 Julia A. Gustavsen
Julia A. Gustavsen Danielle M. Winget
Danielle M. Winget Xi Tian2
Xi Tian2 Curtis A. Suttle
Curtis A. Suttle- 1Department of Earth, Ocean and Atmospheric Sciences, University of British Columbia, Vancouver, BC, Canada
- 2Bioinformatics Graduate Program, Faculty of Science, University of British Columbia, Vancouver, BC, Canada
- 3Departments of Botany, and Microbiology & Immunology, University of British Columbia, Vancouver, BC, Canada
- 4Canadian Institute for Advanced Research, Toronto, ON, Canada
Viruses in the order Picornavirales infect eukaryotes, and are widely distributed in coastal waters. Amplicon deep-sequencing of the RNA dependent RNA polymerase (RdRp) revealed diverse and highly uneven communities of picorna-like viruses in the coastal waters of British Columbia (BC), Canada. Almost 300 000 pyrosequence reads revealed 145 operational taxonomic units (OTUs) based on 95% sequence similarity at the amino-acid level. Each sample had between 24 and 71 OTUs and there was little overlap among samples. Phylogenetic analysis revealed that some clades of OTUs were only found at one site; whereas, other clades included OTUs from all sites. Since most of these OTUs are likely from viruses that infect eukaryotic phytoplankton, and viral isolates infecting phytoplankton are strain-specific; each OTU probably arose from the lysis of a specific phytoplankton taxon. Moreover, the patchiness in OTU distribution, and the high turnover of viruses in the mixed layer, implies continuous infection and lysis by RNA viruses of a diverse array of eukaryotic phytoplankton taxa. Hence, these viruses are likely important elements structuring the phytoplankton community, and play a significant role in nutrient cycling and energy transfer.
Introduction
Viruses are highly abundant and widespread in the oceans (Bergh et al., 1989; Suttle, 2005). Beyond their impacts on host mortality, viruses are significant mediators of biogeochemical processes, horizontal gene transfer, and host community diversity in the oceans (Fuhrman, 1999; Wilhelm and Suttle, 1999; Suttle, 2005). Marine viruses are important pathogens of phytoplankton (Brussaard, 2004), and have been implicated in the termination of blooms (Nagasaki et al., 1994; Schroeder et al., 2003) and with succession in phytoplankton communities (Mühling et al., 2005). Viruses have been characterized that infect a wide variety of phytoplankton such as haptophytes (Bratbak et al., 1993), prasinophytes (Mayer and Taylor, 1979; Brussaard, 2004; Brussaard et al., 2004; Derelle et al., 2008), chlorophytes (Van Etten et al., 1981), diatoms (Shirai et al., 2008), and dinoflagellates (Tomaru et al., 2004). Viruses infecting eukaryotic phytoplankton generally have very narrow host ranges (Short, 2012).
Viruses infecting marine phytoplankton have genomes comprised of double-stranded DNA (dsDNA), single-stranded DNA (ssDNA), double-stranded RNA (dsRNA), and single-stranded RNA (ssRNA) (as reviewed in Short, 2012). Their genomes and particle sizes range from very large dsDNA viruses in the Phycodnaviridae to very small ssDNA and ssRNA viruses belonging to the genus Bacilladnavirus and order Picornavirales, respectively. The order Picornavirales is comprised of positive-sense, ssRNA viruses that infect eukaryotes (Le Gall et al., 2008), including ecologically important marine protists. These viruses are small (25–35 nm), icosahedral, and have a conserved genomic organization that includes a replication area comprised of a type III helicase, a 3C-like proteinase, and a type I RNA dependent RNA polymerase (Sanfaçon et al., 2009). Isolates in the Picornavirales that are pathogens of marine protists infect a wide diversity of hosts including the bloom-forming raphidophyte Heterosigma akashiwo (Tai et al., 2003) (viral family Marnaviridae), the thraustochytrid Aurantiochytrium sp. (Takao et al., 2006; Yokoyama and Honda, 2007) (proposed viral genus Labyrnavirus) and the cosmopolitan diatoms Rhizosolenia setigera (Nagasaki et al., 2004) and Chaetoceros socialis (Tomaru et al., 2009) (proposed viral genus Bacillarnavirus). Viruses in the Picornavirales appear to be common and widely distributed in coastal waters (Culley et al., 2003; Culley and Steward, 2007).
Metagenomic and targeted gene studies are uncovering the diversity of marine RNA viruses. For example, phylogenetic analysis of RNA-dependent RNA polymerase (RdRp) sequences from seawater samples supports a monophyletic marine group within the Picornavirales (Culley et al., 2003, 2014; Culley and Steward, 2007; Tomaru et al., 2009) and several divergent clades within this marine group (Culley et al., 2003, 2014; Culley and Steward, 2007). Additionally, metagenomic analyses reveal that there are numerous sequences from aquatic RNA viruses that cannot be assigned to known taxa (Culley et al., 2006, 2014; Djikeng et al., 2009; Steward et al., 2012). Despite the high diversity of marine RNA viruses (Lang et al., 2009), the spatial and temporal distribution of different phylogenetic groups remains unreported, although there is evidence that the taxonomic structure of marine RNA viral communities is highly uneven. For example, in one sample from a metagenomic study from the coastal waters of British Columbia, 59% of the reads assembled into a single contig, while in a second sample 66% of the reads fell into four contigs, with most falling into two genotypes (Culley et al., 2006). However, with only a few 100 reads in total from the two samples, the coverage of the communities was low. Similarly, RNA viral metagenomic data from a freshwater lake (Djikeng et al., 2009) showed little identical sequence overlap among communities, although there was broad taxonomic similarity over time within a location.
Ecological questions about the distribution of marine viruses over time and space have been examined more extensively in bacteriophages, particularly those infecting cyanobacteria. For example, some data reveal no clear patterns of biogeography in cyanophage isolates locally (Clasen et al., 2013), regionally (Jameson et al., 2011) or more globally (Huang et al., 2010). Other data have shown patterns at a regional scale (Marston et al., 2013), although communities in basins that were connected were most similar and those that were separated by land or current boundaries were the least similar. Other data for marine bacteriophages have shown temporal variability (Chen et al., 2009; Wang et al., 2011; Chow and Fuhrman, 2012; Clasen et al., 2013; Marston et al., 2013). If the dynamics of marine bacteriophages and marine RNA viruses are similar, some RNA viral taxa will persist temporally and spatially, while other taxa will be detected sporadically. To test this hypothesis, we examined two samples, taken 5 months apart at the same location, and three samples taken within hours of each other, but 20 km apart in the same coastal basin.
We used high-throughput 454 pyrosequencing to obtain deep coverage of RdRp amplicon sequences and compare the richness of viruses in the Picornavirales among samples from the coastal waters of British Columbia, Canada. The results revealed a phylogenetically diverse and spatially variable community of viruses, suggesting that taxon-specific lytic events are important in shaping the phytoplankton community.
Materials and Methods
Sampling Locations
To assess viral communities from different coastal habitats, we collected samples from three sites in the Strait of Georgia (49°14.926N 123°35.682W, 49°17.890N 123°43.650W, and 49°23.890N 123°59.706W), and from Jericho Pier (49°16′36.73N, 123°12′05.41W) in British Columbia, Canada) (Figure 1). The Strait of Georgia (SOG) is an estuarine-influenced basin that is on average 22 km across, 222 km long and 150 m deep. The upper 50 m of SOG is where most of the variability in physical and chemical parameters occurs. Jericho Pier (JP) is adjacent to the shoreline, in a well-mixed location with mixed semidiurnal tides.
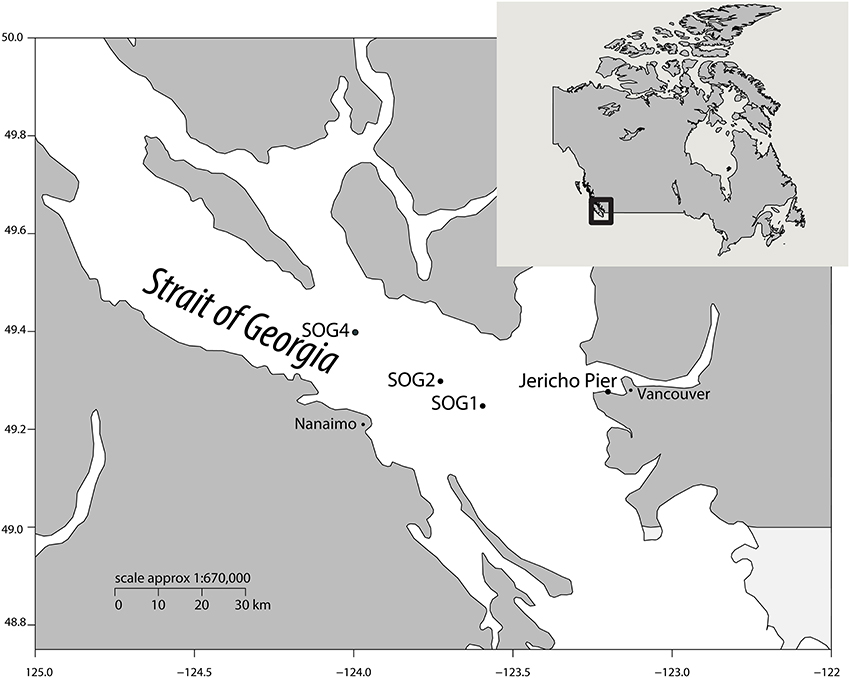
Figure 1. Location of sampling sites. Map showing the location of sampling sites within the Strait of Georgia (SOG) and Jericho Pier, adjacent to Vancouver, British Columbia, Canada. Jericho Pier was sampled in summer (JP-S) and fall (JP-F).
