 James Angus Chandler
James Angus Chandler Rachel M. Liu
Rachel M. Liu Shannon N. Bennett
Shannon N. Bennett- Department of Microbiology, California Academy of Sciences, San Francisco, CA, USA
Mosquitoes, most often recognized for the microbial agents of disease they may carry, harbor diverse microbial communities that include viruses, bacteria, and fungi, collectively called the microbiota. The composition of the microbiota can directly and indirectly affect disease transmission through microbial interactions that could be revealed by its characterization in natural populations of mosquitoes. Furthermore, the use of shotgun metagenomic sequencing (SMS) approaches could allow the discovery of unknown members of the microbiota. In this study, we use RNA SMS to characterize the microbiota of seven individual mosquitoes (species include Culex pipiens, Culiseta incidens, and Ochlerotatus sierrensis) collected from a variety of habitats in California, USA. Sequencing was performed on the Illumina HiSeq platform and the resulting sequences were quality-checked and assembled into contigs using the A5 pipeline. Sequences related to single stranded RNA viruses of the Bunyaviridae and Rhabdoviridae were uncovered, along with an unclassified genus of double-stranded RNA viruses. Phylogenetic analysis finds that in all three cases, the closest relatives of the identified viral sequences are other mosquito-associated viruses, suggesting widespread host-group specificity among disparate viral taxa. Interestingly, we identified a Narnavirus of fungi, also reported elsewhere in mosquitoes, that potentially demonstrates a nested host-parasite association between virus, fungi, and mosquito. Sequences related to 8 bacterial families and 13 fungal families were found across the seven samples. Bacillus and Escherichia/Shigella were identified in all samples and Wolbachia was identified in all Cx. pipiens samples, while no single fungal genus was found in more than two samples. This study exemplifies the utility of RNA SMS in the characterization of the natural microbiota of mosquitoes and, in particular, the value of identifying all microbes associated with a specific host.
Introduction
The diversity of animal-associated microbes, collectively called the microbiota, and their ubiquitous role in host ecology, physiology, and evolution has reached new levels of appreciation with the advent of next-generation sequencing technologies (Foster et al., 2012; McFall-Ngai et al., 2013). Culture-dependent methods are limited in their capacity to capture a full picture of the microorganisms present, as only a minority of microbes are easily culturable under “standard” laboratory conditions (Amann et al., 1995 but see Donachie et al., 2007). Metagenomic PCR circumvents this issue by identifying organisms directly from environmentally acquired nucleic acids, often using taxon-specific primers that target genes such as the ribosomal small subunit (Schmidt et al., 1991; Handelsman et al., 1998). However, this method has known issues related to primer selection, chimera formation, and gene copy number (Ashelford et al., 2006; Kembel et al., 2012; Ghyselinck et al., 2013; Klindworth et al., 2013). Shotgun metagenomic sequencing (SMS), which does not rely upon an initial PCR step, can avoid the limitations of culture-dependent and PCR-based methods (Venter et al., 2004; Haas et al., 2011). Combined with high throughput sequencing technologies, SMS has been successfully used to characterize the microbiota of honeybees (Runckel et al., 2011; Engel et al., 2012), termites (Warnecke et al., 2007), humans (Qin et al., 2010), and a variety of mammals (Muegge et al., 2011). SMS has an added benefit that the data produced is not restricted to a single taxon (i.e., only bacteria or only fungi) and has been used to identify a variety of microorganisms associated with a single host (Runckel et al., 2011). In this study, we use SMS and high throughput sequencing to examine the microbiota of mosquitoes collected in northern California, USA.
Mosquitoes are vectors of many clinically and economically important diseases, such as malaria and dengue (Morens et al., 2004; Fukuda et al., 2011). Additionally, mosquitoes carry many non-pathogenic microbes (Minard et al., 2013) and some of these are prime candidates for symbiont mediated transmission disruption. For example, the intracellular bacterium Wolbachia can affect dengue transmission in Aedes aegypti (Walker et al., 2011) and reduces the titer of West Nile virus in Culex quinquefasciatus (Glaser and Meola, 2010). Genetically modifying bacteria that are naturally associated with mosquitoes is also a promising approach to transmission disruption, as has been shown with Anopheles mosquitoes and malaria (Wang et al., 2012). These studies and others (reviewed in Cirimotich et al., 2011) were made possible through a previous understanding of the mosquito microbiota.
Numerous culture-dependent and culture-independent studies have examined the bacterial communities associated with mosquitoes (reviewed in Minard et al., 2013). Generally, the bacterial taxa Gammaproteobacteria and Firmicutes are major components of the mosquito microbiota, but other groups, such as the Alphaproteobacteria and the Betaproteobacteria, are also present (Minard et al., 2013). Studies investigating the mosquito microbiota have shown that its composition is based on both environmental (such as habitat) and host-intrinsic (such as sex, species, and developmental stage) factors (Minard et al., 2013). The function of the microbiota remains unclear in many cases, although its experimental removal does arrest larval development in three different mosquito species (Coon et al., 2014).
