 Esmaeil Nourani
Esmaeil Nourani Farshad Khunjush
Farshad Khunjush Saliha Durmuş
Saliha Durmuş- 1Department of Computer Science and Engineering, School of Electrical and Computer Engineering, Shiraz University, Shiraz, Iran
- 2School of Computer Science, Institute for Research in Fundamental Sciences (IPM), Tehran, Iran
- 3Computational Systems Biology Group, Department of Bioengineering, Gebze Technical University, Kocaeli, Turkey
Infectious diseases are still among the major and prevalent health problems, mostly because of the drug resistance of novel variants of pathogens. Molecular interactions between pathogens and their hosts are the key parts of the infection mechanisms. Novel antimicrobial therapeutics to fight drug resistance is only possible in case of a thorough understanding of pathogen-host interaction (PHI) systems. Existing databases, which contain experimentally verified PHI data, suffer from scarcity of reported interactions due to the technically challenging and time consuming process of experiments. These have motivated many researchers to address the problem by proposing computational approaches for analysis and prediction of PHIs. The computational methods primarily utilize sequence information, protein structure and known interactions. Classic machine learning techniques are used when there are sufficient known interactions to be used as training data. On the opposite case, transfer and multitask learning methods are preferred. Here, we present an overview of these computational approaches for predicting PHI systems, discussing their weakness and abilities, with future directions.
Introduction
Many studies concerning identification of protein interactions and their associated networks were published (Aloy and Russell, 2003). Most of the previous studies were primarily focused on determining protein-protein interactions (PPIs) within a single organism (intra-species PPI prediction), while the prediction of PPIs between different organisms (inter-species PPI prediction) has recently emerged. Inter-species interactions may take many forms; in this survey, however, we focus on PPIs between pathogens and their hosts. Pathogen-host interaction (PHI) prediction is worthwhile to enlighten the infection mechanisms in the scarcity of experimentally-verified PHI data. Interactions between pathogen and host proteins allow pathogenic microorganisms to manipulate host mechanisms in order to use host capabilities and to escape from host immune responses (Dyer et al., 2010). Therefore, a complete understanding of infection mechanisms through PHIs is crucial for the development of new and more effective therapeutics.
Despite the critical need to improve the PHI knowledge, current progress is not adequate, suffering from scarcity of available experimental PHI data. Reliable experimental methods are time-consuming and expensive, making it unjustifiable to evaluate all possible PHIs. For instance, considering about 26,000 human proteins paired with a few thousands of pathogen proteins lead to millions of protein pairs to test experimentally. Scarce verified interactions are collected within a number of databases like HPIDB (Kumar and Nanduri, 2010), PATRIC (Wattam et al., 2014), PHISTO (Durmuş Tekir et al., 2013), VirHostNet (Navratil et al., 2009), and VirusMentha (Calderone et al., 2014). At this point, computational approaches come to help by predicting putative PHIs. In this paper, we concentrate on these computational studies, which are mandatory for enriching the available data and consequently increasing the pace of research in the field. The methods which were successfully applied specifically for PHI prediction in the literature are categorized based on pathogen-host systems in Table 1.
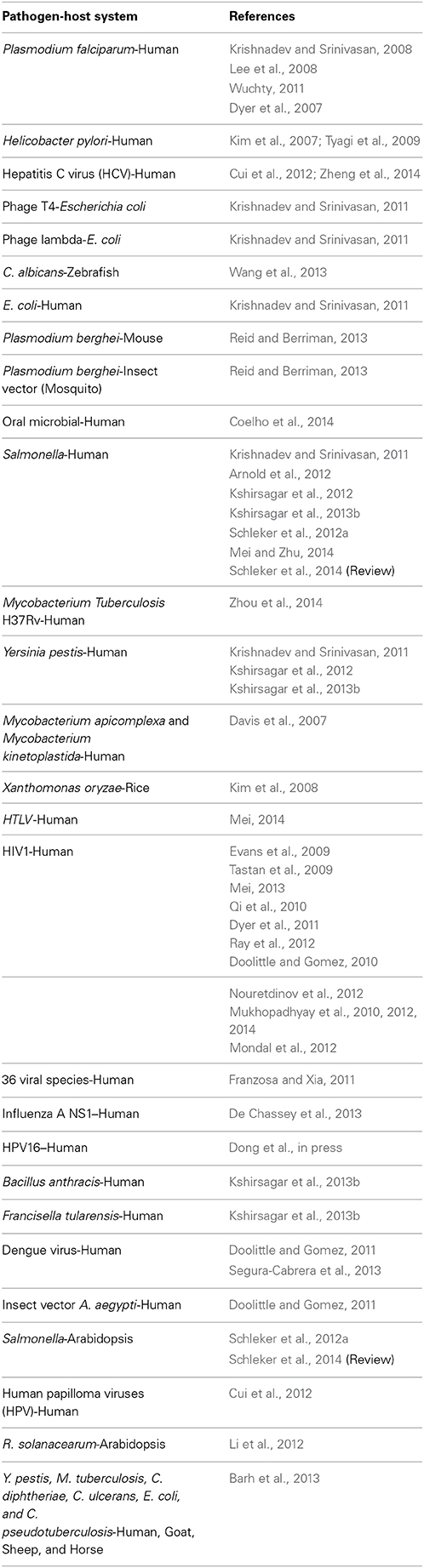
Table 1. Computational studies for prediction of PHIs.
Considering the relative availability of interaction data for HIV-Human system, notable number of studies are dedicated to this pathogen. Some other viral and bacterial pathogens are investigated and human is the main target as the host for investigation. Computational methods for predicting PHIs exploit known protein and domain interactions, and information on sequence of proteins. Network topology measures can complement these data. For instance, targeting hubs and bottleneck proteins in human PPI network by pathogen proteins is a well-accepted idea (Dyer et al., 2008; Durmuş Tekir et al., 2012; Schleker and Trilling, 2013; Zheng et al., 2014), though, they are not the sole targeted proteins (Chen et al., 2012). Classic machine learning methods are valuable remedy for cases where enough data for training are available. However, valuable efforts have recently been performed to apply these techniques for situations suffer from scarcity of known interaction data using machine learning based methods as transfer and multitask learning (Xu et al., 2010; Kshirsagar et al., 2013a,b).
