Ocean. ISME J. 3, 1374–1386. doi: 10.1038/ismej.2009.86
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dalla Rosa, L., Secchi, E. R., Maia, Y. G., Zerbini, N. A., and Heide-Jorgersen, M. P. (2008). Movements of satellite-monitored humpback whales on their feeding ground along the Antarctic Peninsula. Polar Biol. 31, 771–781. doi: 10.1007/s00300-008-0415-2
Dang, H., Li, T., Chen, M., and Huang, G. (2008). Cross-ocean distribution of Rhodobacterales Bacteria as primary surface colonizers in temperate coastal marine waters. App. Environ. Microbiol. 74, 52–60. doi: 10.1128/AEM.01400-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dang, H. Y., Zhu, H., Wang, J., and Li, T. G. (2009). Extracellular hydrolytic enzyme screening of culturable heterotrophic bacteria from deep-sea sediments of the Southern Okinawa Trough. World J. Microbiol. Biotechnol. 25, 71–79. doi: 10.1007/s11274-008-9865-5
DeLong, E. F., and Karl, D. M. (2005). Genomic perspectives in microbial oceanography. Nature 437, 336–342. doi: 10.1038/nature04157
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
DeLong, E. F., Preston, C. M., Mincer, T., Rich, V., Hallam, S. J., Frigaard, N. U., et al. (2006). Community genomics among stratified microbial assemblages in the ocean's interior. Science 311, 496–503. doi: 10.1126/science.1120250
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Duarte, V. S. (2006). Estrutura e Variabilidade Interanual das Massas de Água no Estreito de Bransfield (Antártica) Durante os verões Austrais de 2003 e 2004. Dissertação de Mestrado do Programa de Pós Graduação em Geociências da Universidade Federal do Rio Grande, Rio Grande do Sul, 143.
Edgar, R. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Figueiras, F. G., Estrada, M., López, O., and Arbones, B. (1998). Photosynthetic parameters and primary production in the Bransfield Strait: relationships with mesoscale hydrographic structures. J. Mar. Syst. 17, 129–141. doi: 10.1016/S0924-7963(98)00034-7
Frias-Lopez, J., Shi, Y., Tyson, G. W., Coleman, M. L., Schuster, S. C., Chisholm, S. W., et al. (2008). Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. U.S.A. 105, 3805–3810. doi: 10.1073/pnas.0708897105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garcia, M. A., López, O., Sosédra, J., Espino, M., Gràcia, V., Morrison, G., et al. (1994). Mesoscale variability in the Bransfield Strait region (Antarctica) during austral Summer. Ann. Geophys. 12, 856–867. doi: 10.1007/s00585-994-0856-z
Ghiglione, J. F., and Murray, A. E. (2012). Pronounced summer to winter differences and higher wintertime richness in coastal Antarctic marine bacterioplankton. Environ. Microbiol. 14, 617–629. doi: 10.1111/j.1462-2920.2011.02601.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gordon, A. L., and Huber, B. L. (1995). Warm Weddell deep water west of Maud Rise. J. Geophys. Res. 100, 13747–13753. doi: 10.1029/95JC01361
Grzymski, J. J., Riesenfeld, C. S., Williams, T. J., Dussaq, A. M., Ducklow, H. W., Erickson, M., et al. (2012). A metagenomic assessment of winter and summer bacterioplankton from Antarctica Peninsula coastal surface waters. ISME J. 6, 1901–1915. doi: 10.1038/ismej.2012.31
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iverson, V., Morris, R. M., Frazar, C. D., Berthiaume, C. T., Morales, R. L., and Armbrust, E. V. (2012). Untangling genomes from metagenomes: revealing an uncultured class of marine Euryarchaeota. Science 335, 587–590. doi: 10.1126/science.1212665
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jamieson, R. E., Rogers, A. D., Billett, D. S. M., Smale, D. A., and Pearce, D. A. (2012). Patterns of marine bacterioplankton biodiversity in the surface waters of the Scotia Arc, Southern Ocean. FEMS Microbiol. Ecol. 80, 452–468. doi: 10.1111/j.1574-6941.2012.01313.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kalanetra, K. M., Bano, N., and Hollibaugh, T. (2009). Ammonia-oxidizing Archaea in the Arctic ocean and Antarctic coastal waters. Environ. Microbiol. 11, 2434–2445. doi: 10.1111/j.1462-2920.2009.01974.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Karl, D. M. (2007). Microbial oceanography: paradigms, processes and promise. Nat. Rev. Microbiol. 5, 759–769. doi: 10.1038/nrmicro1749
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucl. Acids Res. 41, 1–11. doi: 10.1093/nar/gks808