As with other animal hosts [e.g., humans (Huffnagle and Noverr, 2013) and Drosophila (Broderick and Lemaitre, 2012)], fungi associated with mosquitoes are relatively understudied compared to their bacterial counterparts. Most previous work investigating the fungi associated with mosquitoes has focused on entomopathogenic fungi and their use in mosquito control (Scholte et al., 2004; de Faria and Wraight, 2007). However, to our knowledge, there are no studies that have used culture-independent techniques to characterize the total fungal communities of natural populations of mosquitoes.
Shotgun metagenomic sequencing has previously been used to investigate the mosquito microbiota. In particular, the viral diversity associated with wild-caught mosquitoes has been characterized and these studies have shown that SMS is sufficient to recover and identify viruses from a variety of taxonomic groups (Ma et al., 2011; Ng et al., 2011; Cook et al., 2013; Chandler et al., 2014). Furthermore, mosquitoes that have been laboratory infected with known mosquito-vectored viruses (such as dengue, yellow fever, or chikungunya) have been subjected to SMS (Bishop-Lilly et al., 2010; Hall-Mendelin et al., 2013). In addition to successful identification of the viruses and mosquitoes, genetic material from both bacteria and fungi were recovered via SMS.
Lab-infected mosquito studies (Bishop-Lilly et al., 2010; Hall-Mendelin et al., 2013) suggest the feasibility of using SMS to identify both viruses and other microorganisms (such as bacteria and fungi) in wild populations of mosquitoes, however, to our knowledge, this has never been done. Because symbiont mediated transmission disruption is an emerging tool in controlling vector-borne diseases (reviewed in Cirimotich et al., 2011; Weiss and Aksoy, 2011), SMS of wild mosquitoes may be particularly informative because it simultaneously identifies both viruses and other symbionts and could uncover any correlation between the two in the same host. This study represents a proof of concept in characterizing microbiota using a taxonomically broad approach that could ultimately prove useful in exposing significant novel interactions between microbes. In particular, we use SMS to characterize the microbiota of seven individual mosquitoes of three different species that were collected from a variety of natural habitats in northern California, USA, which is an area where West Nile virus has been reported. Using RNA-based SMS on the Illumina platform, we identify sequences related to viruses, bacteria, and fungi in each individual. Furthermore, we were able to verify mosquito species identities using SMS data alone. This work exemplifies the utility of SMS to study the natural microbiota of mosquitoes and we hope it prompts future research in this area.
