 Chong Peng
Chong Peng- 1Department of Physics, Tianjin University, Tianjin, China
- 2Key Laboratory of Systems Bioengineering (Ministry of Education), Tianjin University, Tianjin, China
- 3SynBio Research Platform, Collaborative Innovation Center of Chemical Science and Engineering, Tianjin, China
DNA replication, one of the central events in the cell cycle, is the basis of biological inheritance. In order to be duplicated, a DNA double helix must be opened at defined sites, which are called DNA replication origins (ORIs). Unlike in bacteria, where replication initiates from a single replication origin, multiple origins are utilized in the eukaryotic genomes. Among them, the ORIs in budding yeast Saccharomyces cerevisiae and the fission yeast Schizosaccharomyces pombe have been best characterized. In recent years, advances in DNA microarray and next-generation sequencing technologies have increased the number of yeast species involved in ORIs research dramatically. The ORIs in some non-conventional yeast species such as Kluyveromyces lactis and Pichia pastoris have also been genome-widely identified. Relevant databases of replication origins in yeast were constructed, then the comparative genomic analysis can be carried out. Here, we review several experimental approaches that have been used to map replication origins in yeast and some of the available web resources related to yeast ORIs. We also discuss the sequence characteristics and chromosome structures of ORIs in the four yeast species, which can be utilized to improve yeast replication origins prediction.
Introduction
DNA replication is one of the crucial steps for cell cycle. During cell division, accurate and complete duplication of the genome is required to ensure the faithful inheritance of genetic information from one cell generation to the next. To be duplicated, a DNA double helix must be opened at defined sites, termed DNA replication origins (MacAlpine and Bell, 2005; Mechali, 2010; Schepers and Papior, 2010). In general terms, the number of origins (ORIs) in a genome is bound up with the size of the chromosome. Bacterial genomes frequently have a single replication origin, because they usually consist of a small circular chromosome (Gao and Zhang, 2007; Gao et al., 2013; Leonard and Mechali, 2013). In contrast, eukaryotic DNA replication initiates at multiple origins due to their enormous genomic information and the complexity of their chromosome structures (Mechali, 2010). Budding yeast Saccharomyces cerevisiae and the fission yeast Schizosaccharomyces pombe have the best characterized ORIs in eukaryotes. In S. cerevisiae, origin selection is mediated by the formation of a multi-protein complex termed the pre-replicative complex (pre-RC), whose activation leads to DNA unwinding and the assembly of replisomes to carry out DNA synthesis (Bell and Dutta, 2002). Proteins required for pre-RC formation include the origin recognition complex (ORC), the pre-RC assembly factors Cdc6 and Cdt1 and the putative replicative DNA helicase, the MCM2-7 complex (Bell, 2002; Bowers et al., 2004).
Recent advances in DNA microarray technology and next-generation sequencing technologies have brought a dramatic increase in the number of ORIs identified in eukaryotic genomes, such as human (Cadoret et al., 2008; Karnani et al., 2010), mouse (Sequeira-Mendes et al., 2009; Cayrou et al., 2011), Arabidopsis thaliana (Costas et al., 2011), and Drosophila melanogaster (Cayrou et al., 2011; Gao et al., 2012). The ORIs in some non-conventional yeast species such as Kluyveromyces lactis (Liachko et al., 2010) and Pichia pastoris (Liachko et al., 2014) have also been genome-widely identified. Because of the increasing data of eukaryotic ORIs, some secondary databases with comprehensive and intuitive ORIs’ information have been constructed. In this review, we summarize several experimental approaches that have been used to identify replication origins in yeast and list some available web resources relevant to yeast ORIs. In addition, we also discuss the characteristics of ORIs in the four yeast species based on the sequence data in the Database of Eukaryotic ORIs (DeOri), including the significant motifs found by the MEME-ChIP web service, the chromosome structures of ORIs, and the origin replication timing and efficiency features.
Experimental Methods to Identify Yeast Replication Origins
Primal efforts to identify origins across an entire chromosome were two-dimensional gel agarose electrophoresis, which utilized the fact that non-linear DNA molecule does not migrate in gels at the same rate as a linear molecule of equal mass (Bell and Byers, 1983; Brewer and Fangman, 1987). Partially unwound DNA are likely to form only in the vicinity of replication origins, and such structures can be mapped by virtue of being branched. For the relatively low throughput of two-dimensional gel agarose electrophoresis, just a small set of activity origins in the smallest chromosomes in S. cerevisiae were located by this method (Reynolds et al., 1989; Newlon et al., 1993; Friedman et al., 1997; Besnard et al., 2014).
To comprehensively identify the location of origins and characterize the ORIs, microarray-based approaches were developed. The combination of fluorescently labeled DNA and microarray representing all the yeast open reading frames (ORFs) can reveal the replicating details of the DNA sequence. Even though they are time consuming and the resolution may not be ideal, these studies make it possible to locate ORIs genome-widely.