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Könneke, M., Bernhard, A. E., de la Torre, J. R., Walker, C. B., Waterbury, J. B., and Stahl, D. A. (2005). Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 437, 543–546. doi: 10.1038/nature03911
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Letunic, I., and Bork, P. (2007). Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. doi: 10.1093/bioinformatics/btl529
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Letunic, I., and Bork, P. (2011). Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucl. Acids Res. 39. W475–W478. doi: 10.1093/nar/gkr201
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
López-García, P., López-López, A., Moreira, D., and Rodríguez-Valera, F. (2001). Diversity of free-living prokaryotes from a deep-sea site at the Antarctic Polar Front. FEMS Microbiol. Ecol. 36, 193–202. doi: 10.1016/S0168-6496(01)00133-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lozupone, C., and Knight, R. (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luria, C. M., Ducklow, H. W., and Amaral-Zettler, L. (2014). Marine bacterial, archaeal and eukaryotic diversity and community structure on the continental shelf of the Western Antarctic Peninsula. Aquat. Microb. Ecol. 73, 107–121. doi: 10.3354/ame01703
Manganelli, M., Malfatti, F., Samo, T. J., Mitchell, B. G., Wang, H., and Azam, F. (2009). Major role of microbes in carbon fluxes during austral winter in the Southern Drake Passage. PLoS ONE 4:e6941. doi: 10.1371/journal.pone.0006941
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., DeSantias, T. Z., Probst, A., et al. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. doi: 10.1038/ismej.2011.139
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Merbt, S. N., Stahl, D. A., Casamayor, E. O., Martí, E., Nicol, G. W., and Prosser, J. I. (2012). Differential photoinhibition of bacterial and archaeal ammonia oxidation. FEMS Microbiol. Lett. 327, 41–46. doi: 10.1111/j.1574-6968.2011.02457.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murray, A. E., and Grzymski, J. J. (2007). Diversity and genomics of Antarctic marine microorganisms. Philos. Trans. R. Soc. B 362, 2259–2271. doi: 10.1098/rstb.2006.1944
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murray, A. E., Preston, C. M., Massana, R., Taylor, L. T., Blakis, A., Wu, K., et al. (1998). Seasonal and spatial variability of bacterial and archaeal assemblages in the coastal waters near Anvers Island, Antarctica. Appl. Environ. Microbiol. 64, 2585–2595.
Nakagawa, T., Mori, K., Kato, C., Takahashi, R., and Tokuyama, T. (2007). Distribution of cold-adapted ammonia-oxidizing microorganisms in the deep-ocean of the Northeastern Japan Sea. Microbes Environ. 22, 365–372. doi: 10.1264/jsme2.22.365
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O'Hara, R. B., et al. (2013). Vegan: Community Ecology Package. R package version 2.0-9. Available online at: http://CRAN.R-project.org/package=vegan
Parks, D. H., and Beiko, R. G. (2010). Identifying biologically relevant differences between metagenomic communities. Bioinformatics 26, 715–721. doi: 10.1093/bioinformatics/btq041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rappé, M. S., Vergin, K., and Giovannoni, S. J. (2000). Phylogenetic comparisons of a coastal bacterioplankton community with its counterparts in open ocean and freshwater systems. FEMS Microbiol. Ecol. 33, 219–232. doi: 10.1016/S0168-6496(00)00064-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Robertson, R., Visbeck, M., Gordon, A. L., and Fahrbach, E. (2002). Long-term temperature trends in the deep waters of the Weddell Sea. Deep-Sea Res. II 49, 4791–4806. doi: 10.1016/S0967-0645(02)00159-5
Sangrà, P., Gordo, C., Hernández-Arencibia, M., Marrero-Díaz, A., Rodríguez-Santana, A., Stegner, A., et al. (2011). The Bransfield current system. Deep-Sea Res. I 58, 390–402. doi: 10.1016/j.dsr.2011.01.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Santoso, A., England, M. H., and Hirst, A. C. (2006). Circumpolar Deep Water circulation and variability in a coupled climate model. J. Phys. Oceanogr. 36, 1523–1552. doi: 10.1175/JPO2930.1