Sample Collection
On 28 July 2010, a rosette equipped with a Seabird SBE 25 CTD (equipped with Seabird SBE 43 dissolved oxygen sensor, WET Labs WETStar fluorometer, WET Labs C-Star transmissometer, Biospherical Instruments QSP2200PD PAR sensor) and General Oceanics GO-FLO bottles was used to collect 12 L of water from five depths between 2 and 16 m at each of three stations in SOG. The five depths from each station were combined to obtain three integrated water samples. The depths were selected to attempt to encompass the viral diversity at each station, and consisted of a near-surface sample, a sample from the isothermal zone below the mixed layer, and three samples spanning the chlorophyll maximum.
At JP, 60 L of water was pumped from the 1-m depth on 10 July and 12 October 2010. Salinity and temperature at JP were measured using a YSI probe (Yellow Springs, Ohio, USA). For all samples, the water was filtered through 142-mm diameter, 1.2-μm nominal pore-size glass-fiber (GC50 Advantec MFS, Dublin, CA., USA) and 0.22-μm pore-size polyvinyldine (Millipore Bedford, MA, USA) filters. The viral size fraction in the filtrate was concentrated to ~500 mL (viral concentrate) using tangential flow ultrafiltration using a 30 kDa MW prep-scale Spiral Wound TFF-6 cartridge (Millipore) (Suttle et al., 1991).
Phosphate, silicate and nitrate+nitrite concentrations were determined in duplicate 15-mL seawater samples filtered through 0.45-μm pore-size HA filters (Millipore) and stored at −20°C until air-segmented continuous-flow analysis on a AutoAnalyzer 3 (Bran & Luebbe, Norderstedt Germany). Chlorophyll a (Chl a) was determined in triplicate by filtering 100 mL of seawater onto 0.45 μm pore-size HA filters (Millipore), and storing the filters in the dark at −20°C until acetone extraction and then analyzed fluorometrically (Parsons et al., 1984). The average and standard error of the replicates was calculated for each sample.
Nucleic-Acid Extraction and PCR
The viral concentrate was filtered twice through 0.22-μm pore-size Durapore PVDF filters (Millipore) in a sterile Sterivex filter unit (Millipore). The filtrate, containing virus-sized particles, was pelleted by ultracentrifugation (Beckman-Coulter, Brea, California, USA) in a SW40 rotor at 108 000 g for 5 h at 12°C. The pellet was resuspended overnight in 100 μL of supernatant at 4°C. To digest free DNA, the pellets were incubated with 1U/μl DNAse with a final concentration 5 mM MgCl2 for 3 h at room temperature. Nucleic acids were extracted using a Qiamp Viral Minelute spin kit (Qiagen, Hilden, Germany) according to the manufacturer's directions. To remove DNA, the extracted viral pellets were digested with DNase 1 (amplification grade) (Invitrogen, Carlsbad, California, USA) and the reaction was terminated by adding 2.5 mM EDTA (final concentration) and incubating for 10 min at 65°C. Complementary DNA (cDNA) was generated using Superscript III reverse transcriptase (Invitrogen) with random hexamers (50 ng/μl) as per the manufacturer. PCR was performed using primer set MPL-2 for a targeted set of the marine picornavirus-like RdRp (Culley and Steward, 2007). Each reaction mixture (final volume, 50 μl) consisted of 50 ng of cDNA, 1× (final concentration) PCR buffer (Invitrogen), 2 mM MgCl2, 0.2 mM of each deoxynucleoside triphosphate (Bioline, London, UK), 1 μM of each primer, and 1 U Platinum Taq DNA polymerase. The reaction was run in a PCR Express thermocycler (Hybaid, Ashford, UK) with the following conditions: 94°C for 75 s, followed by 40 cycles of denaturation at 94°C for 45 s, annealing at 43°C for 45 s, and extension at 72°C for 60 s and a final extension step of 9 min at 72°C. PCR products were cleaned using the Minelute PCR purification kit (Qiagen).
Library Prep and Pyrosequencing
Libraries for each site were prepared for sequencing using NEBNext DNA Library Prep for 454 kit (New England Biolabs, Ipswich, Massachusetts, USA) following the manufacturer's directions, and using Ampure beads (Beckman-Coulter) for size selection and purification using a bead ratio of 0.8:1 beads:library. PCR amplicons were barcoded and sent for 454 Titanium pyrosequencing (Roche, Basel, Switzerland) at the Génome Québec Innovation Centre at the McGill University (Montreal, QC, Canada).
Sequence Analysis
Sequencing reads were quality trimmed using length settings between 100 and 600 bp, with a maximum of 3 primer mismatches for the specific primer, and denoised using the denoiser algorithm in QIIME (version 1.7) with default settings for Titanium data (Caporaso et al., 2010; Reeder and Knight, 2010). Sequences were checked for chimeras using UCHIME (Edgar, 2010) against a nucleotide database of RdRp sequences built using NCBI-BLAST+ by retrieving nucleotide sequences of the RdRp from Picornavirales viral isolates (accessed: 8 August 2012), and also using the denovo chimera check in UCHIME. The overlap of results from these two methods was defined as chimeric sequences, although none were found in this study. Non-chimeric sequences were queried using BLASTx (Altschul et al., 1990) with an e-value of 1e−3 against the database of RdRp viral isolates. All sequences with hits were retained and all sequences with no hits were then queried against the non-redundant (nr) Genbank database (Benson et al., 2007) using BLASTx with an e-value of 1e−5. All the sequences identified as contaminants or as unknown with only 1 read were removed. Remaining sequences were translated to amino acids using FragGeneScan with the 454_10 training option (Rho et al., 2010). Sequences were grouped into operational taxonomic units (OTUs) using UCLUST at a range of similarities from 50 to 100% (Figure S1) using the original seed sequences (centroids) as the output (Edgar, 2010). A similarity of 95% was chosen for this analysis for the following two main reasons: (1) When the NCBI conserved domain alignment for the RdRp region (all Picornavirales) was analyzed for percent similarity, the only sequences that displayed greater than 95% similarity in this region were strains of the same virus. (2) The sequences from the control libraries (consisting of 1 clone) clustered into 1 sequence at this percentage (see Supplemental methods). Thus clustering at 95% similarity was a way to use biological and sequence-based information to inform our choice of cut-off to collapse strain level variation and as a conservative approach to avoid variation that may be present because of the sequencing platform. Control sequences obtained by cloning and Sanger sequencing (see Supplemental Methods) were used to verify the sequence processing methodology. Raw and processed sequence data were deposited in the NCBI BioProject database ID: PRJNA267690.
Phylogenetic Analysis
All OTUs with less than 5 reads were removed and the remaining OTUs were aligned using profile alignment in Muscle (Edgar, 2004) to seed alignments of viral RdRps from the NCBI Conserved Domain Database (Marchler-Bauer et al., 2011). Sequences from other environmental surveys were clustered in the same manner as the reads in this study (using UCLUST at 95%). The clusters are cluster number followed by the Genbank accession numbers contained in that cluster. 0: 33520549, 33520547, 33520541, 33520533, 33520527, 33520521, 33520519, 33520517, 33520515, 33520513, 33520511, 33520509; 1: 157280772; 2: 33520525; 3: 157280768; 4: 157280770; 5: 568801536, 568801534, 568801530, 568801528, 568801510; 6: 157280786; 7: 157280774; 8: 157280780; 9: 157280788; 10: 568801494, 568801492, 568801488, 568801482, 568801480, 568801474, 568801470, 568801466, 568801464, 568801462, 568801458; 11: 157280776; 12: 157280778; 13: 568801516; 14: 568801616, 568801614, 568801606, 568801588, 568801586, 568801584, 568801582, 568801580, 568801574, 568801572, 568801564, 568801550, 568801548, 568801540, 568801532, 568801522, 568801520, 157280784; 15: 568801508; 16: 568801542, 568801538, 568801518, 568801486, 568801484, 568801478; 17: 568801612, 568801610, 568801608, 568801604, 568801602, 568801600, 568801598, 568801596, 568801594, 568801592, 568801590, 568801578, 568801570, 568801568, 568801552, 157280782; 18: 568801576, 568801566, 568801546, 568801544; 19: 568801562, 568801556; 20: 568801526, 568801524, 568801514, 568801512, 568801506, 568801504, 568801502, 568801500, 568801498, 568801496, 568801490, 568801472, 568801460; 21: 568801476, 568801468; 22: 568801560, 568801558, 568801554; 23: 157280744; 24: 157280758; 25: 157280748; 26: 157280746; 27: 157280742; 28: 157280766; 29: 157280756; 30: 157280762, 157280754, 157280752; 31: 157280750; 32: 157280760; 33: 157280764; 34: 33520545, 33520543, 33520535, 33520531, 33520529; 35: 33520539, 33520537, 33520523, 33520507. Alignments were masked using trimAl with the automatic heuristic (Capella-Gutierrez et al., 2009) and edited manually. ProtTest 3.2 was used for amino-acid model selection (Darriba et al., 2011) before building the initial maximum likelihood phylogenetic tree using FastTree (Price et al., 2010). Final trees were done with RA×ML using sequences belonging to viruses in the Sequiviridae as the outgroup, and the BLOSUM62 amino-acid model with 100 bootstraps (Stamatakis et al., 2008). The tree was visualized in R (R Core Team, 2014) using the ape package (Paradis, 2012) and edited in Figtree (http://tree.bio.ed.ac.uk/software/figtree/).
