 Scott T. Chancey1,2
Scott T. Chancey1,2 Sonia Agrawal3
Sonia Agrawal3 Max R. Schroeder1,2
Max R. Schroeder1,2 Monica M. Farley1,2
Monica M. Farley1,2 Hervé Tettelin3,4†
Hervé Tettelin3,4† David S. Stephens1,2*†
David S. Stephens1,2*†- 1Division of Infectious Diseases, Department of Medicine, Emory University School of Medicine, Atlanta, GA, USA
- 2Laboratories of Microbial Pathogenesis, Department of Veterans Affairs Medical Center, Atlanta, GA, USA
- 3Institute for Genome Sciences, University of Maryland School of Medicine, Baltimore, MD, USA
- 4Department of Microbiology and Immunology, University of Maryland School of Medicine, Baltimore, MD, USA
Macrolide resistance in Streptococcus pneumoniae emerged in the U.S. and globally during the early 1990's. The RNA methylase encoded by erm(B) and the macrolide efflux genes mef(E) and mel were identified as the resistance determining factors. These genes are disseminated in the pneumococcus on mobile, often chimeric elements consisting of multiple smaller elements. To better understand the variety of elements encoding macrolide resistance and how they have evolved in the pre- and post-conjugate vaccine eras, the genomes of 121 invasive and ten carriage isolates from Atlanta from 1994 to 2011 were analyzed for mobile elements involved in the dissemination of macrolide resistance. The isolates were selected to provide broad coverage of the genetic variability of antibiotic resistant pneumococci and included 100 invasive isolates resistant to macrolides. Tn916-like elements carrying mef(E) and mel on the Macrolide Genetic Assembly (Mega) and erm(B) on the erm(B) element and Tn917 were integrated into the pneumococcal chromosome backbone and into larger Tn5253-like composite elements. The results reported here include identification of novel insertion sites for Mega and characterization of the insertion sites of Tn916-like elements in the pneumococcal chromosome and in larger composite elements. The data indicate that integration of elements by conjugation was infrequent compared to recombination. Thus, it appears that conjugative mobile elements allow the pneumococcus to acquire DNA from distantly related bacteria, but once integrated into a pneumococcal genome, transformation and recombination is the primary mechanism for transmission of novel DNA throughout the pneumococcal population.
Introduction
Streptococcus pneumoniae, the pneumococcus, remains a significant risk to human health. Treatment of pneumococcal disease has been hindered by emergence of resistance to the key antibiotics (Klugman and Lonks, 2005). Macrolide resistance in pneumococci emerged throughout the 1990's, a process that has been documented in the Atlanta, Georgia, USA metropolitan area by an ongoing prospective-based surveillance of invasive pneumococcal disease by the Georgia Emerging Infections Program (Farley et al., 2002). A high incidence of efflux-mediated macrolide resistance was observed in serotypes prior to the introduction of the Seven-valent Pneumococcal Conjugate Vaccine (PCV7) in the Atlanta area in late 2000 (Gay et al., 2000; Stephens et al., 2005). PCV7 targets capsular polysaccharides of seven serotypes 4, 6B, 9V, 14, 18C, 19F, and 23F (Stephens et al., 2005). Expansion of these serotypes prior to PCV7 was a major driver of increased macrolide resistance in Atlanta (Stephens et al., 2005).
Most macrolide resistance in S. pneumoniae is conferred by the efflux genes mef(E) and mel or the target-modifying RNA methylase gene erm(B). The mef(E)/mel operon confers the M-phenotype, that is, resistance to 14- and 15-membered macrolides but not lincosamides or streptogramin B (Leclercq and Courvalin, 1991). Erm(B) confers resistance to macrolides, lincosamides and streptogramin B (MLSB antibiotics) which is the MLSB-phenotype (Syrogiannopoulos et al., 2003). The dissemination of macrolide resistance genes in pneumococci has been aided by mobile genetic elements (MGE). The mef(E) and mel genes are located on the Macrolide Genetic Assembly, Mega (Gay and Stephens, 2001). Mega can be 5.5 kb (Mega-1) or 5.4 kb (Mega-2) based on the size of the mef(E)/mel intergenic region. Expression of the mef(E)/mel operon is controlled by transcriptional attenuation and is inducible by 15- and 16-membered macrolides (Ambrose et al., 2005; Wierzbowski et al., 2005). Immediately downstream and convergent to the mef(E)/mel operon are four co-transcribed open reading frames, orfs 3-6. This operon has organization and sequence similarity to a SOS-responsive operon on the pneumococcal conjugative transposons Tn5252 and Tn5253 (Munoz-Najar and Vijayakumar, 1999; Gay and Stephens, 2001). Tn5253, which is Tn5252 with a Tn916-like insertion, is the prototype for a series of Tn5253-like integrative and conjugative element (ICE). It is one of two ICE demonstrated to carry macrolide resistance determinants in pneumococci (Ayoubi et al., 1991; Croucher et al., 2014; Mingoia et al., 2014). The other ICE commonly associated with macrolide resistance is ICESp23FST81 (Croucher et al., 2009).
The Mega element has integrated into at least four loci in the pneumococcal chromosome. Mega insertion classes I–IV were originally identified in US isolates (Gay and Stephens, 2001) and were also observed in Europe (Del Grosso et al., 2006). The class I Mega insertion site is in a homolog of the S. pneumoniae TIGR4 gene SP_1598 encoding a phosphomethylpyrimidine kinase predicted to be involved in thiamine biosynthesis (Gay and Stephens, 2001). Class II insertions are in a DNA-methyladenine glycosidase (SP_0180) and class III are in capA (SP_0103) located in the capsule biosynthesis locus. Class IV is in the RNA-methyltransferase rumA (SP_1029) located at the left flank of the pneumococcal pathogenicity island (PPI-1) (Brown et al., 2001).
In addition to the locations in the pneumococcal chromosome backbone, Mega has integrated into orf6 of Tn916, a 18.0 kb conjugative transposon associated with tetracycline resistance due to the presence of tet(M) (Roberts and Mullany, 2011). Tn916 has been enlisted to disseminate macrolide resistance in many forms. The Tn916-like element carrying Mega inserted into orf6 is Tn2009 (Del Grosso et al., 2004). A Tn916-like element with a 2.8 kb fragment carrying erm(B), named “erm(B) element,” inserted into Tn916 orf20 is Tn6002 (Cochetti et al., 2007) (Figure 1). A Tn916-like element harboring both orf6::Mega and orf20::erm(B) is Tn2010 (Del Grosso et al., 2007) (Figure 1). The fact that the two smaller cassettes are inserted into the same locations in the different Tn916-like elements suggests either a very specific target sequence leading to multiple identical insertions, or that Tn2010 is linked to Tn2009 and Tn6002 through recombination of macrolide resistance-conferring MGE. The same is true for Tn2009, Tn3872, and Tn2017. Tn3872 and Tn2017 are Tn916-like elements with orf9 disrupted by Tn917, a 5.3 kb transposon-related element carrying erm(B) (McDougal et al., 1998). Tn2017 is nearly identical to Tn3872, but also contains Mega integrated into Tn916 orf6 (Del Grosso et al., 2009). The apparent interchangeability of the smaller macrolide resistance elements raises a question regarding the relative roles of conjugation and recombination in the dissemination of macrolide resistance and the evolution in the pneumococcus. S. pneumoniae is a naturally competent bacterium and is extremely proficient at acquiring and incorporating exogenous DNA into its genome (Berge et al., 2002; Claverys et al., 2006; Prudhomme et al., 2006). On the other hand, macrolide resistance-encoding Tn916-like elements appear to be incapable of pneumococcus-to-pneumococcus conjugation (Del Grosso et al., 2004; Zhou et al., 2014). Tn2009 and Tn2010 are non-conjugative while Tn6002 is conjugative but at very low frequencies (Del Grosso et al., 2004, 2007; Cochetti et al., 2007). Conjugative transfer of Mega from S. salivarius, however, has been observed, but only from a host strain with a co-resident conjugative transposon (Santagati et al., 2009). How important is DNA acquisition by conjugation in the pneumococcus, which is so proficient at transformation and recombination?
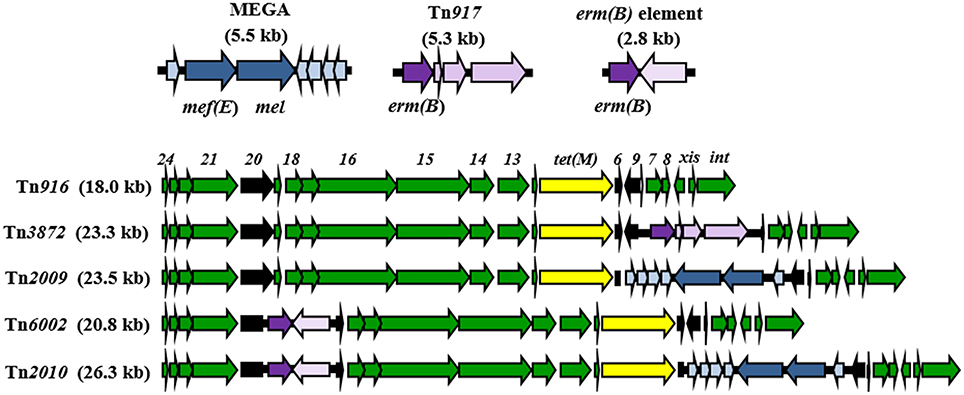
Figure 1. Macrolide resistance determinants associated with mobile elements in pneumococci. Conjugative transposon Tn916 and Tn916-like mosaic elements carrying Mega (blue), and erm(B) on the erm(B) element and Tn917 (purple). The yellow arrows indicate tet(M) encoding tetracycline resistance. Black arrows indicate insertion sites.