There are three widely used microarray-based techniques. (a) By generating a replication timing profile and taking advantage of the fact that ORIs replicate earlier than its neighboring sequences. Methods to differentiate replicated from non-replicated DNA in the progression of DNA replication are diversiform. Both density transfer approach by isotopically labeling of DNA (heavy : light study) and copy number approach by monitoring the change of copy number (Raghuraman et al., 2001; Yabuki et al., 2002; Heichinger et al., 2006) were involved. (b) By identifying pre-replicative complexes at origins of replication using chromatin immunoprecipitation (ChIP). The genome-wide identification of ORC- and MCM-bound sites can reveal the locations of DNA replication origins (Wyrick et al., 2001; Nieduszynski et al., 2006; Xu et al., 2006; Hayashi et al., 2007). (c) By measuring the accumulation of single-stranded DNA (ssDNA) in the presence of a replication-impeding drug, hydroxyurea (HU). This technique makes use of the observation that ssDNA formation is restricted to origins of replication in the checkpoint-deficient mutant rad53 (Feng et al., 2006; Masai et al., 2010).
In recent years, the next-generation sequencing technology has also been combined into replication origins identifying methods. Sequencing of replication intermediates or direct sequencing of short, newly replicated DNA strands can help locate replication origins. Compared with microarray-based approaches, deep-sequencing-based approach is characterized by high efficiency, low cost and high resolution. Some methods can even define replication origin sequences throughout the genome with single-nucleotide resolution. On the other hand, next-generation sequencing technologies exhibit coverage biases, which should be avoided to ensure the accuracy of whole-genome origin maps (Besnard et al., 2014).
ChIP-seq, ChIP followed by direct high-throughput sequencing, is the most representative application (Kharchenko et al., 2008). Xu et al. (2012) identified ORIs in three distantly related fission yeasts, Schizosaccharomyces pombe, Schizosaccharomyces octosporus, and Schizosaccharomyces japonicas at high resolution with a generally applicable deep-sequencing-based approach. They counted the frequency of each region of the genome in S-phase arrested cells by deep sequencing, then produced replication timing profiles by mapping all the sites with increased DNA copy number (Xu et al., 2012). Autonomously replicating sequences ARS-seq followed with miniARS-seq is another sequencing-based method. The most recently updated ORIs in S. cerevisiae and the firstly reported ORIs in P. pastoris are identified with this method (Liachko et al., 2013, 2014). We take P. pastoris for instance here to represent the operation steps of this technique. Liachko et al. (2014) firstly constructed a ∼15 × library of genomic DNA in a non-replicating URA3 shuttle vector, then screened for ARS activity. ARS inserts were amplified by vector-specific Illumina primers and sequenced by paired-end deep sequencing. Short subfragments of ARSs isolated from the initial ARS-seq screen were then constructed as an input library for a follow-up ARS screen. The subsequent usage of miniARS-seq generated a high-resolution map of ARS sites in the P. pastoris genome (Liachko et al., 2014).
In Figure 1, we present DNA replication data from different experimental approaches of chromosome 1 in S. cerevisiae. The data of microarray-based techniques including heavy : light study, copy number study, ORC-ChIP, and MCM-ChIP, as well as ssDNA in HU study were downloaded from the DNA replication origin database OriDB (Nieduszynski et al., 2007). We also mark the ORIs identified by ARS-seq method on the figure (Liachko et al., 2013). Obvious overlaps exist among the different groups of data.
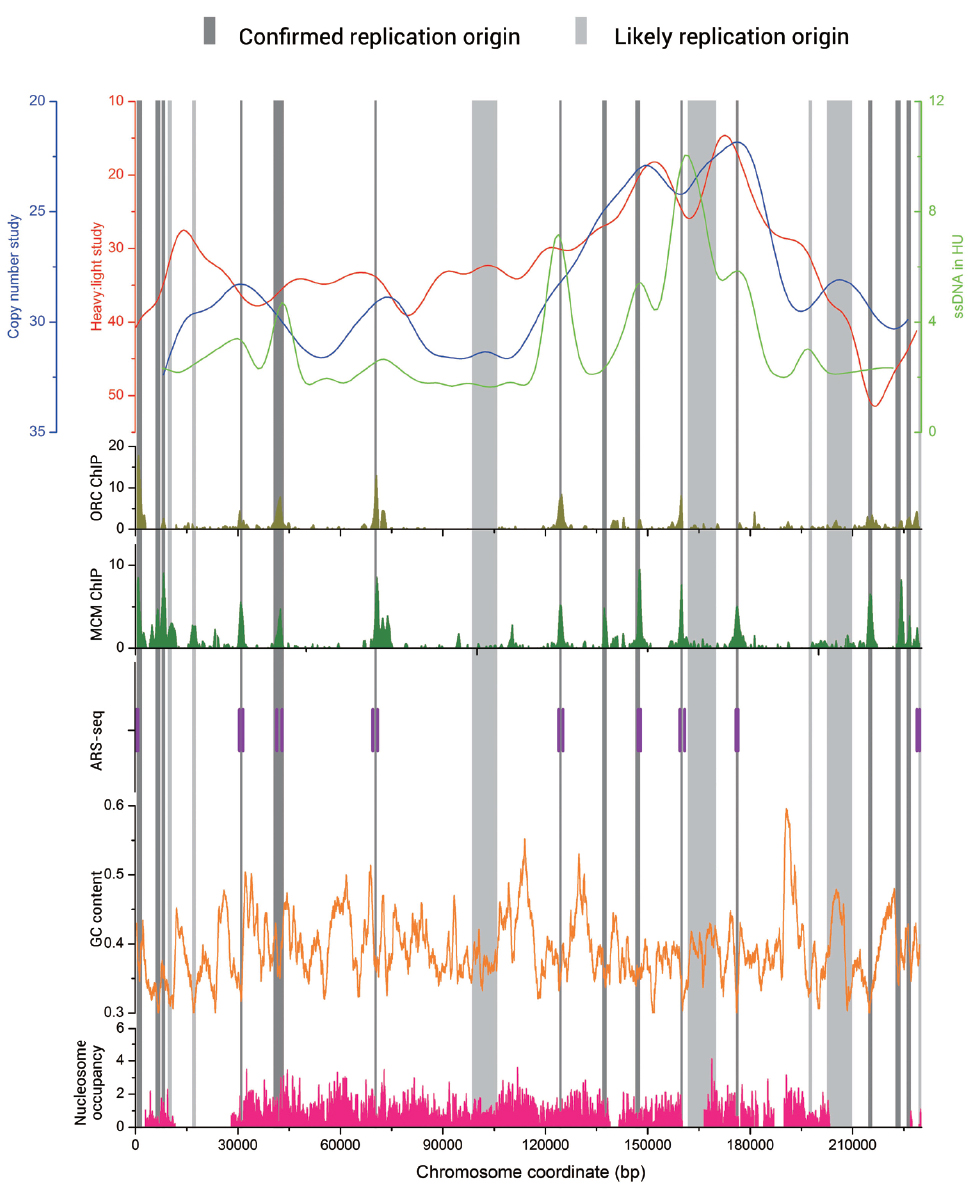