Materials and Methods
Collection and Identification
Several hundred mosquitoes were collected from seven locations in northern California (Pepperwood Preserve, Bolinas, Stinson Beach, San Rafael, Mill Valley, San Francisco, and San Mateo) during March–November 2013 (Figure 1). Locations were chosen to represent a range of habitats, from sylvatic/wild (e.g., Pepperwood Preserve) to urban (e.g., San Francisco), (Table 1). Collections occurred under the permit and permission agreements of the Marin/Sonoma Mosquito and Vector Control District, or on private lands with the owners’ permission.
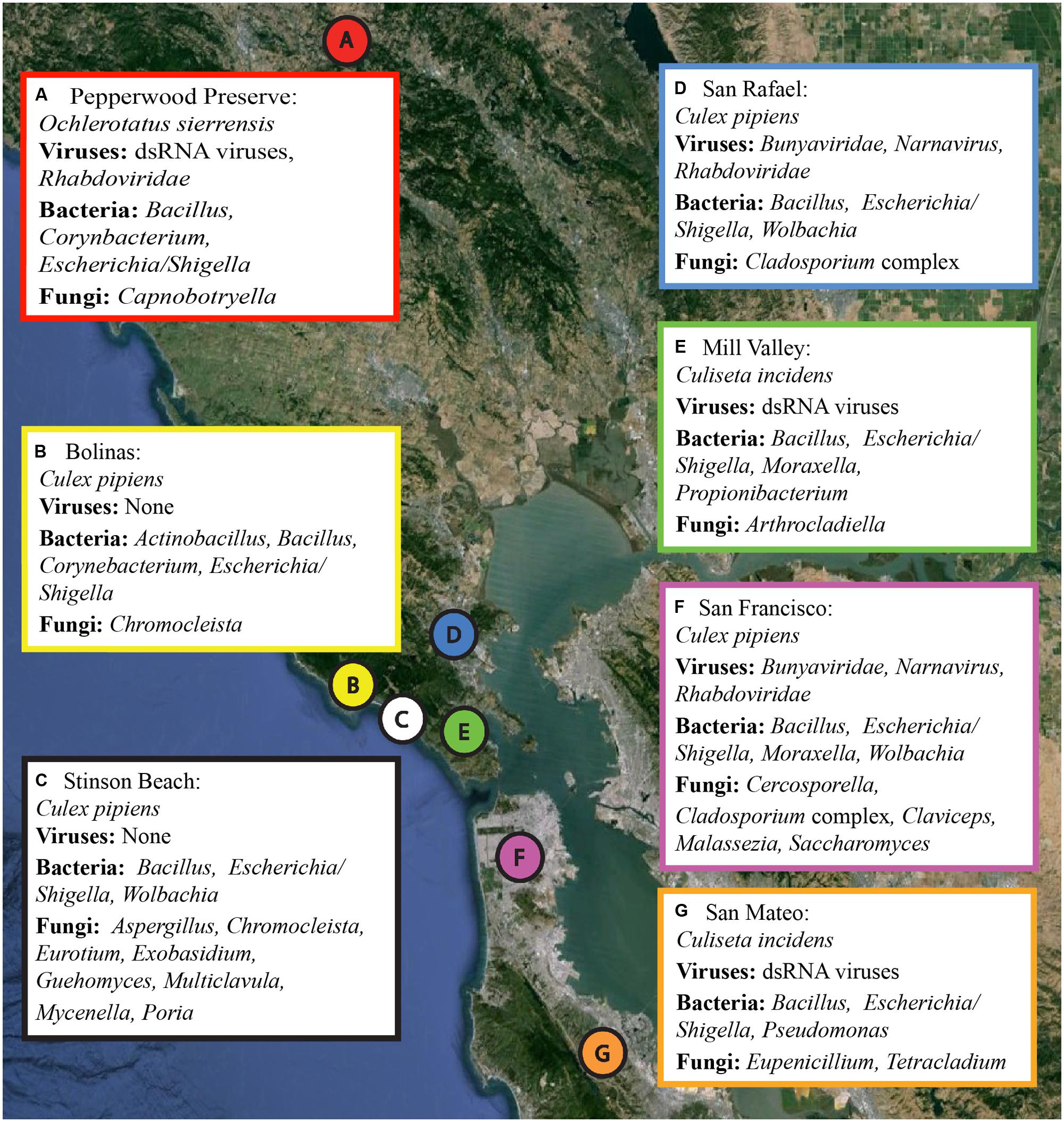
FIGURE 1. Collection locations and a summary of the microbial sequences identified in each sample. Locations A through G correspond to the samples as listed in Table 1. A summary of the viral, bacterial, and fungal sequences identified in each sample is included. Details for each microbial group can be found in Tables 2–4. The map was created using Google Earth Pro, with data from the U.S. Navy, LDEO-Columbia, NSF, NOAA, SIO, NGA, MBARI,and GEBCO.
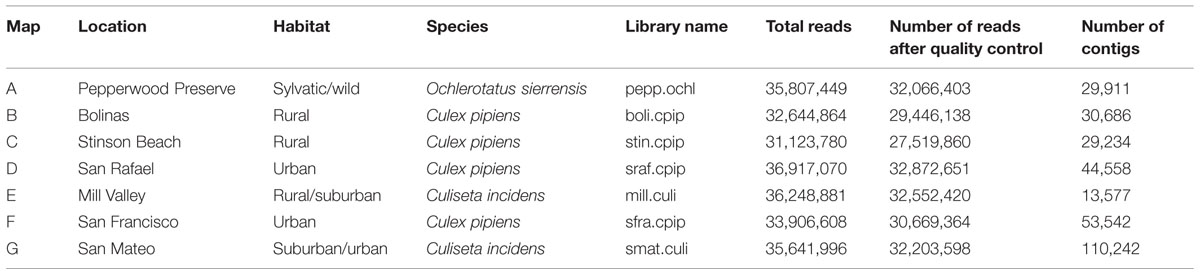
TABLE 1. Sample details.
We employed several methods to collect mosquitoes, including hand nets, gravid traps baited with hay-infused water, or ZumbaTM traps baited with carbon dioxide gas (CO2), heat packs, and SkinLureTM. All samples were frozen within 24–48 h of being trapped and stored at -80°C. We identified mosquitoes morphologically using a dissecting microscope and key (Bohart and Washino, 1978), retaining voucher specimens for each species and trapping event. In several cases, morphological identification was verified by sequence identity for cytochrome oxidase I (COI) using a leg from the voucher specimen.
DNA Extraction, Library Preparation, and Sequencing
From the several hundred mosquitoes collected, one representative individual female from each of the seven different collection sites was selected for SMS sequencing (Table 1). Prior to processing, whole mosquitoes were washed in 70% ethanol, distilled water, and phosphate-buffered saline (PBS) solution to remove external microbes. While we did not explicitly test the final wash for complete removal of external microbes, a similar wash protocol for Drosophila was found to be sufficient for this purpose (Chandler et al., 2011). Washed samples were then individually homogenized in PBS with steel Lysing Matrix I beads (MP Biomedicals).