Statistical Analysis
Generation of rarefaction curves by random resampling of OTU abundances was performed using the vegan package (Oksanen et al., 2013) in R (R Core Team, 2014). Relative abundances were normalized by randomly resampling 10 000 times using vegan (Oksanen et al., 2013), normalizing to the library with the lowest number of reads and then taking the median. Rank-abundance curves were generated with ggplot2 (Wickham, 2009) using the normalized OTU relative abundances. Scripts used in this project are available as part of QIIME and custom user scripts used to process the data are available here: http://github.com/jooolia/RdRp_454_amplicons_Jericho_and_SOG; doi: 10.5281/zenodo.12509
Results
Environmental Parameters
The environmental parameters ranged widely among samples (Figure 2). Chlorophyll a values were lowest at Jericho Pier Fall (JP-F) at 0.16 μg L−1 (± 0.04) and highest at the Strait of Georgia Station 2 (SOG-2) at 3.2 μg L−1 (± 0.4). Silicate values for all samples were similar (range of 25.5 to 37.8 μM), except for Jericho Pier Summer (JP-S) when silicate was lower at 6.2 μM (± 0.01). Phosphate ranged between 0.90 and 1.3 μM at JP-F, SOG-1, and SOG-4, but was lower at SOG-2 (0.58 μM (± 0.03)) and lowest at JP-S (0.06 μM (± 0.04)). Nitrate + nitrite values were more variable than the other nutrients and ranged from 1.35 μM (± 0.75) at JP-S to 14.6 μM (± 0.04) at JP-F. The SOG sites were highly stratified with SOG-4 being the most stratified with a calculated mixed-layer depth of 2 m, while SOG-1 and SOG-2 were similar with a mixed-layer depth of 6 m (Table 1).
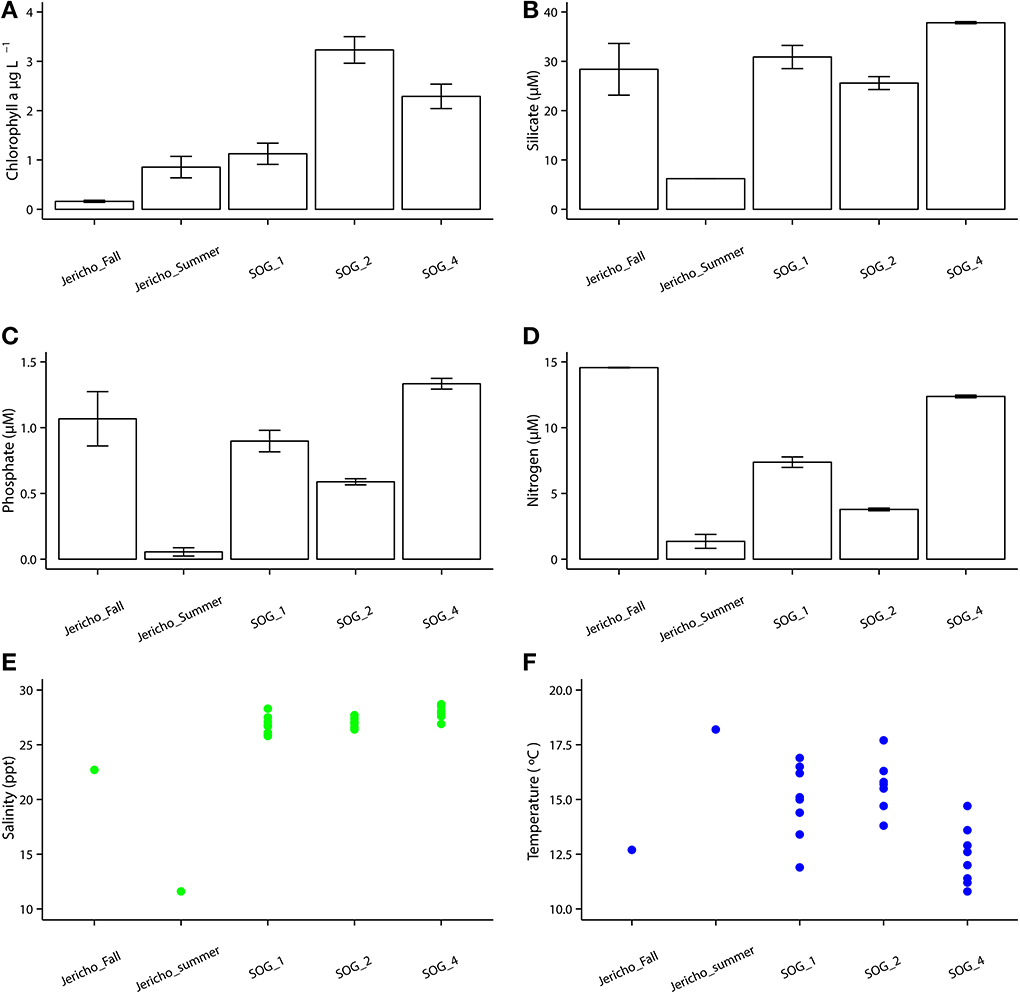
Figure 2. Environmental parameters. (A) Chlorophyll a with standard error of the mean from triplicates. (B) Silicate with standard error of the mean from duplicates. (C) Phosphate with standard error of the mean from duplicates. (D) Nitrate+ nitrite with standard error of the mean from duplicates. (E) Temperature with each point as one GO-FLO bottle (SOG samples) or from total seawater sample (Jericho samples). (F) Salinity with each point representing a seawater sample as one GO-FLO bottle (SOG samples) or from total seawater sample (Jericho samples).
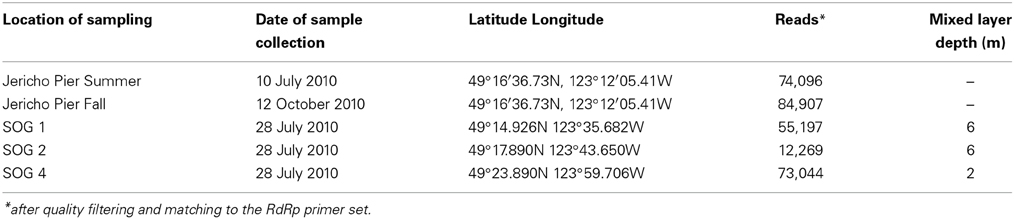
Table 1. Description of samples and resulting sequencing information.
Analysis of RdRp Sequences
After quality filtering to remove homopolymers and contaminating reads, 300 180 reads were recovered from the 5 libraries of RdRp amplicons. At all sites the rarefaction curves plateaued indicating that the depth of sampling was adequate to assess the communities (Figure 3). From these reads, 265 unique OTUs (at 95% similarity) were identified, including 108 singletons. For further analysis OTUs were excluded that did not contain recognizable RdRp motifs (Koonin, 1991; Le Gall et al., 2008), generally did not align well with other RdRp sequences and those that were not present in any sample after normalization.
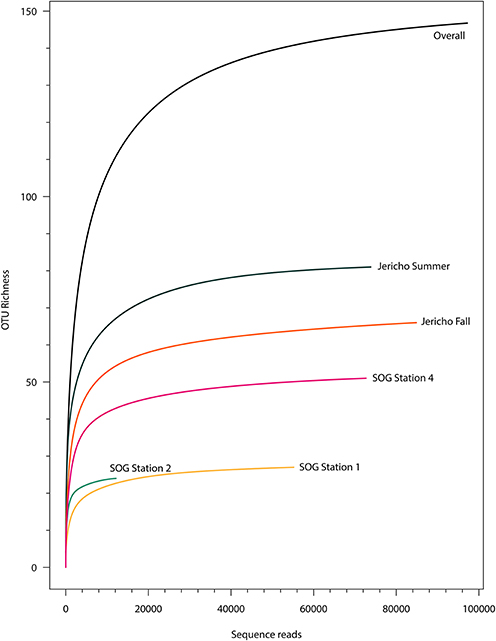
Figure 3. Rarefaction curves. Rarefaction analysis of RdRp amplicons based on Chao1 richness analysis of operational taxonomic units (OTUs) at 95% similarity. Rarefaction curves were resampled using number of reads recovered per library. Rarefaction curves plateau indicating adequate sequencing for these samples.
Using the above criteria there were 145 OTUs identified in all samples, and between 24 to 71 OTUs per site. The Jericho Pier samples had 116 OTUs of which only 10 (8.6%) were shared between sampling times (Figure 4). JP-S had the highest richness with 71 OTUs, of which 59 (83 %) were unique, while JP-F had the second highest richness (49 OTUs), of which 45 (92 %) were unique. The SOG sites together had 64 OTUs, none of which were shared among all sites. SOG-1 and SOG-2 had the lowest number of OTUs (24). SOG-1 had only three OTUs which were unique. However, 21 (33%) were shared between SOG-1 and SOG-4, and 6 (9%) between SOG-2 and SOG-4. The majority of OTUs (75%) from SOG-2 were unique, whereas most OTUs from the other SOG sites were shared with other sites (87% for SOG-1 and 63% for SOG-4).