Figure 1. Graph view of genome-wide data relevant to the replication origins in Saccharomyces cerevisiae chromosome 1. The genome-wide data including heavy: light study (red line), copy number study (blue line), ssDNA in HU study (light green line) are visualized at the top of Figure. The bottom five plots show the genome-wide data of ORC-ChIP (olive bars), MCM-ChIP (green bars), ARS-seq (vertical purple bars), GC content (orange line), and nucleosome occupancy (pink bars), respectively. The replication origin sites are indicated by vertical bars (dark gray for confirmed and light gray for likely).
Databases Relevant to the Study of Yeast Replication Origins
Due to the increasing data of eukaryotic ORIs, developing repositories of these information became feasible and necessary. We list some of the available web resources relevant to DNA replication in yeast, and discuss their contents in this section.
OriDB1 is the most widely used database of DNA replication origins, which is limited to budding yeast (S. cerevisiae) and fission yeast (S. pombe) by present. The data of S. cerevisiae replication origins in OriDB was collated from four microarray-based studies, each of which separately mapped the approximate location of ORIs throughout the yeast genome, and the fifth study that used analysis of phylogenetic conservation and provided another list of origin sites. After amalgamating the data of each study, OriDB produced an integrated list of origin sites. Each proposed origin site is assigned a status (confirmed, likely, or dubious) that indicates the assurance of the site genuinely corresponding to an origin. In 2012, origin sites from S. pombe were collected. OriDB provides lots of assistance to researchers working in the DNA replication field because it brings together comprehensive information which was difficult to access and compare (Nieduszynski et al., 2007; Siow et al., 2012).
DeOri2 was constructed in the year of 2012 and has been updated constantly. When the original version was constructed, DeOri contained replication origins from six eukaryotic organisms. Now the entries have been increased to 173,988 ORIs from eight eukaryotic organisms, including human, mouse, A. thaliana, D. melanogaster, K. lactis, S. pombe, P. pastoris, and S. cerevisiae. We have filtered the replication origin data in the four yeasts for the following sequence analyzing. This database aims to contribute in the comparative genomic analysis of replication origins, and provides some insights into the nature of replication origins on a genome scale (Gao et al., 2012).
DNAReplication3 is a database aimed to provide information and resources for the eukaryotic DNA replication community. Organism-sorted data on replication proteins are presented in this database, and are summarized in the categories of nomenclature, biochemical properties, motifs, interactions, modifications, structure, cell localization and expression, and general comments. Users are also provided with links to recent replication papers, other useful replication websites, and homepages of replication labs. All these functions make this database a valuable tool for the study of eukaryotic DNA replication (Cotterill and Kearsey, 2009).
ReplicationDomain4 is a comparative web-based database for storing, sharing and visualizing DNA replication timing data. Other genome-wide chromatin features as well as comparative information of transcriptional expression are also provided in this database. Replication Domain is also a valuable resource for the scientific community because users not only can download the publicly available microarray data, but also are allowed to upload their own data sets and share them with colleagues prior to providing public access (Weddington et al., 2008).
SGD (Saccharomyces Genome Database, available at http://www.yeastgenome.org/) is a genomic resource of the budding yeast S. cerevisiae. The highest-quality comprehensive information, including the complete S. cerevisiae reference genome DNA sequence, its genes and their products, the phenotypes of its mutants, and the literatures supporting these data, are provided in the SGD project (Cherry et al., 2012). ARSs mentioned in peer-reviewed literatures are also integrated in this database. For each ARS, the details about its sequence, location, relative literatures, and history can be obtained. Users can also use the analysis tools such as BLAST provided in SGD to explore these data.