Samples were prepared for sequencing by first undergoing total RNA isolation, rRNA subtraction, reverse transcription, and random amplification into cDNA libraries. For RNA extraction, we used the MasterPureTM Complete DNA and RNA Purification kit according to the manufacturer’s protocols. After DNase treatment, pellets were resuspended in 30 μl of TE buffer and 1 μl of RiboGuardTM was added to each tube. Next, we performed ribosomal subtraction using Ribo-ZeroTM Gold (magnetic beads, human/rat/mouse kit). While we did not design our experiment to explicitly test the efficacy of Ribo-ZeroTM Gold on the removal of mosquito ribosomal RNA (rRNA), we note that the final percentage of mosquito 28S and 18S rRNA per library was, on average, 29.5% and 2.5%, respectively (data not shown). This was determined using the default settings of Bowtie 2 (Langmead et al., 2009) to map the quality checked reads (see below) to any contigs with a closest match to insect large subunit (LSU) and small subunit (SSU) rRNA.
Samples then underwent random reverse transcription followed by random amplification. Samples were tagged with a molecular ID tag (MID) and pooled equimolar for a single flow cell of sequencing. Sequencing was done on the Illumina HiSeq 2000 at HudsonAlpha Institute for Biotechnology, using an insert size of 100 bp paired-end reads. No water or blank sample was included as a negative control [although this is recommended for future sequencing runs (Salter et al., 2014)]. Raw, unprocessed sequencing reads are available through the NCBI Short Read Database as part of BioProject PRJNA269777.
Sequence Processing and Taxonomy Assignment
An average of 35 million reads were produced per library (details for each library are available in Table 1). Sequences were quality checked and assembled using the A5 pipeline with the metagenome flag (Tritt et al., 2012). A5 combines sequence quality control, adapter trimming, and contig assembly. The SGA software package removes low quality reads and corrects sequencing errors1 and Tagdust removes sequencing adapter contamination (Lassmann et al., 2009). Cleaned sequences are used to build contigs with the IDBA-UD assembler (Peng et al., 2012).
To identify any viral sequences associated with these mosquitoes, contigs were translated into all six frames and any open reading frames (ORFs) longer than 100 amino acids in length were further examined. These ORFs were queried against a custom blast database using the blastp algorithm. We used an e-value cutoff of 1 × 10-3, which is approximately equivalent, for our database, to 30% similarity over 100 amino acids and 20% similarity over 500 amino acids. This custom database contains the entire NCBI non-redundant protein database, the PhAnToMe phage protein database (Aziz et al., 2012), the NCBI Viral RefSeq Database, and the Aedes aegypti, Anopheles gambiae, and Culex quinquefasciatus protein databases (Megy et al., 2012). The rationale for combining databases was to ensure that viral and phage proteins were present, while simultaneously reducing the false positive rate by the inclusion of all possible non-viral or non-phage proteins. While none of the mosquitoes examined in this study have their complete genomes sequenced, we note that this would not reduce our ability to detect viral sequences; rather this simply increases the false positive rate. All ORFs with a closest match to “virus” or “phage” were manually queried to the NCBI website to confirm their identity. Confirmed viral sequences are available on NCBI through the GenBank accession numbers KP642114 to KP642128.
To identify any bacterial and fungal sequences in the datasets, the non-translated contigs were queried using the blastn algorithm (Altschul et al., 1990) to the SILVA SSU and LSU Reference Databases (Release 111; Quast et al., 2013). Contigs with a closest match to “Bacteria” and “Fungi” when queried to the SSU and LSU databases, respectively, and were longer than the 300 bp, were then submitted to Ribosomal Database Project’s (RDP) Classifier for taxonomic assignment (Wang et al., 2007). Contigs above a 90% genus (SSU) or 70% family (LSU) confidence cutoff were considered reliable hits (Tables 3 and 4). All intermediate files, include blast results and fasta files of significant hits are available at http://dx.doi.org/10.6084/m9.figshare.1247641.
To identify the mosquito host taxa, the non-translated contigs were queried to a custom-made database of mosquito COI genes. Matches were manually queried to the NCBI website to confirm their identity. Mosquito COI sequences are available on NCBI through the GenBank accession numbers KP293419 to KP293425.
