 Björn Breidenbach
Björn Breidenbach Ralf Conrad
Ralf Conrad- Department of Biogeochemistry, Max Planck Institute for Terrestrial Microbiology, Marburg, Germany
We studied the resident (16S rDNA) and the active (16S rRNA) members of soil archaeal and bacterial communities during rice plant development by sampling three growth stages (vegetative, reproductive and maturity) under field conditions. Additionally, the microbial community was investigated in two non-flooded fields (unplanted, cultivated with upland maize) in order to monitor the reaction of the microbial communities to non-flooded, dry conditions. The abundance of Bacteria and Archaea was monitored by quantitative PCR showing an increase in 16S rDNA during reproductive stage and stable 16S rRNA copies throughout the growth season. Community profiling by T-RFLP indicated a relatively stable composition during rice plant growth whereas pyrosequencing revealed minor changes in relative abundance of a few bacterial groups. Comparison of the two non-flooded fields with flooded rice fields showed that the community composition of the Bacteria was slightly different, while that of the Archaea was almost the same. Only the relative abundance of Methanosarcinaceae and Soil Crenarchaeotic Group increased in non-flooded vs. flooded soil. The abundance of bacterial and archaeal 16S rDNA copies was highest in flooded rice fields, followed by non-flooded maize and unplanted fields. However, the abundance of ribosomal RNA (active microbes) was similar indicating maintenance of a high level of ribosomal RNA under the non-flooded conditions, which were unfavorable for anaerobic bacteria and methanogenic archaea. This maintenance possibly serves as preparedness for activity when conditions improve. In summary, the analyses showed that the bacterial and archaeal communities inhabiting Philippine rice field soil were relatively stable over the season but reacted upon change in field management.
Introduction
Methane (CH4) is the second most important greenhouse gas after carbon dioxide (CO2) and has a 25 times larger global warming potential than CO2 (Forster et al., 2007). The global budget of atmospheric CH4 is on the order of 500–600 Tg per year (Forster et al., 2007) and rice fields contribute in the range of 25–300 Tg CH4 per year (Chen and Prinn, 2005; Bridgham et al., 2013). Rice production will probably increase, in order to feed an increasing human population (Van Nguyen and Ferrero, 2006) so that CH4 emission from rice fields may also increase in future. In rice fields CH4 is produced as end product of the anaerobic degradation of organic matter by a complex microbial community consisting of hydrolytic and fermentative bacteria, and methanogenic archaea (Zinder, 1993; Conrad, 2007). Flooded rice fields have been used as a model system for studying the functioning of anoxic methanogenic microbial communities (Conrad, 2007, 2009).
In rice fields methane is produced by two major physiological guilds, the acetotrophic and the hydrogenotrophic methanogens. Acetotrophic methanogens dismutate acetate to CH4 and CO2, while the hydrogenotrophic methanogens reduce CO2 with H2 to CH4 (Conrad, 2007). Molecular characterization of 16S rDNA showed a worldwide distribution of methanogens in rice fields (China, Italy, Japan and Philippines) including Methanosarcinaceae, Methanosaetaceae, Methanobacteriales, Methanomicrobiales, and Methanocellales (Großkopf et al., 1998; Ramakrishnan et al., 2001; Wu et al., 2006). The composition of the soil archaeal community changes if temperature is increased (Peng et al., 2008; Conrad et al., 2009) or the rice field soil is treated with organic matter such as rice straw (Conrad and Klose, 2006; Peng et al., 2008). Under field conditions, however, the archaeal communities were usually found to be rather stable even after short term drainage or extended periods of managing rice fields as upland fields (Krüger et al., 2005; Watanabe et al., 2006; Fernandez Scavino et al., 2013). In a recent study of a Korean rice field, numbers of archaea and methanogens changed by less than a factor of two throughout a cropping season (Lee et al., 2014).
In contrast to the archaeal community it has been shown that the bacterial community in rice field soil changes with time after flooding (Noll et al., 2005; Rui et al., 2009). Bacterial communities in irrigated rice fields are described as complex (Asakawa and Kimura, 2008) and differ between oxic and anoxic zones (Shrestha et al., 2007). Additionally, temporal and spatial changes in the composition of the bacterial communities with changing soil conditions were observed (Noll et al., 2005; Shrestha et al., 2009). Variations in relative abundance of dominant phyla under alfalfa-rice crop rotation system were revealed (Lopes et al., 2014) whereas pasture-rice crop rotation showed a rather stable bacterial community composition (Fernandez Scavino et al., 2013).
Moreover, archaeal and bacterial communities in the rhizosphere can be shaped by the plant species (e.g., Grayston et al., 1998; Smalla et al., 2001; Conrad et al., 2008). Several other studies demonstrated that plant type had an effect on soil microbial community structure (Marschner et al., 2001; Smalla et al., 2001; Costa et al., 2006). In addition to plant residues and soil organic matter, rhizodeposits are the major substrate input into soil (Kimura et al., 2004). Rhizodeposits are plant-derived carbon-containing compounds, which are actively secreted via the plant roots or originate from sloughed-off root cells (reviewed by Dennis et al., 2010). Rhizodeposition takes place at the zone around the plant root called rhizosphere which was shown to harbor a specific microbial community (Kowalchuk et al., 2010). Rhizodeposition depends on environmental factors, plant species, type and cultivar as well as plant age (Aulakh et al., 2001; Uren, 2007). The microbial community in the rhizosphere may be influenced by these variations in rhizodeposition.
Therefore, we hypothesized that the microbial community in rice field soil will be influenced by rice plant growth stage. Since a comprehensive seasonal record of resident and active microorganisms was lacking, we investigated the archaeal and bacterial community in the soil under field conditions by sampling three distinct plant growth stages. Additionally, the microbial community was investigated in two fields that were not flooded and were either unplanted or cultivated with upland maize in order to monitor the reaction of the rice specific microbial community to non-flooded conditions and to the presence or absence of maize. The microbial composition and abundance was assessed by fingerprinting with terminal-restriction fragment length polymorphism (T-RFLP) and quantitative PCR (qPCR) targeting the archaeal and bacterial ribosomal 16S rRNA and 16S rDNA. In order to identify changes in the lower taxonomic groups, archaeal and bacterial 16S rRNA was targeted by 454 pyrosequencing. Interestingly, we observed rather stable archaeal and bacterial communities in the soil during rice plant growth but detected more pronounced differences between flooded and non-flooded fields.
Material and Methods
Sampling Site and Sample Processing
The sampling site was located at the International Rice Research Institute (IRRI) in Los Banos, Philippines. Detailed site description can be found in Heinz et al. (2013). Briefly, we studied fields cultivated with irrigated rice throughout one cropping season at the vegetative (February 2012), reproductive (March 2012) and maturity (May 2012) growth phase of the rice plants (variety: NSIC Rc222). Additionally rice fields, which had been drained and were now managed as upland fields cultivating upland maize (variety: Pioneer P3482YR) were sampled after plowing and before maize seeding and fertilization as unplanted and drained rice field (unplanted) and during the reproductive growth phase of maize (maize). The study site was cropped with paddy rice in both wet and dry season over two decades (Weller et al., 2014). This season (dry 2012) was the first season in which the fields were managed as upland maize fields. Fields were operated in triplicates and managed with conventional N-fertilization (rice: seeding 30 kg N/ha, 30 kg P2O5/ha, 30 kg K2O/ha; at 28 and 55 days after seeding (DAS) 50 kg N/ha; maize: seeding 30 kg N/ha, 50 kg P2O5/ha, 30 kg K2O/ha; at 27–29 and 47-50 DAS 50 kg N/ha). In each of these fields we randomly selected three sampling plots of one square meter and sampled one soil core (5 cm diameter) from each plot. Soil cores were always taken from the vicinity of a plant (ca. 10 cm). The soil contained numerous fine roots and thus was most probably influenced by the plant roots. However, no attempts were made to separate a specific rhizospheric soil compartment. Subsequently, soil samples of 5 g were taken from the middle of the core (ca. 10 cm depth), added to 10 mL RNAlater© solution (Life Technologies, Darmstadt, Germany), kept on ice and later stored at −20°C to ensure RNA stability. For further analysis (determination of soil variables), additional samples of 50 g were taken from the same soil core and stored at −20°C.
Determination of Soil Variables
For determination of soil water content small amounts of soil (1–5 g) were dried at 65°C for 3 days. The gravimetric water contents of the samples from fields cultivated with rice or maize and unplanted fields were 42.8 ± 3.5, 34.3 ± 1.2, and 36.0 ± 2.2%, respectively. The pH of the soil was analyzed following the DIN ISO 10390 protocol. Briefly, 3 g of soil was mixed with 0.01 M CaCl2 in a ratio of 1:2.5 and incubated rotating at 25°C for 10 min. Subsequently, the samples were incubated at 25°C for 60 min without agitation and then, after shaking the sample, the pH was measured using a pH meter (pH530 WTW, Weilheim, Germany). The pH values in rice, maize and unplanted fields were pH 6.8, 6.6, and 6.2, respectively. The soil texture was silt loam and the determination was conducted using a laser particle measuring device (LS13320, Beckmann-Coulter, Krefeld, Germany) at the geographic institute of the RTWH Aachen.
Nucleic Acid Extraction
Nucleic acids were extracted following a modified version of the protocol from Bürgmann et al. (2001). Briefly, after removal of RNAlater© solution by centrifugation at 2500 × g for 2 min, 0.5 g of soil were extracted via bead-beating for 45 s at 6 m/s using a FastPrep®-24 (MP Biomedicals, Eschwege, Germany) in the presence of a 850 μl extraction buffer (20 ml 1 M sodium phosphate (pH 8.0), 2.5 g SDS, 10 ml 0.5 M EDTA (pH 8.0) and 2 ml 5 M NaCl). The tube was centrifuged at maximum speed for 5 min at 20°C. Then, 850 μl of phenol/chloroform/isoamylalcohol (25:24:1; Fluka, Sigma-Aldrich, Taufkirchen, Germany) was added to the supernatant and mixed. The bead beating was repeated twice using fresh extraction buffer. After mixing, the tubes were centrifuged for 5 min at maximum speed at 20°C. Then, 800 μl of chloroform/ isoamylalcohol (24:1; Fluka, Sigma-Aldrich, Taufkirchen, Germany) was added to the supernatant. After further centrifugation, 1 ml of precipitation solution (20 g PEG 6000, 16.6 g NaCl) was added to the aqueous supernatant, mixed, and kept at room temperature for 1 h. After centrifugation for 1 h with maximum speed at 4°C the sample was resuspended in 75% ice cold ethanol (Roth, Karlsruhe, Germany) and subsequently centrifuged for 10 min at maximum speed and 4°C. The resulting pellet was air dried and resuspended in 100 μl nuclease free water (Invitrogen, Darmstadt, Germany) and stored at −80°C until analysis. The total nucleic acids in 50 μl aliquot were digested with 37.5 μl nuclease free water (Invitrogen), 2.5 μl RNase-free DNase and 10 μl buffer RDD (Qiagen, Hilden, Germany) at room temperature for 10 min. The digest was then purified using RNeasy kit (Qiagen) following the RNA Cleanup protocol in the manufacturer's instructions. Complete DNA removal was verified by failure to obtain a PCR amplification product of bacterial 16S rDNA with the purified RNA template using the conditions described below. cDNA was synthesized from purified RNA using SuperScript™ III reverse transcriptase (Invitrogen) according to the manufacturer's instructions. Random hexamer primers (50 ng/μl) were used for complete cDNA synthesis which was used for amplification of the archaeal and bacterial 16S rRNA.