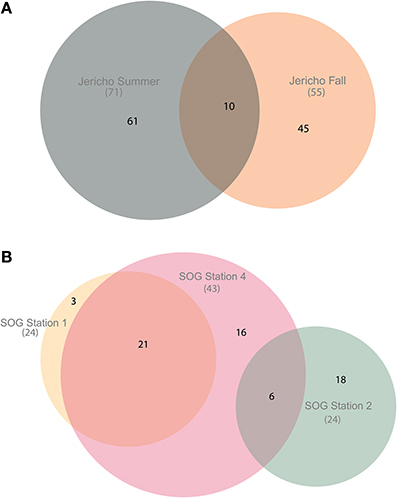
Figure 4. Euler diagrams of normalized RdRp OTUs. (A) Euler diagram of Jericho Pier samples. (B) Euler diagram of SOG samples. The OTUs presented were from reads clustered at 95% similarity, comprise only OTUs that could be aligned to the NCBI CDD RdRp alignment, and contained the RdRp motif C. The diagrams were constructed using the venneuler() algorithm (Wilkinson, 2011). The size of the circles is approximately proportional to the number of OTUs recovered per site. The overlap in the diagram describes OTUs that were found at multiple sites and the non-overlapping areas describe OTUs that were unique to that site.
Rank abundance curves of the viral OTUs showed that at each site most sequences were assigned to only a few OTUs (Figure 5). JP-S had the highest richness but the shallowest slope of these curves, demonstrating more evenness in the abundance of OTUs than at the other sites. SOG-4 and JP-S had similar rank abundance curves that were much shallower that those of SOG-1 and SOG-2 (Figure 5).
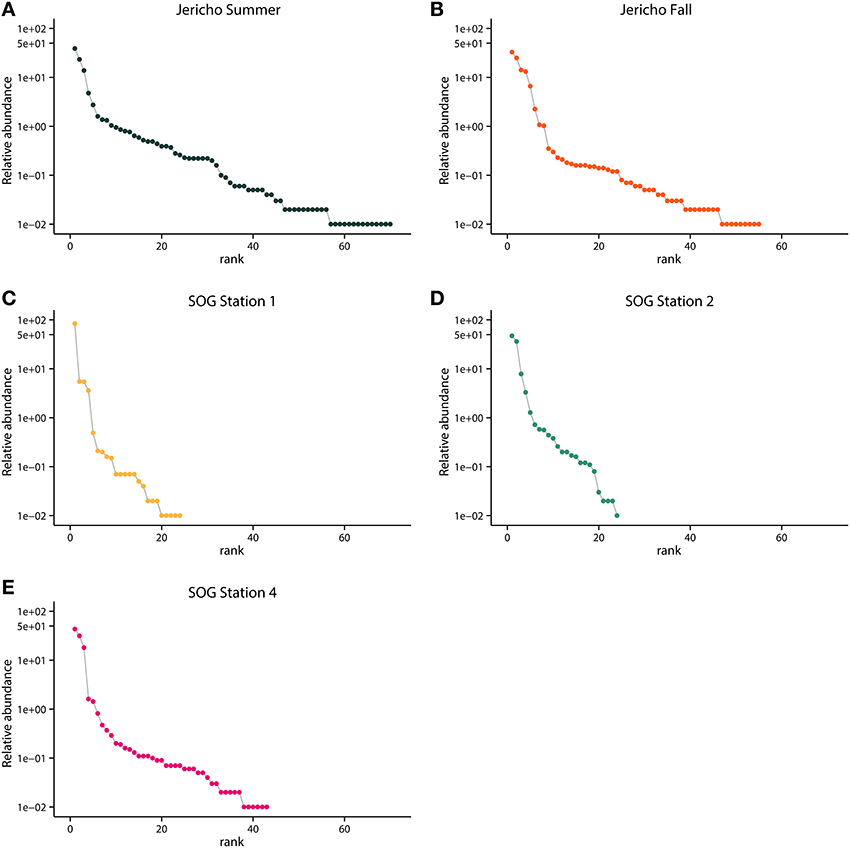
Figure 5. Rank abundance by site. Relative abundance of OTUs in each sample ordered by rank abundance: (A) Jericho Summer, (B) Jericho Fall, (C) SOG Station 1, (D) SOG Station 2, (E) SOG Station 4. OTUs were clustered at 95% amino acid similarity and OTU relative abundances were normalized to the sample with the lowest number of reads.
The OTUs that were observed in more than 5 reads were placed in phylogenetic context using a maximum likelihood RA×ML tree (Stamatakis et al., 2008) with sequences from previous RdRp gene surveys and isolated viruses (Figure 6). OTUs from this study fell within a well-supported clade that includes all the marine isolates belonging to the Picornavirales. Within this group there was a well-supported divide between OTUs grouping in the Marnaviridae clade and those grouping with sequences from viruses infecting diatoms and a thraustochytrid. The overall tree topology is not well supported, although there are a number of well-supported clades containing OTUs from this study and other environmental sequences.
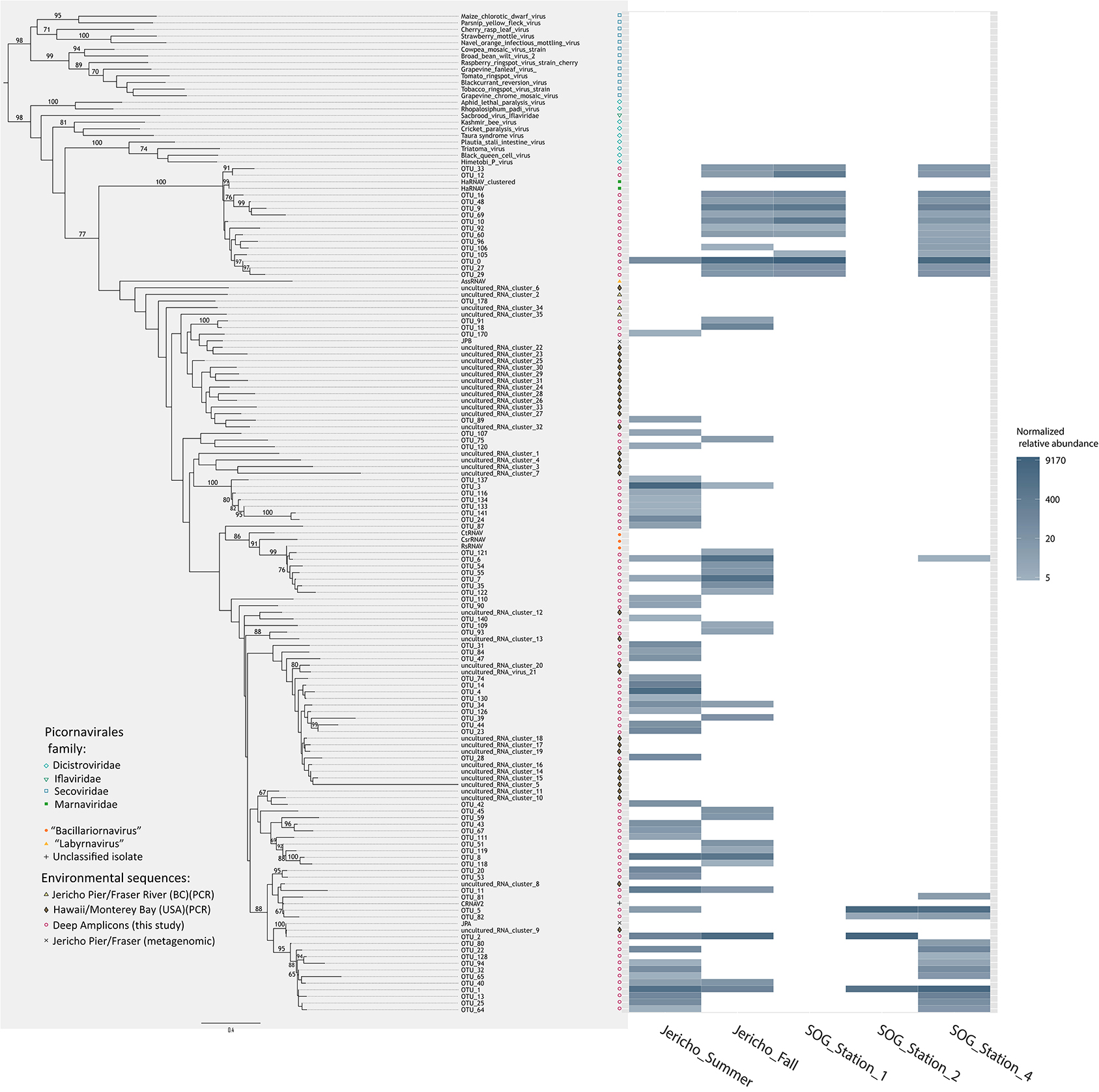
Figure 6. Phylogenetic tree with heatmap. Maximum likelihood (RA×ML) tree of RdRp OTUs that contained more than 5 reads and relevant Picornavirales sequences. Bootstrap values above 65% are labeled. Adjacent to the tips of the tree is a heatmap displaying the relative abundance of each OTU at 95% similarity by site. OTU relative abundances were normalized to the sample with the lowest number of reads.
The Marnaviridae clade had the greatest number of OTUs (10) associated with it; whereas, very few OTUs (only OTUs 89, 107, 75, and 120) from this study were assigned to clades primarily from Hawaii (Culley and Steward, 2007; Culley et al., 2014). No clade contained OTUs from all sites. The Jericho Pier samples were the most phylogenetically diverse (Figure 6, Table 2), and contained OTUs (e.g., OTUs 6, 7, 35, 31, 84, 47, 14, 4, 34, 39, 44, 23, 8, 20) that fell into clades that did not contain OTUs from any of the SOG samples. Some clades contained OTUs from both JP-S and JP-F samples; however, many OTUs within the clades were unique to one Jericho Pier sample. Phylogenetic diversity differed among samples, except for OTUs from the two SOG sites with deeper mixed layers, some of which were present in different clades resulting in similar phylogenetic diversity (Table 2).