Sequence Characteristics of Yeast Replication Origins
In budding yeast S. cerevisiae, replication origins are defined as ARS because they can support the maintenance of a plasmid in growing yeast cells (Stinchcomb et al., 1979). Every replication origin contains a conserved 11-bp motif (sometimes assigned as 17 bp in length) called the ARS consensus sequence (ACS) that is essential for the binding of the initiator protein ORC (Rao and Stillman, 1995; Rowley et al., 1995; Theis and Newlon, 1997). A match to the ACS is essential but not sufficient for origin function. Even though, some bioinformatic algorithms for predicting the location of yeast replication origins have been developed based on ACS. For example, to predict the location of ORIs in the S. cerevisiae genome, Breier et al. (2004) developed an algorithm called Oriscan. This method utilized 268 bp of sequence, including the T-rich ACS and a 3′ A-rich region to identify ORI candidates. It then ranked potential origins by their likelihood of activity. A large proportion of origins in the genome were recognized by Oriscan with near-perfect specificity (Breier et al., 2004). Another computational study made use of the discovery that most replication origin sequences are phylogenetically conserved among closely related Saccharomyces species. It combined motif searches, phylogenetic conservation, and microarray data together to identify replication origin sequences throughout the S. cerevisiae genome (Nieduszynski et al., 2006). Analogously, the ORIs in K. lactis also contain a 50-bp ACS. The difference is that ACS in K. lactis ARSs is both necessary and largely sufficient for ARS activity (Liachko et al., 2010).
Abundant research was also conducted on the replication origins in fission yeast S. pombe, where replication sequences also function as autonomous replicators. However, ORIs in S. pombe do not have recognizable consensus elements but have a 500–1000 bp extended AT-rich structure (Dubey et al., 1994; Clyne and Kelly, 1995). Segurado et al. (2003) identified 384 potential origins by this feature. It was previously believed that replication origins in plant and metazoan are G/C-rich while in yeasts are A/T-rich. However, an industrially important methylotrophic budding yeast, P. pastoris, owed different characteristics in its ORIs compared with other studied yeasts. In this kind of yeast, two different types of ORIs exist simultaneously. In addition to an A/T-rich type more reminiscent of typical budding and fission yeast origins, there is also a G/C-rich type of replication origins associated with transcription start sites (Liachko et al., 2014). We calculate the GC content along S. cerevisiae chromosome 1 with sliding window algorithm (window size: 1000, shift: 20) and present it in Figure 1 by the orange line. This line indicates that GC contents of the ORIs sequences are significantly lower than those of the entire genome sequences. In fact, this status exists in all the four kinds of yeasts, even in P. pastoris, the one includes G/C-rich type of ORIs.
To gain a comprehensive view of the conserved motifs in the origin sequences, we use the MEME-ChIP web service to discovery enriched motifs in the ORI sequences in the four kinds of yeasts. MEME-ChIP web service is designed especially for discovering motifs in the large sets of short DNA sequences (Bailey et al., 2009; Machanick and Bailey, 2011). The motifs we found are displayed in Figure 2A. ORIs in S. cerevisiae, K. lactis, and S. pombe contain AT-rich motifs, whereas GC-rich motifs are found in P. pastoris ORIs. We also construct the phylogenetic tree (Figure 2A) of the four organisms based on the cytochrome c downloaded from NCBI. The tree was constructed using the MEGA6 program (Statistical Method: Maximum Likelihood, Test of Phylogeny: Bootstrap method, No. of Bootstrap Replications: 1000; Tamura et al., 2013). Conserved motifs found in the four yeasts ORIs show no significant correlation with their phylogenetic relationships.