In PPI prediction studies, methods specific for intra-species interactions are usually used. On the other hand, concentrating on the interactions between different organisms is a young branch of this field. The traditional methods cannot be applied here, their adaptation or devising new approaches would be mandatory.
Machine Learning and Data Mining Based Approaches
Applying machine learning techniques to bioinformatics is a well-accepted idea (Baldi and Brunak, 2001), which includes early efforts for PPI predictions (Bock and Gough, 2001). These methods utilize available PPI data as features for training and classifying interacting and non-interacting protein pairs. Both semi-supervised and supervised learning are used for PHI prediction. A Supervised method, which exploits exclusively labeled data, is applied in Tastan et al. (2009) integrating 35 features within eight groups using Random Forest (RF) classifier to deal with noisy and redundant features. The semi-supervised extension of their work is presented in Qi et al. (2010) which discarded 17 attributes from the feature vector that is related to determining 17 HIV-1 proteins. However, they have gained better performance through incorporating likely interactions (called “partially labeled”), which do not have sufficient evidence to be categorized as direct interaction. The same classifier is used as a quality control in Wuchty (2011), where a RF classifier assesses the quality of candidate interactions, obtained by discovering homologous and conserved interactions. The author filters the predicted results based on expression and molecular properties.
Conformal prediction is used in Nouretdinov et al. (2012) and the results are compared with those of Tastan et al. (2009) to assess the predictions. This method evaluates the conformance of new pairs with interacting pairs using a method called non-conformity measure (NCM) which shows distinction measure of an example regarding others. Their approach also allows the user to determine confidence level for prediction.
SVM based approaches as a famous classifier are successfully applied in PHI prediction studies (Kshirsagar et al., 2013a; Mei, 2013). Cui et al. (2012) presents a SVM based approach, which uses a fixed length feature vector, indicating relative frequency of consecutive amino acids in the protein sequence. We categorize the machine learning and data mining based approaches in Figure 1.
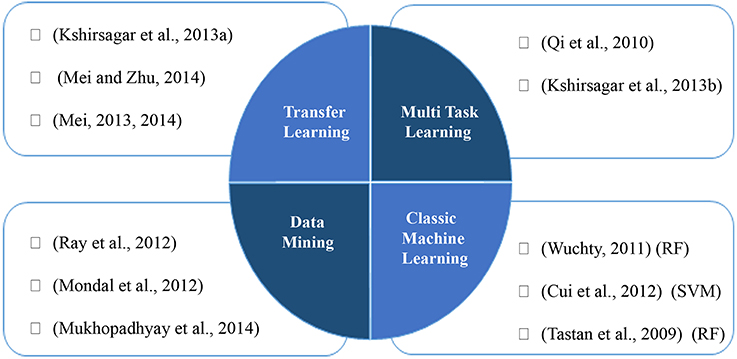
Figure 1. Machine learning and data mining based approaches for prediction of PHIs.
Transfer and Multitask Learning Approaches
One of the promising remedies to tackle the problem of data scarcity is eliciting and transferring data from related domains to desired formulation. Multitask learning uses commonalities among different domains and learn problem simultaneously between them within a shared task formulation, which leads to better performance rather conducting learning task on individual domain. A review paper, Xu and Yang (2011) presents some of the studies utilizing this idea in bioinformatics. For PPI prediction, a method was proposed in Xu et al. (2010) which uses collective matrix factorization originally proposed by Singh and Gordon (2008) to transfer knowledge from a relatively dense PPI network called “source” for predicting new PPIs in a sparse target PPI network. Their goal is to predict intra-species pathogen PPIs as target with the aid of human PPIs as source network through defining a similarity matrix to act as a bridge between them. Another study conducts three different individual classifiers on three GO features (molecular functions, cellular localization, and biological processes) on available protein features and at the same time three classifiers on alternative homolog features to exploit transfer learning. An ensemble classifier produces final result using weighting probability outputs of individual classifiers (Mei, 2013). They applied relatively same idea using a multi instance AdaBoost method to transfer homolog feature as the second instance of proteins (Mei, 2014; Mei and Zhu, 2014). A combination of supervised and semi-supervised approaches is proposed by Qi et al. (2010) through multitask learning. Semi-supervised task on partially positive labels is conducted to improve the supervised classification which trains multi-layer perceptron using labeled data. Another multitask formulation is used in Kshirsagar et al. (2013b) to integrate knowledge from different pathogen-host systems to increase the prediction power of the combined model. Each task is formulated as predicting PHI data between each pathogen and its host. To define similarity between tasks and transfer shared knowledge, they assume that similar pathogens tend to target same biological process in human. In other words, “commonality hypothesis” is introduced that assumes pathway membership of human proteins in positive PHIs should be similar between different tasks. To implement this idea, optimization problem is conducted and dissimilarities are penalized in the objective function. They use transfer learning in Kshirsagar et al. (2013a) for the cases where no known interaction is available by exploiting precisely chosen instances from a source task.
Data Mining Based Approaches
Machine learning based methods which formulate PPI prediction as a classification task use both interacting and non-interacting protein pairs as positive and negative classes, respectively. Constructing negative class is not straightforward due to the fact that there is no experimentally verified non-interacting pair. This has motivated some studies to overcome this problem by removing the need for negative data through using alternative methods (Mukhopadhyay et al., 2010, 2012, 2014; Mondal et al., 2012; Ray et al., 2012). They integrate bi-clustering with association rule mining, utilizing only positive samples to predict virus-human interactions.
Utilized Features
Various studies utilize different sets of biological information through data integration to improve the prediction performance. However, it should be noted that making use of a lot of features without enriching training data may lead to over fitting in the model (Mei, 2014). Table 2 summarizes the utilized features within different studies on PHI prediction, providing all the cataloged feature information is not always possible for all pathogen systems. Furthermore, various features claimed to have different predictive effects in PHI prediction. Outperforming other features was the motivation for some studies to use GO features in PHI prediction (Mei, 2013, 2014) while features extracted from protein sequences, reported as not promising (Yu et al., 2010).