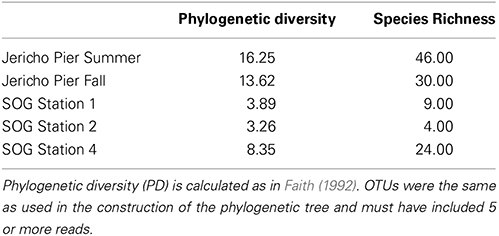
Table 2. Phylogenetic diversity, species richness.
The Strait of Georgia (SOG) sites were sampled within hours of each other, and the water at each site was pooled from multiple depths above, below, and across the chlorophyll maximum. One of the most striking differences among sites was that SOG-1 and SOG -2 had mixed layer depths of 6 m; whereas SOG-4 had a mixed-layer depth of 2 m, and much higher richness and phylogenetic diversity. Sites SOG-1 and SOG-2 had the lowest phylogenetic diversity (Table 2). All the OTUs found at SOG-1 (33, 12, 16, 9, 10, 27, 29) were within the Marnaviridae clade; similarly, all OTUs (5, 82, 2, 1) from SOG-2 were within one distantly related clade. In both cases OTUs from these clades occurred at SOG-4. SOG-2 did not have the high numbers of HaRNAV-related viruses that were found in all other samples.
Discussion
Pyrosequencing of RdRp gene fragments from coastal samples uncovered much greater genetic diversity than in previous gene surveys (Culley et al., 2003, 2014; Culley and Steward, 2007) and revealed many previously unknown taxonomic groups within the Picornavirales. As well, striking differences in the taxonomic richness among samples implies that these viruses infect a wide variety of eukaryotic plankton, but that the mortality imposed on some taxa is highly variable across space and time. Other taxonomic groups within the Picornavirales were more widespread, suggesting that infection of some planktonic taxa is more widespread and persistent. These results and their implications are discussed in detail below.
Expanding the known Diversity of Picornavirales
The high depth of sequencing and limited diversity in each library (Figure 3) gives high confidence that the population structure of RdRp amplicons in each sample has been well characterized (Kemp and Aller, 2004). Although some sequences were closely related to those found in previous studies (Culley et al., 2003; Culley and Steward, 2007) (Figure 6), many OTUs formed new clades. Many OTUs were related to Heterosigma akashiwo RNA virus (HaRNAV) that infects the toxic bloom-forming raphidophyte Heterosigma akashiwo (Tai et al., 2003). HaRNAV is the type virus of the family Marnaviridae (Lang et al., 2004); it has a genome of about 9.1 kb and a high burst size as indicated by the large crystalline arrays of particles in the cytoplasm of infected cells (Tai et al., 2003). HaRNAV was isolated from coastal waters in British Columbia, Canada (Tai et al., 2003) and can remain infectious for many years in sediments (Lawrence and Suttle, 2004). Interestingly, HaRNAV was isolated from the same area as the present study, and appeared ancestral to many of the recovered sequences based on the phylogeny. For example, OTU 0 was most abundant (18,034 reads after rarefaction) and clustered closely with HaRNAV, although the sequence was only 76.4% similar at the amino-acid level. However, other OTUs in the cluster ranged between 55 and 79% similar to HaRNAV, which is low compared to amino-acid similarities of other RNA viruses within a family that usually have greater than 90% aa similarity (Ng et al., 2012).
Distinct Communities Occurred in Different Seasons at the Same Location
While only 8.6% of the OTUs from Jericho Pier were shared between dates, both samples had similar evenness, although the summer sample had greater richness (Figure 5, Table 2). The small overlap in OTUs between sampling dates is not surprising given the very different conditions between July and October (Figure 1), and the dynamic nature of planktonic communities in response to environmental changes. At the same location, dsDNA viruses belonging to the Phycodnaviridae, which infect eukaryotic phytoplankton, varied seasonally based on fingerprint analyses of DNA polymerase gene fragments using denaturing gradient gel electrophoresis; however, some OTUs persisted for extended periods (Short and Suttle, 2003). Similarly, the composition of other aquatic viral communities has been shown to be dynamic although some OTUs persist (Djikeng et al., 2009; Rodriguez-Brito et al., 2010), and in some cases have repeatable seasonal patterns (Chow and Fuhrman, 2012; Clasen et al., 2013; Marston et al., 2013). With only single samples from summer and fall, inferences about dynamics cannot be made from our data.
One of the few taxonomic groups that occurred in both the summer (JP-S) and fall (JP-F) samples from Jericho Pier was related to HaRNAV (Figure 6). There was greater diversity of OTUs in this clade in JP-F, even though JP-S had higher richness and higher phylogenetic diversity overall. This is unlike bacterial and phytoplankton communities that tend to be more diverse in winter (Zingone et al., 2009; Ladau et al., 2013). However, the RdRp primers target a specific subset of the viral community that might not reflect the overall taxonomic diversity.
Based on genome organization and sequence identity, RNA viruses that infect diatoms have been assigned to the genus Bacillarnavirus, that includes Rhizosolenia setigera RNA virus (RsRNAV) (Nagasaki et al., 2004), Chaetoceros tenuissimus Meunier RNA virus (CtenRNAV) (Shirai et al., 2008) and Chaetoceros socialis f. radians RNA virus (CsfrRNAV) (Tomaru et al., 2009). In the JP-F sample, the most relatively abundant cluster grouped with RsRNAV that infects the marine diatom Rhizosolenia setigera (Nagasaki et al., 2004). This corresponded with the highest levels of nitrate + nitrite, which is often associated with high diatom abundances (Zingone et al., 2009); hence, these OTUs are likely associated with viruses infecting diatoms.
Distinct Communities Occurred at Geographically Proximate Sites
Areas of higher habitat diversity, such as stratified water layers, generally have higher biological richness (Klopfer and MacArthur, 1960; Chesson, 2000), and this is consistent with the much higher richness and phylogenetic diversity found at SOG-4, which was the most stratified site and included the most abundant OTUs from SOG-1 and SOG-2. Most OTUs from SOG-1 clustered in the Marnaviridae clade, while most SOG-2 OTUs clustered in a phylogenetically distant clade. Given that we have used very conservative clustering, and that dsDNA viruses infecting phytoplankton are strain specific and have phylogenies that are congruent with their hosts (Clasen and Suttle, 2009; Bellec et al., 2014), and that RNA viruses infecting diatoms and the dinoflagellate, Heterocapsa circularisquama (Nagasaki et al., 2005) are host-specific, it implies that closely related OTUs infect closely related taxa of phytoplankton. Hence, it suggests that the most abundant viruses at these three locations infect different species.
There are few clear patterns in the spatial distribution of viruses in marine waters where geographically distant sites are connected by currents and mixing. The best examples are for cyanophages. For instance, when looking at local variation in cyanophages isolated at sites in Southern New England, 72% of the viral OTUs were shared between at least 2 sites (Marston et al., 2013); however, between Bermuda and Southern New England only 2 OTUs overlapped and they comprised only 0.6% of the isolates. Yet, clear patterns of cyanophage OTU distribution by depth occurred in areas adjacent to the SOG when assessed using community fingerprinting (Frederickson et al., 2003). The biggest differences with depth occurred in stratified water in which some OTUs were present at all depths, while others were only present at specific depths, even though the samples were collected only meters apart (Frederickson et al., 2003). These viruses infect cyanobacteria, as opposed to the picorna-like viruses, which likely infect protistan plankton. Nonetheless, the factors governing the distribution of cyanobacterial and protistan hosts are likely similar; hence, different OTUs would be expected to occur in environments with different vertical structure (stratification) of the water column.
Rank abundance curves showed that SOG-1 and SOG-2 were the least even communities (Figure 5). Overall, at most sites four to five viral OTUs were most abundant (Figure 5) similar to other reports for aquatic viral communities in which a few viruses dominate, but most of the diversity comes from rarer viruses (Angly et al., 2006; Suttle, 2007). Our targeted approach showed that the picornavirus-like virus communities at SOG and JP were dominated by only a few genotypes, supporting previous metagenomic results showing that the OTU distributions of RNA viruses in SOG and JP were highly uneven with little overlap between sites (Culley et al., 2006).
Each OTU Likely Represents a Single Lytic Event
Given that the hosts of marine Picornavirales isolates are protists, and that protists are the most abundant eukaryotes in the sea, it is likely that the majority of OTUs recovered in this study are from viruses that infect these unicellular marine eukaryotes. These eukaryotic communities are highly dynamic and change throughout the year based on environmental and biological factors (Larsen et al., 2004). Since viral infection is usually host specific, the diversity in marine viral communities is a reflection of the underlying diversity of the marine eukaryotic hosts. Moreover, viral propagation is dependent on host encounter rates and is proportional to host-cell abundance (Murray and Jackson, 1992); hence the most abundant taxa will be most likely to encounter and propagate a viral infection, giving the opportunity for rarer species to increase in abundance and promoting diversity (Thingstad, 2000; Winter et al., 2010). Since our study was not over time it is difficult to evaluate whether these data support the Bank model (Breitbart and Rohwer, 2005), however, some taxa were found at one site, but not a similar nearby site, thus these taxa could be present at background levels at some sites and more abundant in others.