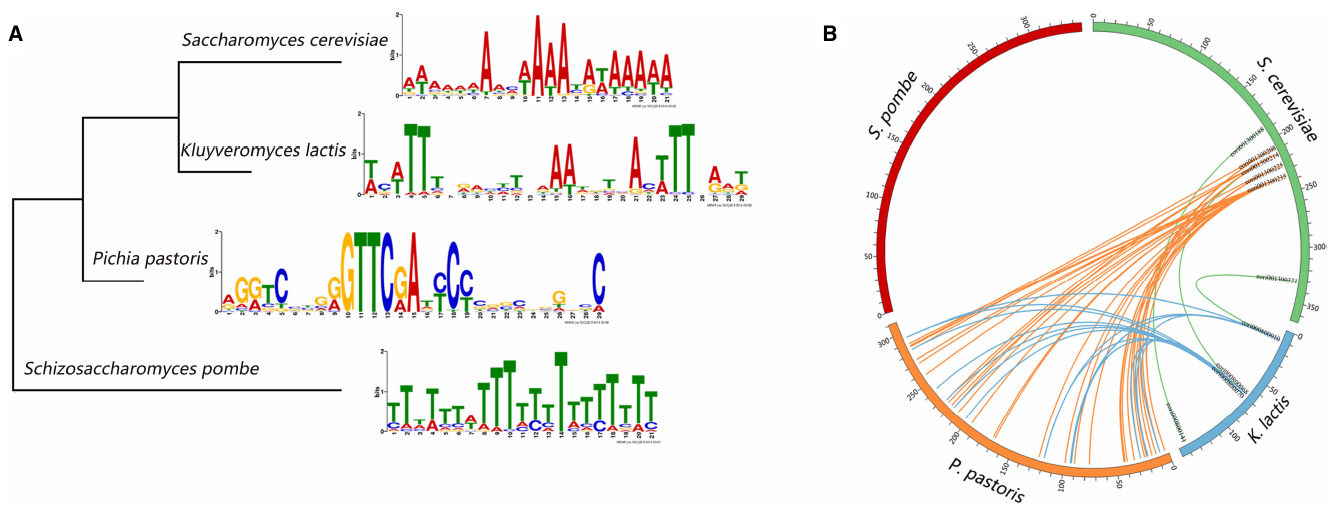
Figure 2. Sequence characteristics of yeast replication origins. (A) The significant motifs found in the replication origin sequences and the phylogenetic tree of the four yeasts. (B) The circos plot of replication origins that share similar sequences. Each number around the circle is the ORI’s serial number in DeOri.
In addition, regions of local similarity in sequences between each pair of organisms are searched by the BLAST program (Altschul et al., 1997). Figure 2B is created by circos (Krzywinski et al., 2009), and shows the ORIs that share similar sequences. Each number around the circle is the ORI’s serial number in DeOri. When two ORIs share similar local regions, a line will be drawn between them. For example, eori001300188, eori001300214, and eori001300331 have local regions similar with eori000800141, eori000800068, and eori000800010, respectively, hence the three pairs of ORIs are connected. No significant similarity is found between sequences in S. pombe ORIs and any other three groups of sequences. This may be caused by the large phylogenetic distance of S. pombe.
A new study suggests that in budding yeast, specific origin sequences are not strictly required for DNA replication in vitro, although they are essential for plasmid replication in vivo. The observation supports the notion that DNA replication specification in budding yeast is not completely dependent on DNA sequences, and epigenetic mechanisms are also important for determining replication origin sites (Gros et al., 2014).
Distribution and Organization of Yeast Replication Origins
Despite the lack of uniform feature of replication origin sequences, ORIs do not randomly locate on chromosome. Indeed, in all the four kinds of yeasts, origins have a significant preference for intergenic regions (Hayashi et al., 2007; Liachko et al., 2010, 2014; Renard-Guillet et al., 2014). We find that the correlation coefficient values (R values) between the chromosome length and replication origins number are 0.956, 0.999, 0.966, and 0.854 for S. cerevisiae, S. pombe, K. lactis, and P. pastoris, respectively, which indicates that longer chromosomes tend to have more ORIs. In addition, ORIs always appear in the nucleosome-free regions (Li et al., 2014; Sherstyuk et al., 2014). We collect the nucleosome occupancy data in S. cerevisiae chromosome 1 (Kaplan et al., 2009) and map it in Figure 1 by pink bars. The nucleosome occupancy scores in ORIs are significantly lower, which agrees well with the above conclusions. An asymmetric pattern of positioned nucleosomes has been verified at origins in both S. cerevisiae and K. lactis (Eaton et al., 2010; Tsai et al., 2014). These nucleosome occupancy information has been successfully used to train a machine learning algorithm to predict the position of active arm origins in the Candida albicans genome (Tsai et al., 2014).
Two other important features of ORIs are origin replication timing and efficiency. Origins are fired at various time throughout the S phase. S. cerevisiae ORIs can be separated into early and late origins. They present different nucleosomal architectures, which are already established in G1 phase. A higher occupancy of nucleosomes and broader nucleosome-depleted region (NDR) features appear in early origins, while late origins display a lower occupancy and tighter NDR (Soriano et al., 2014). In S. pombe, early and late origins tend to distribute separately in large chromosome regions (Hayashi et al., 2007). The dynamics of replication in P. pastoris shows an unexpected difference in replication timing between GC-ARSs and AT-ARSs. GC-rich ORIs replicate remarkably earlier and/or more efficiently than AT-rich ORIs (Liachko et al., 2014). In regard to origin replication efficiency, not all origins are used at each cell cycle. The overall efficiency of origin firing is less than 50% in S. cerevisiae and S. pombe (Friedman et al., 1997; Heichinger et al., 2006). It appears to be that the replication stress presented by different growth conditions affects the number of sites being activated (Tuduri et al., 2010). The flexibility of replication origins may be an obstacle in the thorough genome-wide understanding of ORIs in yeast.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Prof. Chun-Ting Zhang for the invaluable assistance and inspiring discussions. The present work was supported in part by National Natural Science Foundation of China (Grant Nos. 31171238 and 30800642), Program for New Century Excellent Talents in University (No. NCET-12-0396), and the China National 863 High-Tech Program (2015AA020101).