Quantitative Polymerase Chain Reaction
The quantification of archaeal and bacterial 16S rDNA/rRNA was conducted using quantitative polymerase chain reaction (qPCR) using primer combinations Ba519f/Ba907r (Stubner, 2002) for bacterial and Ar364f (Burggraf et al., 1997) / Ar934br (Großkopf et al., 1998) for archaeal genes. The qPCR was conducted in 96-well micro titer plates (BioRad, München, Germany) using an iCycler MyiQ™ (BioRad). Each qPCR reaction contained in a total volume of 25 μl, 1 × SYBRGreen Ready Mix (Sigma), 3 mM MgCl2 (Sigma), 0.25–0.66 μM of each primer and 1 μM FITC (fluorescein thiocyanat; BioRad) as well as 1–2 μl target DNA respectively cDNA. Purity of the used reagents was ensured using negative controls not containing any DNA matrix. The DNA standard prepared from clones containing bacterial or archaeal 16S rDNA in a plasmid insert was applied in a dilution series containing 1 × 107 to 1 × 101 gene copies. The thermal profile used for amplification included 40 to 50 cycles of denaturation at 94°C for 30 s, primer annealing at 50°C (Ba519f /Ba907r) or 66°C (Ar364/Ar934br) for 20–30 s and primer extension at 72°C for 45 s. Afterwards a melting curve from 75 to 95°C (0.2°C s−1) was performed in order to confirm specificity of the real time PCR reaction. The data were analyzed using BioRad IQ5 2.0 Standard Edition Optical System software (Biorad).
Terminal Fragment Length Polymorphism (T-RFLP)
T-RFLP analysis of archaeal and bacterial 16S rRNA and rDNA was conducted based on fractionation of terminal fluorescence-labeled PCR products after use of restriction enzymes as described (Chin et al., 1999) using the primer combination Ar109f (Großkopf et al., 1998)/Ar912rt-FAM (Lueders and Friedrich, 2003) and Ba27f-FAM (Osborne et al., 2005)/Ba907r (Muyzer et al., 1995), respectively. All PCR reactions were performed in a total volume of 50 μl. For amplification each reaction contained 5 × Green GoTaq® Flexi buffer (Promega, Mannheim, Germany), 200 μM deoxy-nucleoside triphosphates (dNTPs; Fermentas, St. Leon-Rot, Germany), 0.5 μM of each primer, 10 μg bovine serum albumin (BSA; Roche, Grenzach, Germany), 1 U GoTaq® Flexi DNA polymerase (Promega) and 1 μl DNA matrix (in most cases diluted to a concentration of 20 ng/μl). All amplifications were carried out in a GenAmp 9700 Thermocycler (Applied Biosystems, Carlsbad CA, USA). The thermal profile used for amplification included 25 to 30 cycles of primer annealing at 52°C for 45 s, primer extension at 72°C for 90 s, and denaturation at 94°C for 45 s. PCR product purification was conducted using the GenElute™ PCR Clean-up kit (Sigma) following the manufacturer's instructions. The purified amplicons were digested by using MspI (cutting side: 5′-C▼CGG-3′, 37°C; Fermentas) for bacterial and TaqI (cutting side: 5′-T▼CGA-3′, 65°C; Fermentas) for archaeal 16S rDNA and rRNA. The fragmented DNA was purified using SigmaSpin™ Post Reaction Clean-Up columns (Sigma) following the manufacturer's instructions. T-RFLP reactions contained 0.2 μl size standard (X-rhodamine MapMarker® 1000, BioVentures, Murfreesboro, USA). Separation was accomplished using capillary electrophoresis in an ABI PRISM 3130 Genetic Analyzer (Applera). Data analysis was conducted using GENESCAN Analysis software 4.0 (Applied Biosystems, Carlsbad, USA). Normalization and standardization of the T-RFLP profiles was done according the method from Dunbar et al. (2001). The relative abundance was calculated from the ratio between the height of the fluorescence signal and the total height of all signals in one sample.
Cloning and Sequencing
A clone library of archaeal 16S rDNA was created for subsequent phylogenetic classification. The pGEM®-T Easy Vector System (Promega) was used. The purified PCR products were ligated into the pGEM®-T Easy Vector according to the manufacturer's instructions. Each ligation reaction contained 5 μl 2X Rapid Ligation Buffer (Promega), 1 μl pGEM®-T Easy Vector (50 ng; Promega), 1 μl T4 DNA ligase (3 Weiss units/μl; Promega) and 50 ng PCR product. Sterile water was added to reach a total volume of 10 μl. The transformation of Escherichia coli JM109 high efficient competent cells (Promega) was carried out according to the manufacturer's instructions. Randomly chosen white colonies were sequenced using Sanger sequencing. The received raw data (electropherograms) were processed using the program Seqman II (DNAStar, Madison, USA). The phylogeny of the archaeal sequences was analyzed using the ARB software (http://www.arb-home.de/). For the archaeal 16S rRNA sequences the public available database was downloaded from SILVA homepage (http://www.arb-silva.de/) and integrated into ARB. Alignment was conducted using the Fast Aligner tool in ARB (Ludwig et al., 2004). The alignment was then manually checked and where necessary corrected. Subsequently, the aligned sequences were calculated into the archaeal 16S rRNA tree under usage of the neighbor-joining algorithm as described in detail by Wu et al. (2006). The restriction sites characteristic for the fragment length of the T-RFs were determined. The T-RFs determined from T-RFLP analyses were assigned to the corresponding clones and their phylogeny. The archaeal 16S rRNA sequences data have been submitted to the GenBank databases under accession numbers: KM463011–KM463082.
454 Pyrosequencing
Tagged pyrosequencing of the bacterial and archaeal community was conducted using primer combinations F515/R806 (Bates et al., 2011) and Arch344F (Casamayor et al., 2002)/A934br (Großkopf et al., 1998), respectively. The forward primers were tagged with a unique 8-base pair barcode. Sequencing of the PCR products was done at the Max Planck Genome Centre in Cologne using a Roche 454 Genome Sequencer GS FLX+. Data analysis was performed using mothur software package version 1.31.2 (http://www.mothur.org/) following the standard operational procedure including sequence quality management (SOP, Schloss et al., 2009). OTU clustering and analysis was conducted using UPARSE pipeline as described by Edgar (2013). Only microbial high-quality sequences with a minimum read length of 200 bp were used. Sequences that did not match the primer sequences and were smaller than 200 bp or contained any ambiguities were excluded from further analysis. After denoising, sequences were aligned against the SILVA bacteria/archaea 16S rDNA database (Schloss et al., 2011; Pruesse et al., 2007). Sequences which were not assigned to bacteria or respectively archaea were discarded. Operational taxonomic units (OTU) were defined using a distance matrix with 3% dissimilarity (Zinger et al., 2011). Further analyses including rarefaction curves, species richness and diversity indices were conducted as described in the SOP pyrosequencing pipeline (Schloss et al., 2011). An overview of the number of sequences retrieved and the accession numbers of the submitted sequences can be found in Tables 2, 3.
Statistical Analysis
Statistical analyses were done in R version 2.14.1 (R Development Core Team, 2011). If necessary, normal distribution was achieved by log-transforming the data. Analysis of variance (ANOVA), PERMANOVA (ADONIS) and canonical correspondence analysis (CCA) were conducted with package vegan version 2.0.5 (Oksanen et al., 2012). All levels of significance were defined at P ≤ 0.05. Ternary plots were created using package vcd version 3.0.3.
Results
Bacterial and Archaeal 16S rDNA/rRNA Copy Numbers
For quantification of bacteria and archaea in rice field soil during rice plant growth we used quantitative PCR (qPCR) targeting the bacterial and archaeal 16S ribosomal RNA (16S rRNA) and their genes (16S rDNA). Copy numbers of bacterial and archaeal 16S rDNA and rRNA were quantified at three different growth stages and in differently cropped fields (Figure 1). Both bacterial and archaeal 16S rDNA copy numbers were highest during rice growth at reproductive stage, whereas the 16S rRNA copy numbers were constant during the whole season (Figures 1A,B). Comparing rice and maize cultivated soils during the reproductive growth phase, the highest copy numbers of 16S rDNA and rRNA were detected in the rice fields (Figures 1C,D). The unplanted fields contained less 16S rDNA copies than the fields cultivated with either rice or maize (Figures 1C,D). However, the numbers of archaeal and bacterial 16S rRNA copies were similar to those in the rice fields (Figures 1C,D) resulting in a high ratio of 16S rRNA/rDNA copies (Figure 2). In contrast, bacterial 16S rRNA copies were lower in the maize field than in the rice cultivated and unplanted fields (Figure 1C). Although the ratio of bacterial and archaeal rRNA/rDNA copies was significantly increased in unplanted fields in comparison to the fields cultivated with either rice or maize across all the replicates sampled, samples from replicate field 7 did not show such increase (Figure 2). The behavior of these particular replicates could not be explained by analyzing possible correlation with soil characteristics (contents of carbon, nitrogen, sulfate, nitrate, water).

Figure 1. Ribosomal 16S rDNA and rRNA copy numbers quantified using qPCR. Abundance of bacterial 16S rDNA and rRNA (A,C) and archaeal 16S rDNA and rRNA (B,D) in rice fields at different plant growth stages (A,B) as well as in fields planted with rice, maize or unplanted at the reproductive growth stage (C,D). Different letters indicate significant difference (mean ± SE, n = 9).
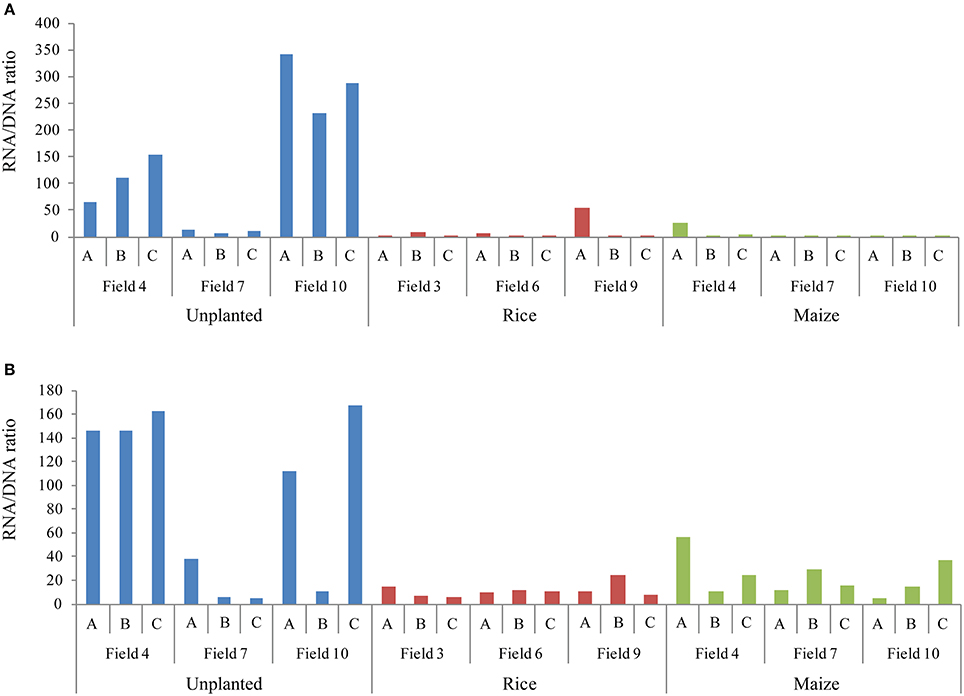
Figure 2. Ratio of ribosomal 16S rRNA and rDNA copy numbers quantified using qPCR. Bacterial (A) and archaeal (B) ratios are shown for each replicate in the unplanted, rice and maize cultivated fields.