It is probable that the most abundant OTUs in these data are from recent lysis of host taxa. An error rate for replication of RNA viruses of about 1 bp mutation per generation (9000 bp genome × 0.0001 error rate per base pair = 1 bp; Holmes, 2009), and a lower-end burst-size estimate of 1000 particles for marine viruses in the Picornavirales that infect protists (Lang et al., 2009), would produce about 1000 different genomes from each lysed cell. For the amplified 500 bp RdRp gene fragment there is a 0.00056% chance of an error in 1 generation, assuming that mutations are distributed evenly in the genome (Sanjuan et al., 2010; Combe and Sanjuán, 2014). Consequently, even with the relatively high error rates of RNA replication, when grouped at 95% similarity at the amino acid level, all of the sequences from a lytic event should fall within a single OTU. The half-life for decay of viral infectivity and particles in the surface mixed layer is typically a few hours (Heldal and Bratbak, 1991; Suttle and Chen, 1992; Noble and Fuhrman, 1997; Bettarel et al., 2009); thus the recovered viral OTUs were likely from recent lytic events. Furthermore, considering the specificity of viruses infecting protists (Short, 2012), each OTU probably stems from viruses infecting a single host taxon. Thus, these data imply that infection of marine protists by viruses in the Picornavirales is not only pervasive, but likely involves a wide diversity of host taxa; hence, these viruses are likely important structuring elements for phytoplankton communities that influence nutrient cycling and energy flow.
Amplicon Deep Sequencing as an Approach for Estimating Viral Diversity
Amplicon deep sequencing is a sensitive and high-resolution approach for examining microbial community dynamics over time and space (Caporaso et al., 2011; Gobet et al., 2012; Gibbons et al., 2013). Careful quality trimming of sequences and removal of singletons is essential for reliable results (Zhou et al., 2011) since errors in sequences will inflate estimates of diversity. With careful data processing and analysis, amplicon deep sequencing is as accurate for assessing community composition and diversity as cloning and Sanger sequencing (Amend et al., 2010), but with much greater depth of coverage of the community.
There are potential biases associated with reverse transcription with random hexamers (which can decrease yield and could inflate diversity) (Zhang and Byrne, 1999), template amplification by PCR (Lee et al., 2012) and with using highly degenerate primers that target a specific part of the community containing many different templates (Culley and Steward, 2007). A danger of the high cycle number can be diversity overestimates which can come from the increasing number of chimeric sequences produced with greater cycle number (Qiu et al., 2001). The sequences were processed with caution considering the high number of PCR cycles employed in this study. Chimera checking denovo was used to look for chimeric sequences originating from two higher abundance reads, and reference-based chimera checking was used a database of RdRps from isolated viruses to correct for this potential error. In addition, a conservative cut-off was used of only OTUs comprising more than 5 reads that aligned to the conserved domain alignment.
Read abundance of OTUs can be considered semi-quantitative and good for comparisons of richness and diversity among samples (but not for absolute counts of genes) (Amend et al., 2010; Pinto and Raskin, 2012; Ibarbalz et al., 2014). Moreover, by using control sequences obtained by cloning and Sanger sequencing alongside pyrosequenced libraries containing the same sequence (Supplemental Methods, Figure S2) we verified that amplicon deep sequencing and our sequence processing methodology recovered accurate environmental viral sequences and non-inflated estimates of richness like in studies for bacterial amplicons (Sogin et al., 2006; Huse et al., 2008; Kirchman et al., 2010; Caporaso et al., 2011) and clinical viral studies (Romano et al., 2013; Watson et al., 2013).
Conclusion
Amplicon deep sequencing of RdRp gene fragments using 454 pyrosequencing revealed the richness and population structure of marine Picornavirales in five coastal samples. The known diversity of viruses in this group was greatly increased with 145 OTUs that differed by at least 5% at the amino-acid level. There were between 24 and 71 OTUs in each sample, with distinct patterns of OTU distribution, richness and diversity among samples. There was little overlap between viral OTUs collected at the same site in summer and fall, and among samples collected 20 km apart on the same day. The high temporal and spatial diversity in RdRp sequences is consistent with viral communities that turnover rapidly, and episodic infection of a wide diversity of protistan hosts. The low overlap in OTUs and phylogenetic diversity among samples implies a dynamic landscape of viral infection and supports the idea that marine picorna-like viruses are important pathogens of marine protists that have an important role in structuring marine planktonic communities, and in nutrient cycling and energy transfer among trophic levels. Ultimately, further study is needed to disentangle the temporal and spatial drivers of these communities.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Thanks to R. Adelshin, A. M. Chan, C. Charlesworth, C. Chénard, R. Cruz, J. Finke, J. M. Labonté, J. Li, T. Nelson, J. P. Payet, E. J. Shelford, M. Vlok, R. A. White III, Q. Zhang, for help with sampling, and sample processing, the crew of the CGCS Vector for facilitating sample collection, and C. Payne for help with sampling, instrumentation and data acquisition, as well as processing of nutrient samples. M. Vlok and R. A. White III contributed to library preparation. Thanks to C. Chénard, C. Chow, J. Finke, T. Heger and E. J. Shelford for insightful comments on the manuscript. Funding was provided by NSERC Discovery and ship-time grants and from the Tula Foundation (Curtis A. Suttle), and NSERC PGS-M and PGS-D scholarships (Julia A. Gustavsen), and a UBC 4-Year Fellowship (Julia A. Gustavsen).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2014.00703/abstract
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Amend, A. S., Seifert, K. A., and Bruns, T. D. (2010). Quantifying microbial communities with 454 pyrosequencing: does read abundance count? Mol. Ecol. 19, 5555–5565. doi: 10.1111/j.1365-294X.2010.04898.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Angly, F. E., Felts, B., Breitbart, M., Salamon, P., Edwards, R. A., Carlson, C., et al. (2006). The marine viromes of four oceanic regions. PLoS Biol. 4:e368. doi: 10.1371/journal.pbio.0040368
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bellec, L., Clerissi, C., Edern, R., Foulon, E., Simon, N., Grimsley, N., et al. (2014). Cophylogenetic interactions between marine viruses and eukaryotic picophytoplankton. BMC Evol. Biol. 14:59. doi: 10.1186/1471-2148-14-59
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Benson, D. A., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., and Wheeler, D. L. (2007). GenBank. Nucleic Acids Res. 35, D21–D25. doi: 10.1093/nar/gkl986
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bergh, Ø., Børsheim, K. Y., Bratbak, G., and Heldal, M. (1989). High abundance of viruses found in aquatic environments. Nature 340, 467–468. doi: 10.1038/340467a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bettarel, Y., Bouvier, T., and Bouvy, M. (2009). Viral persistence in water as evaluated from a tropical/temperate cross-incubation. J. Plankton Res. 31, 909–916. doi: 10.1093/plankt/fbp041
Bratbak, G., Egge, J., and Heldal, M. (1993). Viral mortality of the marine alga Emiliania huxleyi (Haptophyceae) and termination algal blooms. Mar. Ecol. Prog. Ser. 93, 39–48. doi: 10.3354/meps093039
Breitbart, M., and Rohwer, F. (2005). Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 13, 278–284. doi: 10.1016/j.tim.2005.04.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brussaard, C. P. (2004). Viral control of phytoplankton populations—a review. J. Eukaryot. Microbiol. 51, 125–138. doi: 10.1111/j.1550-7408.2004.tb00537.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brussaard, C. P. D., Noordeloos, A. A. M., Sandaa, R.-A., Heldal, M., and Bratbak, G. (2004). Discovery of a dsRNA virus infecting the marine photosynthetic protist Micromonas pusilla. Virology 319, 280–291. doi: 10.1016/j.virol.2003.10.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Capella-Gutierrez, S., Silla-Martinez, J. M., and Gabaldon, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, F., Wang, K., Huang, S., Cai, H., Zhao, M., Jiao, N., et al. (2009). Diverse and dynamic populations of cyanobacterial podoviruses in the Chesapeake Bay unveiled through DNA polymerase gene sequences. Environ. Microbiol. 11, 2884–2892. doi: 10.1111/j.1462-2920.2009.02033.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chesson, P. (2000). Mechanisms of maintenance of species diversity. Annu. Rev. Ecol. Syst. 31, 343–366. doi: 10.1146/annurev.ecolsys.31.1.343
Chow, C.-E. T., and Fuhrman, J. A. (2012). Seasonality and monthly dynamics of marine myovirus communities: marine myovirus community dynamics at SPOT. Environ. Microbiol. 14, 2171–2183. doi: 10.1111/j.1462-2920.2012.02744.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clasen, J., Hanson, C., Ibrahim, Y., Weihe, C., Marston, M., and Martiny, J. (2013). Diversity and temporal dynamics of Southern California coastal marine cyanophage isolates. Aquat. Microb. Ecol. 69, 17–31. doi: 10.3354/ame01613
Clasen, J. L., and Suttle, C. A. (2009). Identification of freshwater Phycodnaviridae and their potential phytoplankton hosts, using DNA pol sequence fragments and a genetic-distance analysis. Appl. Environ. Microbiol. 75, 991–997. doi: 10.1128/AEM.02024-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Combe, M., and Sanjuán, R. (2014). Variation in RNA virus mutation rates across host cells. PLoS Pathog. 10:e1003855. doi: 10.1371/journal.ppat.1003855
