 Markus Gressler1
Markus Gressler1 Matthias Brock
Matthias Brock- 1Microbial Biochemistry and Physiology, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knoell Institute, Jena, Germany
- 2Molecular and Applied Microbiology, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knoell Institute, Jena, Germany
- 3Institute for Microbiology, Friedrich Schiller University, Jena, Germany
Recently, the Aspergillus terreus terrein gene cluster was identified and selected for development of a new heterologous expression system. The cluster encodes the specific transcription factor TerR that is indispensable for terrein cluster induction. To identify TerR binding sites, different recombinant versions of the TerR DNA-binding domain were analyzed for specific motif recognition. The high affinity consensus motif TCGGHHWYHCGGH was identified from genes required for terrein production and binding site mutations confirmed their essential contribution to gene expression in A. terreus. A combination of TerR with its terA target promoter was tested as recombinant expression system in the heterologous host Aspergillus niger. TerR mediated target promoter activation was directly dependent on its transcription level. Therefore, terR was expressed under control of the regulatable amylase promoter PamyB and the resulting activation of the terA target promoter was compared with activation levels obtained from direct expression of reporters from the strong gpdA control promoter. Here, the coupled system outcompeted the direct expression system. When the coupled system was used for heterologous polyketide synthase expression high metabolite levels were produced. Additionally, expression of the Aspergillus nidulans polyketide synthase gene orsA revealed lecanoric acid rather than orsellinic acid as major polyketide synthase product. Domain swapping experiments assigned this depside formation from orsellinic acid to the OrsA thioesterase domain. These experiments confirm the suitability of the expression system especially for high-level metabolite production in heterologous hosts.
Introduction
Aspergillus terreus is a filamentous ascomycete of biotechnological and medical importance, since it produces the primary metabolite itaconic acid (Calam et al., 1939; Klement and Buchs, 2013) and the HMG-CoA reductase inhibitor lovastatin (Alberts et al., 1980; Hutchinson et al., 2000). Besides that, A. terreus can cause life-threatening invasive aspergillosis in immunocompromised patients (Slesiona et al., 2012b), which makes its use in biotechnological applications limited. In previous analyses we searched for secondary metabolite gene clusters that are involved in pigment formation of A. terreus conidia (Zaehle et al., 2014). This was of interest, since preliminary analyses suggested that the pigment in A. terreus differs from that found in other related Aspergillus species (Slesiona et al., 2012a). Coincidentally, we identified the gene cluster producing the metabolite terrein (Zaehle et al., 2014). Terrein is a metabolite with various biological activities, but its phytotoxic potential appears to be at least one of its natural functions and may increase competitiveness of A. terreus in the environment (Zaehle et al., 2014).
Interestingly, terrein (Figure 1A) is produced in large quantities, since short-term cultivation in simple potato dextrose broth resulted in more than 1 g of terrein per liter (Zaehle et al., 2014) and even higher yields have been described for cultivation under more optimized conditions (Xu et al., 2012; Yin et al., 2012). This implies that genes from the terrein gene cluster may be expressed at very high levels.
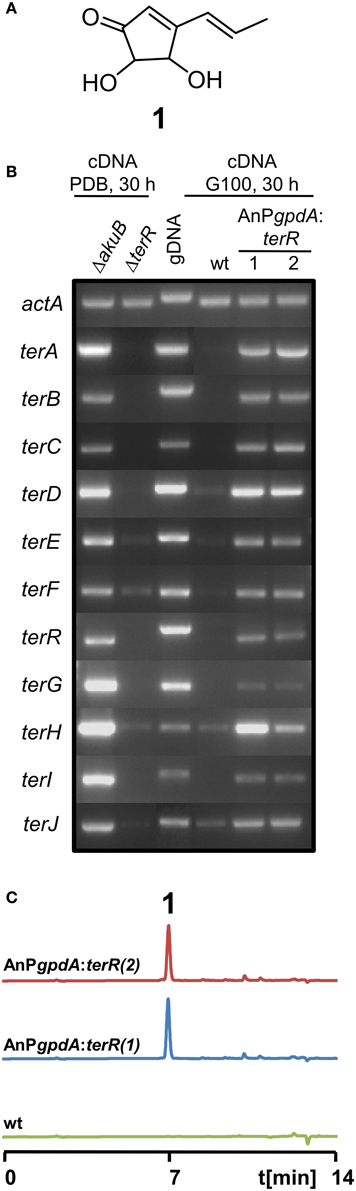
Figure 1. Analysis of terrein cluster induction in dependence of terR expression. (A) Chemical structure of terrein (1). (B) Semiquantitative RT-PCR on terrein cluster genes (terA—terJ) normalized against actA transcript levels. Under naturally inducing conditions (PDB) all genes from the cluster are expressed in A. terreus SBUG844ΔakuB (parental strain for gene deletions), but not in a ΔterR mutant. The wild-type SBUG844 shows no cluster expression under non-inducing conditions (G100), whereas strains constitutively expressing terR under the A. nidulans gpdA promoter (AnPgpdA:terR No. 1 and 2) induce the cluster. (C) HPLC analysis of G100 culture extracts from wild type (wt) and terR overexpressing strains AnPgpdA:terR No. 1 and 2. The metabolite peak for terrein is denoted by “1.”
The terrein cluster contains seven genes responsible for terrein biosynthesis (terA-F, terR), whereby terA encodes a non-reducing polyketide synthase (PKS) that produces polyketides of different chain length namely 2,3-dehydro-6-hydroxymellein (C10), orsellinic acid (C8), and 4-hydroxy-6-methylpyrone (C6). The subsequent formation of the cyclopentenoic structure of terrein is proposed to derive from an oxidative ring contraction of the isocoumarinic precursor 2,3-dehydro-6-hydroxymellein. While the terrein gene cluster contains further co-regulated genes, these genes might be dispensible (terH, terI), because deletion only reduces the final terrein production level or are involved in metabolite export (terG, terJ) (Zaehle et al., 2014). Importantly, the gene cluster encodes its own transcriptional activator at locus tag ATEG_00139, which is called TerR. TerR is a transcriptional activator with a GAL4-type Zn2Cys6 zinc binuclear cluster DNA-binding domain. This type of DNA-binding domain is very common in fungi and is the most common type of DNA-binding domains in transcriptional regulators of secondary metabolite gene clusters from filamentous fungi such as GliZ for gliotoxin biosynthesis from Aspergillus fumigatus (Bok et al., 2006), AflR for aflatoxin biosynthesis from Aspergillus nidulans and Aspergillus flavus (Yu et al., 1996) or ApdR for aspyridone biosynthesis from A. nidulans (Bergmann et al., 2007). A genomic deletion of terR resulted in complete loss of terrein production, suggesting that this transcription factor essentially contributes to cluster expression (Zaehle et al., 2014). However, neither the DNA binding sites recognized by TerR, nor the signals leading to TerR activation have been identified so far. However, if TerR is sufficient to drive expression of cluster genes, we assumed that a combination of TerR together with promoters from its cluster could enable the development of a new heterologous expression system.
Several filamentous fungi have been used for the recombinant production of proteins, among them especially fungi of industrial importance with GRAS status (generally regarded as safe) such as Aspergillus niger, Aspergillus oryzae, Trichoderma reesei, Acremonium chrysogenum, and Penicillium chrysogenum (Sharma et al., 2009). Several post-transcriptional bottlenecks have been reported to limit the production of proteins. These may consist of incorrect protein folding and subsequent degradation, low secretion efficiency, extracellular degradation or hyperglycosylation and several attempts have been made to overcome these limitations (Ward, 2012). However, filamentous fungi have not only been used for protein production, but are also used for the production of lignofuels or metabolic intermediates such as citric acid and the production of secondary metabolites such as antibiotics and other therapeutically useful compounds (Lubertozzi and Keasling, 2009). Despite specific limitations in all expression systems including the codon-adaptation of the target gene and RNA stability, the initial high-level expression of a target gene is the first key step for high production rates.
In general, a strong promoter is used to drive gene expression. In this respect, expression systems frequently rely on endogenous promoters from primary metabolism that are either constitutively active or can be regulated by applying specific inducing or repressing conditions. Examples are alcohol and aldehyde dehydrogenase (alcA; aldA), glucoamylase (glaA), (Taka) amylase (amyA; amyB), glyceraldehyde-3-phosphate dehydrogenase (gpdA), sucrase (sucA), acetamidase (amdS), endoxylanase (exlA), superoxide dismutase (sodA), or cellobiohydrolase I (cbhI) (Sharma et al., 2009; Fleissner and Dersch, 2010). Additionally, multiple integrations of an expression construct frequently increases the overall transcription of the target gene, but a linear increase is mainly limited to the first 5–6 copies (Verdoes et al., 1993). Due to these limitations, an inducible system with a promoter that is strongly activated and produces high transcript levels already in single-copy is generally favored.
Interestingly, although high secondary metabolite production rates have been described for several fungal species, information on the use of the involved regulatory elements to drive recombinant gene expression is limited. To analyze, whether elements from the A. terreus terrein cluster might be suitable for such a new recombinant expression system, we first analyzed, whether TerR is sufficient to drive terrein cluster expression and identified the respective DNA binding sites. With this knowledge, elements from the cluster were tested by reporter gene expression for their performance in the heterologous host A. niger. Finally, the expression system was used to heterologously produce secondary metabolites from A. terreus and A. nidulans in A. niger.
Materials and Methods
Strains and Culture Conditions
Strains used in this study are summarized in Table S1. For maintenance and during transformation procedures, all Aspergillus strains (A. nidulans wild type FGSC A4, A. terreus SBUG844 and its derivatives and all A. niger FGSC A1144 derivatives) were cultivated for 4 days at 37 or 30°C on solid Aspergillus minimal media (AMM) containing 2% agar (http://www.fgsc.net/methods/anidmed.html). When required, either hygromycin B (140 μg/ml, Carl Roth GmbH; Germany), pyrithiamine hydrobromide (0.1 μg/ml) or phleomycin (80 μg/ml) (both Sigma Aldrich; Germany) were added. Conidia were harvested in sterile water from solid media and filtered over 40 μm cell strainers (VWR; Germany). Liquid cultures were generally used in a 50 ml scale and were inoculated with 1 × 106 conidia per ml. Cultures were incubated at 30°C for 48–72 h as specified in the respective experiments. The following liquid media were used: AMM with 100 mM glucose (AMM-G100) or 1% casamino acids (AMM-CA1%), potato dextrose broth (PDB, Sigma Aldrich), and yeast/malt extract medium (YM, 5 g/l peptone, 3 g/l yeast extract, 3 g/l malt extract).