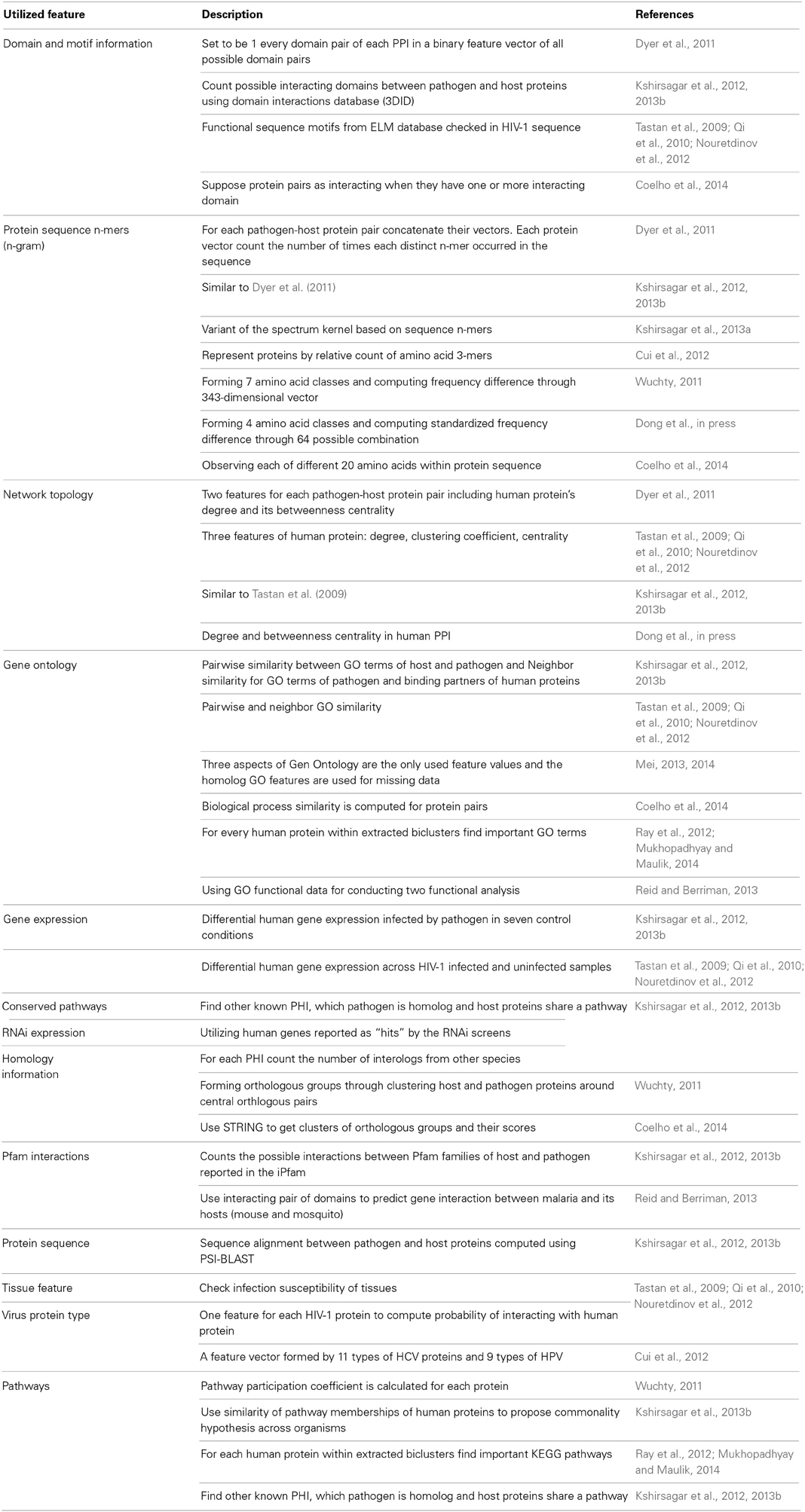
Table 2. Summary of the exploited features for prediction of PHIs.
Handling Missing Data
Applying machine learning methods and specially supervised learning for situations suffer from data scarcity is challenging. Being limited to well-studied pathogen systems like HIV-1 is the consequence of data dependency. Recently, some solutions are proposed to overcome this limitation by offering substituted values for missing data. For instance, in Kshirsagar et al. (2012) two different methods are proposed including information transfer from other species and model-based imputation. First, they rely on homologous proteins data to provide feature values like GO annotations and gene expression data. This contributes a lot and downgrades the missing data significantly. However, for proteins with no available homolog, they have modeled gene expression value distribution. They have compared the proposed “Cross species imputation” with other imputation techniques. The first method is called “RF” which initiates the missing data to mean value and re-estimate it by choosing the nearest leaf node of the created forest. Another intuitive method is choosing the average of the feature values and the last compared method is discarding any pair with missing value which leads to a reduced dataset. Clear improvements are reported in comparison with the listed imputation methods. It should be noted that using solely statistical methods for estimating features like GO values will be hard due to high dimensionality. Mei (2013) uses homolog information when the features of a protein is unavailable. They have designed various experiments to show the performance of substituting homolog features. Pessimistic experiment, which uses only homolog features to train and test without incorporating any base proteins (called “target” in the article), has promising results, indicating that using homolog information is an effective substitute for the target information to tackle the problem of data unavailability.
The Challenge of Non-Interacting PPIs
Since there is no available verified non-interacting PPI to be used for training the model, selecting negative data remains as a challenge for PPI prediction. Some studies try to circumvent the obstacle by using methods which do not require negative samples (Ray et al., 2012). However, ignoring non-interacting patterns may increase the rate of false positives (Mei, 2013). The negative set is not defined in Nouretdinov et al. (2012) and instead they use unknown label for other pairs. Most of the studies which formulate the problem as a classification task, have to construct negative class through randomly sampling the data. The rate of positive to negative class is chosen in different manners to avoid biasing classifier toward wrong predictions. A ratio of 1:100 is chosen in Kshirsagar et al. (2012, 2013b) and Tastan et al. (2009) expecting one interaction pair within 100 random pathogen-host pairs. Mei (2013) chooses the same ratio for negative and positive classes, however proposes different idea for choosing negative samples. They put aside sub-cellular co-localized pairs from the negative class and report better performance in comparison with random sampling. The study in Dyer et al. (2011) conducted experiments with different ratios and 10 randomly chosen sets for each ratio and stated that beside clearly different results for different ratios, variability of randomly selected negative samples for each ratio does not have major effect on the result accuracy.