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Culley, A. I., Lang, A., and Suttle, C. A. (2003). High diversity of unknown picorna-like viruses in the sea. Nature 424, 1054–1057. doi: 10.1038/nature01933
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Culley, A. I., Lang, A., and Suttle, C. A. (2006). Metagenomic analysis of coastal RNA virus communities. Science 312, 1795–1798. doi: 10.1126/science.1127404
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Culley, A. I., Mueller, J. A., Belcaid, M., Wood-Charlson, E. M., Poisson, G., and Steward, G. F. (2014). The characterization of RNA viruses in tropical seawater using targeted PCR and metagenomics. MBio 5:e01210–14–e01210–14. doi: 10.1128/mBio.01210-14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Culley, A. I., and Steward, G. F. (2007). New genera of RNA viruses in subtropical seawater, inferred from polymerase gene sequences. Appl. Environ. Microbiol. 73, 5937–5944. doi: 10.1128/AEM.01065-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Darriba, D., Taboada, G. L., Doallo, R., and Bangor, D. (2011). ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. doi: 10.1093/bioinformatics/btr088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Derelle, E., Ferraz, C., Escande, M.-L., Eycheni,é, S., Cooke, R., Piganeau, G., et al. (2008). Life-cycle and genome of OtV5, a large DNA virus of the pelagic marine unicellular green alga Ostreococcus tauri. PLoS ONE 3:e2250. doi: 10.1371/journal.pone.0002250
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Djikeng, A., Kuzmickas, R., Anderson, N. G., and Spiro, D. J. (2009). Metagenomic analysis of RNA viruses in a fresh water lake. PLoS ONE 4:e7264. doi: 10.1371/journal.pone.0007264
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Faith, D. P. (1992). Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61, 1–10. doi: 10.1016/0006-3207(92)91201-3
Frederickson, C. M., Short, S. M., and Suttle, C. A. (2003). The physical environment affects cyanophage communities in British Columbia inlets. Microb. Ecol. 46, 348–357. doi: 10.1007/s00248-003-1010-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fuhrman, J. A. (1999). Marine viruses and their biogeochemical and ecological effects. Nature 399, 541–548. doi: 10.1038/21119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gibbons, S. M., Caporaso, J. G., Pirrung, M., Field, D., Knight, R., and Gilbert, J. A. (2013). Evidence for a persistent microbial seed bank throughout the global ocean. Proc. Natl. Acad. Sci. U.S.A. 110, 4651–4655. doi: 10.1073/pnas.1217767110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gobet, A., Böer, S. I., Huse, S. M., van Beusekom, J. E., Quince, C., Sogin, M. L., et al. (2012). Diversity and dynamics of rare and of resident bacterial populations in coastal sands. ISME J. 6, 542–553. doi: 10.1038/ismej.2011.132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heldal, M., and Bratbak, G. (1991). Production and decay of viruses in aquatic environments. Mar. Ecol. Prog. Ser. 72, 205–212. doi: 10.3354/meps072205
Huang, S., Wilhelm, S. W., Jiao, N., and Chen, F. (2010). Ubiquitous cyanobacterial podoviruses in the global oceans unveiled through viral DNA polymerase gene sequences. ISME J. 4, 1243–1251. doi: 10.1038/ismej.2010.56
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huse, S. M., Dethlefsen, L., Huber, J. A., Mark Welch, D., Welch, D. M., Relman, D. A., et al. (2008). Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 4:e1000255. doi: 10.1371/journal.pgen.1000255
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ibarbalz, F. M., Pérez, M. V., Figuerola, E. L. M., and Erijman, L. (2014). The bias associated with amplicon sequencing does not affect the quantitative assessment of bacterial community dynamics. PLoS ONE 9:e99722. doi: 10.1371/journal.pone.0099722
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jameson, E., Mann, N. H., Joint, I., Sambles, C., and Mühling, M. (2011). The diversity of cyanomyovirus populations along a North–South Atlantic Ocean transect. ISME J. 5, 1713–1721. doi: 10.1038/ismej.2011.54
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kemp, P. F., and Aller, J. Y. (2004). Estimating prokaryotic diversity: when are 16 s rDNA libraries large enough? Limnol. Oceanogr. Methods 2, 114–125. doi: 10.4319/lom.2004.2.114
Kirchman, D. L., Cottrell, M. T., and Lovejoy, C. (2010). The structure of bacterial communities in the western Arctic Ocean as revealed by pyrosequencing of 16S rRNA genes. Environ. Microbiol. 12, 1132–1143. doi: 10.1111/j.1462-2920.2010.02154.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klopfer, P. H., and MacArthur, R. H. (1960). Niche size and faunal diversity. Am. Nat. 94, 293–300. doi: 10.1086/282130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koonin, E. V. (1991). The phylogeny of RNA-dependent RNA polymerases of positive-strand RNA viruses. J. Gen. Virol. 72, 2197–2206. doi: 10.1099/0022-1317-72-9-2197
Ladau, J., Sharpton, T. J., Finucane, M. M., Jospin, G., Kembel, S. W., O'Dwyer, J., et al. (2013). Global marine bacterial diversity peaks at high latitudes in winter. ISME J. 7, 1669–1677. doi: 10.1038/ismej.2013.37
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lang, A. S., Culley, A. I., and Suttle, C. A. (2004). Genome sequence and characterization of a virus (HaRNAV) related to picorna-like viruses that infects the marine toxic bloom-forming alga Heterosigma akashiwo. Virology 320, 206–217. doi: 10.1016/j.virol.2003.10.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lang, A. S., Rise, M. L., Culley, A. I., and Steward, G. F. (2009). RNA viruses in the sea. FEMS Microbiol. Rev. 33, 295–323. doi: 10.1111/j.1574-6976.2008.00132.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Larsen, A., Flaten, G. A. F., Sandaa, R.-A., Castberg, T., Thyrhaug, R., Erga, S. R., et al. (2004). Spring phytoplankton bloom dynamics in Norwegian coastal waters: microbial community succession and diversity. Limnol. Oceanogr. 49, 180–190. doi: 10.4319/lo.2004.49.1.0180
Lawrence, J. E., and Suttle, C. A. (2004). Effect of viral infection on sinking rates of Heterosigma akashiwo and its implications for bloom termination. Aquat. Microb. Ecol. 37, 1–7. doi: 10.3354/ame037001
Le Gall, O., Christian, P., Fauquet, C. M., King, A. M. Q., Knowles, N. J., Nakashima, N., et al. (2008). Picornavirales, a proposed order of positive-sense single-stranded RNA viruses with a pseudo-T = 3 virion architecture. Arch. Virol. 153, 715–727. doi: 10.1007/s00705-008-0041-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, C. K., Herbold, C. W., Polson, S. W., Wommack, K. E., Williamson, S. J., McDonald, I. R., et al. (2012). Groundtruthing next-gen sequencing for microbial ecology–biases and errors in community structure estimates from PCR amplicon pyrosequencing. PLoS ONE 7:e44224. doi: 10.1371/journal.pone.0044224
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marchler-Bauer, A., Lu, S., Anderson, J. B., Chitsaz, F., Derbyshire, M. K., DeWeese-Scott, C., et al. (2011). CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229. doi: 10.1093/nar/gkq1189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marston, M. F., Taylor, S., Sme, N., Parsons, R. J., Noyes, T. J. E., and Martiny, J. B. H. (2013). Marine cyanophages exhibit local and regional biogeography: biogeography of marine cyanophages. Environ. Microbiol. 15, 1452–1463. doi: 10.1111/1462-2920.12062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mayer, J. A., and Taylor, F. J. R. (1979). A virus which lyses the marine nanoflagellate Micromonas pusilla. Nature 281, 299–301. doi: 10.1038/281299a0
Mühling, M., Fuller, N. J., Millard, A., Somerfield, P. J., Marie, D., Wilson, W. H., et al. (2005). Genetic diversity of marine Synechococcus and co-occurring cyanophage communities: evidence for viral control of phytoplankton. Environ. Microbiol. 7, 499–508. doi: 10.1111/j.1462-2920.2005.00713.x
Murray, A. G., and Jackson, G. A. (1992). Viral dynamics: a model of the effects of size, shape, motion and abundance of single-celled planktonic organisms and other particles. Mar. Ecol. Prog. Ser. 89, 103–116. doi: 10.3354/meps089103
Nagasaki, K., Ando, M., Itakura, S., Imai, I., and Ishida, Y. (1994). Viral mortality in the final stage of Heterosigma akashiwo (Raphidophyceae) red tide. J. Plankton Res. 16, 1595–1599. doi: 10.1093/plankt/16.11.1595
Nagasaki, K., Shirai, Y., Takao, Y., Mizumoto, H., Nishida, K., and Tomaru, Y. (2005). Comparison of genome sequences of single-stranded RNA viruses infecting the bivalve-killing dinoflagellate Heterocapsa circularisquama. Appl. Environ. Microbiol. 71, 8888–8894. doi: 10.1128/AEM.71.12.8888-8894.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nagasaki, K., Tomaru, Y., Katanozaka, N., Shirai, Y., Nishida, K., Itakura, S., et al. (2004). Isolation and characterization of a novel single-stranded RNA virus infecting the bloom-forming diatom Rhizosolenia setigera. Appl. Environ. Microbiol. 70, 704–711. doi: 10.1128/AEM.70.2.704-711.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ng, T. F. F., Marine, R., Wang, C., Simmonds, P., Kapusinszky, B., Bodhidatta, L., et al. (2012). High variety of known and new RNA and DNA viruses of diverse origins in untreated sewage. J. Virol. 86, 12161–12175. doi: 10.1128/JVI.00869-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Noble, R. T., and Fuhrman, J. A. (1997). Virus decay and its causes in coastal waters. Appl. Environ. Microbiol. 63, 77–83.
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O'Hara, R. B., et al. (2013). Vegan: Community Ecology Package. R package version 2.0-10. Available online at: http://CRAN.R-project.org/package=vegan
Paradis, E. (2012). Analysis of Phylogenetics and Evolution with R. New York, NY: Springer. doi: 10.1007/978-1-4614-1743-9
Parsons, T. R., Maita, Y., and Lalli, C. M. (1984). A Manual of Chemical and Biological Methods for Seawater Analysis. New York, NY: Pergamon Press.