Bacterial Expression and Purification of TerR Polypeptides for SPR Analysis
All oligonucleotides used in this study are listed in Table S2. The gene sequences encoding for TerR residues 1–153 (TerR1−153), 43–138 (TerR43−138), and 35–138 (TerR35−138) were amplified by PCR from cDNA of A. terreus wild-type strain SBUG844 that was cultivated for 48 h at 30°C on PDB medium. The following oligonucleotides introducing 5′-NdeI and 3′-HindIII restriction sites were used: P1/2 for TerR1−153, P3/4 for TerR43−138, and P5/4 for TerR35−138. The fragments were cloned into the pET-29a vector (Novagen; Germany). TerR polypeptides were produced by autoinduction in E. coli Rosetta2 (DE3) cells grown at 26°C in 1·l Overnight Express Instant TB Medium (Novagen) in the presence of 1 mM Zn(OAc)2. Fifteen to twenty grams wet cells were collected by centrifugation, resuspended in 200 ml lysis buffer (20 mM HEPES, 150 mM NaCl, 10 μM Zn(OAc)2, 5 mM β-Mercaptoethanol, 1 mM AEBSF, pH 7.5) and disrupted using an Emulsiflex C5 high pressure homogenizer (Avestin; Germany). Cleared cellular extracts were loaded on a SP Sepharose HP (GE Healthcare; Germany) column and eluted with a salt gradient up to 1 M NaCl. Pooled fractions containing TerR1−153, TerR43−138, or TerR35−138 were adjusted to 150 mM NaCl and applied on a Cellufine Sulfate (Millipore; Germany) column that was equilibrated with 20 mM HEPES, 150 mM NaCl, 10 μM Zn(OAc)2, 5 mM β-Mercaptoethanol, pH 7.5, followed by elution with a gradient to 1 M NaCl. Peak fractions were concentrated with an Amicon Ultra-15 10K centrifugal filter device and purified to homogeneity by size exclusion chromatography on a Superdex 75 prep grade column (GE Healthcare) by using 20 mM HEPES, 150 mM NaCl, 10 μM Zn(OAc)2, pH 7.5 as running buffer. TerR proteins were stored in 50% v/v glycerol at −20°C. The absolute molecular mass of TerR35−138 was determined by static light scattering experiments on a miniDawn TREOS monitor in series with an Optilab T-rEX differential refractometer (Wyatt Technology Europe; Germany). TerR35−138 was chromatographed on a Superdex 200 10/300 GL column (GE Healthcare) and molar mass was calculated using ASTRA 6 software (Wyatt Technology Europe).
Surface Plasmon Resonance Measurements
Real-time analyses were performed on a Biacore 2000 system (GE Healthcare) at 25°C. DNA duplexes were produced by annealing complementary 18 bp oligonucleotides using a 5-fold molar excess of the non-biotinylated oligonucleotide. The dsDNA was injected on flow cells of a streptavidin (Sigma Aldrich)-coated CM3 sensor chip at a flow rate of 10 μl/min until the calculated amount of DNA that gives a maximum TerR binding capacity of 50 RU had been bound. TerR proteins were injected in running buffer (10 mM HEPES pH 7.4, containing 150 mM NaCl, 0.005% (v/v) surfactant P20, 5 mM β-Mercaptoethanol and 1 μM ZnCl2) at concentrations from 12.5 to 6400 nM. Sample injection and dissociation times were set to 60 and 120 s at a flow rate of 30 μl/min. Refractive index errors due to bulk solvent effects were corrected with responses from DNA-free flow cell 1 as well as subtracting blank injections. Kinetic raw data were processed and globally fitted with Scrubber 2.0c (BioLogic Software) using a 1:1 interaction model including a mass transport term.
Genetic Manipulation of A. niger and A. terreus
All strains generated in this study are listed in Table S1. The number of genomic integrations of all constructs generated in this study was determined by Southern blot analyses with digoxygenin labeled probes. Blots were developed by chemoluminescence imaging using CDP-star as recommended by the manufacturer (Roche; Germany). For details on specific strain constructions refer to supplementary experimental procedures. In brief, all A. terreus terR expression constructs were amplified from genomic DNA of A. terreus SBUG844. The constructs either contained the native terR promoter or the terR promoter was replaced by the A. nidulans gpdA or the A. oryzae amyB promoter. The terR terminator sequence was maintained in all constructs. Either the pyrithiamine (ptrA) or hygromycin B (hph) resistance cassette was used as a selectable marker in transformations of A. terreus SBUG844, A. niger FGSC A1144, or A. niger FGSC A1144_PterA:lacZ. Transformations were performed as previously described (Zaehle et al., 2014). For β-galactosidase producing reporter strains the lacZ gene from E. coli was fused with different promoters. The following promoters were used: The A. nidulans gpdA promoter, the A. terreus terA promoter in unidirectional (PterA) and bidirectional (PterA/B) orientation, the A. terreus terC promoter and the mutant versions PterCm1 and PterCm2, in which the putative TerR binding site “BS4” was exchanged or mutated. Fusion constructs were cloned into plasmids containing either the ptrA or hph resistance cassette and used for transformation A. terreus SBUG844, A. niger FGSC A1144, or A. niger FGSC A1144_PamyB:terR (P2 strain). For analysis of the bidirectional terA/B promoter a second reporter gene was required and a synthetic codon-optimized tdTomato gene coding for a red fluorescent protein was selected (gene accession KP100262). The lacZ and tdTomato genes were fused in both orientations with PterA/B and cloned in plasmids containing the hph resistance cassette. These reporter plasmids were used for transformation of A. niger FGSC A1144 or A. niger FGSC A1144_PamyB:terR (P2 strain).
Expression of terA and orsA in A. niger P2 Strain and Construction of the Expression Plasmid SM-Xpress
For expression of the A. terreus terA gene and the A. nidulans orsA gene in the A. niger P2 strain (containing the PamyB:terR construct), the terA promotor was either fused to the terA or orsA gene, whereas the respective natural terminator sequences were maintained. The fusion constructs were ligated into an hph containing vector and used for transformation of the A. niger P2 strain. To ease heterologous expression of polyketide synthases in the A. niger P2 strain the expression plasmid SM-Xpress was constructed. A plasmid containing the phleomycin (ble) resistance cassette (AnPgpdA:ble:trpCT) was linearized with EcoRI. A 786 bp fragment of the A. terreus terA promoter was amplified with oligonucleotides P6/7 and fused with a 363 bp trpC terminator (trpCT) from A. terreus (P8/9) and cloned into the EcoRV site of the linearized vector by in vitro recombination using the InFusion HD Cloning Kit (Clonetech laboratories; Germany). Thus, the resulting plasmid “SM-Xpress” contains a phleomycin resistance cassette as selection marker and the fusion of PterA and trpCT, which are separated by a NcoI site. This vector was used for domain swapping experiments of the A. nidulans orsA gene, in which the TE domain of OrsA was replaced by the TE domain from TerA. The orsA gene except its TE domain and the TE domain from terA were PCR amplified and fused with the NcoI restricted plasmid SM-Xpress via in vitro recombination. Plasmids were used for transformation of FGSC A1144_PamyB:terR (P2).
Metabolite Extraction
To analyze cultures for secondary metabolite production, culture broth was extracted with an equal volume of ethylacetate and the procedure was repeated once. Both fractions were combined and evaporated under reduced pressure. Evaporated residues were solved in 1 ml methanol and filtered. Standard metabolite analyses were performed on an Agilent 1100 series HPLC-DAD system coupled with a MSD trap (Agilent Technologies; Germany) operating in alternating ionization mode as previously described (Gressler et al., 2011).
Quantification of Orsellinic Acid
To quantify metabolite production levels from the direct and the coupled expression system orsellinic acid from the TerA PKS was selected. Metabolites were analyzed on an analytical Shimadzu HPLC system equipped with a DAD type SPD-M20A using a Zorbas Eclipse XDB C8 column (4.5 × 150 mm; 5 μm) with H2O + 0.1% formic acid (buffer A) and methanol (buffer B) as solvents. The following gradient was applied: 0–0.5 min = 10% B; 0.5–12 min from 10 to 90% B; 12–14 min from 90 to 100% B; 14–17 min = 100% B; 17–18 min from 100 to 10% B; 18–21 min = 10% B. A standard curve was generated using defined concentrations of purified orsellinic acid (range from 3.9 to 250 μg/ml). Cultures were cultivated for 48 h at 30°C and 200 rpm in AMM-G100+Gln50 liquid medium. Mycelium was collected for dry weight determination and culture supernatants were extracted twice with ethylacetate. Extracts were evaporated, solved in 1 ml methanol and filtered. Different dilutions from each sample in a total volume of 10 μl were loaded to the columns. Peak areas for orsellinic acid at 254 nm were quantified and production levels were calculated against the dry weight biomass.
RNA Isolation, cDNA Synthesis and Semiquantitative PCR
A. terreus SBUG844 and A. niger FGSC A1144 strains were cultivated for 30 or 48 h in PDB, AMM-G100, YM and AMM-CA1% media. Mycelia were ground under liquid nitrogen and RNA was isolated by the RiboPure RNA purification kit (Thermo Scientific; Germany). Residual genomic DNA was removed by the DNA-free kit (Thermo Scientific) and cDNA was synthesized by the Revert Aid Reverse Transcriptase (Thermo Scientific) using anchored oligo dT primers. Semiquantitative PCR was performed as described (Zaehle et al., 2014) previously using oligonucleotides P10–P35. Transcripts were normalized against the actin gene (actA, ATEG_06973) for A. terreus strains and against the glycerinaldehyde-3-phosphate dehydrogenase gene (gpdA, est_fge1_pm_C_70216) for A. niger strains.
Determination of β-Galactosidase Activity and tdTomato Fluorescence Intensity
β-Galactosidase activity was determined as previously described (Gressler et al., 2011). In brief, mycelia from A. niger strains containing lacZ fusion constructs were harvested from cultures grown for 48 h at 30°C in PDB, AMM, YM, or AMM-CA1% media. Mycelia were ground under liquid nitrogen and resuspended in 50 mM MOPS buffer (pH 7.5) with 2 mM MgCl2 and 10 mM β-mercaptoethanol. After centrifugation at 21,000 × g supernatants were used for determination of β-galactosidase activity using o-nitrophenyl-β-galactoside as substrate (Gressler et al., 2011). To detect the fluorescence intensity of A. niger transformants containing tdTomato fusion constructs, cell-free extracts were prepared as described above. After determination of protein concentrations by the Bradford assay (BioRad; Germany) protein concentrations were adjusted to 2.0, 1.0, and 0.5 mg/ml. 200 μl of the dilutions were transferred to a black flat-bottom 96 well plate (Nunc; Germany) and analyzed in a microplate reader (FLUOstar Omega, BMG Labtech; Germany). Plates were shaken for 1 min in double orbital mode and emission was detected at 590 nm using an excitation wavelength of 544 nm (50 flashes/well, top reading, gain 2000). The parental A. niger strains A1144 and P2 that contained no tdTomato gene served as negative controls and were subtracted from the fluorescence values of reporter strains. Specific activities were expressed as fluorescence units per mg protein. To calculate relative expression levels, fluorescence intensities (FI) of reporter strains were normalized against fluorescence intensities of an A. niger strain expressing the tdTomato gene under control of the A. nidulans gpdA promoter using the following formula: Relative fluorescence intensity (rFI) [%] = (FIreporter strain − FIP2)/(FIAnPgpdA − FIWT). All assays were performed in biological and technical triplicates from at least three individual transformants.