In the present study we utilized comparative genomics to identify the mobile elements involved in dissemination of macrolide resistance in a geographically defined population. We sequenced the genomes of 147 pneumococcal isolates, including 131 invasive and carriage isolates collected in Atlanta, Georgia over an 18 year period spanning the pre- and post-conjugative vaccine era (McDougal et al., 1998; Cochetti et al., 2008). The goal of this study was to make a broad survey of the genomic diversity in the pneumococcal clones circulating in the Atlanta metropolitan area in pre- and post-PCV7 eras, and to document the MGE that promoted the dissemination of macrolide resistance genes. By analyzing the genetic organization and the integration sites of the elements in relation to the clonal lineage and serotype of the isolates that harbor them, we hoped to gain a better understanding of the relative roles of conjugation and transformation in the evolution and dissemination of macrolide resistance in S. pneumoniae.
Materials and Methods
Bacterial Strains
Atlanta invasive S. pneumoniae isolates were obtained from the Georgia Emerging Infections Program and were isolated from patients in Atlanta, Georgia as part of an ongoing, prospective, population-based surveillance study (Farley et al., 2002). Macrolide resistant isolates were assayed for the presence of macrolide resistance genes mef(E), mel and erm(B) by PCR as previously described (Stephens et al., 2005). One hundred and twenty one isolates recovered from patients between 1994 and 2011 were non-randomly selected for whole genome sequencing (Table S1). Selection criteria included capsule serotype, antibiotic resistance, macrolide resistance genes, and date of isolation. In addition to macrolide resistance, clindamycin, tetracycline, and chloramphenicol were considered indicators of mobile elements. Sequence type was also considered if available (see below). Thirteen invasive pneumococcal isolates from U.S. states other than Georgia were provided by the Antimicrobial Bacterial Core Surveillance Program and the Centers for Disease Control and Prevention (CDC) (McCormick et al., 2003). These included four isolates from Maryland, two each from Connecticut, Oregon, and Minnesota, and one each from California, New York, and Tennessee. Three invasive isolates, two from Europe and one from the North Carolina, were provided by the Pneumococcal Molecular Epidemiology Network (PMEN) (McGee et al., 2001). Finally, the study was supplemented with 10 carriage isolates collected from the Atlanta population; five each isolated pre-PCV7 (1995) and post-PCV7 (2009) (Sharma et al., 2013). Carriage isolates were collected from nasopharyngeal swabs of healthy children between the ages of 5 months and 5 years old (Sharma et al., 2013). The total number of sequenced pneumococcal genomes was 147 (Table S1).
Characterization of Isolates
The capsule serotype of each isolate was determined by the institution from which it was acquired and verified by analyses of the capsule locus in the genome sequence. Minimal inhibitory concentrations (MIC) of erythromycin, clindamycin, tetracycline, and chloramphenicol were determined by microdilution or Etest (bioMerieux Inc., Durham, NC). The Multi Locus Sequence Type (MLST) of selected isolates was determined by Sanger nucleotide sequencing of PCR products with MLST allele-specific primers as previously described (Enright and Spratt, 1998). The sequence types of the remaining isolates were determined from allelic data extracted from their genomes. Isolates were assigned to a clonal complex based on eBURST analysis of the complete S. pneumoniae MLST database as of September, 2014 (http://pubmlst.org/spneumoniae/).
Genome Sequencing
For each isolate sequenced, two shotgun libraries were constructed for 454 sequencing: one rapid non-paired-end library for coverage and one 3 kb paired-end library for assembly scaffolding. All isolates were sequenced using the 454 GS FLX Titanium to a depth of sequence coverage ranging from 16× to 57×. Shotgun reads were assembled into 4–32 contigs per isolate using Celera Assembler v6.1 (Myers et al., 2000). Genome sequences have been deposited in the GenBank Nucleotide Sequence Database (http://www.ncbi.nlm.nih.gov/) (Table S1) and the PathoSystems Resource Integration Center (PATRIC) (Wattam et al., 2014). Isolates are available through the American Type Culture Collection-managed Biodefense and Emerging Infections Resources repository (http://www.beiresources.org).
Whole Genome Alignment and Phylogenetic Tree Construction
Two high throughput reference-based pipelines were used to perform single nucleotide polymorphism (SNP) discovery and validation in 166 genomes using the TIGR4 genome as a reference (Tettelin et al., 2001). The genomes included the 147 from the present project plus 15 publicly available, closed, pneumococcal genomes and four whole genome shotgun sequences from this project that were later excluded from this study due to the lack of associated meta-data (Table S1). The analysis pipelines made extensive use of Perl (https://www.perl.org) scripts and were implemented within the Ergatis framework (https://github.com/jorvis/ergatis) (Orvis et al., 2010). The alignment-based SNP discovery pipeline named Skirret mainly executes a series of utilities from the MUMmer 3.0 package (http://mummer.sourceforge.net/) (Kurtz et al., 2004) namely nucmer, delta-filter and show-SNPs to determine SNP positions. This pipeline was run on all 166 assembled genomes with a minimum alignment identity of 98%. A k-mer based algorithm called kSNP (http://sourceforge.net/projects/ksnp/) (Gardner and Hall, 2013) was also employed to identify SNPs in the assembled genomes using 41 bp as the length of the k-mer. Next, a BLAST-based validation pipeline was used to merge the SNP panels from the Skirret and kSNP pipelines and 41 bp sequence surrounding each SNP position was extracted from the reference TIGR4 genome. These short sequences were searched against all 166 genomes using all-vs.-all BLASTN searches (e-value cut-off 0.0001) to eliminate false positive SNP calls, only core SNPs in short sequences found in all 166 genomes were selected. Putative recombinogenic regions were not excluded. The set of 10,120 core SNP bases were concatenated from each of the input genomes to create a multi-FASTA file. This file was used for phylogenetic tree construction using the Geneious Basic software version 5.6.4 with neighbor-joining and the Hasegawa, Kishino and Yano (HKY) genetic distance model. The HKY model assumes that every base has a different equilibrium base frequency, and also assumes that transitions evolve at a different rate to transversions. A consensus tree was built based on 100 iterations to provide an estimate for the level of support for each clade in the final tree. We used a 100% consensus support threshold that resulted in a strict consensus tree where the clades included were those that were present in all the trees of the original set.
Identification and Characterization of Mobile Elements
Mega and Mega insertion sites were located by comparing genomes with the 5,511 bp Mega element from strain GA17457 (Data File S1). ICE were identified by BLASTN (Altschul et al., 1997) search of the terminal direct repeats localized at the ends transposons ICESp23FST81 (81.0 kb; Accession no. FM211187), Tn5253 (66.2 kb; Accession no. EU351020) (Iannelli et al., 2014), Tn916 (18.0 kb; Accession no. U49939) and others. Elements were characterized by the int genes they carried. Integrase genes were identified by BLASTN search with query sequences from described ICE including those mentioned above. All query sequences used are supplied as supporting information (Data File S1). Comparisons of composite ICE were visualized with the Artemis Comparison Tool (Carver et al., 2005). Genomes were also searched for antibiotic resistance genes, including mef(E), mef(A), mel(msr(D)), erm(B), erm(A), tet(M) and cat (Data File S1). The nucleotide sequences of each Mega, Tn916-like and composite elements are provided as supporting information (Data Files S2, S3 and S4, respectively).
Results
Whole Genome Comparisons
Isolates of S. pneumoniae were selected from the >13,000 strain collection of the Georgia Emerging Infections Program. Isolates were collected as part of an ongoing, prospective, population-based surveillance of invasive pneumococcal disease (IPD) in the Atlanta metropolitan area (Farley et al., 2002). One hundred and twenty one invasive S. pneumoniae isolates from 1994 to 2011 were non-randomly selected for genome sequencing. Isolates were selected to provide broad coverage of the genotypes existing in the population during the surveillance period, but with a focus on macrolide resistance, with 100 resistant to erythromycin (MIC >1 μg/ml). 74 displayed the M-phenotype as indicated by susceptibility to the lincosamide clindamycin and 24 were also resistant to clindamycin (MLSB-phenotype) (Table S1). In addition to the Atlanta invasive isolates, 10 carriage isolates from Atlanta were included. Also included were 16 strains not from Atlanta. For more detail, please see the Material Methods and supporting information (Table S1). In total 147 pneumococcal genomes were sequenced including 23 different serotypes, 77 multi-locus sequence types (ST), 24 clonal complexes (CC) and two singletons (Table S1).
The 147 isolates from this study and 15 publically available genomes were compared using whole genome single nucleotide polymorphism (SNP) analyses. The resulting phylogenetic tree is shown in Figure 2. The genomes clustered into MLST clonal complexes with the exception of CC156 which was divided into three clades (Figure 2). This may be an example of a limitation of MLST typing as the three clusters appear to be unrelated. CC156 is a large complex consisting of multiple subgroups thus not all were as closely related as the label CC156 implied (Figure S1). For example ST90 has only one allele in common with ST156. However, it was interesting to observe that two of the CC156 subgroups appear to be linked by a common Mega insertion event (see below). Further, the eBURST-predicted subgroups (SG90, SG124 and SG156) are well-established independent clonal complexes (CC90, CC124 and CC156, respectively) indicating that their inclusion into a single clonal complex is an artifact of eBURST and not a true indication of common ancestry.