Bacterial and Archaeal Community Analyzed by T-RFLP
For community profiling we used T-RFLP targeting the bacterial and archaeal 16S rDNA and rRNA. To identify parameters which significantly explain the variance in the microbial community, canonical correspondence analysis (CCA) was performed. Field management (rice, maize, unplanted), growth stage (vegetative, reproductive, maturity) and gravimetric water content were identified to significantly affect the microbial community explaining 6–23% of the variance (Figures 3A–D). The pure effect of each factor is shown in Supplement Table 1, with field management explaining 12–16%, growth stage 11–23% and gravimetric water content 6–12% of the variance. Although these factors were significant, the resident bacterial (Supplement Figure 1) and the archaeal (Figure 4) community composition did not change significantly during rice plant growth (ADONIS, P > 0.05). The non-flooded fields (unplanted and maize) also revealed a bacterial and archaeal community composition that was not significantly different from the rice field community (ADONIS, P > 0.05). Only the archaeal T-RF of 186 bp significantly increased during maize cultivation in comparison to the rice fields (Figure 4; ANOVA, P < 0.05). The more active bacterial and archaeal communities (16S rRNA) showed only minor variations in relative abundance of T-RFs during rice plant growth and in the unplanted and maize fields. These variations were not statistically significant (ADONIS, P > 0.05).
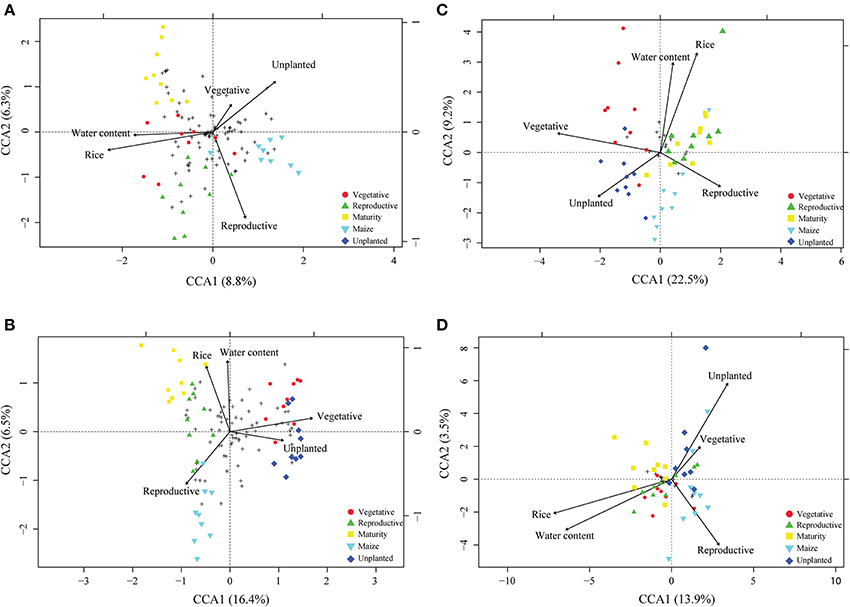
Figure 3. Canonical correspondence analysis (CCA) biplot of T-RFLP based on bacterial and archaeal communities. T-RFLP based communities of bacterial 16S rDNA and rRNA (A,B) as well as archaeal 16S rDNA and rRNA (C,D) are displayed. Arrows indicate the direction and relative importance (arrow lengths) of environmental variables associated with bacterial and archaeal community structures, respectively. Solely the environmental variables significantly influencing the model were displayed (ANOVA p < 0.05). Circle, triangle and square symbols, respectively, represent vegetative, reproductive and maturity growth phase of rice. Inverted triangle and diamante symbols, respectively, characterize samples originating from maize and unplanted fields while crosses represent T-RFs.
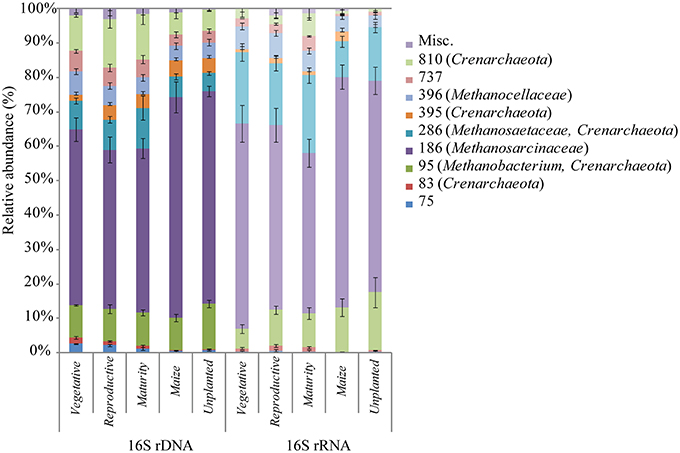
Figure 4. Histograms of the relative abundance of T-RFs obtained from T-RFLP analysis of archaeal 16S rDNA and rRNA during rice plant growth. Terminal restriction fragment sizes and affiliated clone taxonomy are given in brackets. Bars represent standard errors of n = 9.
The archaeal T-RFs were assigned to different archaeal lineages by sequence analysis (Table 1). The assignment was based on a clone library of 16S rDNA containing 72 randomly selected clones retrieved from soil cultivated with rice and maize. The major T-RFs of 95, 186, 286, 396, and 810 bp were assigned as Methanobacteriales, Methanosarcinaceae, Methanosaetaceae, Methanocellales, and Miscellaneous Crenarchaeota respectively (Table 1). Some additional T-RFs of minor relative abundance were detected at 75, 308, 611, 671, 682, 695, 737, and 771 bp (most of them are too minor to be shown in Figure 4), which were not represented in the clone library, and therefore could not be assigned. A few clones, which were solely found in the clone library but not in the T-RFLP analysis, were assigned as Miscellaneous Crenarchaeota (97, 263, 333, 656, 725 bp).
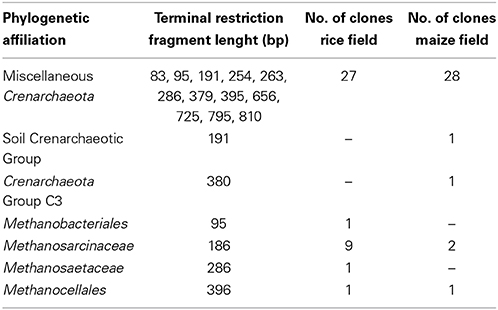
Table 1. Lengths of distinct terminal restriction fragments (T-RFs) of different archaeal 16S rDNA clones obtained rice and maize cultivated Philippine rice field soil and affiliation with a distinct phylogenetic lineage by 16S rDNA sequence analysis of the clones.
Pyrosequencing of Bacterial 16S rDNA and rRNA
Pyrosequencing targeting the bacterial 16S rDNA and rRNA was conducted in order to indentify the resident and the active bacterial phylotypes in the Philippine rice field soil and to monitor the influence of plant growth stage on the bacterial community composition. Therefore, triplicate samples were sequenced for each growth stage resulting in 3468 to 11,311 high quality sequences of rDNA as well as 2062 to 10,275 sequences of rRNA (Table 2). For bacterial rDNA, the most dominant phylum was Proteobacteria (23–32%) followed by Acidobacteria (16–20%). Other important bacterial phyla were Chloroflexi (8–10%), Verrucomicrobia (5–6%), Firmicutes (4–5%), Actinobacteria (2–3%), Planctomycetes (2%), and Cyanobacteria (1–3%) (Supplement Figures 2A–E). The bacterial community composition did not change dramatically during the rice growing season (Supplement Figures 2A–C). Comparison of the dominant OTUs retrieved at different rice plant growth stages showed a uniform distribution over the season (Figure 5A). Only OTUs with a minor relative abundance were distinct for a particular growth stage, e.g., OTU 396 identified as Anaeromyxobacter, which was only found at the vegetative growth stage (Figure 5A). Comparison of unplanted, maize and rice cultivated fields showed more pronounced differences among bacterial OTUs. The OTUs number 1 (Spartobacteria), 4 (Unclassified) and 7 (Acidobacteria Gp25) were more abundant in the unplanted and in the upland maize fields than in the rice fields, while OTU number 9 (Deltaproteobacteria) was relatively more abundant in the rice fields (Figure 5C). Additionally, unplanted fields as well as fields cultivated with upland maize showed significantly less Proteobacteria in comparison to rice fields (Supplement Figures 2A,D,E). The lower relative abundance of the Proteobacteria was due to the lower abundance of Geobacteraceae. Otherwise, however, the bacterial community composition was not affected by the rice growth stage or the type of crop (ADONIS, P > 0.005).
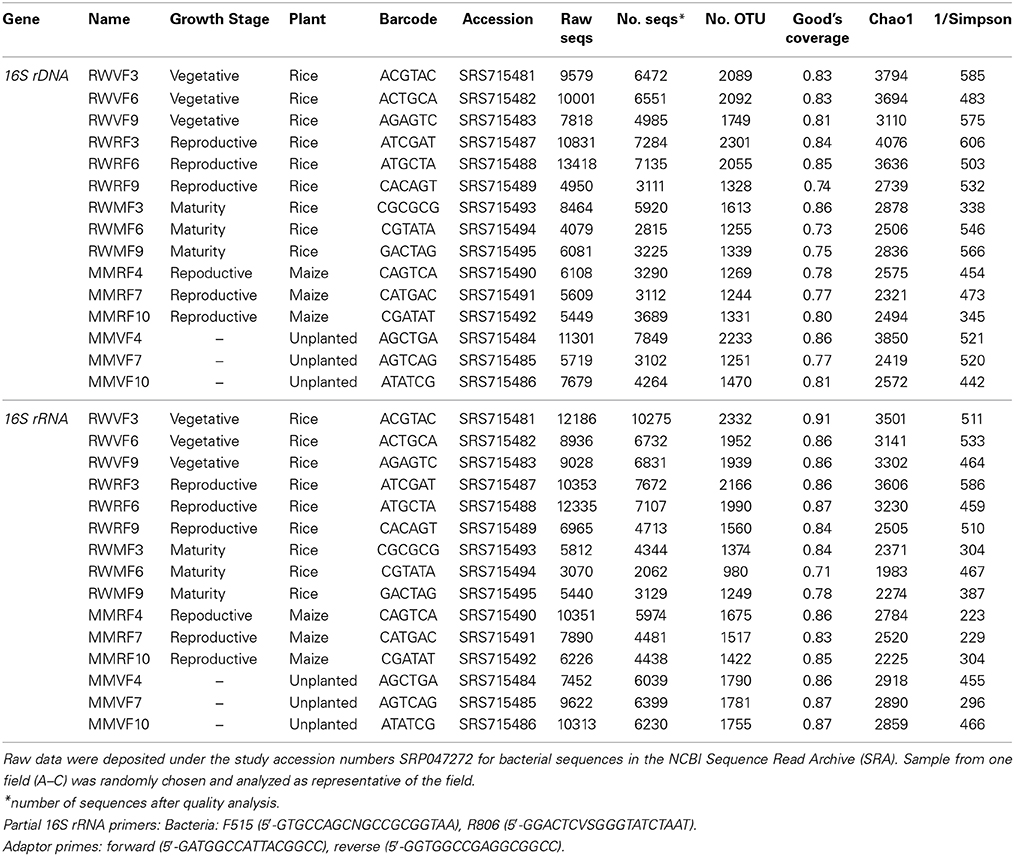
Table 2. Number of bacterial sequences before and after quality management, barcode, number of OTUs, coverage, Chao1 and inverted Simpson index of the environmental samples analyzed by 454-pyrosquencing.