Accession Numbers
The DDBJ/EMBL/GenBank nucleotide sequence database accession number for the Aspergillus codon-optimized tdTomato gene reported in this paper is KP100262.
Results and Discussion
The Transcription Factor TerR is Essential for Expression of Terrein Cluster Genes
Previously, we have shown that A. terreus produces large quantities of the metabolite terrein (Figure 1A) and we discovered that the responsible gene cluster contains its own transcription factor TerR, which is encoded at locus tag ATEG_00139 (Zaehle et al., 2014). Gene deletion analysis revealed that TerR is indispensable for terrein production in A. terreus, since terR deletion resulted in complete loss of terrein production during cultivation on inducing potato dextrose broth (PDB) (Zaehle et al., 2014). To confirm that TerR is directly involved in the activation of the gene cluster, comparative semiquantitative RT-PCR analyses between A. terreus wild type and terR mutant (ΔterR) were performed. Under inducing conditions, all genes from the cluster spanning the region from terA – terJ (locus tags ATEG_00145—ATEG_00135) were actively transcribed in the wild type (Figure 1B). While all analyzed genes were strictly dependent on TerR for induction, terE, F and H showed some background expression in the ΔterR background. Thus, TerR seems to act as a transcriptional activator with special importance for transcription of the key polyketide synthase TerA, which shares a bi-directional promoter with the terB gene. Interestingly, among the 11 genes spanning the cluster, six of them (terA/B; terE/F, and terH/I) share bi-directional promoters, implying that either multiple putative TerR binding sites are found in these promoters or that single binding sites can activate expression in both directions. However, the control of gene expression from bi-directional promoters is frequently found in fungal secondary metabolite gene clusters. As examples, in the aflatoxin gene cluster from Aspergillus parasiticus 12 out of 25 genes share a bi-directional promoter (Yu et al., 2004) and in the gliotoxin gene cluster from Aspergillus fumigatus (Forseth et al., 2011) 8 out of 12 genes are controlled by shared promoter sequences. Furthermore, similar to our results on TerR-dependency for terrein cluster activation, the Zn2Cys6 binuclear transcription factor gliZ from the gliotoxin cluster is essential for activation of cluster genes (Bok et al., 2006).
TerR is Sufficient for Activation of Terrein Cluster Genes
In A. terreus terrein is only produced under specific environmental conditions, suggesting that transcription and activation of terR is itself under the control of other transcription factors that mediate environmental signals toward terrein production. Thus, to analyze, whether TerR production under non-inducing conditions is sufficient to stimulate terrein production in A. terreus, we aimed in the exchange of the native terR promoter against the constitutively active gpdA promoter from Aspergillus nidulans (Punt et al., 1990). The resulting PgpdA:terR fusion construct was used to transform an A. terreus wild-type strain. Indeed, when transformants were grown on complete glucose minimal medium, which is a repressive condition for terrein production in A. terreus, semiquantitative RT-PCR revealed induction of all cluster genes and high amounts of terrein were produced (Figures 1B,C). Thus, terR expression in A. terreus wild type is strictly dependent on specific inducing signals, but once expressed, TerR is sufficient to induce all genes required for terrein production. Similarly, the transcription factors ApdR (Bergmann et al., 2007), AflR (Liu and Chu, 1998), and GliZ (Bok et al., 2006) have been shown to induce expression of the respective cluster when overproduced in the homologous host. Therefore, our data indicate that promoter elements of the terrein cluster contain specific DNA binding sites that are recognized by TerR.
The TerR Zinc Binuclear Cluster DNA-Binding Domain has a Monomeric Structure
Since TerR was able to induce all genes required for terrein synthesis, we expected that conserved DNA binding sites are present in the promoter regions of genes spanning the terrein cluster. TerR belongs to the family of transcriptional activators with a GAL4-type Zn2Cys6 zinc binuclear cluster DNA-binding domain. This type of transcription factors is one of the most abundant transcriptional regulators present in fungi and involved in the regulation of primary and secondary metabolic processes (MacPherson et al., 2006). Their DNA-binding domain is generally located at the N-terminus of the protein and is sufficient to recognize its target sequence (Todd et al., 1998). Additionally, most GAL4-type transcription factors contain a coiled-coil domain near the N-terminus that is involved in dimerization and results in the recognition of consensus repeats (Fitzgerald et al., 2006). Interestingly, no coiled-coil domain is detected in the N-terminus of TerR, instead RADAR (Rapid Automatic Detection and Alignment of Repeats in protein sequences) analysis (Heger and Holm, 2000) detected an unusual amino acid repeat at positions 97–111 and 121–135 with unknown function. Thus, for analysis of the TerR subunit composition and identification of DNA-binding sites we first selected an N-terminal peptide of 153 amino acids (TerR1−153) that was cloned into an expression vector with or without N-terminal His-tag. Unfortunately, regardless the peptide version (tagged or untagged) and the purification procedure, we were unable to obtain a monodispersed protein fraction and MALDI-TOF MS/MS analysis showed co-purification of N- and C-terminal degradation products (Figure 2B). A similar phenomenon was observed during purification of the transcriptional activator AlcR of the ethanol utilization pathway in A. nidulans (Felenbok et al., 1988; Cahuzac et al., 2001). An AlcR1−197 fragment enclosing the DNA-binding domain could not be purified to homogeneity (Lenouvel et al., 1997). Therefore, we truncated the TerR domain to a TerR35−138 fragment (Figure 2A). This protein fragment was purified to >98% homogeneity as judged by SDS-gel electrophoresis (Figure 2B) and analytical size exclusion chromatography. Multiangel static light scattering analysis revealed a molar mass of 11.65 kDa (Figure 2C), demonstrating that the purified protein exclusively consisted of monomers in solution (theoretical molar mass of 11.35 kDa). Thus, besides the transcriptional activator AlcR, TerR seems to form another example of GAL4-type transcription factors with monomeric solution structure.
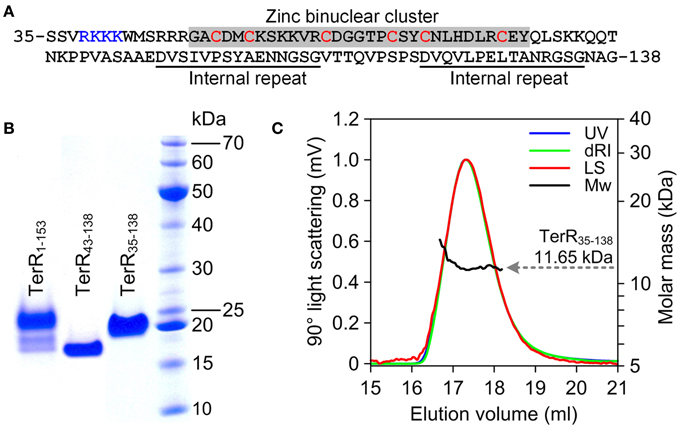
Figure 2. Characterization of recombinant TerR polypeptides used in this study. (A) Amino acid sequence of TerR35-138 used for SPR interaction analysis with specific DNA fragments. The N-terminal basic stretch at positions 38 to 47 and the zinc cluster region are highlighted. (B) SDS-PAGE analysis of purified TerR fragments as indicated at the specific lanes. (C) Analysis of oligomeric state of TerR35-138 in solution as determined via size exclusion chromatography and multiangle static light scattering. The light scattering signal (LS) is shown overlaid with the calculated molar mass (Mw) across the elution profile as monitored by the absorbance at 280 nm (UV) and changes of the refractive index (dRI).
TerR Recognizes CGG Direct Repeat Consensus Sequences in the Terrein Cluster
To identify putative TerR binding sites in the terrein gene cluster an in silico prediction of candidates constituting regulatory DNA motifs was performed on the intergenic regions of terA—terJ (Figure 3A) using the SCOPE motif finder suite (Chakravarty et al., 2007). A SPACER bipartite motif with the direct CGG half-site repeat consensus 5′-CGGHHHBNCGG-3′ (Figure 3Q) was identified that was present in all promoter regions of the cluster, with the exception of terG, terI, and terH, whereby the latter genes were dispensable for terrein production (Zaehle et al., 2014). Interestingly, the intergenic region that acts as a bidirectional promoter of terA and terB contained three putative binding sites that were annotated as BS1, BS2, and BS3 (Figure 3A). To analyze recognition of these binding sites by the TerR35−138 fragment, protein: DNA real-time surface plasmon resonance (SPR) biosensor interaction analyses were performed. For this purpose, oligonucleotides with a four-nucleotide overhang at the 5′-CGG half-site and a three-nucleotide overhang at the 3′-CGG half-site were deduced. Biotinylated oligonucleotides were hybridized with complementary unbiotinylated anti-strands and immobilized on streptavidin-coated CM3 sensor chips to give a maximum response (Rmax) of 50 resonance units (RU) when bound by a single monomeric TerR35−138 domain. Kinetic SPR binding responses of TerR35−138 to BS1-BS3 fitted with KD-values ranging between 0.5 and 0.7 μM. Interestingly, while steady state binding analysis showed that only one monomer binds to the BS1 sequence (Figure 3B, Table 1), BS2 and BS3 revealed that a significant fraction of DNA duplexes were bound by two monomers at these binding sites (Figures 3C,F).