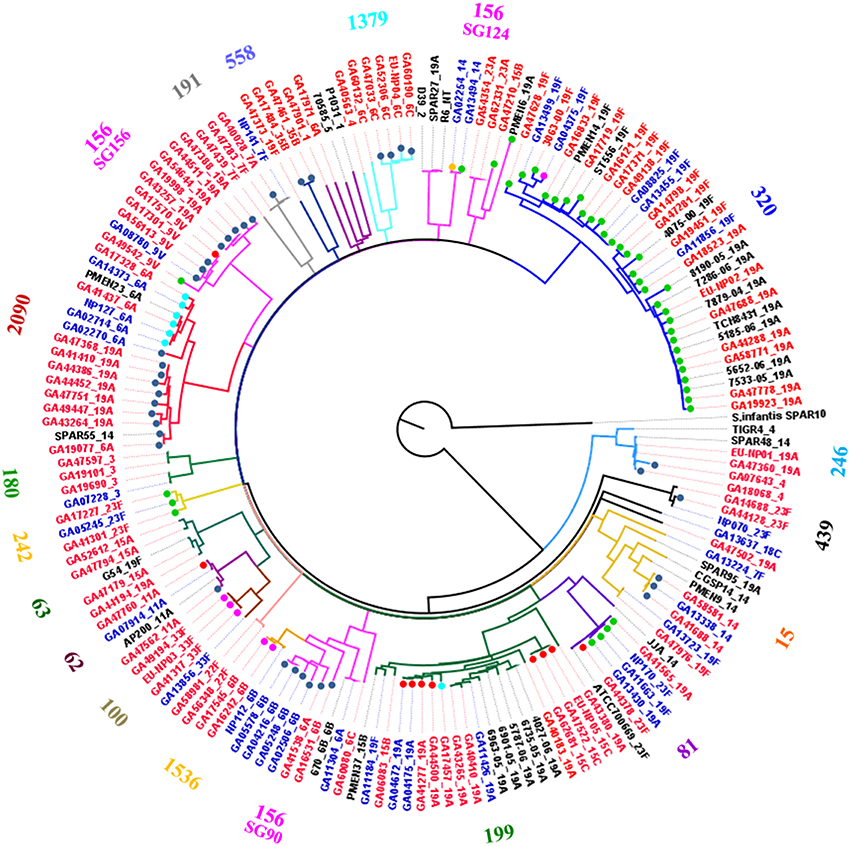
Figure 2. Whole genome SNP-derived phylogenetic tree. Whole genome single nucleotide polymorphism analysis of 166 genomes, including 147 from the present study and nine publically available closed genomes, using the TIGR4 genome as reference. The tree was rooted using S. infantis SPAR10 as the outgroup. Numbers on the perimeter indicate the MLST clonal complex of the isolates and are colored the same as the branches leading to the isolates in each complex. The clonal complex of each sequence type was defined as the founder ST of a clonal group, as predicted by eBURST analyses of the entire S. pneumoniae MLST databased accessed in September, 2014 (http://pubmlst.org/spneumoniae/). Isolate names and serotype labels colored blue indicates that the strain was isolated prior to the introduction of PCV7 in 2000. Red colored names and serotypes indicate the strains were isolated in 2000 or later. Colored dots indicate a Mega insertion in the isolate and are color coded by Mega insertion site, light blue, class I; dark blue, class II; red, class III; pink, class IV; green, class V (Tn916 orf6); and gold, novel insertion site.
Distribution of the Macrolide Resistance-Encoding Element Mega
Of the Atlanta invasive isolates, 86 contained Mega integrated into the pneumococcal chromosome backbone or nested within larger mobile elements. Mega was found in 17 serotypes, 16 clonal complexes and two singletons (Table 1). Of these, 48 carried the 5,511 kb Mega type 1 (Mega-1) with a 119 bp mef(E)/mel intergenic region and 38 carried the 5,412 kb Mega type 2 (Mega-2) with a 99 bp deletion in the intergenic region (Table 1). The genome of GA07643 contained a previously unidentified 5,617 bp Mega variant, designated Mega-3, characterized by a 107 bp tandem duplication that included the first 90 bp of mel and 23 bp upstream containing its ribosomal binding site (RBS). Analyses of the 454 raw reads revealed that both copies of the repeated sequence were contained on single reads and the tandem duplication region had greater than 40× sequence coverage. This confirmed that the duplication was not an assembly error. The authentic duplication resulted in an intergenic region that contained the 119 bp that immediately follows mef(E) in Mega-1, followed by a 38 codon ORF that includes the first 30 amino acids of Mel. The ORF had the mel RBS and appeared suitable for protein expression. Theoretically this could have a polar effect of mel translation, however, GA07643 had an erythromycin MIC of >16 μg/ml so it does not appear to have had a negative impact on resistance (Table S1). The full-length copy of mel overlapped the 3′ end of the duplicated ORF by 10 bp and maintained the native mel RBS. There was no correlation between the size of the mef(E)/mel intergenic region and the level of efflux-mediated erythromycin expressed by that strain (data not shown).
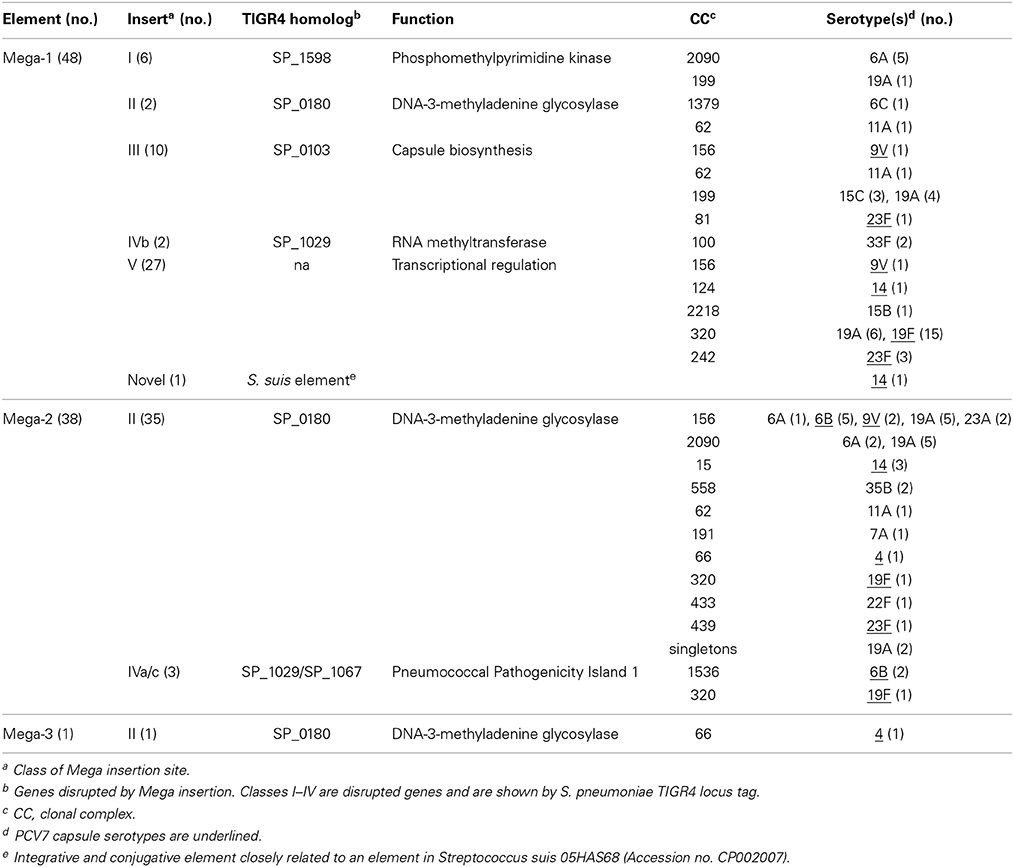
Table 1. Insertion sites of Mega.
Mega was found inserted into each of the four previously identified insertion sites within the pneumococcal chromosome backbone (classes I-IV) (Gay and Stephens, 2001), in Tn916-like elements (class V), and in one novel site (Table 1). Six genomes contained Mega integrated into the class I insertion site, five of which were clonal (6A, CC2090) indicating clonal dissemination (Figure 2, light blue dots; Table 1). The exception was GA17457, a serotype 19A, CC199 isolate that has been used as the genetic background for studying the regulation of mef(E) and mel (Zähner et al., 2010; Chancey et al., 2011). GA17457 was closely related to four isolates in CC199 that contained Mega-1 in class III sites (Mega-1.III) and was the only CC199 strain to have Mega-1 integrated into class I (Mega-1.I) (Figure 2). Class II Mega insertions were identified in 38 genomes (Table 1). The elements inserted into this site include Mega-1 (n = 2), Mega-2 (n = 35) and the novel Mega-3 in strain GA07643 (Table 1). Mega-1.III was found in ten genomes in diverse backgrounds: five serotypes and four clonal complexes (Table 1). Mega class III insertions clustered mainly in CC199 but single isolates containing Mega-1.III were identified in CC156, CC81, and CC62 (Figure 2). Five genomes contained Mega-1 or Mega-2 inserted into the class IV site which was divided into three subclasses, IVa, IVb, and IVc (see below).