Homology Based Approaches
The rationale behind this type of methods is the expectation of conserved interactions between a pair of proteins which have interacting homologs in another species. The conserved interaction is called as “Interolog.” The simple method of identifying Interologs is as follows: Consider a template PPI pair (a, b) in a source species, find the homolog a′ in the host and the homolog b′ in the pathogen, conclude that (a′, b′) interact. Simplicity and clear biological basis are the main advantages of these methods. However, homology to known interactions is not sufficient for evaluating the biological evidence of the predicted results. Different filtering techniques should be considered for assessing the feasibility of the interactions under an in vivo condition and consequently decreasing the false positives.
A homology detection method using template PPI databases, DIP (Salwinski et al., 2004) and iPfam (Finn et al., 2014), is published in Krishnadev and Srinivasan (2008) to predict PHI pairs. Searching the sequences of host and pathogen proteins within two template databases are conducted to find a superset of all interactions which are physically and structurally compatible. These potential interactions are refined within two additional filtering steps, to detect biologically feasible interactions including integration of expression and sub-cellular localization data. The authors have applied the same procedure for different pathogens in their subsequent works (Tyagi et al., 2009; Krishnadev and Srinivasan, 2011).
Another research uses the conceptually same approach by exploiting sequence similarity augmented with domain-domain interaction detection (Schleker et al., 2012a). They have two compressive reviews of the computational approaches predicting Salmonella-Host interactions (Schleker et al., 2012b, 2014), which include comparing Salmonella-Human and Salmonella-Plant interaction predictions.
Homolog knowledge can be used indirectly as a remedy for data scarcity and data unavailability by homolog knowledge transfer. Mei (2013) uses homolog information (features) when the features of a protein is unavailable. They have designed different experiments to show the performance of substituting homology features. Pessimistic experiment, which uses only homology features for train and test without incorporating any base proteins (called as “target” in the article) has promising results, indicating that using homolog information is an effective substitute for the target information to tackle the problem of data unavailability.
Another research uses high confidence intra-species PPIs to detect Interologs using ortholog information (Lee et al., 2008). The assumption is that when two orthologous groups are shared between more than two species, there will be a potential Interolog between those orthologous groups. The potential interactions are filtered using gene ontology annotations followed by pathogen sequence filtering based on the presence or absence of translocational signals to refine the predictions. The notable point is negligible intersection of the predicted interactions with those of the reported predictions in Dyer et al. (2007) due to applying different techniques and datasets for same pathogen-host system.
Zhou et al. (2014) introduces the “stringent homology” which does not rely only on intra-species template PPIs to discover interologs and make use of two different organisms as the source of template PPIs to predict PHIs. They also claim that it is not only for the targeted host proteins which tend to be hub in their own PPI network and this is also true about targeting pathogen proteins.
The most important obstacle for using homology based methods is scarcity of available homolog information. For instance, the number of interologs within bacterial PPIs are not dignificant (Kshirsagar et al., 2013b) demonstrating that we cannot rely only on homolog information for every situation without being cautious about data availability. Clearly, it is reasonable to predict more genomic and proteomic data will be available in the future and consequently more accurate homologs are identified paving the way of studying less-known pathogens. Table 3 summarizes the published research for predicting PHIs based on homology information.

Table 3. Homology based approaches for prediction of PHIs.
Structure Based Approaches
A number of studies are based on structural similarities and use template PPIs to detect similar interacting pairs within host and pathogen proteins. Preliminary ideas presented in Davis et al. (2007) called comparative modeling and was based on their prior work (Davis et al., 2006). Their method starts with a set of host and pathogen proteins and then sequence matching procedures are used to determine the similarities between the host or pathogen proteins with known structure or known interaction protein partners. Sequence similarity score is only used when structure information is unavailable as a statistical potential assessment, to predict interacting partners. Filtering the set of potential interactions is the last step which is performed using the biological contexts of proteins and a network-level filter. The outcome of this process is decreasing the potential PHIs by about five orders of magnitude. The main drawback of this method is that finding high similarity between pathogen proteins and proteins with known structure is not guaranteed for all pathogen proteins. Therefore, unavailability of the spatial structural information would restrict the applicability of this method. Furthermore, they have only the ability to collect limited number of benchmark PPIs from literature to evaluate their prediction performance.
Authors in Franzosa and Xia (2011) claim to significantly reduce the rate of false positives by presenting virus-human structural interaction network, in which, each PPI is associated with a high confidence 3D structural model. Applicability of the method is limited to human-human and virus-human PPIs for which 3D structural models are available. The method starts with extracting human interacting pairs from PDB and followed by mapping virus proteins to them by sequence similarity. They emphasize the importance of constructing a high-resolution, 3D structural view of pathogen-host and within-host PPI networks to discover new principles of PHIs through their review paper in Franzosa et al. (2012).
Another research developed a map of interactions between HIV-1 and human proteins based on protein structural similarity (Doolittle and Gomez, 2010). A comparison of known crystal structures is performed to measure structural similarity between host and pathogen proteins. Human proteins which have high structural similarity to a HIV protein are identified and their known interacting partners are determined as targets. The assumption is that HIV proteins have the same interactions as their human peers. These predicted results refined by two filtering steps using data from the recent RNAi screens and cellular co-localization information. They apply the same method for developing an interaction network between Dengue virus and its hosts (Doolittle and Gomez, 2011). Again, with a similar idea those proteins with comparable structures share interaction partners. The work suffers from the lack of assessment data in a way that, very limited number of used benchmark PPIs are specific to the viral pathogen. Table 4 summarizes the conducted research for predicting PHIs based on structural data.