Footnotes
- ^ http://cerevisiae.oridb.org/
- ^ http://tubic.tju.edu.cn/deori/
- ^ http://www.dnareplication.net/
- ^ http://www.replicationdomain.org
References
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Bell, L., and Byers, B. (1983). Separation of branched from linear DNA by two-dimensional gel electrophoresis. Anal. Biochem. 130, 527–535. doi: 10.1016/0003-2697(83)90628-0
Bell, S. P. (2002). The origin recognition complex: from simple origins to complex functions. Genes Dev. 16, 659–672. doi: 10.1101/gad.969602
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bell, S. P., and Dutta, A. (2002). DNA replication in eukaryotic cells. Annu. Rev. Biochem. 71, 333–374. doi: 10.1146/annurev.biochem.71.110601.135425
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Besnard, E., Desprat, R., Ryan, M., Kahli, M., Aladjem, M. I., and Lemaitre, J.-M. (2014). Best practices for mapping replication origins in eukaryotic chromosomes. Curr. Protoc. Cell Biol. 64, 22.18.21–22.18.13. doi: 10.1002/0471143030.cb2218s64
Bowers, J. L., Randell, J. C. W., Chen, S. Y., and Bell, S. P. (2004). ATP hydrolysis by ORC catalyzes reiterative Mcm2-7 assembly at a defined origin of replication. Mol. Cell 16, 967–978. doi: 10.1016/j.molcel.2004.11.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Breier, A. M., Chatterji, S., and Cozzarelli, N. R. (2004). Prediction of Saccharomyces cerevisiae replication origins. Genome Biol. 5:R22. doi: 10.1186/gb-2004-5-4-r22
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brewer, B. J., and Fangman, W. L. (1987). The localization of replication origins on ARS plasmids in S. cerevisiae. Cell 51, 463–471. doi: 10.1016/0092-8674(87)90642-8
Cadoret, J. C., Meisch, F., Hassan-Zadeh, V., Luyten, I., Guillet, C., Duret, L., et al. (2008). Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proc. Natl. Acad. Sci. U.S.A. 105, 15837–15842. doi: 10.1073/pnas.0805208105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cayrou, C., Coulombe, P., Vigneron, A., Stanojcic, S., Ganier, O., Peiffer, I., et al. (2011). Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res. 21, 1438–1449. doi: 10.1101/gr.121830.111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cherry, J. M., Hong, E. L., Amundsen, C., Balakrishnan, R., Binkley, G., Chan, E. T., et al. (2012). Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 40, D700–D705. doi: 10.1093/nar/gkr1029
Clyne, R. K., and Kelly, T. J. (1995). Genetic analysis of an ARS element from the fission yeast Schizosaccharomyces pombe. EMBO J. 14, 6348–6357.
Costas, C., Sanchez, M. D., Stroud, H., Yu, Y., Oliveros, J. C., Feng, S., et al. (2011). Genome-wide mapping of Arabidopsis thaliana origins of DNA replication and their associated epigenetic marks. Nat. Struct. Mol. Biol. 18, 395–400. doi: 10.1038/nsmb.1988
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cotterill, S., and Kearsey, S. E. (2009). DNAReplication: a database of information and resources for the eukaryotic DNA replication community. Nucleic Acids Res. 37, D837–D839. doi: 10.1093/nar/gkn726
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dubey, D. D., Zhu, J., Carlson, D. L., Sharma, K., and Huberman, J. A. (1994). Three ARS elements contribute to the ura4 replication origin region in the fission yeast, Schizosaccharomyces pombe. EMBO J. 13, 3638–3647.
Eaton, M. L., Galani, K., Kang, S., Bell, S. P., and Macalpine, D. M. (2010). Conserved nucleosome positioning defines replication origins. Genes Dev. 24, 748–753. doi: 10.1101/gad.1913210
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Feng, W. Y., Collingwood, D., Boeck, M. E., Fox, L. A., Alvino, G. M., Fangman, W. L., et al. (2006). Genomic mapping of single-stranded DNA in hydroxyurea-challenged yeasts identifies origins of replication. Nat. Cell Biol. 8, 148–155. doi: 10.1038/ncb1358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Friedman, K. L., Brewer, B. J., and Fangman, W. L. (1997). Replication profile of Saccharomyces cerevisiae chromosome VI. Genes Cells 2, 667–678. doi: 10.1046/j.1365-2443.1997.1520350.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gao, F., Luo, H., and Zhang, C.-T. (2012). DeOri: a database of eukaryotic DNA replication origins. Bioinformatics 28, 1551–1552. doi: 10.1093/bioinformatics/bts151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gao, F., Luo, H., and Zhang, C.-T. (2013). DoriC 5.0: an updated database of oriC regions in both bacterial and archaeal genomes. Nucleic Acids Res. 41, D90–D93. doi: 10.1093/nar/gks990
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gao, F., and Zhang, C. T. (2007). DoriC: a database of oriC regions in bacterial genomes. Bioinformatics 23, 1866–1867. doi: 10.1093/bioinformatics/btm255
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gros, J., Devbhandari, S., and Remus, D. (2014). Origin plasticity during budding yeast DNA replication in vitro. EMBO J. 33, 621–636. doi: 10.1002/embj.201387278