Comparison of the nucleotide sequences flanking Mega inserted into each site revealed conserved sequences upstream of the Mega insertion site that may indicate a target sequence (Figure 3). The potential consensus was 5′ TTTCCNCAA 3′ and was located approximately six bp upstream of Mega in all sites (Figure 3). The six base pairs flanking Mega on either side resembled coupling sequences as described for conjugative transposons such as Tn916 (Figure 3). The coupling sequences and Mega flanking sequences illustrated in Figure 3 were representative of all Mega elements inserted into the respective sites. This implies that the Mega integrated into each site were descended from a single, or very few, ancestral transposition event(s). An example of this can be seen in the whole genome phylogeny (Figure 2). The class II Mega insertion was present in almost all CC156 isolates (Figure 2, dark blue dots). Genomes from isolates of this large complex clustered into three clades correlating to CC156 subgroups 90, 124, and 156 (Figure 2). Almost all genomes in subgroups 90 and 156 contained Mega (Figure 2). All 10 subgroup 156 genomes contained Mega; eight Mega-2, class II, one Mega-1 class III insertion and one integration into orf6 of Tn916 (Table 1; Figure 2). Further, CC2090, which branches from CC156, has two clades in one of which nine of nine isolates contain Mega class II. The other branch of CC2090 contains almost all isolates with Mega class I indicating a second conjugation event in this complex (Figure 2). Again this strongly implies that Mega inserted into the class II site early in the history of these lineages.
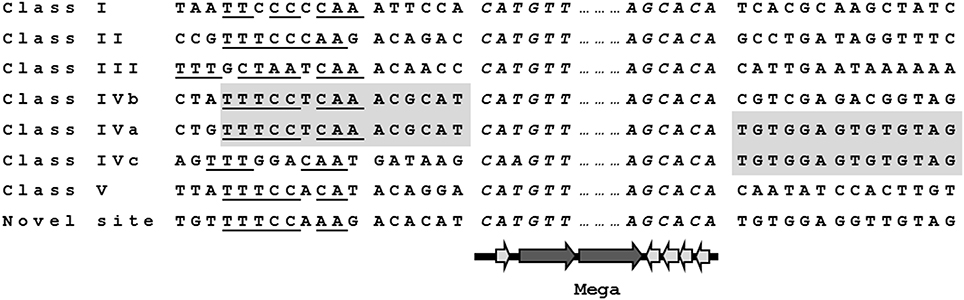
Figure 3. Mega insertion sites. Target sites of the mobile element Mega encoding macrolide efflux genes mef(E) and mel. Putative six base coupling sequences (C.S.) are shown adjacent to both ends of Mega (italics). Underlined letters indicate conserved nucleotides that may be the consensus recognition sequence 5′ TTTCCNCAA 3′. Class IV was composed of three distinct, subclasses, IVa, IVb, and IVc. Class IVb and IVa share the same left junction and IVa and IVc same right junction (shaded).
The Mega class IV insertion site, as previously described (Gay and Stephens, 2001), was near the 3′ end of rumA (SP_1029, TIGR4 annotation) that encoded a tRNA methyltransferase and was located at the left end of the Pneumococcal Pathogenicity Island-1 (PPI-1) (Brown et al., 2004) (Figure 4). Large deletions and multiple MGEs were associated with Mega integrated into this site, giving rise to the three class IV subclasses (Table 1). Starting with the simplest, the class IVb insertion contained Mega-1 integrated into rumA leaving Mega flanked by the fragments of the gene (Figure 4). Two class IVb isolates were identified, all serotype 33F, CC100 (Table 1). The class IVa insertion contained Mega-2 integrated into the same location in rumA and had an identical sequence to the left junction of Mega in class IVb (Figure 4). However, the downstream remnant of rumA was deleted along with the entire 30.7 kb PPI-1 (SP_1029 to SP_1067) (Figure 4). The island was replaced by Mega-2 and a 1,801 bp insertion sequence ISSmi2 found in S. mitis strain B6 (Accession no. FN568063.1) (Figure 4). The class IVc Mega-2 insertion was similar to class IVa in that PPI-1 was deleted and ISSmi2 was located at the right junction with the chromosome (Figure 4). However, a 41,995 bp transposon-like sequence most closely related to ICESz1 from S. equi subspecies zooepidemicus strain H70 (Accession no. FM204884) (Holden et al., 2009) was inserted between the rumA insertion site and the left end of Mega-2. Thus, the class IVc Mega site organization consists of, in tandem, a transposon previously undefined in pneumococci, Mega-2 and ISSmi2. The Atlanta invasive isolate GA17545 (6B, CC1536) contained the only class IVc Mega insertion identified (Tables S1, S2). Class IVa isolates are also typically 6B, CC100, however, a single Atlanta invasive isolate, GA04375, with serotype 19F, and in CC320 was also found to contain Mega-2.IVa (Table 1; Table S1). GA04375 was isolated in 1995 and was the earliest isolated CC320 in the genome collection and only one of two CC320 that contained Mega inserted into the pneumococcal chromosome backbone. The other was 3063-00, an invasive 19F, CC320 collected in 1999 in Tennessee (Table S1, S2). The serotype 14, CC156 isolate GA02254 from 1994 contained Mega-1 inserted into an unidentified transposon-like sequence and was most closely related to a sequence in S. suis 05HAS68 (Accession no. CP002007). Twenty-seven genomes contained Mega-1 inserted into orf6 of a Tn916-like element (class V) (Table 1). Tn916-like elements are discussed further in the next section. These data suggest that PPI-1 is a hotspot for integration by recombination and conjugation. This locus could be an entry point for non-pneumococcal DNA to integrate into the pneumococcal genome. Conversely, the conservation of the target site in rumA across non-pneumococcal streptococci makes it possible that pneumococcus has been the donor and a non-pneumococcal streptococcus the recipient. The high degree of recombination in the pneumococcus makes it difficult to determine directionality of gene flow, but it likely can flow in both directions. With just these few isolates, we showed evidence for HGT with three non-pneumococcal streptococci; S. equi, S. mitis, and S. suis.
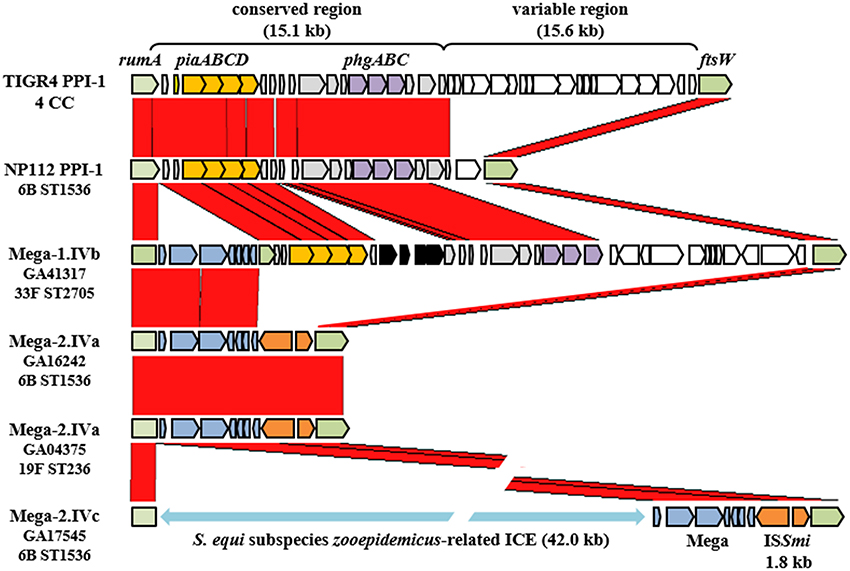
Figure 4. Variations of the Pneumococcal Pathogenicity Island-1 associated with Mega. The Pneumococcal Pathogenicity Island-1 in TIGR4 extends from rumA (SP_1029) to ftsW (SP_1065). Strain NP112 is closely related to GA16242 and GA17545, the Mega-2.IVa and IVc strains, but contains the conserved region of PPI-1. Red blocks represent regions of homology. Arrows indicate genes. Green, PPI-1 borders; yellow, iron uptake operon piaABCD; purple, osmotic stress tolerance operon phgABC; gray, PPI-1 conserved region genes not otherwise labeled; white, PPI-1 variable region genes; blue, Mega; orange, ISSmi1. The light blue double arrow indicates the presence of an undefined transposon-like sequence from S. equi subspecies zooepidemicus.
Tn916-Like Elements Carrying Macrolide Resistance Determinants
The genomes were searched for Tn916-like elements by BLASTN using the conserved ends of Tn916 (Data File S1). Of the 86 macrolide resistant invasive isolates from Atlanta, 53 contained a full-length Tn916-like element (Table S3). A macrolide resistance determinant was nested within the Tn916 element in all but six, which instead contained Tn916 and an unlinked Mega-2.II (n = 5) or Mega-1.III (n = 1) (Table S3). Four distinct Tn916-like elements containing one or two macrolide resistance determinants were identified: Tn2009, Tn2010, Tn6002, and Tn3872 (Figure 1; Table 2). Tn2009 was found in 15 Atlanta invasive isolates spanning six CC and six serotypes (Table S3). Tn6002 was carried by nine Atlanta invasive isolates including five CC (and a singleton) and seven serotypes (Table S3). Tn2010 was found in 13 Atlanta invasive isolates, two Atlanta carriage isolates, and eight non-Atlanta invasive isolates (Table 2; Table S3).