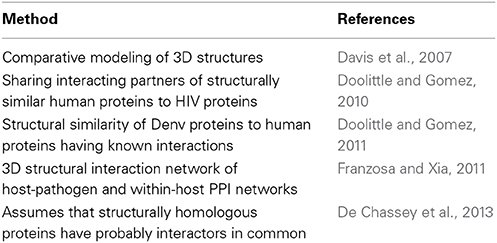
Table 4. Structure based approaches for prediction of PHIs.
Domain and Motif Based Approaches
The idea of exploiting domains as building blocks of proteins for predicting PPIs is well-studied for single organisms (Wojcik and Schächter, 2001; Pagel et al., 2004) regarding the fact that domains are the mediators of interactions. The approach presented in Dyer et al. (2007) is one of the pioneer published research for predicting PHIs. However, small list of interactions are presented and their biological relevance are not strongly evaluated. To predict interactions between host and pathogen proteins, they present an algorithm that integrates protein domain profiles with interactions between proteins from the same organism. For every pair of functional domains (d, e) which is present in protein pair (g, h) respectively, the probability of interacting (g, h) is assessed using Bayesian statistics. To apply this idea to a pathogen-host system, they identify domains in every host and pathogen proteins and compute the interaction probability for each pair of host and pathogen proteins that contain at least one domain. Assuming Mg as the set of domains contained in protein g the interaction probability of proteins (g, h) is computed as:
The authors have published another study which uses domain profiles as features in supervised machine learning for predicting interactions in HIV-Human system.
A similar knowledge source is chosen in Kim et al. (2007) which makes use of domain information from InterProScan (Quevillon et al., 2005). They predict PPIs using PreDIN (Kim et al., 2002) and PreSPI (Han et al., 2004) algorithms based on domain information. A study for prediction of interacting proteins of rice and Xanthomonas oryzae pathovar oryzae (Xoo) also uses domain information (Kim et al., 2008). They presented XooNET which provides about 3500 possible interaction pairs as well as the graphical visualizations of the interaction networks.
The work in Arnold et al. (2012) presents a method to predict and rank bacteria-human PPIs based on domain-domain interactions. They collect a list of Pfam domains and bacterial-human proteins which contains one of the listed domains. Then the data was searched for experimentally verified effectors or their homologs in another bacteria. The result is the possible interactions between Salmonella effectors and host proteins.
Not all pathogen systems are appropriate for applying the mentioned domain based approaches, since domains and the related information are not available for all pathogens. For instance, information on domains and the related statistics are not available for a considerable number of the HIV-1 proteins. Regarding this limitation, the work in Evans et al. (2009) concentrates on protein interactions based on short eukaryotic linear motifs (ELMs) for HIV-1 proteins interacting with human protein counter domains (CDs). They do not accept the idea of having relatively weak link among motif/domain bindings and the actual virus-host PPIs which is presented in Tastan et al. (2009). They predict two kinds of interactions for each virus protein, including direct human protein targets (called H1) which bind to virus via a human CD and a virus ELM and the second type includes indirect interactions in which, host proteins that their normal interactions with H1 proteins are potentially disrupted by competition with an HIV-1 protein. Table 5 summarizes the conducted research for predicting PHIs based on domain and motif knowledge.

Table 5. Domain and motif based approaches for prediction of PHIs.
Performance Evaluation
The lack of gold standard PHI data and the complexity of PHI mechanisms lead to a hard assessment phase, in a way that predicted interactions are rarely supported by a biological basis. Some studies validate their results by measuring the shared interactions with other published materials (Mukhopadhyay et al., 2012, 2014; Segura-Cabrera et al., 2013). Here we focus on computational metrics which are widely used in publications to evaluate the accuracy of their results, which are shown in Table 6.
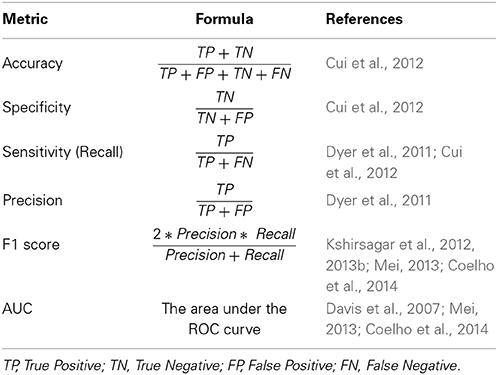
Table 6. Popular evaluation metrics used for PHI prediction.
Conclusions
Inter-species PPI predictions have gained more popularity in recent years. Computational methods may have important roles in paving the way for experimental PHI verifications by highlighting the high potential interactions and limiting the experimental scope which lead to expense reduction and probably the rapid knowledge development. In this paper, we reviewed the studies which directly focused on computationally PHI prediction. Published approaches are categorized based on pathogen-host and the method they utilize. Clearly some pathogen systems are well studied and targeted in more research regarding the availability of the required data. HIV-1 is the most distinguished pathogen which studied specifically using data-requiring machine learning methods. Therefore, the most important challenge for computationally prediction of PHIs, is the lack of available verified interactions and the relevant feature information in most of the pathogens systems. Data unavailability and scarcity refer to verified interacting PPIs, lack of verified non-interacting protein pairs and missing feature information for proteins. Recent studies have found a new source of data to overcome these limitations. Knowledge transfer from related pathogen systems has shown to be an effective remedy, even for situations with no available interactions. These methods enlighten a promising future direction for establishing computational methods which are augmented with additional transferred knowledge.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aloy, P., and Russell, R. B. (2003). InterPreTS: protein interaction prediction through tertiary structure. Bioinformatics (Oxford, England) 19, 161–162. doi: 10.1093/bioinformatics/19.1.161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arnold, R., Boonen, K., Sun, M. G. F., and Kim, P. M. (2012). Computational analysis of interactomes: current and future perspectives for bioinformatics approaches to model the host-pathogen interaction space. Methods 57, 508–518. doi: 10.1016/j.ymeth.2012.06.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baldi, P., and Brunak, S. (2001). Bioinformatics: the Machine Learning Approach. Cambridge: MIT press.