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hayashi, M., Katou, Y., Itoh, T., Tazumi, M., Yamada, Y., Takahashi, T., et al. (2007). Genome-wide localization of pre-RC sites and identification of replication origins in fission yeast. EMBO J. 26, 1327–1339. doi: 10.1038/sj.emboj.7601585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heichinger, C., Penkett, C. J., Bähler, J., and Nurse, P. (2006). Genome-wide characterization of fission yeast DNA replication origins. EMBO J. 25, 5171–5179. doi: 10.1038/sj.emboj.7601390
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaplan, N., Moore, I. K., Fondufe-Mittendorf, Y., Gossett, A. J., Tillo, D., Field, Y., et al. (2009). The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458, 362–366. doi: 10.1038/nature07667
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Karnani, N., Taylor, C. M., Malhotra, A., and Dutta, A. (2010). Genomic study of replication initiation in human chromosomes reveals the influence of transcription regulation and chromatin structure on origin selection. Mol. Biol. Cell 21, 393–404. doi: 10.1091/mbc.E09-08-0707
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kharchenko, P. V., Tolstorukov, M. Y., and Park, P. J. (2008). Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat. Biotechnol. 26, 1351–1359. doi: 10.1038/nbt.1508
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leonard, A. C., and Mechali, M. (2013). DNA replication origins. Cold Spring Harb. Perspect. Biol. 5:a010116. doi: 10.1101/cshperspect.a010116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, W.-C., Zhong, Z.-J., Zhu, P.-P., Deng, E.-Z., Ding, H., Chen, W., et al. (2014). Sequence analysis of origins of replication in the Saccharomyces cerevisiae genomes. Front. Microbiology 5:574. doi: 10.3389/fmicb.2014.00574
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liachko, I., Bhaskar, A., Lee, C., Chung, S. C. C., Tye, B.-K., and Keich, U. (2010). A comprehensive genome-wide map of autonomously replicating sequences in a naive genome. PLoS Genet. 6:e1000946. doi: 10.1371/journal.pgen.1000946
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liachko, I., Youngblood, R. A., Keich, U., and Dunham, M. J. (2013). High-resolution mapping, characterization, and optimization of autonomously replicating sequences in yeast. Genome Res. 23, 698–704. doi: 10.1101/gr.144659.112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liachko, I., Youngblood, R. A., Tsui, K., Bubb, K. L., Queitsch, C., Raghuraman, M. K., et al. (2014). GC-rich DNA elements enable replication origin activity in the methylotrophic yeast Pichia pastoris. PLoS Genet. 10:e1004169. doi: 10.1371/journal.pgen.1004169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
MacAlpine, D., and Bell, S. (2005). A genomic view of eukaryotic DNA replication. Chromosome Res. 13, 309–326. doi: 10.1007/s10577-005-1508-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Machanick, P., and Bailey, T. L. (2011). MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697. doi: 10.1093/bioinformatics/btr189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Masai, H., Matsumoto, S., You, Z., Yoshizawa-Sugata, N., and Oda, M. (2010). Eukaryotic chromosome DNA replication: where, when, and how? Annu. Rev. Biochem. 79, 89–130. doi: 10.1146/annurev.biochem.052308.103205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mechali, M. (2010). Eukaryotic DNA replication origins: many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 11, 728–738. doi: 10.1038/nrm2976
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Newlon, C. S., Collins, I., Dershowitz, A., Deshpande, A. M., Greenfeder, S. A., Ong, L. Y., et al. (1993). Analysis of replication origin function on chromosome III of Saccharomyces cerevisiae. Cold. Spring Harb. Symp. Quant. Biol. 58, 415–423.
Nieduszynski, C. A., Hiraga, S., Ak, P., Benham, C. J., and Donaldson, A. D. (2007). OriDB: a DNA replication origin database. Nucleic Acids Res. 35, D40–D46. doi: 10.1093/nar/gkl758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nieduszynski, C. A., Knox, Y., and Donaldson, A. D. (2006). Genome-wide identification of replication origins in yeast by comparative genomics. Genes Dev. 20, 1874–1879. doi: 10.1101/gad.385306
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Raghuraman, M. K., Winzeler, E. A., Collingwood, D., Hunt, S., Wodicka, L., Conway, A., et al. (2001). Replication dynamics of the yeast genome. Science 294, 115–121. doi: 10.1126/science.294.5540.115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rao, H., and Stillman, B. (1995). The origin recognition complex interacts with a bipartite DNA binding site within yeast replicators. Proc. Natl. Acad. Sci. U.S.A. 92, 2224–2228. doi: 10.1073/pnas.92.6.2224
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Renard-Guillet, C., Kanoh, Y., Shirahige, K., and Masai, H. (2014). Temporal and spatial regulation of eukaryotic DNA replication: from regulated initiation to genome-scale timing program. Semin. Cell Dev. Biol. 30, 110–120. doi: 10.1016/j.semcdb.2014.04.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reynolds, A. E., Mccarroll, R. M., Newlon, C. S., and Fangman, W. L. (1989). Time of replication of ARS elements along yeast chromosome III. Mol. Cell. Biol. 9, 4488–4494.