Viral Phylogenetic Analysis and Coverage Estimation
Phylogenetic analysis was performed by comparing the complete and contiguous sequence of each identified viral ORFs to related taxa. Alignments were performed using MAFFT v7.058 and the E-INS-i algorithm (Katoh and Standley, 2013). This alignment algorithm is suitable for sequences that contain multiple conserved regions embedded in long unalignable regions2. Bayesian analysis was performed using MrBayes v3.1.2 (Ronquist and Huelsenbeck, 2003). The substitution model was determined by allowing MrBayes to sample across the fixed amino acid rate matrices. For each ORF, two independent chains were run for 1,000,000 generations. The resulting average standard deviation of split frequencies is indicated in the caption to each figure. Tracer v1.5.0 was used to confirm the stationarity of log likelihoods (Rambaut et al., 2013) and the first 25% of the 10,000 total trees were discarded. Results were visualized using FigTree v1.4.0 (Rambaut, 2012; Figures 2–5). All trees are available at http://dx.doi.org/10.6084/m9.figshare.1247641. In each case, the alignments contain highly divergent regions without homologs in all taxa, which are encoded with gaps to represent missing data. To confirm these gap sections do not significantly affect reconstruction, we repeated all phylogenetic analyses using only sites that contain data from at least 50% of the taxa. Phylogenetic reconstruction using MrBayes on these shortened alignments found very similar topologies as presented in Figures 2–5, and did not change any conclusions (phylogenetic trees resulting from shortened alignments available at http://dx.doi.org/10.6084/m9.figshare.1247641).
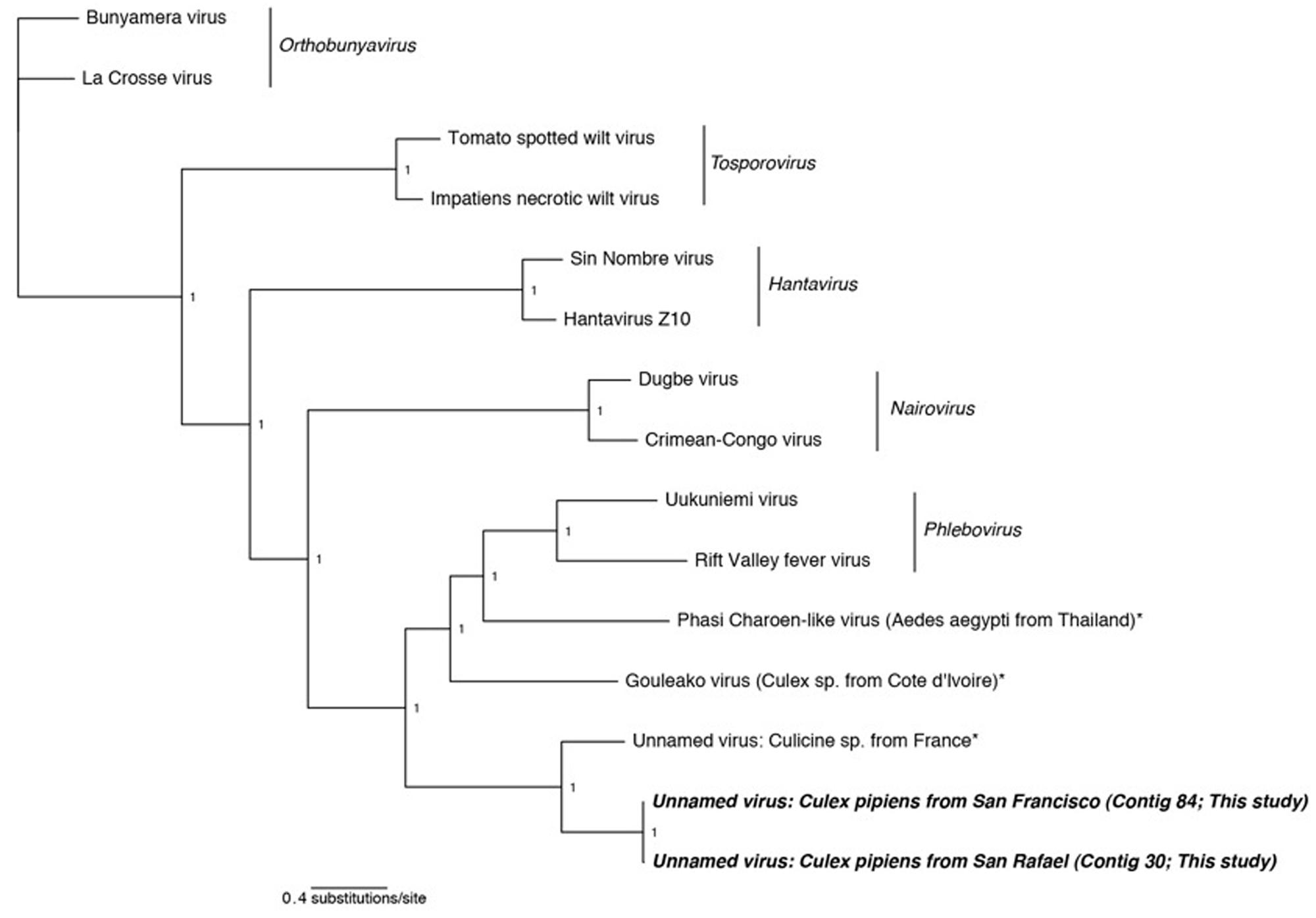
FIGURE 2. Phylogenetic history of all genera in the Bunyaviridae including sequences from this study. This consensus phylogeny was generated in MrBayes (Ronquist and Huelsenbeck, 2003) based on two independent chains run for one million generations to convergence. The average standard deviation of split frequencies was 0.000008. Posterior node probabilities are shown at nodes. Branch lengths are scaled to substitutions/site. Viral sequences uncovered in this study are labeled as such. Closely related mosquito-associated viruses are marked with an asterisk. The accession numbers of all sequences are available at http://dx.doi.org/10.6084/m9.figshare.1247641.