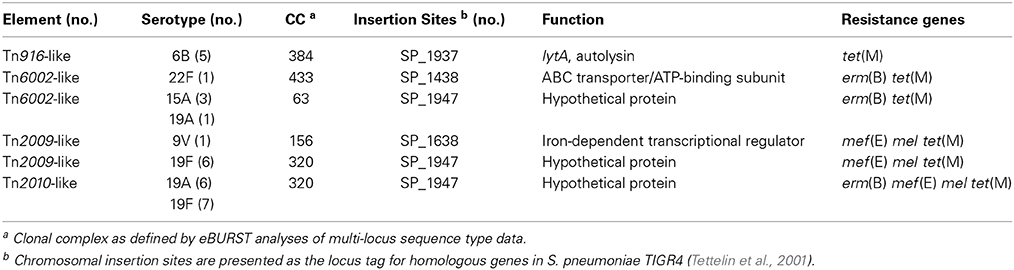
Table 2. Tn916 and Tn916-like elements inserted directly into the chromosome backbone of invasive S. pneumoniae isolates from Atlanta, GA.
To better understand the evolutionary relationships between these elements, the insertion site of each was determined. Tn916-like elements were integrated into four unique loci in the pneumococcal chromosome backbone and into several loci within Tn5253-like elements (Figure 5). Every Tn2010 element discovered was inserted into SP_1947 (TIGR4 annotation) encoding a hypothetical secreted protein (Table 2; Figure 5). This is the same location where Tn2010 has been previously reported to be integrated (Zhou et al., 2014). Six of 15 Tn2009 elements and four of nine Tn6002 elements found in invasive Atlanta isolates were inserted into the same locus as Tn2010 (Table 2). The 6-base coupling sequences were identical for all elements in this locus suggesting that the elements found integrated into this locus were derived from a single transposition event (Figure 5). This further implies that the macrolide resistance elements Mega-1, Tn917 and erm(B) were independently acquired by a Tn916 residing in the SP_1947 locus. In addition, elements in this locus were identified in two clonal complexes (CC320, CC156) indicating transformation and recombination of the element in this locus between clonal complexes.

Figure 5. Insertion sites of Tn916-like elements. Insertion sites within composite elements. The terminal sequences of Tn916 are shown in italics. Coupling sequences are underlined. Plain text indicates the sequences of ICE flanking the Tn916-like element. (R) indicates that the Tn916-like element is in the reverse orientation. The ORFs that flank the Tn916-like elements are indicated adjacent to the ICE sequences on either side.
Tn2009 was also identified integrated into SP_1638 (TIGR4), an iron-dependent transcriptional regulator, in a single serotype 9V, CC156 isolate from 2007. The fact that no element was identified in this locus from an earlier collected isolate may indicate that this integration event occurred more recently than the SP_1947 integration. Likewise, a single isolate (22F, CC433) from 2010 contained Tn6002 integrated into the ABC transporter gene SP_1438. Five serotype 6B, CC384 isolates contained Tn916 integrated into lytA (SP_1937) encoding an autolysin (Table 2). Each of these closely related isolates contained unlinked Mega-2.II (Table S2). The other Tn916-like elements identified were integrated into a composite ICE resembling Tn5253 and are discussed below.
Composite ICE Containing Macrolide Resistance Determinants
Tn916-like elements with macrolide resistance determinants have been associated with large conjugative transposons also known as ICE (Henderson-Begg et al., 2009; Mingoia et al., 2014). To determine if ICE have been involved in macrolide resistance dissemination in Atlanta, the genomes of 100 macrolide resistant invasive pneumococcal isolates from Atlanta were searched by BLASTN for the conserved ends of known ICE (Data File S1). The genomes of 15 macrolide resistant isolates contained composite ICE-like elements that ranged in size from approximately 57 to 88 kb (Table 3). Comparison of the integrase genes encoded on the ICE revealed two distinct families of conjugative transposons. Nine belonged to the Tn5253 family and six belonged to the ICESp23FST81 family (Table 3). Each of the Tn5253-family transposons was inserted into the pneumococcal chromosome backbone in the gene for ribobiogenesis GTPase, rbgA (SP_1155, TIGR4 annotation) involved in assembly of 23S ribosomes. Downstream of Tn5253-like elements in this locus is the immunoglobulin A1 protease gene iga as has been described previously (Croucher et al., 2009). Each of the ICESp23FST81-like elements was inserted into the chromosome backbone gene rplL (SP_1354) which encodes ribosomal protein L7/L12 (Croucher et al., 2009). The correlation of ICE family and insertion sites reflects the specificity of the integrases IntTn5253 and IntICESp23FST81 (Mingoia et al., 2007; Wyres et al., 2013).
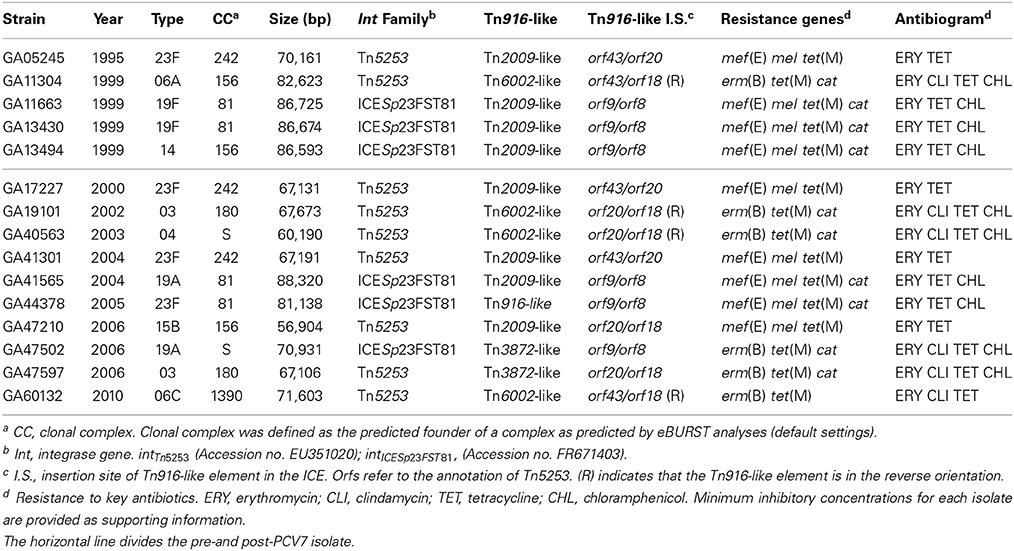
Table 3. Atlanta invasive isolates containing composite ICE encoding macrolide resistance.
The five ICESp23FST81-like elements contained Tn916-like insertions, including a Tn916-like, a Tn3872-like and four Tn2009-like elements (Table 3). Each of the smaller elements was integrated between orf9 and orf8 of Tn5253 and contained identical flanking and coupling sequences, indicating that each was derived from a single conjugation event and subsequent transfer of Mega-1 between Tn916 and Tn2009 (Figure 6). GA44378 containing Tn916 integrated into the ICESp23FST81-like element was macrolide resistant due to an unlinked Mega-1.III insertion (Table 3; Table S2). ICESp23FST81 was originally described in a 23F, ST81 isolate (Croucher et al., 2009). Five of six ICESp23FST81 family elements were also in CC81, however only two were serotype 23F (Table 3). The others were serotype 19A (n = 1) and 19F (n = 2) and represent capsule switching from a PCV7 vaccine serotype to a non-vaccine serotype and a different vaccine serotype, respectively. This conclusion is supported by the close relationship between the isolates, all but one of which was also serotype 23F and CC81 (Figure 6). The lone exception was GA13494, a serotype 14, CC156 isolate that contained Tn2009, indicating horizontal transfer of the composite element between the clones (Figure 6). These were isolated over a 9-year period spanning the pre-PCV7 and post-PCV7 eras (1995–2004). This demonstrated clonal expansion and the long-term stability of the clone and the Tn5253-like element harboring Tn2009.
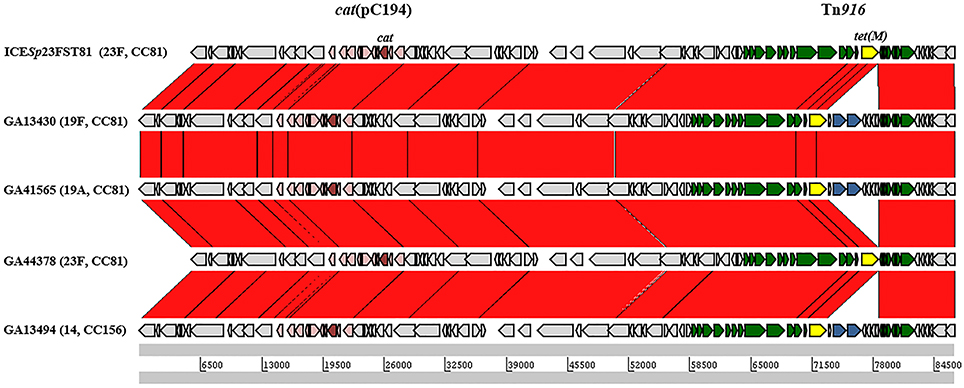
Figure 6. Comparison of ICESp23FST81-like elements encoding macrolide resistance. The location of the Tn916-like element in each is indicated by the Tn5253 genes shown as arrows above each track. Green arrows, Tn916-genes; Blue arrows, mef(E) and mel; yellow arrows, tet(M); pink arrows, integrated plasmid pC194; red arrows, cat.