Rowley, A., Cocker, J. H., Harwood, J., and Diffley, J. F. (1995). Initiation complex assembly at budding yeast replication origins begins with the recognition of a bipartite sequence by limiting amounts of the initiator, ORC. EMBO J. 14, 2631–2641.
Schepers, A., and Papior, P. (2010). Why are we where we are? Understanding replication origins and initiation sites in eukaryotes using ChIP-approaches. Chromosome Res. 18, 63–77. doi: 10.1007/s10577-009-9087-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Segurado, M., De Luis, A., and Antequera, F. (2003). Genome-wide distribution of DNA replication origins at A+ T-rich islands in Schizosaccharomyces pombe. EMBO Rep. 4, 1048–1053. doi: 10.1038/sj.embor.7400008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sequeira-Mendes, J., Diaz-Uriarte, R., Apedaile, A., Huntley, D., Brockdorff, N., and Gomez, M. (2009). Transcription initiation activity sets replication origin efficiency in mammalian cells. PLoS Genet. 5:e1000446. doi: 10.1371/journal.pgen.1000446
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sherstyuk, V. V., Shevchenko, A. I., and Zakian, S. M. (2014). Epigenetic landscape for initiation of DNA replication. Chromosoma 123, 183–199. doi: 10.1007/s00412-013-0448-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Siow, C. C., Nieduszynska, S. R., Muller, C. A., and Nieduszynski, C. A. (2012). OriDB, the DNA replication origin database updated and extended. Nucleic Acids Res. 40, D682–D686. doi: 10.1093/nar/gkr1091
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Soriano, I., Morafraile, E. C., Vazquez, E., Antequera, F., and Segurado, M. (2014). Different nucleosomal architectures at early and late replicating origins in Saccharomyces cerevisiae. BMC Genomics 15:791. doi: 10.1186/1471-2164-15-791
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stinchcomb, D., Struhl, K., and Davis, R. (1979). Isolation and characterisation of a yeast chromosomal replicator. Nature 282, 39–43. doi: 10.1038/282039a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Theis, J. F., and Newlon, C. S. (1997). The ARS309 chromosomal replicator of Saccharomyces cerevisiae depends on an exceptional ARS consensus sequence. Proc. Natl. Acad. Sci. U.S.A. 94, 10786–10791. doi: 10.1073/pnas.94.20.10786
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsai, H.-J., Baller, J. A., Liachko, I., Koren, A., Burrack, L. S., Hickman, M. A., et al. (2014). Origin replication complex binding, nucleosome depletion patterns, and a primary sequence motif can predict origins of replication in a genome with epigenetic centromeres. mBio 5, e01703–e01714. doi: 10.1128/mBio.01703-14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tuduri, S., Tourriere, H., and Pasero, P. (2010). Defining replication origin efficiency using DNA fiber assays. Chromosome Res. 18, 91–102. doi: 10.1007/s10577-009-9098-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weddington, N., Stuy, A., Hiratani, I., Ryba, T., Yokochi, T., and Gilbert, D. M. (2008). ReplicationDomain: a visualization tool and comparative database for genome-wide replication timing data. BMC Bioinformatics 9:530. doi: 10.1186/1471-2105-9-530
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wyrick, J. J., Aparicio, J. G., Chen, T., Barnett, J. D., Jennings, E. G., Young, R. A., et al. (2001). Genome-wide distribution of ORC and MCM proteins in S. cerevisiae: high-resolution mapping of replication origins. Science 294, 2357–2360. doi: 10.1126/science.1066101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, J., Yanagisawa, Y., Tsankov, A. M., Hart, C., Aoki, K., Kommajosyula, N., et al. (2012). Genome-wide identification and characterization of replication origins by deep sequencing. Genome Biol. 13:R27. doi: 10.1186/gb-2012-13-4-r27
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, W., Aparicio, J. G., Aparicio, O. M., and Tavaré, S. (2006). Genome-wide mapping of ORC and Mcm2p binding sites on tiling arrays and identification of essential ARS consensus sequences in S. cerevisiae. BMC Genomics 7:276. doi: 10.1186/1471-2164-7-276
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yabuki, N., Terashima, H., and Kitada, K. (2002). Mapping of early firing origins on a replication profile of budding yeast. Genes Cells 7, 781–789. doi: 10.1046/j.1365-2443.2002.00559.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: DNA replication, replication origin, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Kluyveromyces lactis, Pichia pastoris
Citation: Peng C, Luo H, Zhang X and Gao F (2015) Recent advances in the genome-wide study of DNA replication origins in yeast. Front. Microbiol. 6:117. doi: 10.3389/fmicb.2015.00117
Received: 08 December 2014; Accepted: 29 January 2015;
Published online: 19 February 2015.
Edited by:
John R. Battista, Louisiana State University and Agricultural and Mechanical College, USAReviewed by:
Suleyman Yildirim, Istanbul Medipol University, TurkeyAbd El-Latif Hesham, Assiut University, Egypt
Copyright © 2015 Peng, Luo, Zhang and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Gao, Department of Physics, Tianjin University, No. 92 Weijin Road, Nankai District, Tianjin 300072, China e-mail:Zmdhb0B0anUuZWR1LmNu