Pinto, A. J., and Raskin, L. (2012). PCR biases distort bacterial and archaeal community structure in pyrosequencing datasets. PLoS ONE 7:e43093. doi: 10.1371/journal.pone.0043093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490. doi: 10.1371/journal.pone.0009490
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qiu, X., Wu, L., Huang, H., McDonel, P. E., Palumbo, A. V., Tiedje, J. M., et al. (2001). Evaluation of PCR-Generated chimeras, mutations, and heteroduplexes with 16S rRNA gene-based cloning. Appl. Environ. Microbiol. 67, 880–887. doi: 10.1128/AEM.67.2.880-887.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
R Core Team (2014). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available online at: http://www.R-project.org/
Reeder, J., and Knight, R. (2010). Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat. Methods 7, 668–669. doi: 10.1038/nmeth0910-668b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rho, M., Tang, H., and Ye, Y. (2010). FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res. 38:e191. doi: 10.1093/nar/gkq747
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rodriguez-Brito, B., Li, L., Wegley, L., Furlan, M., Angly, F., Breitbart, M., et al. (2010). Viral and microbial community dynamics in four aquatic environments. ISME J. 4, 739–751. doi: 10.1038/ismej.2010.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Romano, C. M., Lauck, M., Salvador, F. S., Lima, C. R., Villas-Boas, L. S., Araújo, E. S. A., et al. (2013). Inter- and intra-host viral diversity in a large seasonal DENV2 outbreak. PLoS ONE 8:e70318. doi: 10.1371/journal.pone.0070318
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sanfaçon, H., Wellink, J., Gall, O., Karasev, A., Vlugt, R., and Wetzel, T. (2009). Secoviridae: a proposed family of plant viruses within the order Picornavirales that combines the families Sequiviridae and Comoviridae, the unassigned genera Cheravirus and Sadwavirus, and the proposed genus Torradovirus. Arch. Virol. 154, 899–907. doi: 10.1007/s00705-009-0367-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sanjuan, R., Nebot, M. R., Chirico, N., Mansky, L. M., and Belshaw, R. (2010). Viral mutation rates. J. Virol. 84, 9733–9748. doi: 10.1128/JVI.00694-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schroeder, D. C., Oke, J., Hall, M., Malin, G., and Wilson, W. H. (2003). Virus succession observed during an Emiliania huxleyi bloom. Appl. Environ. Microbiol. 69, 2484–2490. doi: 10.1128/AEM.69.5.2484-2490.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shirai, Y., Tomaru, Y., Takao, Y., Suzuki, H., Nagumo, T., and Nagasaki, K. (2008). Isolation and characterization of a single-stranded RNA virus infecting the marine planktonic diatom Chaetoceros tenuissimus Meunier. Appl. Environ. Microbiol. 74, 4022–4027. doi: 10.1128/AEM.00509-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Short, S. M. (2012). The ecology of viruses that infect eukaryotic algae: Algal virus ecology. Environ. Microbiol. 14, 2253–2271. doi: 10.1111/j.1462-2920.2012.02706.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Short, S. M., and Suttle, C. A. (2003). Temporal dynamics of natural communities of marine algal viruses and eukaryotes. Aquat. Microb. Ecol. 32, 107–119. doi: 10.3354/ame032107
Sogin, M. L., Morrison, H. G., Huber, J. A., Welch, D. M., Huse, S. M., Neal, P. R., et al. (2006). Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc. Natl. Acad. Sci. U.S.A. 103, 12115–12120. doi: 10.1073/pnas.0605127103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stamatakis, A., Hoover, P., and Rougemont, J. (2008). A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771. doi: 10.1080/10635150802429642
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steward, G. F., Culley, A. I., Mueller, J. A., Wood-Charlson, E. M., Belcaid, M., and Poisson, G. (2012). Are we missing half of the viruses in the ocean? ISME J. 7, 672–679. doi: 10.1038/ismej.2012.121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suttle, C. A. (2007). Marine viruses—major players in the global ecosystem. Nat. Rev. Microbiol. 5, 801–812. doi: 10.1038/nrmicro1750
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suttle, C. A. (2005). Viruses in the sea. Nature 437, 356–361. doi: 10.1038/nature04160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suttle, C. A., Chan, A. M., and Cottrell, M. T. (1991). Use of ultrafiltration to isolate viruses from seawater which are pathogens of marine phytoplankton. Appl. Environ. Microbiol. 57, 721–726.
Suttle, C. A., and Chen, F. (1992). Mechanisms and rates of decay of marine viruses in seawater. Appl. Environ. Microbiol. 58, 3721–3729.
Tai, V., Lawrence, J. E., Lang, A. S., Chan, A. M., Culley, A. I., and Suttle, C. A. (2003). Characterization of HaRNAV, a single-stranded RNA virus causing lysis of Heterosigma akashiwo (Raphidophyceae). J. Phycol. 39, 343–352. doi: 10.1046/j.1529-8817.2003.01162.x
Takao, Y., Mise, K., Nagasaki, K., Okuno, T., and Honda, D. (2006). Complete nucleotide sequence and genome organization of a single-stranded RNA virus infecting the marine fungoid protist Schizochytrium sp. J. Gen. Virol. 87, 723–733. doi: 10.1099/vir.0.81204-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thingstad, T. F. (2000). Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol. Oceanogr. 45, 1320–1328. doi: 10.4319/lo.2000.45.6.1320
Tomaru, Y., Katanozaka, N., Nishida, K., Shirai, Y., Tarutani, K., Yamaguchi, M., et al. (2004). Isolation and characterization of two distinct types of HcRNAV, a single-stranded RNA virus infecting the bivalve-killing microalga Heterocapsa circularisquama. Aquat. Microb. Ecol. 34, 207–218. doi: 10.3354/ame034207
Tomaru, Y., Takao, Y., Suzuki, H., Nagumo, T., and Nagasaki, K. (2009). Isolation and characterization of a single-stranded RNA virus infecting the bloom-forming diatom Chaetoceros socialis. Appl. Environ. Microbiol. 75, 2375–2381. doi: 10.1128/AEM.02580-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Etten, J. L., Meints, R. H., Burbank, D. E., Kuczmarski, D., Cuppels, D. A., and Lane, L. C. (1981). Isolation and characterization of a virus from the intracellular green alga symbiotic with Hydra viridis. Virology 113, 704–711. doi: 10.1016/0042-6822(81)90199-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, K., Wommack, K. E., and Chen, F. (2011). Abundance and distribution of Synechococcus spp. and cyanophages in the Chesapeake Bay. Appl. Environ. Microbiol. 77, 7459–7468. doi: 10.1128/AEM.00267-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watson, S. J., Welkers, M. R. A., Depledge, D. P., Coulter, E., Breuer, J. M., de Jong, M. D., et al. (2013). Viral population analysis and minority-variant detection using short read next-generation sequencing. Philos. Trans. R. Soc. B Biol. Sci. 368:20120205. doi: 10.1098/rstb.2012.0205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wilhelm, S. W., and Suttle, C. A. (1999). Viruses and nutrient cycles in the sea. Bioscience 49, 781–788. doi: 10.2307/1313569
Wilkinson, L. (2011). Venneuler: Venn and Euler Diagrams. Available online at: http://CRAN.R-project.org/package=venneuler
Winter, C., Bouvier, T., Weinbauer, M. G., and Thingstad, T. F. (2010). Trade-offs between competition and defense specialists among unicellular planktonic organisms: the “killing the winner” hypothesis revisited. Microbiol. Mol. Biol. Rev. 74, 42–57. doi: 10.1128/MMBR.00034-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yokoyama, R., and Honda, D. (2007). Taxonomic rearrangement of the genus Schizochytrium sensu lato based on morphology, chemotaxonomic characteristics, and 18S rRNA gene phylogeny (Thraustochytriaceae, Labyrinthulomycetes): emendation for Schizochytrium and erection of Aurantiochytrium and Oblongichytrium gen. nov. Mycoscience 48, 199–211. doi: 10.1007/S10267-006-0362-0
Zhang, J., and Byrne, C. (1999). Differential priming of RNA templates during cDNA synthesis markedly affects both accuracy and reproducibility of quantitative competitive reverse-transcriptase PCR. Biochem. J. 337, 231–241. doi: 10.1042/0264-6021:3370231
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, J., Wu, L., Deng, Y., Zhi, X., Jiang, Y.-H., Tu, Q., et al. (2011). Reproducibility and quantitation of amplicon sequencing-based detection. ISME J. 5, 1303–1313. doi: 10.1038/ismej.2011.11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: RNA viruses, phytoplankton mortality, viral ecology, pyrosequencing, viral diversity, Picornavirales
Citation: Gustavsen JA, Winget DM, Tian X and Suttle CA (2014) High temporal and spatial diversity in marine RNA viruses implies that they have an important role in mortality and structuring plankton communities. Front. Microbiol. 5:703. doi: 10.3389/fmicb.2014.00703
Received: 06 October 2014; Accepted: 26 November 2014;
Published online: 15 December 2014.
Edited by:
Bas E. Dutilh, Radboud University Medical Center, NetherlandsReviewed by:
Alexander Culley, Université Laval, CanadaJennifer R. Brum, University of Arizona, USA
Copyright © 2014 Gustavsen, Winget, Tian and Suttle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Curtis A. Suttle, Department of Earth, Ocean and Atmospheric Sciences, University of British Columbia, 2020 - 2207 Main Mall, Vancouver, BC V6T 1Z4, Canada e-mail:c3V0dGxlQHNjaWVuY2UudWJjLmNh
†Present address: Danielle M. Winget, Department of Biology, Seattle Pacific University, Seattle, WA, USA