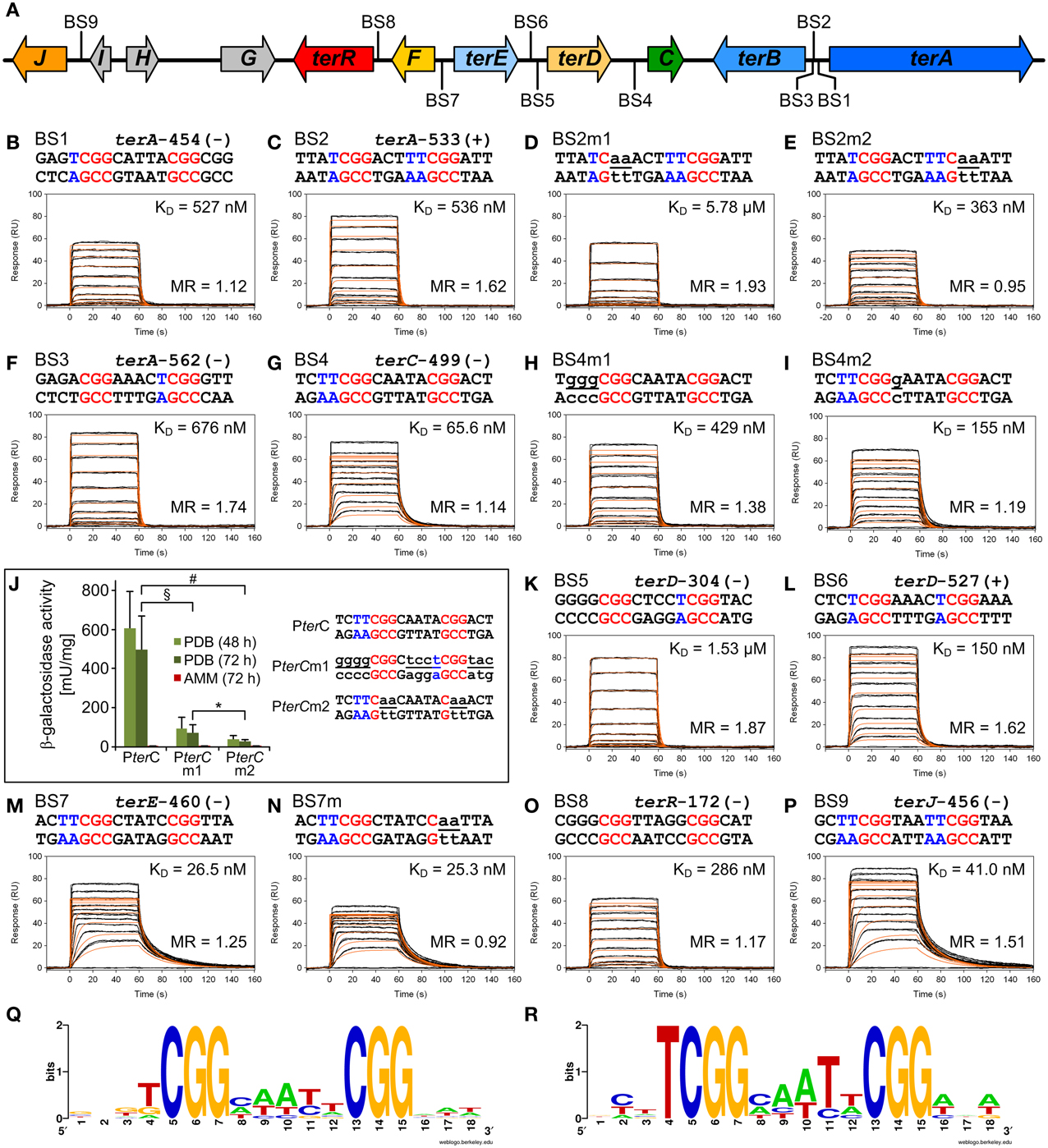
Figure 3. In vitro TerR binding to a consensus motif identified in promoters of the A. terreus terrein biosynthesis gene cluster and in vivo verification of in vitro data. (A) Schematic presentation of the terrein gene cluster. Intergenic positions of the consensus TerR-binding motif identified by the SCOPE motif finder suite (Q) are annotated with BS1–9. (B–I,K–P) Real-time in vitro SPR interaction analysis of TerR35-138 with DNA containing the predicted natural or mutated (denoted by the letter “m”) binding sites from promoters of terrein cluster genes. Sequences of DNA duplexes used for SPR analysis are shown on top of the sensorgrams. Numbers represent the CGG direct repeat motif positions relative to the start of the open reading frame. CGG half-sites and 5′-flanking thymidines are highlighted in red and blue. Substituted nucleotides relative to the wild-type sequence are underlined and shown in lowercase letters. TerR35-138 binding responses from triplicate injections of different concentrations (black lines) are overlaid with the best fit derived from a 1:1 interaction model including a mass transport term (red lines). Dissociation constants (KD) are plotted inside the sensorgrams. For details on the specific binding motifs refer to the main text. (J) In vivo verification of the PterC binding motif BS4. PterC:lacZ reporter strains were grown for 48 and 72 h on PDB or for 72 h on AMM and β-galactosidase activity was determined. Highest activity is observed with the native terC promoter (PterC), whereas activity is significantly reduced by replacement of the native binding site (BS4) against the low affinity BS5 from the terD promoter (PterCm1) or when the two CGG half-sites of BS4 are replaced by CAA triplets (PterCm2). No activity for any reporter is detected on AMM. Statistical significance (p < 0.01) was calculated by the student's unpaired t-test using data from at least three independent transformants measured in biological triplicates. Error bars represent SEM. §PterC vs. PterCm1; #PterC vs. PterCm2; *PterCm1 vs. PterCm2, all significances for PDB, but not AMM. (R) WebLogo sequence consensus motif of the experimentally mapped high affinity TerR binding sites BS1, BS2, BS4, BS6, BS7, and BS9.
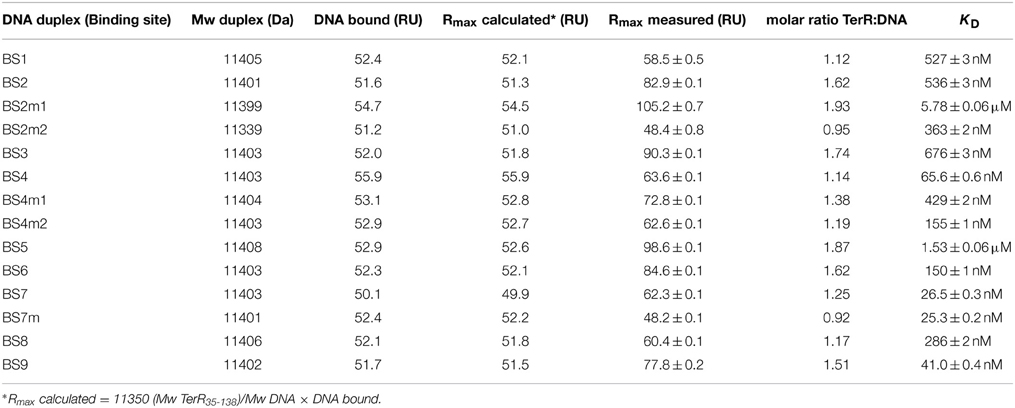
Table 1. Dissociation constants and stoichiometry of analyzed TerR35-138:DNA interactions.
To analyze the specificity for the predicted motif, BS2 was selected for mutations in the first and second CGG half-sites. Mutation of the 5′-CGG to CAA led to a predominant loss of TerR35−138 binding (Figure 3D). In contrast, mutation of the 3′-CGG to CAA altered the KD from 536 to 363 nM, but simultaneously reduced the number of TerR35−138 units that bind to the sequence (Figure 3E). While 1.6 monomers bound to the native sequence, only 0.95 monomers were found to bind at the mutated sequence. A similar phenomenon was observed when the second CGG half-site was mutated in BS7 (Figures 3M,N, Table 1). This implies that the 5′-CGG half-site is required for high affinity binding, whereas the 3′-CGG half-site attracts a second monomer to the motif. To confirm this assumption and to define a consensus sequence, we tested all other binding sites that were predicted by SCOPE using SPR analysis (Figure 3). Indeed, all predicted binding sites were recognized by TerR35−138, whereby KD values ranged from 26 nM to 1.5 μM with either a tendency to bind only one or two TerR35−138 fragments. In this respect, binding of a second monomer seems to be favored when the last nucleotide of the five base pair spacer is a thymidine (BS2, BS3, BS5, BS6, and BS9, Figure 3). In contrast, motifs, in which the spacer between the two CGG half-sites was reduced to four instead of five base pairs as well as motifs that harbor inverted CGG half-sites were not recognized by TerR (data not shown). We additionally observed that sequences that contained one or two “T” nucleotides in front of the 5′-CGG half-site had a tendency for high-affinity binding (BS1, BS2, BS4, BS6, BS7, BS9). To analyze the importance of these residues, we mutated the two “T” nucleotides in BS4 into “G” nucleotides (BS4m1; Figure 3H). Indeed, the affinity of the mutated binding site decreased by a factor of 6.5. In contrast, mutation of “C” into “G” directly behind the 5′-CGG half-site only showed a minor effect on affinity (Figure 3I).
A Basic Stretch at the TerR N-terminus Promotes High Affinity DNA Binding
The selected TerR35–138 fragment contained a basic stretch at positions 38–47 that is followed by the predicted DNA-binding domain. In this respect, TerR appears to possess similar features as the regulator of ethanol utilization AlcR from Aspergillus nidulans, which also recognizes DNA-binding sites in monomeric form as mentioned in Section “The TerR Zinc Binuclear Cluster DNA-Binding Domain has a Monomeric Structure.” For AlcR it has been shown that a basic sequence at the N-terminus is important for high affinity DNA binding, since it strengthens the interaction of the monomer with the phosphate backbone (Nikolaev et al., 1999). In order to elucidate the importance of this basic stretch in TerR, we generated a TerR43−138 fragment that lacked four basic amino acids present in the TerR35−138 fragment (Figure 2). When the TerR43−138 fragment was tested by SPR analysis on selected binding sites, a similar order of binding affinity was observed as for the TerR35−138 fragment. However, KD-values increased by a factor of 7–22, confirming that the basic stretch is essential for tight binding to the DNA phosphate backbone (Figure S2). Additionally, while some binding motifs showed the tendency to bind two TerR35−138 fragments, the TerR43−138 fragment exclusively recognized these motifs as a single monomer (Table 2). This implies that recognition of the second CGG half-site is not only dependent on the sequence motif, but also on the general affinity of the TerR fragment toward DNA binding.
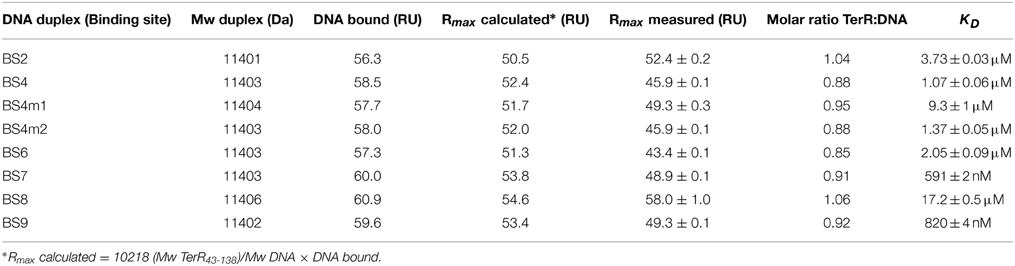
Table 2. Dissociation constants and stoichiometry of analyzed TerR35-138:DNA interactions.
Thus, comparison of the different features of the TerR fragments and sequence analyses lead to the following conclusions: (i) A TerR1−153 fragment is unstable in vitro, but the basic stretch at the N-terminus is required for high affinity DNA binding. (ii) The 5′-CGG half-site is of major importance for sequence recognition and binding. (iii) High affinity binding sites are characterized by a “T” before the 5′-CGG half-site. Similarly (iv) when the last nucleotide of the spacer is a “T,” there is a tendency for a second monomer to bind to the 3′-CGG half-site, but only in the presence of the basic amino acid stretch in the TerR domain. (v) The consensus sequence for TerR high affinity binding sites is 5′- TCGGHHWYHCGG –3′ (Figure 3R).