Barh, D., Gupta, K., Jain, N., Khatri, G., León-Sicairos, N., Canizalez-Roman, A., et al. (2013). Conserved host-pathogen PPIs. Globally conserved inter-species bacterial PPIs based conserved host-pathogen interactome derived novel target in C. pseudotuberculosis, C. diphtheriae, M. tuberculosis, C. ulcerans, Y. pestis, and E. coli targeted by Piper betel compounds. Integr. Biol. 5, 495–509. doi: 10.1039/c2ib20206a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bock, J. R., and Gough, D. A. (2001). Predicting protein–protein interactions from primary structure. Bioinformatics 17, 455–460. doi: 10.1093/bioinformatics/17.5.455
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Calderone, A., Licata, L., and Cesareni, G. (2014). VirusMentha: a new resource for virus-host protein interactions. Nucleic Acids Res. 43, D588–D592. doi: 10.1093/nar/gku830
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, K.-C., Wang, T.-Y., and Chan, C. (2012). Associations between HIV and human pathways revealed by protein-protein interactions and correlated gene expression profiles. PloS ONE 7:e34240. doi: 10.1371/journal.pone.0034240
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Coelho, E. D., Arrais, J. P., Matos, S., Pereira, C., Rosa, N., Correia, M. J., et al. (2014). Computational prediction of the human-microbial oral interactome. BMC Syst. Biol. 8, 24. doi: 10.1186/1752-0509-8-24
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cui, G., Fang, C., and Han, K. (2012). Prediction of protein-protein interactions between viruses and human by an SVM model. BMC Bioinformatics 13 (Suppl. 7): S5. doi: 10.1186/1471-2105-13-S7-S5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Davis, F. P., Barkan, D. T., Eswar, N., McKerrow, J. H., and Sali, A. (2007). Host pathogen protein interactions predicted by comparative modeling. Protein Sci. 16, 2585–2596. doi: 10.1110/ps.073228407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Davis, F. P., Braberg, H., Shen, M.-Y., Pieper, U., Sali, A., and Madhusudhan, M. S. (2006). Protein complex compositions predicted by structural similarity. Nucleic Acids Res. 34, 2943–2952. doi: 10.1093/nar/gkl353
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Chassey, B., Meyniel-Schicklin, L., Aublin-Gex, A., Navratil, V., Chantier, T., André, P., et al. (2013). Structure homology and interaction redundancy for discovering virus-host protein interactions. EMBO Rep. 14, 938–944. doi: 10.1038/embor.2013.130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dong, Y., Kuang, Q., Dai, X., Li, R., Wu, Y., Leng, W., et al. (in press). Improving the understanding of pathogenesis of human papillomavirus 16 via mapping protein-protein interaction network. Biomed Res. Int. 890381. doi: 10.1155/2014/890381
Doolittle, J. M., and Gomez, S. M. (2010). Structural similarity-based predictions of protein interactions between HIV-1 and Homo sapiens. Virol. J. 7:82. doi: 10.1186/1743-422X-7-82
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Doolittle, J. M., and Gomez, S. M. (2011). Mapping protein interactions between Dengue virus and its human and insect hosts. PLoS Negl. Trop. Dis. 5:e954. doi: 10.1371/journal.pntd.0000954
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Durmuş Tekir, S., Cakir, T., and Ulgen, K. Ö. (2012). Infection strategies of bacterial and viral pathogens through pathogen-human protein-protein interactions. Front. Microbiol. 3:46. doi: 10.3389/fmicb.2012.00046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Durmuş Tekir, S., Çakır, T., Ardiç, E., Sayılırbaş, A. S., Konuk, G., Konuk, M., et al. (2013). PHISTO: pathogen-host interaction search tool. Bioinformatics (Oxford, England) 29, 1357–1358. doi: 10.1093/bioinformatics/btt137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dyer, M., Murali, T. M., and Sobral, B. W. (2011). Supervised learning and prediction of physical interactions between human and HIV proteins. Infect. Genet. Evol. 11, 917–923. doi: 10.1016/j.meegid.2011.02.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dyer, M. D., Murali, T. M., and Sobral, B. W. (2007). Computational prediction of host-pathogen protein-protein interactions. Bioinformatics (Oxford, England) 23, i159–i166. doi: 10.1093/bioinformatics/btm208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dyer, M. D., Murali, T. M., and Sobral, B. W. (2008). The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathogens 4:e32. doi: 10.1371/journal.ppat.0040032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dyer, M. D., Neff, C., Dufford, M., Rivera, C. G., Shattuck, D., Bassaganya-Riera, J., et al. (2010). The human-bacterial pathogen protein interaction networks of Bacillus anthracis, Francisella tularensis, and Yersinia pestis. PloS ONE 5:e12089. doi: 10.1371/journal.pone.0012089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Evans, P., Dampier, W., Ungar, L., and Tozeren, A. (2009). Prediction of HIV-1 virus-host protein interactions using virus and host sequence motifs. BMC Med. Genomics 2:27. doi: 10.1186/1755-8794-2-27
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Finn, R. D., Miller, B. L., Clements, J., and Bateman, A. (2014). iPfam: a database of protein family and domain interactions found in the Protein Data Bank. Nucleic Acids Res. 42, D364–D373. doi: 10.1093/nar/gkt1210
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Franzosa, E. A., Garamszegi, S., and Xia, Y. (2012). Toward a three-dimensional view of protein networks between species. Front. Microbiol. 3:428. doi: 10.3389/fmicb.2012.00428
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Franzosa, E. A., and Xia, Y. (2011). Structural principles within the human-virus protein-protein interaction network. Proc. Natl. Acad. Sci. U.S.A. 108, 10538–10543. doi: 10.1073/pnas.1101440108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Han, D.-S., Kim, H.-S., Jang, W.-H., Lee, S.-D., and Suh, J.-K. (2004). PreSPI: a domain combination based prediction system for protein-protein interaction. Nucleic Acids Res. 32, 6312–6320. doi: 10.1093/nar/gkh972
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, J.-G., Park, D., Kim, B.-C., Cho, S.-W., Kim, Y. T., Park, Y.-J., et al. (2008). Predicting the interactome of Xanthomonas oryzae pathovar oryzae for target selection and DB service. BMC Bioinformatics 9:41. doi: 10.1186/1471-2105-9-41
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, W. K., Kim, K., Lee, E., Marcotte, E. M., Kim, H., and Suh, J. (2007). Identification of disease specific protein interactions between the gastric cancer causing pathogen, H. pylori, and Human Hosts using protein network modeling and gene chip analysis. Gastric Cancer 1, 179–187.