Schofield, O., Ducklow, H. W., Martinson, D. G., Meredith, M. P., Moline, M. A., and Fraser, W. R. (2010). How do polar marine ecosystems respond to rapid climate change? Science 328, 1520–1523. doi: 10.1126/science.1185779
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sheik, C. S., Jain, S., and Dick, G. J. (2014). Metabolic flexibility of enigmatic SAR324 revealed through metagenomics and metatranscriptomics. Environ. Microbiol. 16, 304–317. doi: 10.1111/1462-2920.12165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smetacek, V., and Nicol, S. (2005). Polar ocean ecosystems in a changing world. Nature 437, 362–368. doi: 10.1038/nature04161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sogin, M. L., Morrison, H. G., Huber, J. A., Welch, D. M., Huse, S. M., Neal, P. R., et al. (2006). Microbial diversity in the deep sea and the underexplored rare biosphere. Proc. Natl. Acad. Sci. U.S.A. 103, 12115–12120. doi: 10.1073/pnas.0605127103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Swan, B. K., Martinez-Garcia, M., Preston, C. M., Sczyrba, A., Woyke, T., Lamy, D., et al. (2011). Potential for chemolithoautotrophy among ubiquitous Bacteria lineages in the dark ocean. Science 333, 1296–1300. doi: 10.1126/science.1203690
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tokarczyk, R. (1987). Classification of water masses in the Bransfield Strait and Southern part of the Drake Passage using a method of statistical multidimensional analysis. Pol. Polar Res. 8, 333–336.
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wilkins, D., Sheree, Y., Williams, T. J., Allen, M. A., Brown, M. V., DeMaere, M. Z., et al. (2013b). Key microbial drivers in Antarctic aquatic environments. FEMS Microbiol. Rev. 37, 303–335. doi: 10.1111/1574-6976.12007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wilkins, D., van Sebille, E., Rintoul, S. R., Lauro, F. M., and Cavicchioli, R. (2013a). Advection shapes Southern Ocean microbial assemblages independent of distance and environment effects. Nat. Commun. 4, 2457. doi: 10.1038/ncomms3457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Williams, T. J., Long, E., Evans, F., Demaere, M. Z., Lauro, F. M., Raftery, M. J., et al. (2012). A metaproteomic assessment of winter and summer bacterioplankton from Antarctic Peninsula coastal surface waters. ISME J. 6, 1883–1900. doi: 10.1038/ismej.2012.28
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wuchter, C., Abbas, B., Coolen, M. J. L., Herfort, L., van Bleijswijk, J., Timmers, P., et al. (2006). Archaeal nitrification in the ocean. Proc. Natl. Acad. Sci. U.S.A. 103, 12317–12322. doi: 10.1073/pnas.0600756103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yooseph, S., Nealson, K. H., Rusch, D. B., McCrow, J. P., Dupont, C. L., Kim, M., et al. (2010). Genomic and functional adaptation in surface ocean planktonic prokaryotes. Nature 468, 60–66. doi: 10.1038/nature09530
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: Antarctica, pyrosequencing, microbial community structure, environmental factors, microbial oceanography, climate change
Citation: Signori CN, Thomas F, Enrich-Prast A, Pollery RCG and Sievert SM (2014) Microbial diversity and community structure across environmental gradients in Bransfield Strait, Western Antarctic Peninsula. Front. Microbiol. 5:647. doi: 10.3389/fmicb.2014.00647
Received: 08 September 2014; Accepted: 07 November 2014;
Published online: 16 December 2014.
Edited by:
Hongyue Dang, Xiamen University, ChinaReviewed by:
Barbara J. Campbell, Clemson University, USAHugh Ducklow, Columbia University in the City of New York, USA
Copyright © 2014 Signori, Thomas, Enrich-Prast, Pollery and Sievert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alex Enrich-Prast, Laboratory of Biogeochemistry, Department of Ecology, Institute of Biology, Universidade Federal do Rio de Janeiro, Cidade Universitária s/n, Rio de Janeiro 68020, Brazil e-mail:YWVwcmFzdEBiaW9sb2dpYS51ZnJqLmJy;
Stefan M. Sievert, Department of Biology, Stanley W. Watson Laboratory for Biogeochemistry, Woods Hole Oceanographic Institution, Mail Stop 52, Woods Hole, MA 02543, USA e-mail:c3NpZXZlcnRAd2hvaS5lZHU=
†Present address: François Thomas, Laboratoire Interdisciplinaire des Environnements Continentaux, UMR7360 CNRS-Université de Lorraine, Vandoeuvre-les-Nancy, France
‡These authors have contributed equally to this work.