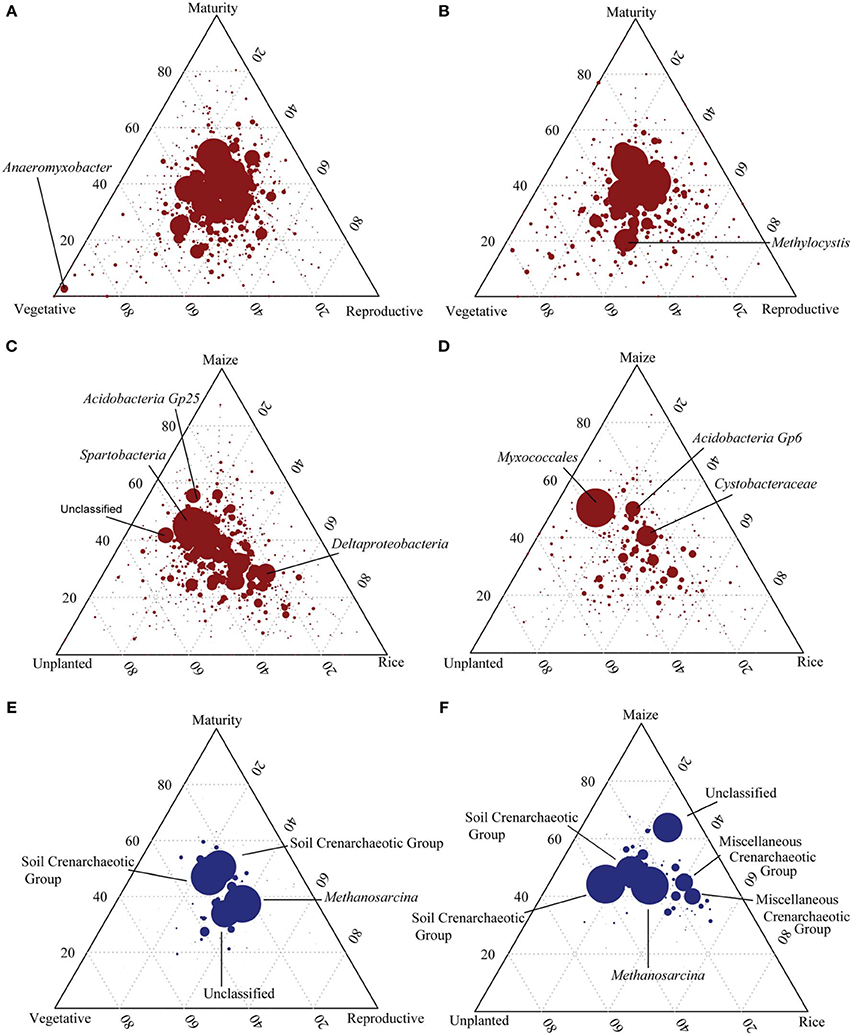
Figure 5. Ternary plots showing the distribution of bacterial and archaeal 16S rDNA and rRNA based OTUs. Axes represent rice plant growth stages (vegetative, reproductive and maturity) (A,B,E) as well as unplanted and maize cultivated fields in comparison to rice cultivated fields (C,D,F) and the percentage of reads associated with each sample for each OTU. Bacterial 16S rDNA (red, A,C) and 16S rRNA (B,D) as well as archaeal 16S rDNA (blue, E,F) are displayed. Each circle represents an individual OTU while its size indicates number of reads associated. The position of each OTU is determined by the contribution of the sample type to the total count (n = 3).
The sequences of ribosomal RNA presumably represent the more active bacterial community. This community was composed of the same phyla as the ribosomal gene-based community, but exhibited a different composition (Supplement Figures 2F–J). The most dominant phyla within the bacterial rRNA community were Proteobacteria (36–40%) followed by Acidobacteria (14–18%). Other important bacterial phyla were Chloroflexi (3–4%), Verrucomicrobia (4–6%), Firmicutes (3–4%), Actinobacteria (2–4%), Planctomycetes (5–6%), and Cyanobacteria (3–9%) (Supplement Figures 2F–J). At the phylum level the bacterial community was not significantly different between the different plant growth stages (ADONIS, P > 0.005). Nevertheless, specific bacterial groups changed in abundance during rice plant growth. Only OTUs with relatively low abundance were characteristic for individual growth stages, whereas the dominant OTUs were equally distributed and observed at all three growth stages. Only OTU number 4 (Methylocystis) was more prominent at the vegetative and reproductive than at the maturity stage (Figure 5B). Additionally, Verrucomicrobia and Bacteroidia increased from vegetative to reproductive growth stage, Cyanobacteria decreased (data not shown). Comparison of the ribosomal OTUs in unplanted, maize and rice-cultivated soils showed more pronounced preferences (Figure 5D). The most dominant OTU number 1 (Myxococcales) was preferentially associated with the non-flooded fields (unplanted, maize) while OTU number 3 (Acidobacteria Gp6) and 2 (Cystobacteraceae) were found in all field types. Additionally unplanted fields showed higher Bacteroidetes and Sphingobacteria than the rice fields (ANOVA, p < 0.05). Less Verrucomicrobia were detected in the unplanted fields in comparison with rice and maize fields.
The analysis of presence and absence of individual OTUs based on rRNA revealed that the OTUs detected in all fields (core OTUs) constituted 70, 74, and 71% of the relative abundance in rice, maize and unplanted fields, respectively. Direct comparison of the rice and the unplanted fields showed similar distribution of bacterial lineages in core, shared and unique OTUs (Figure 6). The relative abundance of core OTUs assigned as Deltaproteobacteria and unique OTUs assigned as Armantimonadetes, Bacteroidetes, Alphaproteobacteria, and Gammaproteobacteria was increased in the unplanted fields (Figure 6).
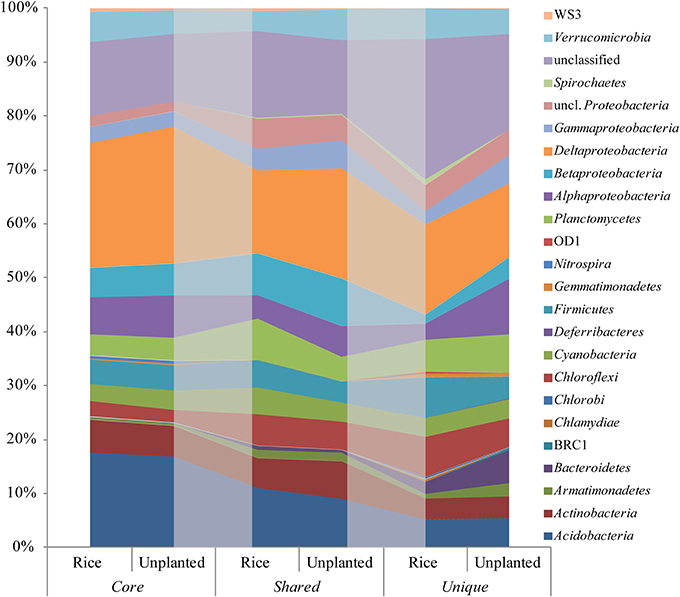
Figure 6. OTU based relative sequence abundance of bacterial phyla based on 16S rRNA in the rice and unplanted fields. OTUs detected in all fields (core), only in unplanted and rice cultivated fields (shared) and OTUs only detected in rice or unplanted fields (unique) were presented. The shaded areas serve visualization and have no special meaning.
Pyrosequencing of Archaeal 16S rDNA and rRNA
Analogously to the bacterial community, the archaeal community was analyzed using pyrosequencing targeting archaeal 16S rDNA and rRNA. Sequencing resulted in 1326 to 9290 high quality sequences of rDNA as well as 532 to 1962 sequences of rRNA (Table 3). For archaeal rDNA, the major taxa with a relative abundance of >2% in at least one sample are shown in Supplement Figure 3A. The most dominant class was Methanomicrobia (40–50% relative abundance) followed by Soil Crenarchaeotic Group (20–34%), Misc. Crenarchaeotic Group (17–23%), and Methanobacteria (2–6%). The class Methanomicrobia was further subdivided into orders and families. The most dominant archaeal order was Methanosarcinales with the families Methanosarcinaceae, Methanosaetaceae, and the group GOM Arc I (Supplement Figure 3A). Comparison of the dominant OTUs retrieved at different rice plant growth stages showed a uniform distribution over the season (Figure 5E). Most dominant OTUs were assigned as Soil Crenarchaeotic Group and Methanosarcina and their distribution did not change in composition during rice plant growth (Figure 5E). Several trends, albeit not significant, are worth mentioning. Thus, the relative abundance of Methanobacteriaceae, Methanosarcinaceae, Methanosaetaceae, and Methanocellaceae was relatively high in the reproductive stage, while that of GOM Arc I was relatively low (Supplement Figure 3A). The relative abundance of the genera Methanolinea and Candidatus Methanoregula decreased from vegetative to later growth stages.
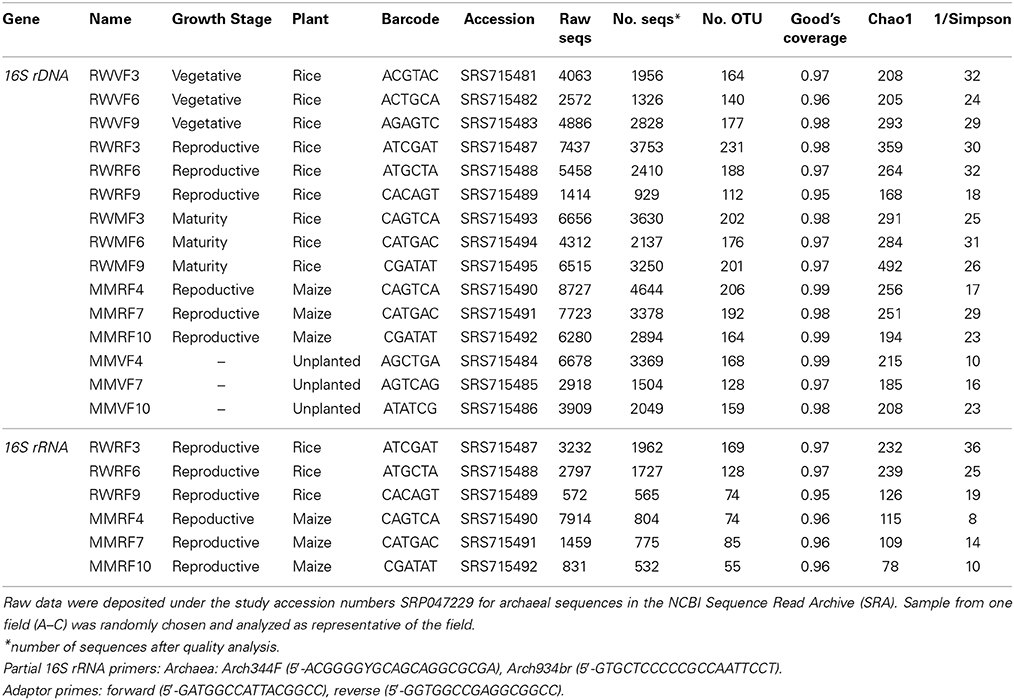
Table 3. Number of archaeal sequences before and after quality management, barcode, number of OTUs, coverage, Chao1 and inverted Simpson index of the environmental samples analyzed by 454-pyrosquencing.