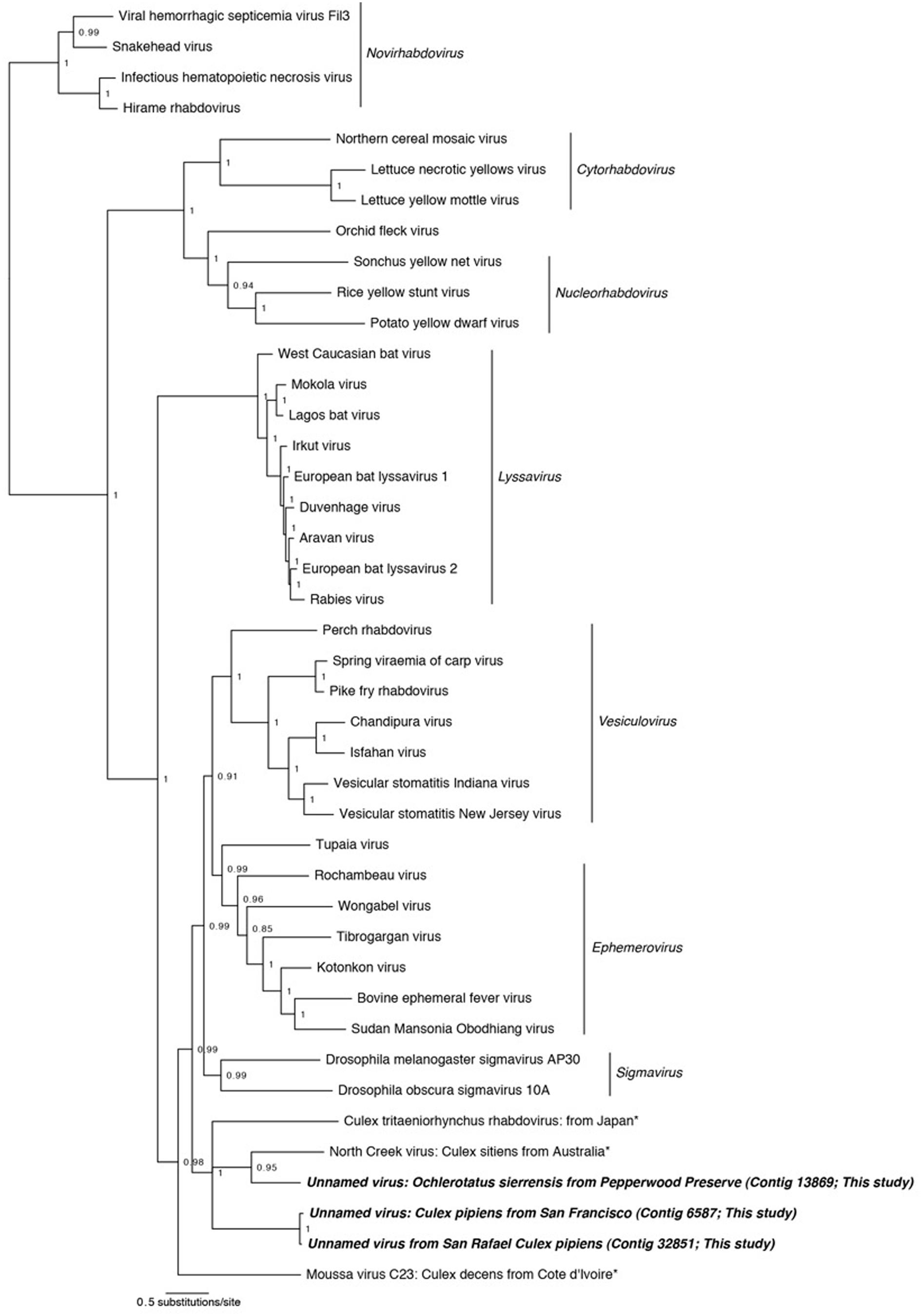
FIGURE 3. Phylogenetic history of the Rhabdoviruses including sequences from this study. This consensus phylogeny was generated in MrBayes (Ronquist and Huelsenbeck, 2003) based on two independent chains run for one million generations to convergence. The average standard deviation of split frequencies was 0.002810. Posterior node probabilities are shown at nodes. Branch lengths are scaled to substitutions/site. Viral sequences uncovered in this study are labeled as such. Closely related mosquito-associated viruses are marked with an asterisk. The accession numbers of all sequences are available at http://dx.doi.org/10.6084/m9.figshare.1247641.