In vitro Binding Affinity of TerR35−138 is Reflected by in vivo Promoter Activity
The in vitro analyses of the TerR35−138 fragment showed that BS4 reflects a high affinity binding site with a KD of 66 nM, but tendency for single monomer binding (Figure 3G). BS4 is the only confirmed binding site in the intergenic region of terC (ATEG_00143, Figure 3A), which is no longer expressed when terR is deleted (Figure 1B). Thus, we were interested, whether the in vitro binding properties of the predicted binding sites reflect promoter activation in vivo. To address this question, we generated three reporter constructs. The first construct contained the native terC promoter fused with the E. coli lacZ gene (PterC:lacZ). For the second construct, BS4 from the terC promoter was exchanged with BS5 and also fused with the lacZ gene resulting in PterCm1:lacZ. BS5 is one of two binding sites in the intergenic region of terD (ATEG_00142), which showed a 23 times reduced TerR35−138 binding affinity in vitro, but a tendency for binding two monomers (Figure 3K). Finally, for the last construct, the two CGG half-sites of BS4 were replaced by CAA in PterC and fused to the lacZ gene resulting in PterCm2:lacZ. These mutations in both CGG half-sites were assumed to lead to a complete loss of binding site recognition. The A. terreus wild-type strain was transformed with the three constructs and several independent transformants with single genomic integrations were selected for analysis of β-galactosidase activity under inducing and non-inducing conditions (Figure 3J). As expected, none of the transformants revealed β-galactosidase activity when cells were grown under non-inducing conditions (AMM). In contrast, activity significantly increased, when cells were grown on PDB medium. Here, β-galactosidase activity was at least 6 times higher from strains containing the native BS4 site compared to strains containing the less affined BS5 site (PterCm1). Moreover, compared to the native promoter, the conversion of both CCG half-sites to CAA (PterCm2) reduced promoter activity by a factor 17. Thus, the binding sites predicted from in silico and in vitro analyses are indeed important for promoter activation by TerR and in vitro parameters reflect the in vivo situation.
TerR Activates the terA Promoter in the Heterologous Host A. niger, but Requires a Promoter Exchange
In order to generate a heterologous expression system using regulatory elements from the terrein gene cluster, we selected the filamentous fungus A. niger, which is frequently used in biotechnological applications (Sharma et al., 2009). Here, an analysis was required that tested the possibility of TerR mediated induction of terrein cluster promoters in the heterologous host. Thus, to analyze, whether TerR is sufficient to activate terA in A. niger, different β-galactosidase reporter strains were generated (Figure 4 and Figure S1). First, the promoter of terA (PterA), which contained the three DNA binding sites BS1-3 was fused with the E. coli lacZ gene and transferred to A. niger. After cultivation on different media, β-galactosidase activity hardly exceeded the background level, indicating that the terA promoter is not recognized by A. niger transcription factors that are present under the applied conditions (Figure 4A and Figure S3). Unfortunately, when terR under its native promoter was additionally introduced into the PterA:lacZ strain, again no β-galactosidase activity was detected (Figure S3). This indicated that either (i) TerR is not able to activate the terA promoter in the heterologous system or that (ii) the native promoter of TerR is not recognized in A. niger. To test these assumptions, we analyzed the expression of terR by semiquantitative PCR. Indeed, no terR transcript was detected, which confirms that specific regulatory elements from A. terreus are required to drive activation of terR expression (Figure S3). However, to show that TerR can also activate the terA promoter in a heterologous host, terR was fused with the constitutively active glyceraldehyde-3-phosphate dehydrogenase promoter (PgpdA) from A. nidulans and the resulting PgpdA:terR construct was introduced into the A. niger strain with the PterA:lacZ reporter. Indeed, a strong β-galactosidase activity was detected, which confirmed the specific recognition of terA promoter elements by TerR even in a heterologous system (Figure 4A). Additionally, β-galactosidase activity doubled in a strain that contained two copies of the PgpdA:terR constructs, indicating that TerR levels are the rate limiting step in terA promoter activation (Figure 4A). In conclusion, the native terR promoter is not stimulated and transcribed in A. niger, but when TerR is produced under control of an active promoter, this transcription factor is sufficient to stimulate expression of terA also in the heterologous system.
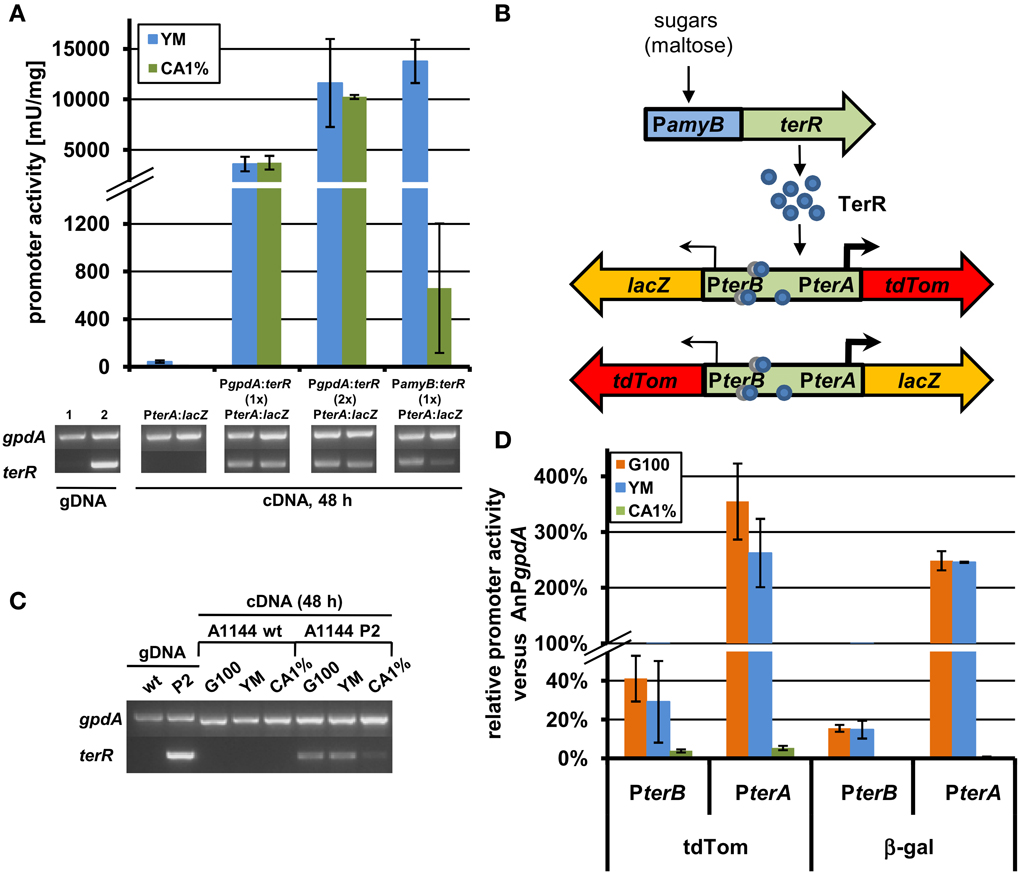
Figure 4. Analysis of recombinant terR and terA expression in A. niger by semiquantitative RT-PCR, LacZ, and tdTomato reporter activity. (A) Semiquantitative RT-PCR (lower panels) and β-galactosidase activity (upper panel) of A. niger A1144_PterA:lacZ either without terR or co-expressing the terR gene under control of the A. nidulans gpdA promoter (PgpdA:terR, PterA:lacZ; with one (1×) or two (2×) copies of PgpdA:terR) or the A. oryzae amyB promoter (PamyB:terR, PterA:lacZ). Expression of terA requires terR co-expression and is dependent on terR expression levels. A. niger gpdA was used as reference gene in semiquantitative RT-PCR analyses. Lane 1, control genomic DNA (gDNA) of A. niger PterA:lacZ; Lane 2, control gDNA of A. niger PamyB:terR, PterA:lacZ. (B) Scheme of the terR dependent expression system using the bi-directional promoter of terA and terB. After sugars induced expression of terR, the TerR protein (blue circles) recognizes the three bindings sites in the bi-directional terA/B promoter and activates the expression of reporter genes (lacZ and tdTomato) presumably in different strength (indicated by arrows). (C) Semiquantitative RT-PCR analysis of wild type A. niger A1144 and A1144_PamyB:terR (P2) expressing terR under control of the amyB promoter. terR is strongly expressed on PamyB inducing media (G100 and YM), but hardly detectable on CA1% medium. (D) Relative promoter activity of PterA and PterB from the reporter system described in (C). The parental strain P2 and the reporter strains P2_PterA:lacZ_PterB:tdTom and P2_PterB:lacZ_PterA:tdTom were cultivated for 48 h on G100, YM and CA1% and β-galactosidase activity and fluorescence intensity from cell-free extracts were determined. All values were normalized against reporter activities from the A. nidulans gpdA promoter (AnPgpdA:lacZ and AnPgpdA:tdTom) that were used as 100% reference values. Each experiment was carried out with three independent mutants in biological and technical triplicates.
Control of terR Expression Under the Inducible amyB Promoter Allows Regulated Gene Expression from the Bi-Directional terA/B Promoter
Experiments in Section “TerR Activates the terA Promoter in the Heterologous Host A. niger, but Requires a Promoter Exchange” revealed that control of terR expression under the gpdA promoter leads to constitutive activation of the terA promoter. In order to regulate expression, the amyB promoter was selected, since it was previously shown to be highly active in the presence of maltose or glucose, but only shows low background activity on sugar-free media such as 1% casamino acids (CA medium) (Ward, 2012; Zaehle et al., 2014). Indeed when the A. niger strain carrying the PterA:lacZ reporter was transformed with a PamyB:terR construct, high β-galactosidase activity was obtained when cells were cultivated on maltose containing YM medium, whereas activity after cultivation in CA medium was approximately 20 times lower (Figure 4A). This was also reflected by terR expression levels that were highly abundant on YM medium, but hardly detected on CA medium (Figure 4A). This again confirms that high terR expression levels are crucial for strong activation of target promoters. This also indicates that expression from promoters of the terrein cluster can be regulated by modifying the production level of the TerR regulator.