Comparison of unplanted, maize and rice cultivated fields showed again a uniform distribution of the OTUs (Figure 5F). However, the OTUs assigned as Misc. Crenarchaeotic Group and unclassified were more abundant in the planted fields (rice, maize), while the unclassified OTU was more associated with the maize field. Similar to T-RFLP analysis an increase of Methanosarcinaceae in the maize fields and unplanted fields was observed, but not statistically significant (Supplement Figure 3A). In fact, no significant changes in the archaeal community composition were observed (ADONIS, P > 0.05).
The sequences of ribosomal RNA were composed of the same archaeal lineages as the ribosomal gene-based community, but exhibited different relative abundances (Supplement Figure 3B). The most dominant class was Methanomicrobia (38–61% relative abundance) followed by Soil Crenarchaeotic Group (18–31%), Misc. Crenarchaeotic Group (4%), and Methanobacteria (2–6%). The archaeal community composition was not significantly different between fields cultivated with rice and maize on class, order and family level (ADONIS, P > 0.005). Nevertheless, specific archaeal groups changed in abundance. A significant increase of Soil Crenarchaeotic Group in the maize fields was observed (Supplement Figure 3B). Within Soil Crenarchaeotic Group Candidatus Nitrososphaera was higher in maize cultivated fields (data not shown). Only the top 30 OTUs representing up to 80% of all sequences were used for analysis (Figure 7). Again, there was a trend that methanogenic archaeal lineages (Methanosarcina, Methanosaeta, Methanocella, Methanobacterium) were more abundant in the rice than in the maize cultivated soil, and that non methanogenic groups (Soil Crenarchaeotic Group, GOM Arc I, Misc. Crenarchaeotic Group, Candidatus Nitrososphaera) were in particular observed in the maize field, but the trend was not statistically significant.
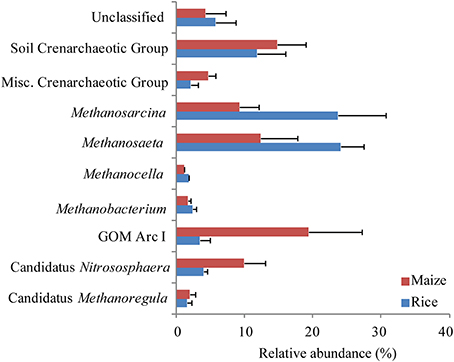
Figure 7. Relative abundance of the archaeal OTUs in rice field soil. Based on relative abundance top 30 OTUs derived from pyrosequencing of the archaeal 16S rRNA were grouped according to their phylogenetic assignment. OTUs were monitored in rice (blue) and maize (red) fields. Columns represent mean and bars standard errors of n = 3.
Discussion
Bacterial and Archaeal Communities at Different Rice Growth Stages
Total bacterial and archaeal 16S rDNA copy numbers increased during reproductive growth stage indicating growth. Recently, Lee et al. (2014) likewise described in a Korean rice field an increase in bacterial and archaeal copy numbers during rice plant growth followed by a decrease at plant maturity. Although the microbial numbers in our Philippine soil were one order of magnitude higher than in the Korean soil (Lee et al., 2014), in both soils the microbial numbers changed by only a factor of two over the season. A similar observation was made by Itoh et al. (2013) showing a moderate increase of the resident microbes during rice plant growth under flooded conditions in a Japanese rice field. Textbook knowledge tells that plants secrete a complex mixture of organic and inorganic compounds (rhizodeposits) via their root system. Several studies showed an increase in root exudation with rice plant growth reaching a maximum at reproductive stage and decreasing again toward maturity of the rice plants (Aulakh et al., 2001; Lu et al., 2002; Watanabe et al., 2004; Pump and Conrad, 2014). Therefore, it is likely that the modest increase from vegetative to reproductive and the decrease toward maturity of the bacterial and archaeal abundance is driven by root exudation.
In contrast to the change in the resident (16S rDNA) populations, the numbers of the active populations (16S rRNA) were stable and did not show seasonal dynamics. Studies comprehensively covering the resident and active archaeal as well as bacterial communities in rice fields during plant growth are hardly available (Itoh et al., 2013). The detection of ribosomal RNA is equivalent to that of ribosomes, which are indicative for actively dividing or actively metabolizing microbial cells (Blazewicz et al., 2013). Therefore, changes in the 16S rRNA pool caused by growing cells can be superimposed by fluctuations in the amount of active but non-growing cells. Additionally, it was shown that different taxa can have different numbers of 16S rDNA copies (e.g., Sukenik et al., 2012). Interestingly, the standard errors of the abundance of both active bacterial and archaeal populations were higher in the vegetative and reproductive plant growth phase and decreased with plant maturity. This may be an indication for the influence of root exudation affecting microbial activity. All together the data indicate that the bacterial and archaeal communities were composed of active and growing cells being enhanced during the reproductive growth stage possibly due to root exudation.
The composition of both the resident (16S rDNA) and the active (16S rRNA) bacterial community revealed only minor changes with the rice plant growth stages. Among the resident bacteria, only OTUs with negligible relative abundance were found to be specific for a particular plant growth stage. E.g., an OTU identified as Anaeromyxobacter was specific for the vegetative growth stage. Anaeromyxobacter spp. are known as iron reducers and are possibly important for carbon and iron dynamics in the rice rhizosphere (Ratering and Schnell, 2001; Treude et al., 2003). In rice fields oxidants like ferric iron are rapidly reduced (Conrad et al., 2014), but may be regenerated when plants allow O2 release from their roots, thus as during vegetative plant growth (Liesack et al., 2000). Among the active bacteria, Cyanobacteria were highest during the early vegetative growth phase possibly caused by the previous field preparation (i.e., puddling) mixing sun-exposed soil parts into the bulk. The increase in relative abundance of active Verrucomicrobia and Bacteroidia during reproductive plant growth may be a consequence of their ecophysiology, which is playing a role in carbon degradation (Sugano et al., 2005; Tanahashi et al., 2005; Kikuchi et al., 2007; Rui et al., 2009). A member of Verrucomicrobia, i.e., Opitutus terrae, was isolated from a paddy rice field as potential polysaccharolytic and saccharolytic and capable of hydrogen production (Chin et al., 1999, 2001). Bacteroidia are known key players in decomposition of rice plant residue (Weber et al., 2001; Akasaka et al., 2003) and Bacteroidetes prominent heterotrophs in rice field soil including a propionate-producing fermentative representative (Akasaka et al., 2003). The OTU based analysis showed that the methanotrophic Methylocystis became prominent before plant maturity. Methylocystis, a type-II methanotroph, has commonly been found in rice field soil (Murase and Frenzel, 2007; Shrestha et al., 2010). Methanotrophs are dependent on their primary substrate methane and oxygen. Oxygen was probably released by the roots during the reproductive growth phase of the rice plant. For example Gilbert and Frenzel (1998) reported radial oxygen loss by roots of up to 6 weeks old rice plants.
The T-RFLP analysis in the Philippine rice fields showed a relatively constant composition of the archaeal community over the season. Similar results had been obtained in an Italian rice field (Krüger et al., 2005). Our pyrosequencing data indicated an increase in relative abundance of the dominating methanogens (Methanosarcinaceae, Methanosaetaceae, Methanobacteriaceae, and Methanocellaceae) during reproductive growth stage, but this increase was statistically not significant. Within the order of Methanosarcinales GOM Arc I species were notably detected under all tested conditions and decreased during reproductive stage. GOM Arc I was formerly known as ANME-2d caused by phylogenetic relation to the anaerobic methanotrophs ANME-2 (Mills et al., 2005; Martinez et al., 2006). Nevertheless, the role of GOM Arc I in the methane biogeochemistry is still unclear (Lloyd et al., 2006; Knittel and Boetius, 2009). We speculate that the importance of these organisms which were previously detected in relatively high numbers in South Korean rice field soil (Ahn et al., 2014) has been underestimated and strengthen the need to identify their function in methane cycling.
All together rhizodeposition and oxygen release seemed to increase growth and activity of specific bacterial and archaeal lineages during the reproductive growth stage. However, changes over the season were only small and the resident and active microbial communities remained relatively conserved.
Bacterial and Archaeal Communities in Flooded and Non-Flooded Fields
Rotation of the cultivated crop from paddy rice (flooded) to upland maize (non-flooded) changes the field conditions dramatically. In our study, we were dealing with flooded rice fields and non-flooded unplanted fields, which were then planted with maize, but kept under non-flooded conditions. Anaerobic degradation of organic matter to CH4 is only possible if the bulk of the soil is anoxic, such as in flooded fields. In non-flooded fields no or comparatively little anaerobic microbial activity is expected. Indeed, compared to the flooded fields CH4 emission was only minor from the non-flooded fields (Weller et al., 2014). Therefore, living conditions of obligately anaerobic microorganisms, such as methanogenic archaea and many fermenting bacteria were restricted.
The abundances of resident bacteria and archaea (16S rDNA) were lowest in unplanted fields, whereas they were highest in the flooded rice fields and intermediate in the non-flooded maize fields. The microbial populations apparently increased in number when the non-flooded fields were planted with maize, but did not reach the same level as in the flooded rice fields. Hence, microbial abundance was apparently affected by both flooding and the presence of vegetation. The low microbial abundance in unplanted fields was possibly due to the absence of release of organic material from roots and/or lack of fertilization, which allowed the microbes to grow to some extent in the maize fields. In these oxic soils, however, the number of microbes remained lower than in the anoxic flooded fields. Surprisingly, the abundance of ribosomal RNA, being indicative for active microbes, was in the same range for both unplanted and planted fields and for both non-flooded maize and flooded rice fields. Therefore, we assume that the microbial cells in non-flooded unplanted soil, and to some extend also the maize field soil, contained more ribosomal RNA than those in the flooded rice field soil. The rather high ratio of rRNA/rDNA in non-flooded fields was observed in most replicate samples, but there were a few replicates, which behaved differently. High ratios of rRNA/rDNA have also been observed in non-flooded Japanese rice fields but not been further discussed (Watanabe et al., 2007). However, numbers of rRNA decreasing with drainage have also been observed in a Japanese rice field (Itoh et al., 2013). At a first glance high ratios of rRNA/rDNA seem surprising, since anaerobic microorganisms should be less active in the unplanted and maize fields than in the flooded rice fields. However, it has been shown that even dormant cells harbor measureable amounts of 16S rRNA and that in some cases the 16S rRNA amount can even be significantly higher than in vegetative cells (Chambon et al., 1968; Sukenik et al., 2012). The maintenance of a high level of ribosomal RNA under unfavorable conditions is interpreted as preparedness for activity when conditions improve. Hence, we assume that numbers of anaerobic microorganisms decreased when the flooded rice fields were turned into non-flooded maize fields, but at the same time increased the cellular levels of rRNA (presumably ribosomes) as a stress response and possibly to be prepared for new flooding.