In the other composites identified, the Tn916-like element was inserted between orf43 and orf20 of Tn5253. Tn3872 was inserted in this locus in GA47597, a serotype 3, CC180 isolate from 2006 (Table 3). Tn3872 was also inserted downstream of orf20 and contained the same flanking DNA sequences, however, the right junction was flanked by orf18. This indicated that GA47597, a serotype 3, CC180 isolate from 2006, also contained Tn3872 which had a sequence identical to GA47502 flanking the left junction of Tn3872 (Figure 6). Tn6002 was also found flanked by orf20 on the left and orf18 on the right in two isolates (Table 3; Figure 5). Like GA47597, GA19101 was a serotype CC180 isolate suggesting a common integration event for a Tn916-like element (Figure 6). However, Tn6002 in both of these isolates was integrated in the opposite orientation from Tn2009 and Tn3872 (Figure 6). This suggests separate conjugation events (one for each orientation) followed by homologous recombination of one junction to create a chimeric insertion site (i.e., orf20/orf18). In both isolates with Tn3872, the element was integrated into orf20/orf18 in the same orientation as Tn2009 in this location (Table 3). Again, Tn2009 and Tn6002 in this location had identical coupling sequences indicating a single event integration event of a Tn916-like element followed by acquisition of nested macrolide resistance elements by homologous recombination. Finally, orf43/orf18 was identified as the site of insertion of Tn6002 in four isolates (Table 2). Tn5253 is also flanked by these genes and with conserved coupling sequences, indicating that each is descended from a single conjugation event followed by integration of the erm(B) element between orf43 and orf20. These data suggest that de novo integration Tn916-like elements into pneumococcal chromosome backbones or ICE was infrequent relative to the rate of horizontal transfer by transformation and recombination.
Discussion
The genome of S. pneumoniae is extremely plastic due to its natural competency and possession of numerous MGE. While transformation and conjugation are both involved in shaping the pneumococcal chromosome, the diversity of macrolide resistance elements, both independent and integrated into larger conjugative elements, raises the question of the relative role of each mechanism in the dissemination of macrolide resistance in pneumococci. Recombination events involving various macrolide resistance determinants were evidenced by an apparent interchangeability of Mega, the erm(B) element and Tn917 in Tn916-like elements. The conversion of Tn6002 and Tn6003 (containing aminoglycoside resistance gene aphA-3) has been reported (Palmieri et al., 2012). These elements in turn were integrated into the chromosome backbone or were inserted into larger integrative and conjugative transposons. In this study, comparative genomics were used to assess the elements existing in a geographically defined location before and after the introduction of PCV7, to better understand the complex history of macrolide dissemination.
We identified Mega inserted in the four previously described locations in the pneumococcal chromosome backbone (Gay and Stephens, 2001) and inserted into orf6 of Tn916-like elements. We also found a novel insertion site within an undefined transposon-like sequence similar to an element in S. suis. It is not clear if Mega integrated into the element before or after it made the cross-species horizontal transfer into the pneumococcus. Either way, it is an apparent example of pneumococci acquiring novel DNA from a non-pneumococcal streptococci, likely by conjugation event introducing the S. suis element into pneumococci or by Mega inserting into a novel locus in pneumococci. We also identified a transposon-like element most closely related to ICESz1 from S. equi subsp. zooepidemicus. This element was located in the Pneumococcal Pathogenicity Island 1 (PPI-1) and was associated with Mega. However, the possibility of S. suis and S. equi having acquired these elements from the pneumococcus cannot be disregarded. In fact, there is no data to suggest that these elements cannot flow equally in both directions.
PPI-1 appears to be a hotspot for recombination and/or integration of MGE. Not only was Mega inserted in PPI-1 but it appeared that integration of Mega at this location created, or retained, target sites for mobile elements. The Mega class IVb insertion contains Mega with no deletion of the target site. This may have been the original integrated structure. Subsequently, each flank of Mega-1.IVb may have been dislocated by insertion of additional MGE. Downstream of Mega in classes IVa and IVc was an insertion sequence, ISSm1, originally identified in S. mitis (Denapaite et al., 2010). Interestingly, ISSmi precisely replaced the variable region of the pathogenicity island, which was a remnant of a Tn5252-like ICE (Brown et al., 2001). The variability at this locus and the presence of multiple integrated MGE-related sequences suggested this to be a hotspot for recombination or a common target site for conjugative elements. Upstream of Mega in class IVc was a large unidentified conjugative transposon-like sequence related most closely to a transposon-like sequence from S. equi subspecies zooepidemicus. The undefined element was integrated into the exact location within rumA as Mega and was integrated immediately upstream of Mega resulting in three MGEs linked in tandem: the unidentified transposon, Mega, and ISSmi1. Both undefined pneumococcal transposons are currently under investigation to identify their origin, understand where they came from, how they moved, what cargo they carry and their ability to transfer by conjugation from pneumococci to pneumococci and non-pneumococcal streptococci.
Closed circle intermediates of excised Tn916-like elements are formed by binding of semi-complementary coupling sequences creating a heteroduplex. This process produces novel coupling sequences with each integration event. Based on the observation that coupling sequences are altered upon integration, we used predicted coupling sequences as indicators of unique integration events. Mega and erm(B) often integrate in Tn916-like elements (Del Grosso et al., 2004, 2007, 2011). In the present study we identified many of the previously reported macrolide resistant Tn916-like elements in the Atlanta population including Tn2009, Tn2010, Tn6002, and Tn3872. The mechanism of conjugation of Tn916 is well-understood. The Tn916 integrase is promiscuous, that is, targeting sequences that are A-T rich, but not sequence specific (Rudy et al., 1997; Rocco and Churchward, 2006). Tn916-like transposons excise imperfectly from a donor strain to carry some flanking genomic bases and create a closed-circle intermediate with a mismatched junction (Caparon and Scott, 1989). The resulting overlapping region is the coupling sequence for subsequent integration, and thus each transposition event may create a new coupling sequence (Lu and Churchward, 1995). These facts and the observation by our laboratory and others that Tn916-like elements are found integrated within relatively few sites in the pneumococcus and with little variation in the coupling sequences, suggest that conjugation into and between pneumococci is relatively infrequent compared to transformation and recombination events.
ICE appear to be well-established in the pneumococcal population. They were present during a rapid emergence of macrolide resistant invasive pneumococcal disease during the early to mid-1990's (Wyres et al., 2013) in the United States. In addition, ICE have been reported in isolates from the 1960's (Wyres et al., 2013). ICE were identified in invasive and carriage pneumococcal isolates in Atlanta (Table 3). The ICE carrying the shorter macrolide resistance elements were of two types as has been reported previously (Roberts and Kreth, 2014). The first was a group of Tn5253-like elements containing different combinations of Mega, the erm(B) element, and Tn917 also carrying erm(B). All of these elements were integrated into the same location in the pneumococcal chromosome backbone: the ribobiogenesis GTPase, rbgA. The second group was similar to ICESp23FST81 and were all inserted into the ribosome protein encoding gene rplL. The conservation of insertion sites reflects the specificity of different integrases encoded on each element. Because of the specificity of these integrases, it is to be expected that they would integrate into the exact location repeatedly. Therefore, we can make no conclusions on the frequency of transfer of the element into or out of pneumococci.
Comparison of the sequence types and serotypes of the pneumococcal isolates carrying macrolide resistance determinants showed closely related isolates with variable macrolide resistance elements indicating horizontal transfer. For example, in Figure 6 GA41565 and GA44378 are both CC81, but the former is serotype 19A and the latter is 23F. These isolates cluster together in the whole genome analyses further indicating their similarity. Both contain ICESp23FST81-like ICE that are nearly identical and both have a Tn916-like element with no macrolide resistance determinant, while GA41565 contains Tn2009. This indicates the acquisition or loss of Mega by recombination. Analyses of the insertion sites suggested that conjugative insertion events of the elements into the pneumococcus are infrequent leading to the conclusion that these elements have been largely disseminated by lateral gene transfer through transformation and homologous recombination. Because homologous recombination requires homology between donor and host DNAs, gene flow between closely related isolates may occur more frequently than between distantly related clones. However, the presence of macrolide elements in composite transposons and the wide dissemination of composite transposons may aid in horizontal gene transfer between distant clones by creating 50–90 kb regions of homology that act as landing sites for the small nested elements carrying macrolide resistance determinants. These additional regions of homology may have aided in the dissemination of macrolide resistance not only because of the binding sites created by the composite elements, but also because of the multidrug resistance nature of the chimeric genetic elements. For example, macrolide resistance dissemination is aided by the non-macrolide antibiotic resistance determinants tetracycline and chloramphenicol due to soft selective sweeps of macrolide elements linked to cat and tet(M) genes carried on composite mobile elements.
The 7-valent Pneumococcal Conjugate Vaccine PCV7 was introduced into Atlanta in late 2000. We analyzed the genomic data in respect to the pre-vaccine and post-vaccine years. There appeared to be little effect on the types of elements harboring macrolide resistance determinants, however, the sequence types and serotypes harboring these elements were changed significantly. Some examples of this can be seen in the whole genome phylogenetic analyses where blue text indicated isolation in the pre-vaccine era and red indicated post-vaccine isolation (Figure 2). Note that in clonal complex 320, all isolates from before the vaccine were serotype 19F, a PCV7 serotype (Figure 2). After the vaccine was introduced serotype 19A, a non-PCV7 serotype, appeared. The shift to 19A was concomitant with the appearance of Tn2010, which was not detected in the pre-vaccine era. Interestingly, Tn2010 was detected in only one chromosome backbone site and was not found in any ICE (Table 2). Likewise, CC2090 appears to have switched from the vaccine-associated serotype 6A to the non-vaccine serotype 19A (Figure 2). Every serotype 19A CC2090 strain was isolated after the introduction of the vaccine (Figure 2).