The selected terrein cluster promoter is assumed to depict a bi-directional promoter, because it separates the reading frames of terA and terB that are transcribed in opposite directions as already described in Section “The Transcription Factor TerR is Essential for Expression of Terrein Cluster Genes.” Interestingly, it has been shown that in the bi-directional promoters of penicillin biosynthesis from A. nidulans (acvA/ipnA) (Brakhage, 1997) and cephalosporin biosynthesis from Acremonium chrysogenum (pcbAB/pcbC) (Menne et al., 1994) transcription rate is favored in one direction. To test transcriptional activation from the terA/B promoter, the lacZ gene and a codon-optimized tdTomato gene encoding a red fluorescent protein were selected as reporters in both reading directions (Figure 4B). An A. niger strain carrying a single copy of the PamyB:terR construct (P2 strain) that shows the expected expression pattern of terR under inducing and non-inducing conditions (Figure 4C), was used as recipient strain for the different reporter constructs. Four to eight single copy transformants from each construct were analyzed in a pre-screening approach to test for variations in the expression pattern due to positioning effects (Minetoki et al., 1998; Liu et al., 2003; Blumhoff et al., 2013). All transformants revealed a similar tendency of expression levels (±25% from average). Therefore, three independent transformants were selected from each construct and strains were cultivated in biological triplicates for determination of the average promoter activation rate in comparison to the gpdA control promoter. Selected transformants with single copy integration were grown on glucose, YM or CA medium and screened for β-galactosidase activity and fluorescence intensity from cell-free extracts. All measurements were background corrected against extracts from an A. niger strain without reporter integration. Reporter activities were normalized against activities obtained from expression of the respective reporters under control of the A. nidulans gpdA promoter (Figure 4D).
Results from both reporters show that (i) terA/B promoter activity in both directions is strongly induced on glucose and YM medium, whereas activity remained near background values when cells were grown on non-inducing CA medium. (ii) Reporter activity in direction of the terA gene exceeded that of the gpdA promoter by up to three times, whereas activity in terB direction only reached 20–40% of the control value. Thus, transcription in the direction of the polyketide synthase gene terA exceeds that of terB by 8–14-fold. Additionally, the relative accumulation of TdTomato was slightly higher than that of the β-galactosidase, which could be due to increased stability of the codon-optimized TdTomato gene. In conclusion, these results indicate that terA/B promoter activation is directly dependent on the level of the TerR regulator.
Previous approaches to develop high-level expression systems used strongly expressed and, preferentially, inducible promoter elements for direct control of target gene expression. Examples are the thiamine promoter (PthiA) for expression in A. oryzae (Shoji et al., 2005), the xylose induced xyl1 promoter in A. chrysogenum (Blatzer et al., 2014), synthetic promoters containing the human estrogen receptor (hERα) response elements in A. nidulans and A. niger (Pachlinger et al., 2005) or the Escherichia coli tetracycline resistance operon (Tet-on system) in A. fumigatus and A. niger (Vogt et al., 2005; Meyer et al., 2011). Here, we identified that the activity of the artificial promoter that controls terR expression is amplified via TerR-dependent activation of the terA promoter. This assumption derives from the following observation: In previous studies we have shown that the gpdA and amyB promoter display similar activation levels on G100 medium (Zaehle et al., 2014). Thus, in case there is no signal amplification, reporter activity from the coupled system should be equivalent to PgpdA-driven activity. However, the analyses performed here show that reporter activity increased by a factor >2 when PamyB controls terR expression and the reporter is under terA control. Therefore, the coupled system of terrein cluster elements leads to an amplification of the strength of the promoter that is used to regulate terR expression. We assume that activity of promoters that are even stronger than PamyB could be amplified in our new coupled system. Although it might be expected that a saturation of the terA promoter may occur at a specific TerR level, this could either be compensated by increasing the number of terR binding sites in the terA promoter or by adding additional copies of the fusion of PterA with the gene of interest. However, a preliminary analysis of multicopy integrants with the PterA:lacZ construct revealed that expression levels run into saturation at approximately five integrations.
The PamyB:terR/PterA System is Suitable for High-Level Heterologous Secondary Metabolite Production
Due to the high reporter activities obtained from the terA promoter in combination with the PamyB:terR control element, we were interested in the use of this system for the heterologous production of secondary metabolites. In a previous study, we expressed the terrein synthase gene terA under direct control of the amyB promoter in A. niger (Zaehle et al., 2014). Sufficient quantities of the TerA metabolites orsellinic acid (4), 6,7-dihydroxymellein (3) and 4-hydroxy-6-methylpyrone (2) were produced (for molecular structures refer to Figure 5E). However, we assumed that metabolite production in the heterologous system could strongly exceed the quantity of metabolites compared to the previous construct. To confirm this hypothesis, the P2 strain with the single copy of the PamyB:terR integration was transformed with the A. terreus terA gene under control of its native terA promoter. Transformants were cultivated in glucose minimal medium, which was selected since HPLC profiles of the P2 strain showed no significant background metabolite production under this condition (Figure 5A). The metabolic profile of transformants was directly compared with that of a PamyB:terA strain that was cultivated under the same conditions (Figures 5B,C). Indeed, although all three metabolites were detected in similar ratios as detected from the PamyB:terA strain, the amount of metabolites produced by the new expression system strongly exceeded that of the former strain. In order to quantify the increase of metabolite production in the coupled system, we selected three PamyB:terA single copy and three PamyB:terA double copy integrants in the A. niger A1144 genetic background (Zaehle et al., 2014) and four single copy and two double copy PterA:terA integrants from the P2 strain. Transformants were cultivated in AMM-G100+Gln50 for 48 h, supernatants were extracted and orsellinic acid concentration was quantified by HPLC and normalized against the mycelial dry weight (Figure S4). Although production rates varied among independent transformants, the single and double copy strains with the PamyB:terA construct produced on average 0.37 (±0.19) and 0.43 (±0.13) mg orsellinic acid per gram of dried mycelium. Single copy PterA:terA integrants produced on average 1.16 (±0.64) and double copy integrants 3.20 (±1.69) mg of orsellinic acid per gram of dried mycelium. This results in an approximated 3-fold increase in the coupled expression system for single copy and a 7.5-fold increase for the double copy integrants in comparison to the direct expression system under control of the amyB promoter. Although increases in production rates may be metabolite dependent, these results reflect the increased transcription rate from the coupled expression system as shown in Figure 4D and confirms the suitability of the system for heterologous expression.
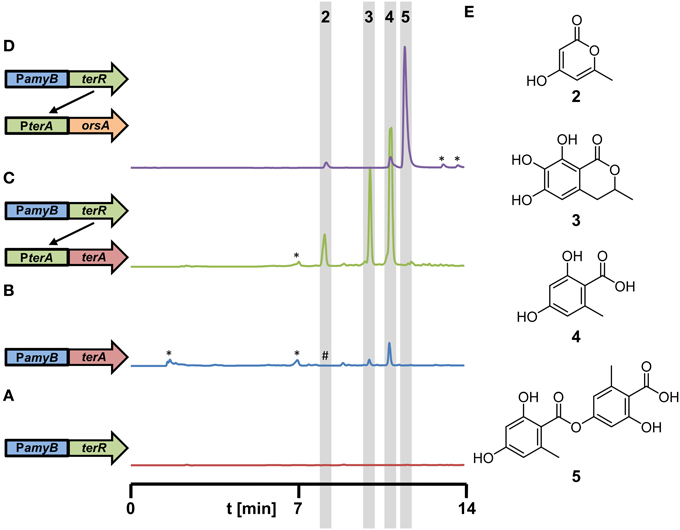
Figure 5. Metabolite detection from culture filtrates of recombinant A. niger strains expressing different polyketide synthases under control of the terR/PterA expression system. All strains were grown for 48 h in AMM-G100 medium with 20 mM glutamine as nitrogen source. (A–D) HPLC profiles from (A) A1144_PamyB:terR (P2, parental control strain), (B) A1144_PamyB:terA (control strain) with conventional direct terA expression, (Zaehle et al., 2014), (C) P2_PterA:terA (coupled system), and (D) P2_PterA:orsA (coupled system). A schematic drawing of the integrated constructs is given on the left next to the respective profile. (E) Structures of metabolites identified in the extracts are: 4-hydroxy-6-methylpyrone (2), 6,7-dihydroxymellein (3), orsellinic acid (4), and lecanoric acid (5). *Indicate non-reproducible metabolite peaks; #refers to (2) that is only visualized by loading concentrated extracts of the PamyB:terA transformants.
In another approach, we selected the orsA PKS gene from A. nidulans for heterologous expression in A. niger. Previous analyses have shown that the orsA gene is induced during the cocultivation of A. nidulans with the bacterium Streptomyces hygroscopicus (Schroeckh et al., 2009). Structure elucidation of the metabolites produced by OrsA required a 14 liter cocultivation of both organisms to obtain sufficient amounts of metabolites. Interestingly, orsellinic acid and lecanoric acid, a depside of two orsellinic acid molecules, were identified. However, it remained unclear, which of the two products is the major metabolite produced by OrsA and whether OrsA is directly performing the transesterification of two orsellinic acid molecules (Schroeckh et al., 2009). Therefore, we generated a PterA:orsA construct that was transferred into the A. niger P2 strain and single copy integrants were tested for product formation in 50 ml culture scale. Ethylacetate extracts revealed one major and two minor metabolites (Figure 5D) that were identified by HRESI-MS as lecanoric acid (5), orsellinic acid (4) and 4-hydroxy-6-methylpyrone (2) and the identity was confirmed by reference against known standards and 1H and 13C NMR spectra of purified lecanoric acid (Figure S5 and supplementary experimental procedures). Since lecanoric acid was by far the most prominent metabolite produced by all independent transformants, it can be assumed that the thioesterase (TE)-domain of OrsA performs the transesterification of the p-hydroxyl-group of one orsellinic acid molecule with the carboxyl-group of a second molecule rather than releasing orsellinic acid by simple hydrolysis. A similar mechanism of depside formation from orsellinic acid-derived metabolites might be present in lichen-forming fungi in which lecanoric acid and derivatives thereof constitute major secondary metabolites (Parrot et al., 2015). However, whether the involved polyketide synthases perform depside formations has not yet been characterized in detail (Armaleo et al., 2011).
A New Expression Vector Enables Rapid Cloning and Modification of Secondary Metabolite Genes
In order to ease the use of the heterologous expression system, we constructed a plasmid that allows rapid generation of expression constructs called SM-Xpress. For this purpose, we used a plasmid with a phleomycin resistance cassette in which the ble gene was controlled by the A. nidulans gpdA promoter. Subsequently, the 786 bp terA promoter and a 363 bp fragment of the A. terreus trpC terminator were amplified by PCR and integrated into the plasmid by in vitro recombination. This resulted in plasmid “SM-Xpress” that contained a NcoI restriction site that separated promoter and terminator and allowed insertion of the gene of interest by restriction ligation or in vitro recombination (Figure 6A). Resulting overexpression plasmids are suitable for direct transformation of the A. niger P2 strain that contains the PamyB:terR construct.