However, there may be additional explanations for the high ratio of rRNA/rDNA in the non-flooded soil. For example, during field preparation and after drainage soil structure gets disturbed, and this process may cause death of microbes by breaking up the cells. The nutrients of the dead cells become then available for the surviving microbes and may thus enhance their activity. The soil texture was a silt loam, which is characterized by a high water holding capacity. The high water holding capacity may have allowed the maintenance of anaerobic microniches with active populations of anaerobic microorganisms. Finally, drainage may have allowed an increase of the soil temperature, thus promoting the activity of the overall community which inhabits the anaerobic microniches.
Interestingly, the microbial community compositions were not much different between flooded and non-flooded fields. Although CCA analysis of the community based on T-RFLP revealed some differences in composition, the variance on the two CCA axes were less than 16%. Only the analysis by 454 pyrosequencing unveiled some changes in relative abundance of few bacterial groups but no dramatic community shifts. In the present study these bacterial lineages can be grouped due to their ecophysiology. Spartobacteria and Sphingobacteria were both described as aerobes and increased in their relative abundance in the non-flooded fields possibly due to decreasing water level and concomitant increased oxygen exposure. The first isolate of Spartobacteria was described as an aerobic heterotrophic bacterium able to grow on saccharide components of plant biomass (Sangwan et al., 2004). Additionally, some members of the Sphingobacteria were described as aerobes, while others are anaerobes or facultative anaerobes suggesting a dependence on oxygen levels (Janssen, 2006). The second group of bacterial lineages (Bacteroidetes and Acidobacteria) increasing in non-flooded fields is associated with their ability to sustain low substrate conditions and to degrade complex organic compounds under anaerobic conditions. For instance Bacteroidetes were frequently detected during rice plant residue decomposition (e.g., Weber et al., 2001; Rui et al., 2009) and have the ability to grow on various complex carbon substrates (Kirchman, 2002). Although Acidobacteria are widely distributed and highly abundant in soil environments little is known about their ecology (Lee and Cho, 2009). Various observations suggest that the chemo-organotrophic and oligotrophic Acidobacteria are adapted to low substrate availability highlighted by slow growth rates (e.g., Davis et al., 2005, 2011) and are able to decompose complex carbon compounds like xylan, cellulose and pectin (Eichorst et al., 2011). The last bacterial lineage more pronounced in the non-flooded fields was Myxococcales. Iron reducing Anaeromyxobacter are members of Myxococcales and represented the maturity of the order in the present study. During drainage regeneration of inorganic electron acceptors like ferric iron occurs. Therefore, it is likely that iron reducers out of Myxococcales are supported in their competition with methanogens for electron acceptors like hydrogen and acetate (Ratering and Conrad, 1998). In summary, the bacterial groups in the unplanted fields were characterized by their abilities to grow under oxic conditions and to degrade complex carbon substrates.
Similarly, the archaeal community composition was quite similar in non-flooded and flooded fields. This observation is consistent with previous studies showing that crop rotation including upland crop management affected archaeal communities only little (Watanabe et al., 2006, 2011; Fernandez Scavino et al., 2013). However, we observed a significant increase in relative abundance of Methanosarcinaceae in upland maize fields by T-RFLP analysis and a non-significant increase by pyrosequencing. In Japanese rice fields Methanosarcinales were a major group under both flooded and drainage conditions (Watanabe et al., 2009; Itoh et al., 2013). Methanosarcina spp. together with Methanocella spp. have also been found in dry ecosystems, such as upland soils and desert biological soil crusts (Nicol et al., 2003; Angel et al., 2012; Conrad et al., 2012; Aschenbach et al., 2013). These species possess a relatively large number of genes coding for oxygen-detoxifying enzymes (Erkel et al., 2006), thus probably allowing them to survive exposition to oxygen in dry soils (Angel et al., 2011, 2012). Therefore, it is likely that Methanosarcina spp. survived relatively well when the flooded rice fields were turned into non-flooded maize fields, thus increasing their relative abundance among the other archaea.
The Soil Crenarchaeotic Group also showed a relatively high abundance in the non-flooded maize fields. The ecophysiology of Crenarchaeota is largely unknown (Pester et al., 2011), although Thaumarchaeota, with potential for ammonia oxidation are found as a dominant archaeal group in aerated soils (e.g., Nicol et al., 2003). An upland pasture in Uruguay was reported to be dominated by Crenarchaeota/Thaumarchaeota, which decreased in relative abundance as soon as the soil was turned into a pasture-rice crop rotation (Fernandez Scavino et al., 2013). The predominance of Methanosarcinaceae and Soil Crenarchaeotic Group in non-flooded soils emphasize their capability to withstand temporal desiccation and oxygen stress.
Conclusion
The bacterial and archaeal abundance and activity only moderately changed during rice growth most likely by the influence of rice plants and their root exudation. However, neither archaeal nor bacterial community composition changed much suggesting good adaptation to the conditions in the rice field. By contrast, the change from flooded rice to non-flooded cropping caused a comparatively stronger change in the microbial community composition, which however, was also not very dramatic. The relatively minor effect of change to non-flooded cropping was probably caused by the fact that the microbial communities in the rice field soil were historically adapted to regular drainage. This adaptation was also seen by the maintenance of a high ratio of ribosomal RNA per gene copy, being equivalent to a high number of ribosomes per cell, indicating a preparedness for change between unfavorable non-flooded to favorable flooded conditions for the methanogenic archaea and anaerobic bacteria resident in the rice field soil. The similarity in composition together with the statistically significant increase in ribosomal numbers imply that it was not so much specific members of the communities that regulated their ratios of rRNA/rDNA, but the communities in general that reacted upon the change from flooded to non-flooded state. We conclude that methods reducing greenhouse gas emission from rice fields like mid-season drainage and crop rotation (Wassmann et al., 2000; Li et al., 2006; Pittelkow et al., 2014) will have only little immediate effect on the bacterial and archaeal communities and thus, allow their function to be largely conserved over unfavorable periods.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work has been funded as part of the ICON consortium (BR2238/9-1). We are thankful to Martin B. Blaser for his organizational work, support during sample collection and valuable comments on the study design. We thank Peter Frenzel for valuable comments on the study design and his support during data analysis. Furthermore, we thank the International Rice Research Institute and Reiner Wassmann for providing research space and support during sample collection. We thank Mary Louise Mendoza, Eugene Aquino and Jerico Stefan Bigornia for sample collection. We are thankful to Franziska B. Brandt for valuable comments on the manuscript. We thank MPGC in Cologne for access to the sequencing facility and support.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2014.00752/abstract
References
Ahn, J. H., Choi, M. Y., Kim, B. Y., Lee, J. S., Song, J., Kim, G. Y., et al. (2014). Effects of water-saving irrigation on emissions of greenhouse gases and prokaryotic communities in rice paddy soil. Microb. Ecol. 68, 271–283. doi: 10.1007/s00248-014-0371-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Akasaka, H., Izawa, T., Ueki, K., and Ueki, A. (2003). Phylogeny of numerically abundant culturable anaerobic bacteria associated with degradation of rice plant residue in Japanese paddy field soil. FEMS Microbiol. Ecol. 43, 149–161. doi: 10.1111/j.1574-6941.2003.tb01054.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Angel, R., Claus, P., and Conrad, R. (2012). Methanogenic archaea are globally ubiquitous in aerated soils and become active under wet anoxic conditions. ISME J. 6, 847–862. doi: 10.1038/ismej.2011.141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Angel, R., Matthies, D., and Conrad, R. (2011). Activation of methanogenesis in arid biological soil crusts despite the presence of oxygen. PLoS ONE 5:e20453. doi: 10.1371/journal.pone.0020453
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Asakawa, S., and Kimura, M. (2008). Comparison of bacterial community structures at main habitats in paddy field ecosystem based on DGGE analysis. Soil. Biol. Biochem. 40, 1322–1329. doi: 10.1016/j.soilbio.2007.09.024
Aschenbach, K., Conrad, R., Řeháková, K., Doležal, J., Janatková, K., and Angel, R. (2013). Methanogens at the top of the world: occurrence and potential activity of methanogens in newly deglaciated soils in high-altitude cold deserts in the Western Himalayas. Front. Microbio. 4:359. doi: 10.3389/fmicb.2013.00359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aulakh, M. S., Wassmann, R., Bueno, C., Kreuzwieser, J., and Rennenberg, H. (2001). Characterization of root exudates at different growth stages of ten rice (Oryza sativa L.) cultivars. Plant Biol. 3, 139–148. doi: 10.1055/s-2001-12905
Bates, S. T., Cropsey, G. W., Caporaso, J. G., Knight, R., and Fierer, N. (2011). Bacterial communities associated with the lichen symbiosis. Appl. Environ. Microbiol. 77, 1309–1314. doi: 10.1128/AEM.02257-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blazewicz, S. J., Barnard, R. L., Daly, R. A., and Firestone, M. K. (2013). Evaluating rRNA as an indicator of microbial activity in environmental communities: limitations and uses. ISME J. 7, 2061–2068. doi: 10.1038/ismej.2013.102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bridgham, S. D., Cadillo−Quiroz, H., Keller, J. K., and Zhuang, Q. (2013). Methane emissions from wetlands: biogeochemical, microbial, and modeling perspectives from local to global scales. Glob. Change Biol. 19, 1325–1346. doi: 10.1111/gcb.12131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Burggraf, S., Huber, H., and Stetter, K. O. (1997). Reclassification of the crenarchaeal orders and families in accordance with 16S rRNA sequence data. Int. J. Syst. Evol. Microbiol. 47, 657–660.
Bürgmann, H., Pesaro, M., Widmer, F., and Zeyer, J. (2001). A strategy for optimizing quality and quantity of DNA extracted from soil. J. Microbiol. Meth. 45, 7–20. doi: 10.1016/S0167-7012(01)00213-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Casamayor, E. O., Massana, R., Benlloch, S., Øvreås, L., Díez, B., Goddard, V. J., et al. (2002). Changes in archaeal, bacterial and eukaryal assemblages along a salinity gradient by comparison of genetic fingerprinting methods in a multipond solar saltern. Environ. Microbiol. 4, 338–348. doi: 10.1046/j.1462-2920.2002.00297.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chambon, P., DuPraw, E. J., and Kornberg, A. (1968). Biochemical studies of bacterial sporulation and germination. J. Biol. Chem. 243, 5101–5109.
Chen, Y. H., and Prinn, R. G. (2005). Atmospheric modeling of high-and low-frequency methane observations: importance of interannually varying transport. J. Geophys. Res. 110:D10303. doi: 10.1029/2004JD005542
Chin, K.-J., Hahn, D., Hengstmann, U., Liesack, W., and Janssen, P. H. (1999). Characterization and identification of numerically abundant culturable bacteria from the anoxic bulk soil of rice paddy microcosms. Appl. Environ. Microbiol. 65, 5042–5049.