In conclusion, macrolide resistance dissemination in pneumococci has been greatly influenced by conjugation and transformation. Because Mega and Tn916-like element integration events appear infrequent, transformation and homologous recombination appear to be the primary means of horizontal transfer into the pneumococcus. With limited sites for insertion, the element has multiplied its capacity to disseminate throughout the pneumococcal population by co-opting the insertion sites of other elements. Thus, conjugation allows acquisition of DNA from more distantly related bacteria. Once in a pneumococcal genome, if beneficial and selected, the conjugated transposon is subsequently disseminated by transformation throughout the pneumococcal population.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Wendy Baughman, Emily Crispell and the Georgia Emerging Infections Program for providing the invasive pneumococcal isolates from Georgia for genome sequencing. We are also grateful to Leslie McGee, the Pneumococcal Molecular Epidemiology Network and the Centers for Disease Control Streptococcus Laboratory for providing invasive isolates and Pekka Nourti and Shabnam Jain for providing nasopharyngeal isolates. This study was funded in part with an R01 grant from the National Institutes of Health (R01 AI070829 (to David S. Stephens) and with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under contract number HHSN272200900009C. The funders had no role in study design, data collection and analysis, or preparation of the manuscript, but signed off on the publication after we sent them a copy for review.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2015.00026/abstract
References
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ambrose, K. D., Nisbet, R., and Stephens, D. S. (2005). Macrolide efflux in Streptococcus pneumoniae is mediated by a dual efflux pump (mel and mef) and is erythromycin inducible. Antimicrob. Agents Chemother. 49, 4203–4209. doi: 10.1128/AAC.49.10.4203-4209.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ayoubi, P., Kilic, A. O., and Vijayakumar, M. N. (1991). Tn5253, the pneumococcal omega (cat tet) BM6001 element, is a composite structure of two conjugative transposons, Tn5251 and Tn5252. J. Bacteriol. 173, 1617–1622.
Berge, M., Moscoso, M., Prudhomme, M., Martin, B., and Claverys, J.-P. (2002). Uptake of transforming DNA in Gram-positive bacteria: a view from Streptococcus pneumoniae. Mol. Microbiol. 45, 411–421. doi: 10.1046/j.1365-2958.2002.03013.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brown, J., Gilliland, S., and Holden, D. (2001). A Streptococcus pneumoniae pathogenicity island encoding an ABC transporter involved in iron uptake and virulence. Mol. Microbiol. 40, 572–585. doi: 10.1046/j.1365-2958.2001.02414.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brown, J. S., Gilliland, S. M., Spratt, B. G., and Holden, D. W. (2004). A locus contained within a variable region of pneumococcal pathogenicity Island 1 contributes to virulence in mice. Infect. Immun. 72, 1587–1593. doi: 10.1128/IAI.72.3.1587-1593.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caparon, M. G., and Scott, J. R. (1989). Excision and insertion of the conjugative transposon Tn916 involves a novel recombination mechanism. Cell 59, 1027–1034. doi: 10.1016/0092-8674(89)90759-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carver, T. J., Rutherford, K. M., Berriman, M., Rajandream, M.-A., Barrell, B. G., and Parkhill, J. (2005). ACT: the Artemis Comparison Tool. Bioinformatics (Oxford, England) 21, 3422–3423. doi: 10.1093/bioinformatics/bti553
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chancey, S. T., Zhou, X., Zähner, D., and Stephens, D. S. (2011). Induction of efflux-mediated macrolide resistance in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 55, 3413–3422. doi: 10.1128/AAC.00060-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Claverys, J.-P., Prudhomme, M., and Martin, B. (2006). Induction of competence regulons as a general response to stress in gram-positive bacteria. Annu. Rev. Microbiol. 60, 451–475. doi: 10.1146/annurev.micro.60.080805.142139
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cochetti, I., Tili, E., Mingoia, M., Varaldo, P. E., and Montanari, M. P. (2008). erm(B)-Carrying elements in tetracycline-resistant pneumococci and correspondence between Tn1545 and Tn6003. Antimicrob. Agents Chemother. 52, 1285–1290. doi: 10.1128/AAC.01457-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cochetti, I., Tili, E., Vecchi, M., Manzin, A., Mingoia, M., Varaldo, P. E., et al. (2007). New Tn916-related elements causing erm(B)-mediated erythromycin resistance in tetracycline-susceptible pneumococci. J. Antimicrob. Chemother. 60, 127–131. doi: 10.1093/jac/dkm120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Croucher, N. J., Hanage, W. P., Harris, S. R., McGee, L., van der Linden, M., de Lencastre, H., et al. (2014). Variable recombination dynamics during the emergence, transmission and ‘disarming’ of a multidrug-resistant pneumococcal clone. BMC Biol. 12:49. doi: 10.1186/1741-7007-12-49
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Croucher, N. J., Walker, D., Romero, P., Lennard, N., Paterson, G. K., Bason, N. C., et al. (2009). Role of conjugative elements in the evolution of the multidrug-resistant pandemic clone Streptococcus pneumoniae Spain23F ST81. J. Bacteriol. 191, 1480–1489. doi: 10.1128/JB.01343-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Del Grosso, M., Camilli, R., Barbabella, G., Blackman Northwood, J., Farrell, D. J., and Pantosti, A. (2011). Genetic resistance elements carrying mef subclasses other than mef(A) in Streptococcus pyogenes. Antimicrob. Agents Chemother. 55, 3226–3230. doi: 10.1128/AAC.01713-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Del Grosso, M., Camilli, R., Iannelli, F., Pozzi, G., and Pantosti, A. (2006). The mef(E)-carrying genetic element (Mega) of Streptococcus pneumoniae: insertion sites and association with other genetic elements. Antimicrob. Agents Chemother. 50, 3361–3366. doi: 10.1128/AAC.00277-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Del Grosso, M., Camilli, R., Libisch, B., Fuzi, M., and Pantosti, A. (2009). New composite genetic element of the Tn916 family with dual macrolide resistance genes in a Streptococcus pneumoniae isolate belonging to clonal complex 271. Antimicrob. Agents Chemother. 53, 1293–1294. doi: 10.1128/AAC.01066-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Del Grosso, M., Northwood, J. G. E., Farrell, D. J., and Pantosti, A. (2007). The macrolide resistance genes erm(B) and mef(E) are carried by Tn2010 in dual-gene Streptococcus pneumoniae isolates belonging to clonal complex CC271. Antimicrob. Agents Chemother. 51, 4184–4186. doi: 10.1128/AAC.00598-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Del Grosso, M., Scotto d'Abusco, A., Lannelli, F., Pozzi, G., and Pantosti, A. (2004). Tn2009, a Tn916-like element containing mef(E) in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 48, 2037–2042. doi: 10.1128/AAC.48.6.2037-2042.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Denapaite, D., Bruckner, R., Nuhn, M., Reichmann, P., Henrich, B., Maurer, P., et al. (2010). The genome of Streptococcus mitis B6–what is a commensal? PLoS ONE 5:e9426. doi: 10.1371/journal.pone.0009426
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Enright, M. C., and Spratt, B. G. (1998). A multilocus sequence typing scheme for Streptococcus pneumoniae: identification of clones associated with serious invasive disease. Microbiology 144, 3049–3060. doi: 10.1099/00221287-144-11-3049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Farley, M. M., Baughman, W., and Arnold, K. E. (2002). The Georgia emerging infections program: monitoring trends in invasive pneumococcal disease. J. Med. Assoc. Ga. 91, 20–23.
Gardner, S. N., and Hall, B. G. (2013). When whole-genome alignments just won't work: kSNP v2 software for alignment-free SNP discovery and phylogenetics of hundreds of microbial genomes. PLoS ONE 8:e81760. doi: 10.1371/journal.pone.0081760
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gay, K., Baughman, W., Miller, Y., Jackson, D., Whitney, C. G., Schuchat, A., et al. (2000). The emergence of Streptococcus pneumoniae resistant to macrolide antimicrobial agents: a 6-year population-based assessment. J. Infect. Dis. 182, 1417–1424. doi: 10.1086/315853
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gay, K., and Stephens, D. S. (2001). Structure and dissemination of a chromosomal insertion element encoding macrolide efflux in Streptococcus pneumoniae. J. Infect. Dis. 184, 56–65. doi: 10.1086/321001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Henderson-Begg, S. K., Roberts, A. P., and Hall, L. M. C. (2009). Diversity of putative Tn5253-like elements in Streptococcus pneumoniae. Int. J. Antimicrob. Agents 33, 364–367. doi: 10.1016/j.ijantimicag.2008.10.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Holden, M. T. G., Heather, Z., Paillot, R., Steward, K. F., Webb, K., Ainslie, F., et al. (2009). Genomic evidence for the evolution of Streptococcus equi: host restriction, increased virulence, and genetic exchange with human pathogens. PLoS Pathog. 5:e1000346. doi: 10.1371/journal.ppat.1000346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iannelli, F., Santoro, F., Oggioni, M. R., and Pozzi, G. (2014). Nucleotide sequence analysis of integrative conjugative element Tn5253 of Streptococcus pneumoniae. Antimicrob. Agents Chemother. 58, 1235–1239. doi: 10.1128/AAC.01764-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klugman, K., and Lonks, J. (2005). Hidden epidemic of macrolide-resistant pneumococci. Emerging Infect. Dis. 11, 802–807. doi: 10.3201/eid1106.050147
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leclercq, R., and Courvalin, P. (1991). Intrinsic and unusual resistance to macrolide, lincosamide, and streptogramin antibiotics in bacteria. Antimicrob. Agents Chemother. 35, 1273–1276. doi: 10.1128/AAC.35.7.1273
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lu, F., and Churchward, G. (1995). Tn916 target DNA sequences bind the C-terminal domain of integrase protein with different affinities that correlate with transposon insertion frequency. J. Bacteriol. 177, 1938–1946.