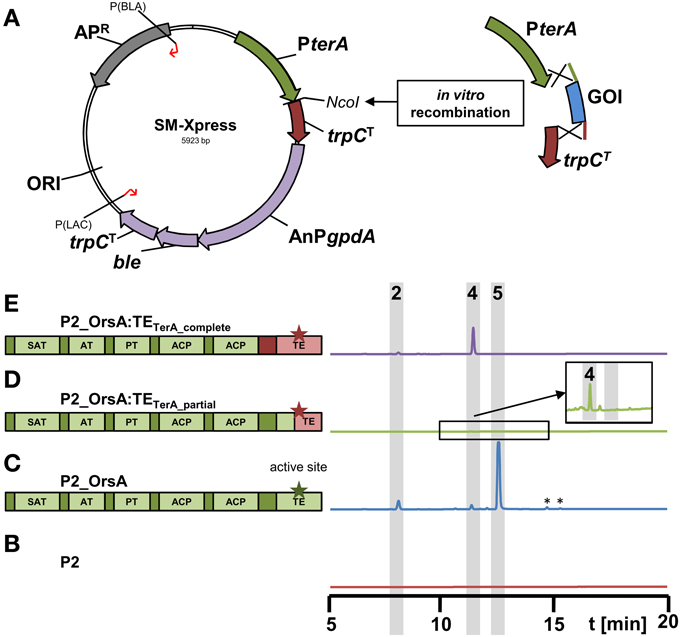
Figure 6. Scheme of the new expression vector SM-Xpress and its use for rapid cloning of polyketide synthases in domain swapping experiments. (A) The plasmid SM-Xpress contains the native terA promoter and a trpC terminator and allows cloning of a gene of interest (GOI) by restriction cloning or in vitro recombinantion into the NcoI site. Due to a phleomycin resistance cassette (AnPgpdA:ble:trpCT), the plasmid is suitable for direct transformation of the A. niger P2 strain expressing terR under PamyB control. (B–E) HPLC profiles of culture filtrates from A. niger P2 strains grown for 40 h in AMM-G100 + Gln50 and expressing different versions of the original A. nidulans orsA gene or chimeric versions with exchanged thioesterase (TE) domains as depicted in the schemes on the left. Green parts originate from orsA and pink parts from A. terreus terA. Asterisks indicate the active sites of the TE domains. (B) P2 control strain (PamyB:terR), (C) P2_A1144_PterA:orsA, (D) P2_PterA:orsA:TEterA_partial and (E) P2_PterA:orsA:TEterA_complete. Indicated peaks correspond to 4-hydroxy-6-methylpyrone (2), orsellinic acid (4) and lecanoric acid (5). Note that a partial exchange of the TE domain (D) leads to a chimeric protein that hardly releases products and only trace amounts of orsellinic acid are detected (magnified inlay). *Indicate non-reproducible metabolite peaks.
To test this system, we went back to the orsA gene from A. nidulans. As stated in Section “The PamyB:terR/PterA System is Suitable for High-Level Heterologous Secondary Metabolite Production,” OrsA predominantly produced lecanoric acid and we hypothesized that the depside formation from two orsellinic acid molecules is mediated by the thioesterase domain (TE) of this enzyme. In general, TE domains are important for product release, but can also perform an interketide esterification (cross-coupling) between the carboxyl-function of the polyketide and an alcohol, whereby in a recent example of melleolide formation in Armillaria mellea the hydroxyl group derives from a sesquiterpene (Lackner et al., 2013). To confirm that the depside formation in lecanoric acid is specific for the OrsA TE domain, we performed an in vitro recombination between the orsA gene lacking its TE domain and the TE domain from A. terreus terA that only releases monomers of its products (Figures 5C,D and Zaehle et al., 2014). In this approach, two different fusions were made: (i) the complete TE domain starting directly 3′ of ACP2 was removed from orsA and fused with the complete TE domain of terA (orsA:TEterA_complete; Figure 6E). (ii) The TE domain from orsA was exchanged at a conserved region of the active site of the TE domain (orsA:TEterA_partial; Figure 6D). The A. niger P2 strain was transformed with the fusion constructs and transformants were analyzed for their metabolic profile (Figures 6B-E). The partial exchange of the active site only resulted in trace amounts of orsellinic acid (Figure 6D), indicating that this exchange led to loss of function of the TE domain. Additionally, it indicates that a functional TE domain is required for product release. This is in agreement with other investigations that showed that TE domains are required for efficient product release from PKS enzymes, whereby the specific product release mechanism is mainly directed by the TE domain (Du and Lou, 2010; Xu et al., 2013).
In contrast to the chimeric protein with partial exchange of the TE domain at the active site, A. niger transformants expressing the chimeric orsA with the complete terA domain exclusively produced orsellinic acid in high yields and lecanoric acid was no longer detected (Figure 6E). Due to these results we provide the first experimental evidence that the depside formation in lecanoric acid is attributed to the TE domain of orsA. This opens a new avenue for domain swapping experiments, in which the orsA TE domain could be used for fusions with other polyketide synthases to generate new depsides. Furthermore, these results not only confirm the suitability of our expression system for rapid analysis of products formed from secondary metabolite gene clusters, but also shows that the system provides a tool for rapid combinatory domain swapping experiments.
Conclusions and Outlook
In this study we confirmed specificity of the transcriptional activator TerR from the terrein gene cluster for its target promoters. Furthermore, we were able to identify DNA-binding motifs recognized by TerR. Interestingly, TerR predominantly binds as a monomer to a single CGG half-site, but a second CGG motif that is separated by a five nucleotide spacer can be bound by a second monomer. High affinity sites with tendency for binding of two monomers are characterized by the consensus motif TCGGHHWYHCGGH. At least one site with this high affinity motif is present in all promoters of genes required for terrein synthesis. Interestingly, no high-affinity consensus motifs are detected in terG, terH, and terI that are dispensable for terrein production, but show the same TerR dependent regulation pattern. Thus, single half-site motifs may be present in these promoters, but these are difficult to predict by bioinformational methods. On the contrary, the terA promoter contains three CGG direct repeat motifs and especially expression of the terA gene is strongly activated by TerR. The terC promoter contains only a single, but high affinity binding motif and its expression level is much lower than that from terA. Therefore, the number and probably also the distance of the motifs relative to the transcriptional start point appear important for high-level transcriptional activation. To confirm this assumption, future studies will replace all binding sites in the terA promoter by motifs from other promoters (such as BS6 and BS7). Furthermore, the number of binding sites will be increased to analyze the effect on the promoter activation potential.
Although there might be space for further optimization of the system, heterologous expression analyses in A. niger clearly showed that a combination of TerR with its terA target promoter is highly efficient and leads to an amplification of the expression level of the promoter that controls terR expression. Thus, additional promoters will be used to control terR expression to analyze the maximum amplification rates that can be obtained. Another important aspect is the transfer of the system to other fungi of industrial importance. While preliminary analyses showed that the system also works in other Aspergillus species, we will study the functionality of the system in yeasts such as Saccharomyces cerevisiae and various Basidiomycetes that are amenable to genetic modifications.
Last but not least, our proof of principle studies for heterologous expression of secondary metabolite genes by the TerR/PterA coupled system demonstrate an easy and rapid method for production of secondary metabolites. The vector SM-Xpress allows fast cloning of genes and eases domain swapping experiments. By this approach, we were able to show that the TE domain of OrsA performs a depside formation and future studies will use this domain for generating chimera with other polyketide synthases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We greatly acknowledge Sylke Fricke for assistance in cloning and purification of TerR DNA-binding domains, Daniela Hildebrandt for assistance in cloning of fungal reporter constructs, Andrea Perner for HR-MS and Heike Heinecke for recording 1H- and 13C-NMR spectra of secondary metabolites. This work was supported by grant BR 2216/4-1 from the German Science Foundation (DFG) and internal funding from the Hans-Knoell-Institute (HKI).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2015.00184/abstract
References
Alberts, A. W., Chen, J., Kuron, G., Hunt, V., Huff, J., Hoffman, C., et al. (1980). Mevinolin: a highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc. Natl. Acad. Sci. U.S.A. 77, 3957–3961. doi: 10.1073/pnas.77.7.3957
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Armaleo, D., Sun, X., and Culberson, C. (2011). Insights from the first putative biosynthetic gene cluster for a lichen depside and depsidone. Mycologia 103, 741–754. doi: 10.3852/10-335
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bergmann, S., Schumann, J., Scherlach, K., Lange, C., Brakhage, A. A., and Hertweck, C. (2007). Genomics-driven discovery of PKS-NRPS hybrid metabolites from Aspergillus nidulans. Nat. Chem. Biol. 3, 213–217. doi: 10.1038/nchembio869
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Blatzer, M., Gsaller, F., Abt, B., Schrettl, M., Specht, T., and Haas, H. (2014). An endogenous promoter for conditional gene expression in Acremonium chrysogenum: the xylan and xylose inducible promoter xyl1(P.). J. Biotechnol. 169, 82–86. doi: 10.1016/j.jbiotec.2013.11.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Blumhoff, M., Steiger, M. G., Marx, H., Mattanovich, D., and Sauer, M. (2013). Six novel constitutive promoters for metabolic engineering of Aspergillus niger. Appl. Microbiol. Biotechnol. 97, 259–267. doi: 10.1007/s00253-012-4207-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bok, J. W., Chung, D., Balajee, S. A., Marr, K. A., Andes, D., Nielsen, K. F., et al. (2006). GliZ, a transcriptional regulator of gliotoxin biosynthesis, contributes to Aspergillus fumigatus virulence. Infect. Immun. 74, 6761–6768. doi: 10.1128/IAI.00780-06
Brakhage, A. A. (1997). Molecular regulation of penicillin biosynthesis in Aspergillus (Emericella) nidulans. FEMS Microbiol. Lett. 148, 1–10. doi: 10.1111/j.1574-6968.1997.tb10258.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cahuzac, B., Cerdan, R., Felenbok, B., and Guittet, E. (2001). The solution structure of an AlcR-DNA complex sheds light onto the unique tight and monomeric DNA binding of a Zn(2)Cys(6) protein. Structure 9, 827–836. doi: 10.1016/S0969-2126(01)00640-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Calam, C. T., Oxford, A. E., and Raistrick, H. (1939). Studies in the biochemistry of micro-organisms: itaconic acid, a metabolic product of a strain of Aspergillus terreus Thom. Biochem. J. 33, 1488–1495.