Chin, K. J., Liesack, W., and Janssen, P. H. (2001). Opitutus terrae gen. nov., sp. nov., to accommodate novel strains of the division “Verrucomicrobia” isolated from rice paddy soil. Int. J. Syst. Evol. Micr. 51, 1965–1968. doi: 10.1099/00207713-51-6-1965
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Conrad, R. (2007). Microbial ecology of methanogens and methanotrophs. Adv. Agron. 96, 1–63. doi: 10.1016/S0065-2113(07)96005-8
Conrad, R. (2009). The global methane cycle: recent advances in understanding the microbial processes involved. Environ. Microbiol. Rep. 1, 285–292. doi: 10.1111/j.1758-2229.2009.00038.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Conrad, R., Claus, P., Chidthaisong, A., Lu, Y., Fernandez Scavino, A., Liu, Y., et al. (2014). Stable carbon isotope biogeochemistry of propionate and acetate in methanogenic soils and lake sediments. Org. Geochem. 73, 1–7. doi: 10.1016/j.orggeochem.2014.03.010
Conrad, R., and Klose, M. (2006). Dynamics of the methanogenic archaeal community in anoxic rice soil upon addition of straw. Eur. J. Soil. Sci. 57, 476–484. doi: 10.1111/j.1365-2389.2006.00791.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Conrad, R., Klose, M., Lu, Y., and Chidthaisong, A. (2012). Methanogenic pathway and archaeal communities in three different anoxic soils amended with rice straw and maize straw. Front. Microbiol. 3:4. doi: 10.3389/fmicb.2012.00004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Conrad, R., Klose, M., and Noll, M. (2009). Functional and structural response of the methanogenic microbial community in rice field soil to temperature change. Environ. Microbiol. 11, 1844–1853. doi: 10.1111/j.1462-2920.2009.01909.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Conrad, R., Klose, M., Noll, M., Kemnitz, D., and Bodelier, P. L. E. (2008). Soil type links microbial colonization of rice roots to methane emission. Glob. Change Biol. 14, 657–669. doi: 10.1111/j.1365-2486.2007.01516.x
Costa, R., Götz, M., Mrotzek, N., Lottmann, J., Berg, G., and Smalla, K. (2006). Effects of site and plant species on rhizosphere community structure as revealed by molecular analysis of microbial guilds. FEMS Microbiol. Ecol. 56, 236–249. doi: 10.1111/j.1574-6941.2005.00026.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Davis, K. E., Joseph, S. J., and Janssen, P. H. (2005). Effects of growth medium, inoculum size, and incubation time on culturability and isolation of soil bacteria. Appl. Environ. Microbiol. 71, 826–834. doi: 10.1128/AEM.71.2.826-834.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Davis, K. E., Sangwan, P., and Janssen, P. H. (2011). Acidobacteria, Rubrobacteridae and Chloroflexi are abundant among very slow−growing and mini−colony−forming soil bacteria. Environ. Microbiol. 13, 798–805. doi: 10.1111/j.1462-2920.2010.02384.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dennis, P. G., Miller, A. J., and Hirsch, P. R. (2010). Are root exudates more important than other sices of rhizodeposits in structuring rhizosphere bacterial communities? FEMS Microbiol. Ecol. 72, 313–327. doi: 10.1111/j.1574-6941.2010.00860.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dunbar, J., Ticknor, L. O., and Kuske, C. R. (2001). Phylogenetic specificity and reproducibility and new method for analysis of terminal restriction fragment profiles of 16S rRNA genes from bacterial communities. Appl. Environ. Microbiol. 67, 190–197. doi: 10.1128/AEM.67.1.190-197.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eichorst, S. A., Kuske, C. R., and Schmidt, T. M. (2011). Influence of plant polymers on the distribution and cultivation of bacteria in the phylum Acidobacteria. Appl. Environ. Microbiol. 77, 586–596. doi: 10.1128/AEM.01080-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Erkel, C., Kube, M., Reinhardt, R., and Liesack, W. (2006). Genome of Rice Cluster I archaea—the key methane producers in the rice rhizosphere. Science 313, 370–372. doi: 10.1126/science.1127062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fernandez Scavino, A., Ji, Y., Pump, J., Klose, M., Claus, P., and Conrad, R. (2013). Structure and function of the methanogenic microbial communities in Uruguayan soils shifted between pasture and irrigated rice fields. Environ. Microbiol. 15, 2588–2602. doi: 10.1111/1462-2920.12161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Forster, P., Ramaswamy, P., Artaxo, P., Berntsen, T., Betts, R., Fahey, D. W., et al. (2007). “Changes in atmospheric constituents and in radiative forcing,” in Climate Change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, eds S. Solomon, D. Qin, M. Manning, Z. Chen, M. Marquis, K. B. Averyt et al. (Cambridge: Cambridge University Press), 129–234.
Gilbert, B., and Frenzel, P. (1998). Rice roots and CH4 oxidation: the activity of bacteria, their distribution and the microenvironment. Soil. Biol. Biochem. 30, 1903–1916. doi: 10.1016/S0038-0717(98)00061-3
Grayston, S. J., Wang, S., Campbell, C. D., and Edwards, A. C. (1998). Selective influence of plant species on microbial diversity in the rhizosphere. Soil. Biol. Biochem. 30, 369–378. doi: 10.1016/S0038-0717(97)00124-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Großkopf, R., Janssen, P. H., and Liesack, W. (1998). Diversity and structure of the methanogenic community in anoxic rice paddy soil microcosms as examined by cultivation and direct 16S rRNA gene sequence retrieval. Appl. Environ. Microbiol. 64, 960–969.
Heinz, E., Kraft, P., Buchen, C., Frede, H. G., Aquino, E., and Breuer, L. (2013). Set up of an automatic water quality sampling system in irrigation agriculture. Sensors 14, 212–228. doi: 10.3390/s140100212
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Itoh, H., Ishii, S., Shiratori, Y., Oshima, K., Otsuka, S., Hattori, M., et al. (2013). Seasonal transition of active bacterial and archaeal communities in relation to water management in paddy soils. Microbes Environ. 28, 370–380. doi: 10.1264/jsme2.ME13030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Janssen, P. H. (2006). Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 72, 1719–1728. doi: 10.1128/AEM.72.3.1719-1728.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kikuchi, H., Watanabe, T., Jia, Z., Kimura, M., and Asakawa, S. (2007). Molecular analyses reveal stability of bacterial communities in bulk soil of a Japanese paddy field: estimation by denaturing gradient gel electrophoresis of 16S rRNA genes amplified from DNA accompanied with RNA. Soil Sci. Plant Nutr. 53, 448–458. doi: 10.1111/j.1747-0765.2007.00177.x
Kimura, M., Murase, J., and Lu, Y. (2004). Carbon cycling in rice field ecosystems in the context of input, decomposition and translocation of organic materials and the fates of their end products (CO2 and CH4). Soil. Biol. Biochem. 36, 1399–1416. doi: 10.1016/j.soilbio.2004.03.006
Kirchman, D. L. (2002). The ecology of Cytophaga–Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 39, 91–100. doi: 10.1016/S0168-6496(01)00206-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Knittel, K., and Boetius, A. (2009). Anaerobic oxidation of methane: progress with an unknown process. Annu. Rev. Microbiol. 63, 311–334. doi: 10.1146/annurev.micro.61.080706.093130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kowalchuk, G. A., Yergeau, E., Leveau, J. H. J., Sessitsch, A., and Bailey, M. (2010). “Plant-associated microbial communities,” in Environmental Molecular Microbiology, eds W.-T. Liu and J. K. Jansson (Poole: Caister Academic Press), 131–148.
Krüger, M., Frenzel, P., Kemnitz, D., and Conrad, R. (2005). Activity, structure and dynamics of the methanogenic archaeal community in a flooded Italian rice field. FEMS Microbiol. Ecol. 51, 323–331. doi: 10.1016/j.femsec.2004.09.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, H. J., Kim, S. Y., Kim, P. J., Madsen, E. L., and Jeon, C. O. (2014). Methane emission and dynamics of methanotrophic and methanogenic communities in a flooded rice field ecosystem. FEMS Microbiol. Ecol. 88, 195–212. doi: 10.1111/1574-6941.12282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, S. H., and Cho, J. C. (2009). Distribution patterns of the members of phylum acidobacteria in global soil samples. J. Microbiol. Biotech. 19, 1281–1287. doi: 10.4014/jmb.0904.4017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, C. S., Salas, W., DeAngelo, B., and Rose, S. (2006). Assessing alternatives for mitigating net greenhouse gas emissions and increasing yields from rice production in China over the next twenty years. J. Environ. Qual. 35, 1554–1565. doi: 10.2134/jeq2005.0208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liesack, W., Schnell, S., and Revsbech, N. P. (2000). Microbiology of flooded rice paddies. FEMS Microbiol. Rev. 24, 625–645. doi: 10.1111/j.1574-6976.2000.tb00563.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lloyd, K. G., Lapham, L., and Teske, A. (2006). An anaerobic methane-oxidizing community of ANME-1b archaea in hypersaline Gulf of Mexico sediments. Appl. Environ. Microbiol. 72, 7218–7230. doi: 10.1128/AEM.00886-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lopes, A. R., Manaia, C. M., and Nunes, O. C. (2014). Bacterial community variations in an alfalfa−rice rotation system revealed by 16S rRNA gene 454−pyrosequencing. FEMS Microbiol. Ecol. 87, 650–663. doi: 10.1111/1574-6941.12253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lu, Y., Watanabe, A., and Kimura, M. (2002). Contribution of plant-derived carbon to soil microbial biomass dynamics in a paddy rice microcosm. Biol. Fert. Soils 36, 136–142. doi: 10.1007/s00374-002-0504-2
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Buchner, A., et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lueders, T., and Friedrich, M. W. (2003). Evaluation of PCR amplification bias by terminal restriction fragment length polymorphism analysis of small-subunit rRNA and mcrA genes by using defined template mixtures of methanogenic pure cultures and soil DNA extracts. Appl. Environ. Microbiol. 69, 320–326. doi: 10.1128/AEM.69.1.320-326.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marschner, P., Yang, C. H., Lieberei, R., and Crowley, D. E. (2001). Soil and plant specific effects on bacterial community composition in the rhizosphere. Soil. Biol. Biochem. 33, 1437–1445. doi: 10.1016/S0038-0717(01)00052-9
Martinez, R. J., Mills, H. J., Story, S., and Sobecky, P. A. (2006). Prokaryotic diversity and metabolically active microbial populations in sediments from an active mud volcano in the Gulf of Mexico. Environ. Microbiol. 8, 1783–1796. doi: 10.1111/j.1462-2920.2006.01063.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mills, H. J., Martinez, R. J., Story, S., and Sobecky, P. A. (2005). Characterization of microbial community structure in Gulf of Mexico gas hydrates: comparative analysis of DNA- and RNA-derived clone libraries. Appl. Environ. Microbiol. 71, 3235–3247. doi: 10.1128/AEM.71.6.3235-3247.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murase, J., and Frenzel, P. (2007). A methane−driven microbial food web in a wetland rice soil. Environ. Microbiol. 9, 3025–3034. doi: 10.1111/j.1462-2920.2007.01414.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muyzer, G., Teske, A., Wirsen, C. O., and Jannasch, H. W. (1995). Phylogenetic relationship of Thiomicrospira species and their identification in deep-sea hydrothermal vent samples by denaturing gradient gel electrophoresis of 16S rDNA fragments. Arch. Microbiol. 164, 165–172. doi: 10.1007/BF02529967