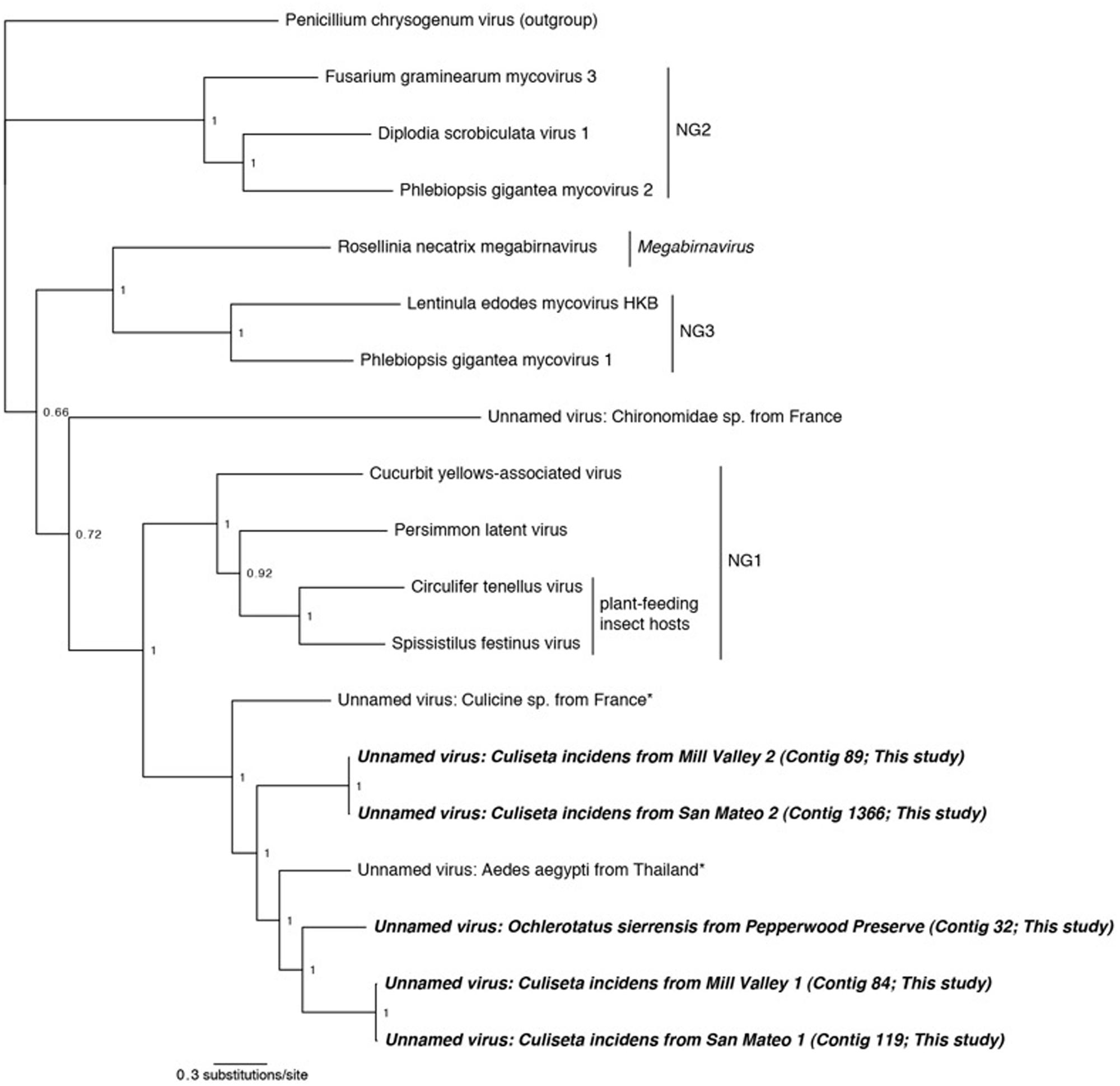
FIGURE 4. Phylogenetic history of select dsRNA viruses including sequences from this study and Chandler et al. (2014). This consensus phylogeny was generated in MrBayes (Ronquist and Huelsenbeck, 2003) based on two independent chains run for one million generations to convergence. The average standard deviation of split frequencies was 0.000948. Posterior node probabilities are shown at nodes. Branch lengths are scaled to substitutions/site. Viral sequences uncovered in this study are labeled as such. Closely related mosquito-associated viruses are marked with an asterisk. As in Ito et al. (2013), Penicillium chrysogenum virus is designated the outgroup. Proposed genera (Ito et al., 2013) are designated NG1, NG2, and NG3. The accession numbers of all sequences are available at http://dx.doi.org/10.6084/m9.figshare.1247641.