McCormick, A. W., Whitney, C. G., Farley, M. M., Lynfield, R., Harrison, L. H., Bennett, N. M., et al. (2003). Geographic diversity and temporal trends of antimicrobial resistance in Streptococcus pneumoniae in the United States. Nat. Med. 9, 424–430. doi: 10.1038/nm839
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McDougal, L. K., Tenover, F. C., Lee, L. N., Rasheed, J. K., Patterson, J. E., Jorgensen, J. H., et al. (1998). Detection of Tn917-like sequences within a Tn916-like conjugative transposon (Tn3872) in erythromycin-resistant isolates of Streptococcus pneumoniae. Antimicrob. Agents Chemother. 42, 2312–2318.
McGee, L., McDougal, L., Zhou, J., Spratt, B. G., Tenover, F. C., George, R., et al. (2001). Nomenclature of major antimicrobial-resistant clones of Streptococcus pneumoniae defined by the pneumococcal molecular epidemiology network. J. Clin. Microbiol. 39, 2565–2571. doi: 10.1128/JCM.39.7.2565-2571.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mingoia, M., Morici, E., Morroni, G., Giovanetti, E., Del Grosso, M., Pantosti, A., et al. (2014). Tn5253 Family Integrative and Conjugative Elements Carrying mef(I) and catQ Determinants in Streptococcus pneumoniae and Streptococcus pyogenes. Antimicrob. Agents Chemother. 58, 5886–5893. doi: 10.1128/AAC.03638-14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mingoia, M., Vecchi, M., Cochetti, I., Tili, E., Vitali, L. A., Manzin, A., et al. (2007). Composite structure of Streptococcus pneumoniae containing the erythromycin efflux resistance gene mef(I) and the chloramphenicol resistance gene catQ. Antimicrob. Agents Chemother. 51, 3983–3987. doi: 10.1128/AAC.00790-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Munoz-Najar, U., and Vijayakumar, M. (1999). An operon that confers UV resistance by evoking the SOS mutagenic response in streptococcal conjugative transposon Tn5252. J. Bacteriol. 181, 2782–2788.
Myers, E. W., Sutton, G. G., Delcher, A. L., Dew, I. M., Fasulo, D. P., Flanigan, M. J., et al. (2000). A whole-genome assembly of Drosophila. Science 287, 2196–2204. doi: 10.1126/science.287.5461.2196
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Orvis, J., Crabtree, J., Galens, K., Gussman, A., Inman, J. M., Lee, E., et al. (2010). Ergatis: a web interface and scalable software system for bioinformatics workflows. Bioinformatics (Oxford, England) 26, 1488–1492. doi: 10.1093/bioinformatics/btq167
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Palmieri, C., Mingoia, M., Massidda, O., Giovanetti, E., and Varaldo, P. E. (2012). Streptococcus pneumoniae transposon Tn1545/Tn6003 changes to Tn6002 due to spontaneous excision in circular form of the erm(B)- and aphA3-containing macrolide-aminoglycoside-streptothricin (MAS) Element. Antimicrob. Agents Chemother. 56, 5994–5997. doi: 10.1128/AAC.01487-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prudhomme, M., Attaiech, L., Sanchez, G., Martin, B., and Claverys, J. P. (2006). Antibiotic stress induces genetic transformability in the human pathogen Streptococcus pneumoniae. Science 313, 89–92. doi: 10.1126/science.1127912
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roberts, A. P., and Kreth, J. (2014). The impact of horizontal gene transfer on the adaptive ability of the human oral microbiome. Front. Cell. Infect. Microbiol. 4:124. doi: 10.3389/fcimb.2014.00124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roberts, A. P., and Mullany, P. (2011). Tn916-like genetic elements: a diverse group of modular mobile elements conferring antibiotic resistance. FEMS Microbiol. Rev. 35, 856–871. doi: 10.1111/j.1574-6976.2011.00283.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rocco, J. M., and Churchward, G. (2006). The integrase of the conjugative transposon Tn916 directs strand- and sequence-specific cleavage of the origin of conjugal transfer, oriT, by the endonuclease Orf20. J. Bacteriol. 188, 2207–2213. doi: 10.1128/JB.188.6.2207-2213.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rudy, C. K., Scott, J. R., and Churchward, G. (1997). DNA binding by the Xis protein of the conjugative transposon Tn916. J. Bacteriol. 179, 2567–2572.
Santagati, M., Lupo, A., Scillato, M., Di Martino, A., and Stefani, S. (2009). Conjugal mobilization of the Mega element carrying mef(E) from Streptococcus salivarius to Streptococcus pneumoniae. FEMS Microbiol. Lett. 290, 79–84. doi: 10.1111/j.1574-6968.2008.01408.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sharma, D., Baughman, W., Holst, A., Thomas, S., Jackson, D., Beall, B., et al. (2013). Pneumococcal carriage and invasive disease in children before introduction of the 13-valent conjugate vaccine: comparison with the era before 7-valent conjugate vaccine. Pediatr. Infect. Dis. J. 32, e45–e53. doi: 10.1097/INF.0b013e3182788fdd
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stephens, D. S., Zughaier, S. M., Whitney, C. G., Baughman, W. S., Barker, L., Gay, K., et al. (2005). Incidence of macrolide resistance in Streptococcus pneumoniae after introduction of the pneumococcal conjugate vaccine: population-based assessment. The Lancet 365, 855–863. doi: 10.1016/S0140-6736(05)71043-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Syrogiannopoulos, G. A., Grivea, I. N., Ednie, L. M., Bozdogan, B., Katopodis, G. D., Beratis, N. G., et al. (2003). Antimicrobial susceptibility and macrolide resistance inducibility of Streptococcus pneumoniae carrying erm(A), erm(B), or mef(A). Antimicrob. Agents Chemother. 47, 2699–2702. doi: 10.1128/AAC.47.8.2699-2702.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tettelin, H., Nelson, K. E., Paulsen, I. T., Eisen, J. A., Read, T. D., Peterson, S., et al. (2001). Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293, 498–506. doi: 10.1126/science.1061217
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wattam, A. R., Abraham, D., Dalay, O., Disz, T. L., Driscoll, T., Gabbard, J. L., et al. (2014). PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 42, D581–D591. doi: 10.1093/nar/gkt1099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wierzbowski, A. K., Boyd, D., Mulvey, M., Hoban, D. J., and Zhanel, G. G. (2005). Expression of the mef(E) gene encoding the macrolide efflux pump protein increases in Streptococcus pneumoniae with increasing resistance to macrolides. Antimicrob. Agents Chemother. 49, 4635–4640. doi: 10.1128/AAC.49.11.4635-4640.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wyres, K. L., van Tonder, A., Lambertsen, L. M., Hakenbeck, R., Parkhill, J., Bentley, S. D., et al. (2013). Evidence of antimicrobial resistance-conferring genetic elements among pneumococci isolated prior to 1974. BMC Genomics 14:500. doi: 10.1186/1471-2164-14-500
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zähner, D., Zhou, X., Chancey, S. T., Pohl, J., Shafer, W. M., and Stephens, D. S. (2010). Human antimicrobial peptide LL-37 induces mefE/mel-mediated macrolide resistance in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 54, 3516–3519. doi: 10.1128/AAC.01756-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, W., Yao, K., Zhang, G., Yang, Y., Li, Y., Lv, Y., et al. (2014). Mechanism for transfer of transposon Tn2010 carrying macrolide resistance genes in Streptococcus pneumoniae and its effects on genome evolution. J. Antimicrob. Chemother. 69, 1470–1473. doi: 10.1093/jac/dku019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: mobile genetic elements, transposons, integrative and conjugative elements, macrolides, antibiotic resistance, Streptococcus pneumoniae
Citation: Chancey ST, Agrawal S, Schroeder MR, Farley MM, Tettelin H and Stephens DS (2015) Composite mobile genetic elements disseminating macrolide resistance in Streptococcus pneumoniae. Front. Microbiol. 6:26. doi: 10.3389/fmicb.2015.00026
Received: 07 October 2014; Accepted: 08 January 2015;
Published online: 09 February 2015.
Edited by:
Claudio Palmieri, Polytechnic University of Marche, ItalyReviewed by:
Agnese Lupo, University of Bern, SwitzerlandKelly L. Wyres, IBM Research - Australia, Australia
Copyright © 2015 Chancey, Agrawal, Schroeder, Farley, Tettelin and Stephens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David S. Stephens, Division of Infectious Diseases, Department of Medicine, Emory University School of Medicine, Atlanta, GA, USA; Laboratories of Microbial Pathogenesis, Department of Veterans Affairs Medical Center, Atlanta, GA, USA e-mail:ZHN0ZXAwMUBlbW9yeS5lZHU=
†These authors have contributed equally to this work.