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicol, G. W., Glover, L. A., and Prosser, J. I. (2003). The impact of grassland management on archaeal community structure in upland pasture rhizosphere soil. Environ. Microbiol. 5, 152–162. doi: 10.1046/j.1462-2920.2003.00399.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Noll, M., Matthies, D., Frenzel, P., Derakshani, M., and Liesack, W. (2005). Succession of bacterial community structure and diversity in a paddy soil oxygen gradient. Environ. Microbiol. 7, 382–395. doi: 10.1111/j.1462-2920.2005.00700.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oksanen, J., Blanchet, G. F., Kindt, R., Legendre, R., Minchin, P. R., O'Hara, R. B., et al. (2012). vegan: Community Ecology Package ver. 2.0–5. Available online at: http://cran.r-project.org/web/packages/vegan/index.html
Osborne, C. A., Galic, M., Sangwan, P., and Janssen, P. H. (2005). PCR−generated artefact from 16S rRNA gene−specific primers. FEMS Microbiol. Lett. 248, 183–187. doi: 10.1016/j.femsle.2005.05.043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peng, J. J., Lü, Z., Rui, J., and Lu, Y. H. (2008). Dynamics of the methanogenic archaeal community during plant residue decomposition in an anoxic rice field soil. Appl. Environ. Microbiol. 74, 2894–2901. doi: 10.1128/AEM.00070-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pester, M., Schleper, C., and Wagner, M. (2011). The Thaumarchaeota: an emerging view of their phylogeny and ecophysiology. Curr. Opin. Microbiol. 14, 300–306. doi: 10.1016/j.mib.2011.04.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pittelkow, C. M., Adviento−Borbe, M. A., Kessel, C., Hill, J. E., and Linquist, B. A. (2014). Optimizing rice yields while minimizing yield−scaled global warming potential. Glob. Change Biol. 20, 1382–1393. doi: 10.1111/gcb.12413
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pump, J., and Conrad, R. (2014). Rice biomass production and carbon cycling in 13CO2 pulse-labeled microcosms with different soils under submerged conditions. Plant Soil 384, 213–229. doi: 10.1007/s11104-014-2201-y
Ramakrishnan, B., Lueders, T., Dunfield, P. F., Conrad, R., and Friedrich, M. W. (2001). Archaeal community structures in rice soils from different geographical regions before and after initiation of methane production. FEMS Microbiol. Ecol. 37, 175–186. doi: 10.1111/j.1574-6941.2001.tb00865.x
Ratering, S., and Conrad, R. (1998). Effects of short−term drainage and aeration on the production of methane in submerged rice soil. Glob. Change Biol. 4, 397–407. doi: 10.1046/j.1365-2486.1998.00162.x
Ratering, S., and Schnell, S. (2001). Nitrate−dependent iron (II) oxidation in paddy soil. Environ. Microbiol. 3, 100–109. doi: 10.1046/j.1462-2920.2001.00163.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
R Development Core Team. (2011). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Vienna. Available online at: http://www.R-project.org/
Rui, J., Peng, J., and Lu, Y. (2009). Succession of bacterial populations during plant residue decomposition in rice field soil. Appl. Environ. Microbiol. 75, 4879–4886. doi: 10.1128/AEM.00702-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sangwan, P., Chen, X., Hugenholtz, P., and Janssen, P. H. (2004). Chthoniobacter flavus gen. nov., sp. nov., the first pure-culture representative of subdivision two, Spartobacteria classis nov., of the phylum Verrucomicrobia. Appl. Environ. Microbiol. 70, 5875–5881. doi: 10.1128/AEM.70.10.5875-5881.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schloss, P. D., Gevers, D., and Westcott, S. L. (2011). Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 6:e27310. doi: 10.1371/journal.pone.0027310
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shrestha, M., Shrestha, P. M., Frenzel, P., and Conrad, R. (2010). Effect of nitrogen fertilization on methane oxidation, abundance, community structure, and gene expression of methanotrophs in the rice rhizosphere. The ISME J. 4, 1545–1556. doi: 10.1038/ismej.2010.89
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shrestha, P. M., Kube, M., Reinhardt, R., and Liesack, W. (2009). Transcriptional activity of paddy soil bacterial communities. Environ. Microbiol. 11, 960–970. doi: 10.1111/j.1462-2920.2008.01821.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shrestha, P. M., Noll, M., and Liesack, W. (2007). Phylogenetic identity, growth-response time and rRNA operon copy number of soil bacteria indicate different stages of community succession. Environ. Microbiol. 9, 2464–2474. doi: 10.1111/j.1462-2920.2007.01364.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smalla, K., Wieland, G., Buchner, A., Zock, A., Parzy, J., Kaiser, S., et al. (2001). Bulk and rhizosphere soil bacterial communities studied by denaturing gradient gel electrophoresis: plant-dependent enrichment and seasonal shifts revealed. Appl. Environ. Microbiol. 67, 4742–4751. doi: 10.1128/AEM.67.10.4742-4751.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stubner, S. (2002). Enumeration of 16S rDNA of Desulfotomaculum lineage 1 in rice field soil by real-time PCR with SybrGreen™ detection. J. Microbiol. Meth. 50, 155–164. doi: 10.1016/S0167-7012(02)00024-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sugano, A., Tsuchimoto, H., Tun, C. C., Asakawa, S., and Kimura, M. (2005). Succession and phylogenetic profile of eubacterial communities in rice straw incorporated into a rice field: estimation by PCR-DGGE analysis. Soil Sci. Plant Nutr. 51, 51–60. doi: 10.1111/j.1747-0765.2005.tb00006.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sukenik, A., Kaplan-Levy, R. N., Welch, J. M., and Post, A. F. (2012). Massive multiplication of genome and ribosomes in dormant cells (akinetes) of Aphanizomenon ovalis-porum (Cyanobacteria). ISME J. 6, 670–679. doi: 10.1038/ismej.2011.128
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tanahashi, T., Murase, J., Matsuya, K., Hayashi, M., Kimura, M., and Asakawa, S. (2005). Bacterial communities responsible for the decomposition of rice straw compost in a Japanese rice paddy field estimated by DGGE analysis of amplified 16S rDNA and 16S rRNA fragments. Soil Sci. Plant Nutr. 51, 351–360. doi: 10.1111/j.1747-0765.2005.tb00040.x
Treude, N., Rosencrantz, D., Liesack, W., and Schnell, S. (2003). Strain FAc12, a dissimilatory iron−reducing member of the Anaeromyxobacter subgroup of Myxococcales. FEMS Microbiol. Ecol. 44, 261–269. doi: 10.1016/S0168-6496(03)00048-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Uren, N. C. (2007). “Types, amounts, and possible functions of compounds released into the rhizosphere by soil-grown plants,” in The Rhizosphere. Biochemistry and Organic Substances at the Soil-Plant Interface, eds R. Pinton, Z. Varanini, and P. Nannipieri (New York, NY: Marcel Dekker), 1–21. doi: 10.1201/9781420005585.ch1
Van Nguyen, N., and Ferrero, A. (2006). Meeting the challenges of global rice production. Paddy Water Environ. 4, 1–9. doi: 10.1007/s10333-005-0031-5
Wassmann, R., Lantin, R. S., Neue, H. U., Buendia, L. V., Corton, T. M., and Lu, Y. (2000). Characterization of methane emissions from rice fields in Asia. III. Mitigation options and future research needs. Nutr. Cycl. Agroecosys. 58, 23–36. doi: 10.1023/A:1009874014903
Watanabe, A., Machida, N., Takahashi, K., Kitamura, S., and Kimura, M. (2004). Flow of photosynthesized carbon from rice plants into the paddy soil ecosystem at different stages of rice growth. Plant Soil 258, 151–160. doi: 10.1023/B:PLSO.0000016545.36421.bc
Watanabe, T., Kimura, M., and Asakawa, S. (2006). Community structure of methanogenic archaea in paddy field soil under double cropping (rice-wheat). Soil Biol. Biochem. 38, 1264–1274. doi: 10.1016/j.soilbio.2005.09.020
Watanabe, T., Kimura, M., and Asakawa, S. (2007). Dynamics of methanogenic archaeal communities based on rRNA analysis and their relation to methanogenic activity in Japanese paddy field soils. Soil Biol. Biochem. 39, 2877–2887. doi: 10.1016/j.soilbio.2007.05.030
Watanabe, T., Kimura, M., and Asakawa, S. (2009). Distinct members of a stable methanogenic archaeal community transcribe mcrA genes under flooded and drained conditions in Japanese paddy field soil. Soil Biol. Biochem. 41, 276–285. doi: 10.1016/j.soilbio.2008.10.025
Watanabe, T., Wang, G., Lee, C. G., Murase, J., Asakawa, S., and Kimura, M. (2011). Assimilation of glucose-derived carbon into methanogenic archaea in soil under unflooded condition. Appl. Soil Ecol. 48, 201–209. doi: 10.1016/j.apsoil.2011.03.005
Weber, S., Stubner, S., and Conrad, R. (2001). Bacterial populations colonizing and degrading rice straw in anoxic paddy soil. Appl. Environ. Microbiol. 67, 1318–1327. doi: 10.1128/AEM.67.3.1318-1327.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weller, S., Kraus, D., Ayag, K. R. P., Wassmann, R., Butterbach-Bahl, K., and Kiese, R. (2014). Methane and nitrous oxide emissions from rice and maize production in diversified rice cropping systems. Nutr. Cycl. Agroecosys. doi: 10.1007/s10705-014-9658-1. (in press).
Wu, X. L., Friedrich, M. W., and Conrad, R. (2006). Diversity and ubiquity of thermophilic methanogenic archaea in temperate anoxic soils. Environ. Microbiol. 8, 394–404. doi: 10.1111/j.1462-2920.2005.00904.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zinder, S. H. (1993). “Physiological ecology of methanogens,” in Methanogenesis, ed J. G. Ferry (New York, NY: Springer), 128–206.
Zinger, L., Amaral-Zettler, L. A., Fuhrman, J. A., Horner-Devine, M., Huse, S. M., Welch, D. B., et al. (2011). Global patterns of bacterial beta-diversity in seafloor and sea-water ecosystems. PLoS ONE 6:e24570. doi: 10.1371/journal.pone.0024570
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: methanogenic microbial community, rice field, pyrosequencing, 16S rRNA, season, crop rotation
Citation: Breidenbach B and Conrad R (2015) Seasonal dynamics of bacterial and archaeal methanogenic communities in flooded rice fields and effect of drainage. Front. Microbiol. 5:752. doi: 10.3389/fmicb.2014.00752
Received: 07 November 2014; Accepted: 11 December 2014;
Published online: 08 January 2015.
Edited by:
Paul Bodelier, Netherlands Institute of Ecology (NIOO-KNAW), NetherlandsCopyright © 2015 Breidenbach and Conrad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ralf Conrad, Department of Biogeochemistry, Max Planck Institute for Terrestrial Microbiology, Karl-von-Frisch-Strasse 10, D-35043 Marburg, Germany e-mail:Y29ucmFkQG1waS1tYXJidXJnLm1wZy5kZQ==