Kim, W. K., Park, J., and Suh, J. K. (2002). Large scale statistical prediction of protein-protein interaction by potentially interacting domain (PID) pair. Genome Inform. 13, 42–50. doi: 10.1.1.105.8455
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krishnadev, O., and Srinivasan, N. (2008). A data integration approach to predict host-pathogen protein-protein interactions: application to recognize protein interactions between human and a malarial parasite. In Silico Biol. 8, 235–250. doi: 10.1016/j.ijbiomac.2011.01.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krishnadev, O., and Srinivasan, N. (2011). Prediction of protein-protein interactions between human host and a pathogen and its application to three pathogenic bacteria. Int. J. Biol. Macromol. 48, 613–619. doi: 10.1016/j.ijbiomac.2011.01.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kshirsagar, M., Carbonell, J., and Klein-Seetharaman, J. (2012). Techniques to cope with missing data in host-pathogen protein interaction prediction. Bioinformatics 28, i466–i472. doi: 10.1093/bioinformatics/bts375
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kshirsagar, M., Carbonell, J., and Klein-Seetharaman, J. (2013a). “Multisource transfer learning for host-pathogen protein interaction prediction in unlabeled tasks,” in A Workshop at the Annual Conference on Neural Information Processing Systems (NIPS 2013), NIPSWorkshop on Machine Learning for Computational Biology (Lake Tahoe, NV), 3–6.
Kshirsagar, M., Carbonell, J., and Klein-Seetharaman, J. (2013b). Multitask learning for host-pathogen protein interactions. Bioinformatics 29, i217–i226. doi: 10.1093/bioinformatics/btt245
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kumar, R., and Nanduri, B. (2010). HPIDB–a unified resource for host-pathogen interactions. BMC Bioinformatics 11 (Suppl. 6):S16. doi: 10.1186/1471-2105-11-S6-S16
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, S., Chan, C., Tsai, C., and Lai, J. (2008). Ortholog-based protein-protein interaction prediction and its application to inter-species interactions. BMC Bioinformatics 9:S11. doi: 10.1186/1471-2105-9-S12-S11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, Z.-G., He, F., Zhang, Z., and Peng, Y.-L. (2012). Prediction of protein-protein interactions between Ralstonia solanacearum and Arabidopsis thaliana. Amino Acids 42, 2363–2371. doi: 10.1007/s00726-011-0978-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mei, S. (2013). Probability weighted ensemble transfer learning for predicting interactions between HIV-1 and human proteins. PloS ONE 8:e79606. doi: 10.1371/journal.pone.0079606
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mei, S. (2014). Computational reconstruction of proteome-wide protein interaction networks between HTLV retroviruses and Homo sapiens. BMC Bioinformatics 15:245. doi: 10.1186/1471-2105-15-245
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mei, S., and Zhu, H. (2014). AdaBoost based multi-instance transfer learning for predicting proteome-wide interactions between Salmonella and human proteins. PloS ONE 9:e110488. doi: 10.1371/journal.pone.0110488
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mondal, K. C., Pasquier, N., Mukhopadhyay, A., Pereira, C., Maulik, U., and Tettamanzi, A. G. B. (2012). “Prediction of protein interactions on HIV-1–human Ppi data using a novel closure-based integrated approach,” in Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (Vilamoura), 164–173.
Mukhopadhyay, A., and Maulik, U. (2014). Network-based study reveals potential infection pathways of hepatitis-C leading to various diseases. PloS ONE 9:e94029. doi: 10.1371/journal.pone.0094029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mukhopadhyay, A., Maulik, U., and Bandyopadhyay, S. (2012). A novel biclustering approach to association rule mining for predicting HIV-1-human protein interactions. PloS ONE 7:e32289. doi: 10.1371/journal.pone.0032289
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mukhopadhyay, A., Maulik, U., Bandyopadhyay, S., and Eils, R. (2010). “Mining association rules from HIV-human protein interactions,” in 2010 International Conference on Systems in Medicine and Biology (Kharagpur), 344–348. doi: 10.1109/ICSMB.2010.5735401
Mukhopadhyay, A., Ray, S., and Maulik, U. (2014). Incorporating the type and direction information in predicting novel regulatory interactions between HIV-1 and human proteins using a biclustering approach. BMC Bioinformatics 15:26. doi: 10.1186/1471-2105-15-26
Navratil, V., de Chassey, B., Meyniel, L., Delmotte, S., Gautier, C., André, P., et al. (2009). VirHostNet: a knowledge base for the management and the analysis of proteome-wide virus-host interaction networks. Nucleic Acids Res. 37, D661–D668. doi: 10.1093/nar/gkn794
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nouretdinov, I., Gammerman, A., Qi, Y., Klein-Seetharaman, J., and Learning, C. (2012). Determining confidence of predicted interactions between HIV-1 and human proteins using conformal method. Pac. Symp. Biocomput. 311, 311–322. doi: 10.1142/9789814366496_0030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pagel, P., Wong, P., and Frishman, D. (2004). A domain interaction map based on phylogenetic profiling. J. Mol. Biol. 344, 1331–1346. doi: 10.1016/j.jmb.2004.10.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qi, Y., Tastan, O., Carbonell, J. G., Klein-Seetharaman, J., and Weston, J. (2010). Semi-supervised multi-task learning for predicting interactions between HIV-1 and human proteins. Bioinformatics 26, i645–i652. doi: 10.1093/bioinformatics/btq394
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Quevillon, E., Silventoinen, V., Pillai, S., Harte, N., Mulder, N., Apweiler, R., et al. (2005). InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120. doi: 10.1093/nar/gki442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ray, S., Mukhopadhyay, A., and Maulik, U. (2012). “Predicting annotated HIV-1 – human PPIs using a biclustering approach to association rule mining,” in 2012 Third International Conference on Emerging Applications of Information Technology (EAIT) (Kolkata), 3–6.