Chakravarty, A., Carlson, J. M., Khetani, R. S., and Gross, R. H. (2007). A novel ensemble learning method for de novo computational identification of DNA binding sites. BMC Bioinform. 8:249. doi: 10.1186/1471-2105-8-249
Du, L., and Lou, L. (2010). PKS and NRPS release mechanisms. Nat. Prod. Rep. 27, 255–278. doi: 10.1039/b912037h
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Felenbok, B., Sequeval, D., Mathieu, M., Sibley, S., Gwynne, D. I., and Davies, R. W. (1988). The ethanol regulon in Aspergillus nidulans: characterization and sequence of the positive regulatory gene alcR. Gene 73, 385–396. doi: 10.1016/0378-1119(88)90503-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fitzgerald, M. X., Rojas, J. R., Kim, J. M., Kohlhaw, G. B., and Marmorstein, R. (2006). Structure of a Leu3-DNA complex: recognition of everted CGG half-sites by a Zn2Cys6 binuclear cluster protein. Structure 14, 725–735. doi: 10.1016/j.str.2005.11.025
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fleissner, A., and Dersch, P. (2010). Expression and export: recombinant protein production systems for Aspergillus. Appl. Microbiol. Biotechnol. 87, 1255–1270. doi: 10.1007/s00253-010-2672-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Forseth, R. R., Fox, E. M., Chung, D., Howlett, B. J., Keller, N. P., and Schroeder, F. C. (2011). Identification of cryptic products of the gliotoxin gene cluster using NMR-based comparative metabolomics and a model for gliotoxin biosynthesis. J. Am. Chem. Soc. 133, 9678–9681. doi: 10.1021/ja2029987
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gressler, M., Zaehle, C., Scherlach, K., Hertweck, C., and Brock, M. (2011). Multifactorial induction of an orphan PKS-NRPS gene cluster in Aspergillus terreus. Chem. Biol. 18, 198–209. doi: 10.1016/j.chembiol.2010.12.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Heger, A., and Holm, L. (2000). Rapid automatic detection and alignment of repeats in protein sequences. Proteins 41, 224–237. doi: 10.1002/1097-0134(20001101)41:2<224::AID-PROT70>3.0.CO;2-Z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hutchinson, C. R., Kennedy, J., Park, C., Kendrew, S., Auclair, K., and Vederas, J. (2000). Aspects of the biosynthesis of non-aromatic fungal polyketides by iterative polyketide synthases. Antonie Van Leeuwenhoek 78, 287–295. doi: 10.1023/A:1010294330190
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Klement, T., and Buchs, J. (2013). Itaconic acid–a biotechnological process in change. Bioresour. Technol. 135, 422–431. doi: 10.1016/j.biortech.2012.11.141
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lackner, G., Bohnert, M., Wick, J., and Hoffmeister, D. (2013). Assembly of melleolide antibiotics involves a polyketide synthase with cross-coupling activity. Chem. Biol. 20, 1101–1106. doi: 10.1016/j.chembiol.2013.07.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lenouvel, F., Nikolaev, I., and Felenbok, B. (1997). In vitro recognition of specific DNA targets by AlcR, a zinc binuclear cluster activator different from the other proteins of this class. J. Biol. Chem. 272, 15521–15526. doi: 10.1074/jbc.272.24.15521
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, B. H., and Chu, F. S. (1998). Regulation of aflR and its product, AflR, associated with aflatoxin biosynthesis. Appl. Environ. Microbiol. 64, 3718–3723.
Liu, L., Liu, J., Qiu, R. X., Zhu, X. G., Dong, Z. Y., and Tang, G. M. (2003). Improving heterologous gene expression in Aspergillus niger by introducing multiple copies of protein-binding sequence containing CCAAT to the promoter. Lett. Appl. Microbiol. 36, 358–361. doi: 10.1046/j.1472-765X.2003.01321.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lubertozzi, D., and Keasling, J. D. (2009). Developing Aspergillus as a host for heterologous expression. Biotechnol. Adv. 27, 53–75. doi: 10.1016/j.biotechadv.2008.09.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
MacPherson, S., Larochelle, M., and Turcotte, B. (2006). A fungal family of transcriptional regulators: the zinc cluster proteins. Microbiol. Mol. Biol. Rev. 70, 583–604. doi: 10.1128/MMBR.00015-06
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Menne, S., Walz, M., and Kuck, U. (1994). Expression studies with the bidirectional pcbAB-pcbC promoter region from Acremonium chrysogenum using reporter gene fusions. Appl. Microbiol. Biotechnol. 42, 57–66. doi: 10.1007/BF00170225
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meyer, V., Wanka, F., Van Gent, J., Arentshorst, M., Van Den Hondel, C. A., and Ram, A. F. (2011). Fungal gene expression on demand: an inducible, tunable, and metabolism-independent expression system for Aspergillus niger. Appl. Environ. Microbiol. 77, 2975–2983. doi: 10.1128/AEM.02740-10
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Minetoki, T., Kumagai, C., Gomi, K., Kitamoto, K., and Takahashi, K. (1998). Improvement of promoter activity by the introduction of multiple copies of the conserved region III sequence, involved in the efficient expression of Aspergillus oryzae amylase-encoding genes. Appl. Microbiol. Biotechnol. 50, 459–467. doi: 10.1007/s002530051321
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nikolaev, I., Cochet, M. F., Lenouvel, F., and Felenbok, B. (1999). A single amino acid, outside the AlcR zinc binuclear cluster, is involved in DNA binding and in transcriptional regulation of the alc genes in Aspergillus nidulans. Mol. Microbiol. 31, 1115–1124. doi: 10.1046/j.1365-2958.1999.01250.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pachlinger, R., Mitterbauer, R., Adam, G., and Strauss, J. (2005). Metabolically independent and accurately adjustable Aspergillus sp. expression system. Appl. Environ. Microbiol. 71, 672–678. doi: 10.1128/AEM.71.2.672-678.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Parrot, D., Peresse, T., Hitti, E., Carrie, D., Grube, M., and Tomasi, S. (2015). Qualitative and spatial metabolite profiling of lichens by a LC-MS approach combined with optimised extraction. Phytochem. Anal. 26, 23–33. doi: 10.1002/pca.2532
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Punt, P. J., Dingemanse, M. A., Kuyvenhoven, A., Soede, R. D., Pouwels, P. H., and Van Den Hondel, C. A. (1990). Functional elements in the promoter region of the Aspergillus nidulans gpdA gene encoding glyceraldehyde-3-phosphate dehydrogenase. Gene 93, 101–109. doi: 10.1016/0378-1119(90)90142-E
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schroeckh, V., Scherlach, K., Nutzmann, H. W., Shelest, E., Schmidt-Heck, W., Schuemann, J., et al. (2009). Intimate bacterial-fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc. Natl. Acad. Sci. U.S.A. 106, 14558–14563. doi: 10.1073/pnas.0901870106
Sharma, R., Katoch, M., Srivastava, P. S., and Qazi, G. N. (2009). Approaches for refining heterologous protein production in ?lamentous fungi. World J. Microbiol. Biotechnol. 25, 2083–2094. doi: 10.1007/s11274-009-0128-x
Shoji, J. Y., Maruyama, J., Arioka, M., and Kitamoto, K. (2005). Development of Aspergillus oryzae thiA promoter as a tool for molecular biological studies. FEMS Microbiol. Lett. 244, 41–46. doi: 10.1016/j.femsle.2005.01.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Slesiona, S., Gressler, M., Mihlan, M., Zaehle, C., Schaller, M., Barz, D., et al. (2012a). Persistence versus escape: Aspergillus terreus and Aspergillus fumigatus employ different strategies during interactions with macrophages. PLoS ONE 7:e31223. doi: 10.1371/journal.pone.0031223
Slesiona, S., Ibrahim-Granet, O., Olias, P., Brock, M., and Jacobsen, I. D. (2012b). Murine infection models for Aspergillus terreus pulmonary aspergillosis reveal long-term persistence of conidia and liver degeneration. J. Infect. Dis. 205, 1268–1277. doi: 10.1093/infdis/jis193
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Todd, R. B., Andrianopoulos, A., Davis, M. A., and Hynes, M. J. (1998). FacB, the Aspergillus nidulans activator of acetate utilization genes, binds dissimilar DNA sequences. Embo J. 17, 2042–2054. doi: 10.1093/emboj/17.7.2042
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Verdoes, J. C., Punt, P. J., Schrickx, J. M., Van Verseveld, H. W., Stouthamer, A. H., and Van Den Hondel, C. A. (1993). Glucoamylase overexpression in Aspergillus niger: molecular genetic analysis of strains containing multiple copies of the glaA gene. Transgenic Res. 2, 84–92. doi: 10.1007/BF01969381
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vogt, K., Bhabhra, R., Rhodes, J. C., and Askew, D. S. (2005). Doxycycline-regulated gene expression in the opportunistic fungal pathogen Aspergillus fumigatus. BMC Microbiol. 5:1. doi: 10.1186/1471-2180-5-1
Ward, O. P. (2012). Production of recombinant proteins by filamentous fungi. Biotechnol. Adv. 30, 1119–1139. doi: 10.1016/j.biotechadv.2011.09.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, B., Yin, Y., Zhang, F., Li, Z., and Wang, L. (2012). Operating conditions optimization for (+)-terrein production in a stirred bioreactor by Aspergillus terreus strain PF-26 from marine sponge Phakellia fusca. Bioprocess Biosyst. Eng. 35, 1651–1655. doi: 10.1007/s00449-012-0735-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, Y., Espinosa-Artiles, P., Schubert, V., Xu, Y. M., Zhang, W., Lin, M., et al. (2013). Characterization of the biosynthetic genes for 10,11-dehydrocurvularin, a heat shock response-modulating anticancer fungal polyketide from Aspergillus terreus. Appl. Environ. Microbiol. 79, 2038–2047. doi: 10.1128/AEM.03334-12
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yin, Y., Xu, B., Li, Z., and Zhang, B. (2012). Enhanced production of (+)-terrein in fed-batch cultivation of Aspergillus terreus strain PF26 with sodium citrate. World J. Microbiol. Biotechnol. 29, 441–446. doi: 10.1007/s11274-012-1196-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yu, J., Chang, P. K., Ehrlich, K. C., Cary, J. W., Bhatnagar, D., Cleveland, T. E., et al. (2004). Clustered pathway genes in aflatoxin biosynthesis. Appl. Environ. Microbiol. 70, 1253–1262. doi: 10.1128/AEM.70.3.1253-1262.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yu, J. H., Butchko, R. A., Fernandes, M., Keller, N. P., Leonard, T. J., and Adams, T. H. (1996). Conservation of structure and function of the aflatoxin regulatory gene aflR from Aspergillus nidulans and A. flavus. Curr. Genet. 29, 549–555. doi: 10.1007/BF02426959
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zaehle, C., Gressler, M., Shelest, E., Geib, E., Hertweck, C., and Brock, M. (2014). Terrein biosynthesis in Aspergillus terreus and its impact on phytotoxicity. Chem. Biol. 21, 719–731. doi: 10.1016/j.chembiol.2014.03.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: Aspergillus niger, secondary metabolites, transcription factor, DNA-binding motif, reporter strains, thioesterase domain
Citation: Gressler M, Hortschansky P, Geib E and Brock M (2015) A new high-performance heterologous fungal expression system based on regulatory elements from the Aspergillus terreus terrein gene cluster. Front. Microbiol. 6:184. doi: 10.3389/fmicb.2015.00184
Received: 19 December 2014; Paper pending published: 26 January 2015;
Accepted: 19 February 2015; Published: 16 March 2015.
Edited by:
Ana Lúcia Leitão, Universidade Nova de Lisboa, PortugalCopyright © 2015 Gressler, Hortschansky, Geib and Brock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthias Brock, Microbial Biochemistry and Physiology, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knoell Institute, Beutenbergstr. 11a, 07745 Jena, GermanybWF0dGhpYXMuYnJvY2tAaGtpLWplbmEuZGU=