Reid, A. J., and Berriman, M. (2013). Genes involved in host – parasite interactions can be revealed by their correlated expression. Nucleic Acids Res. 41, 1508–1518. doi: 10.1093/nar/gks1340
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Salwinski, L., Miller, C. S., Smith, A. J., Pettit, F. K., Bowie, J. U., and Eisenberg, D. (2004). The database of interacting proteins: 2004 update. Nucleic Acids Res. 32, D449–D451. doi: 10.1093/nar/gkh086
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schleker, S., Garcia-Garcia, J., Klein-Seetharaman, J., and Oliva, B. (2012a). Prediction and comparison of Salmonella-human and Salmonella-Arabidopsis interactomes. Chem. Biodiver. 9, 991–1018. doi: 10.1002/cbdv.201100392
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schleker, S., Kshirsagar, M., and Klein-seetharaman, J. (2014). Comparing human-Salmonella with plant-Salmonella proteinprotein interaction predictions. Front. Microbiol. 5:552. doi: 10.3389/fmicb.2014.00552
Schleker, S., Sun, J., Raghavan, B., Srnec, M., Müller, N., Koepfinger, M., et al. (2012b). The current Salmonella – host interactome. Proteomics Clin. Appl. 6, 117–133. doi: 10.1002/prca.201100083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schleker, S., and Trilling, M. (2013). Data-warehousing of protein-protein interactions indicates that pathogens preferentially target hub and bottleneck proteins. Front. Microbiol. 4:51. doi: 10.3389/fmicb.2013.00051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Segura-Cabrera, A., García-Pérez, C. A., Guo, X., and Rodríguez-Pérez, M. A. (2013). A viral-human interactome based on structural motif-domain interactions captures the human infectome. PloS ONE 8:e71526. doi: 10.1371/journal.pone.0071526
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Singh, A. P., and Gordon, G. J. (2008). “Relational learning via collective matrix factorization,” in Proceedings of the 14th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (Lasvegas, NV), 650–658. doi: 10.1145/1401890.1401969
Tastan, O., Qi, Y., Carbonell, J. G., and Klein-Seetharaman, J. (2009). Prediction of interactions between HIV-1 and human proteins by information integration. Pac. Symp. Biocomput. 14, 516–527.
Tyagi, N., Krishnadev, O., and Srinivasan, N. (2009). Prediction of protein-protein interactions between Helicobacter pylori and a human host. Mol. Biosyst. 5, 1630–1635. doi: 10.1039/b906543c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, Y.-C., Lin, C., Chuang, M.-T., Hsieh, W.-P., Lan, C.-Y., Chuang, Y.-J., et al. (2013). Interspecies protein-protein interaction network construction for characterization of host-pathogen interactions: a Candida albicans-zebrafish interaction study. BMC Syst. Biol. 7:79. doi: 10.1186/1752-0509-7-79
Wattam, A. R., Abraham, D., Dalay, O., Disz, T. L., Driscoll, T., Gabbard, J. L., et al. (2014). PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 42, D581–D591. doi: 10.1093/nar/gkt1099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wojcik, J., and Schächter, V. (2001). Protein-protein interaction map inference using interacting domain profile pairs. Bioinformatics 17, S296–S305. doi: 10.1093/bioinformatics/17.suppl_1.S296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wuchty, S. (2011). Computational prediction of host-parasite protein interactions between P. falciparum and H. sapiens. PloS ONE 6:e26960. doi: 10.1371/journal.pone.0026960
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, Q., Xiang, E. W., and Yang, Q. (2010). “Protein-protein interaction prediction via collective matrix factorization,” in 2010 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) (Hong Kong), 62–67. doi: 10.1109/BIBM.2010.5706537
Xu, Q., and Yang, Q. (2011). A survey of transfer and multitask learning in bioinformatics. J. Comput. Sci. Eng. 5, 257–268. doi: 10.5626/JCSE.2011.5.3.257
Yu, J., Guo, M., Needham, C. J., Huang, Y., Cai, L., and Westhead, D. R. (2010). Simple sequence-based kernels do not predict protein-protein interactions. Bioinformatics 26, 2610–2614. doi: 10.1093/bioinformatics/btq483
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zheng, L.-L., Li, C., Ping, J., Zhou, Y., Li, Y., and Hao, P. (2014). The domain landscape of virus-host interactomes. Biomed Res Int. 2014:867235. doi: 10.1155/2014/867235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, H., Gao, S., Nguyen, N., Fan, M., and Jin, J. (2014). Stringent homology-based prediction of H. sapiens-M. tuberculosis H37Rv protein-protein interactions. Biol. Dir. 9, 1–30. doi: 10.1186/1745-6150-9-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: protein-protein interaction, pathogen-host interaction (PHI), computational PHI prediction, machine learning, data mining
Citation: Nourani E, Khunjush F and Durmuş S (2015) Computational approaches for prediction of pathogen-host protein-protein interactions. Front. Microbiol. 6:94. doi: 10.3389/fmicb.2015.00094
Received: 01 December 2014; Accepted: 26 January 2015;
Published online: 24 February 2015.
Edited by:
Evangelos Giamarellos-Bourboulis, University of Athens, Medical School, GreeceReviewed by:
Magdalena Chirila, Iuliu Hatieganu University of Medicine and Pharmacy, RomaniaMalgorzata Anna Mikaszewska-Sokolewicz, The Medical University of Warsaw, Poland
Copyright © 2015 Nourani, Khunjush and Durmuş. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Farshad Khunjush, Department of Computer Science and Engineering, School of Electrical and Computer Engineering, Zand Avenue, Shiraz 71348 - 51154, Iran e-mail:a2h1bmp1c2hAc2hpcmF6dS5hYy5pcg==