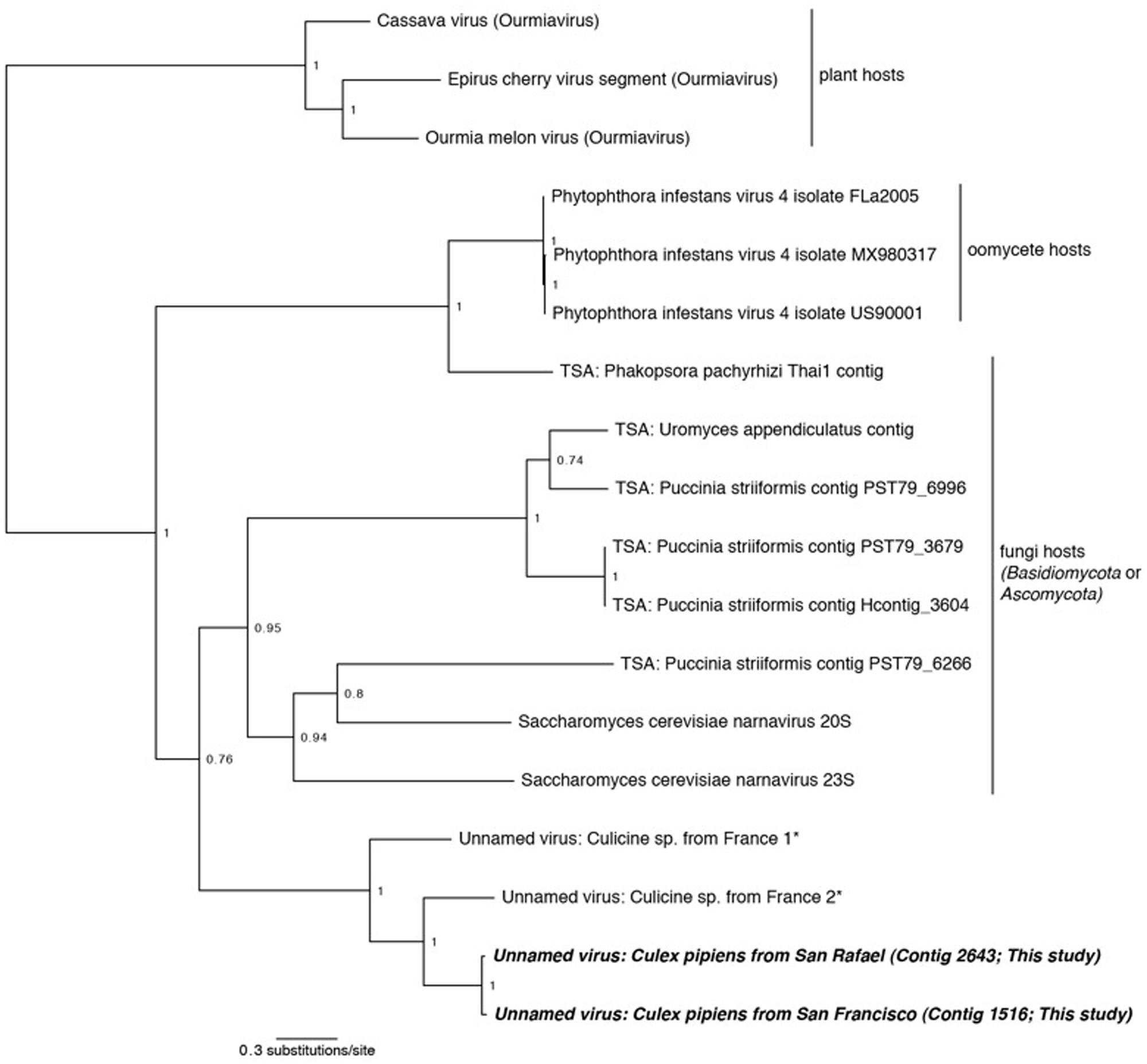
FIGURE 5. Phylogenetic history of the Narnaviruses including sequences from this study. This consensus phylogeny was generated in MrBayes (Ronquist and Huelsenbeck, 2003) based on two independent chains run for one million generations to convergence. Ourmiaviruses, which share the RNA-dependent RNA polymerase through a genomic rearrangement, are included as the outgroup. The average standard deviation of split frequencies was 0.004048. Posterior node probabilities are shown at nodes. Branch lengths are scaled to substitutions/site. Viral sequences uncovered in this study are labeled as such. Closely related mosquito-associated viruses are marked with an asterisk. Sequences labeled TSA were uncovered in the NCBI Transcriptome Shotgun Assembly database (Cook et al., 2013). The accession numbers of all sequences are available at http://dx.doi.org/10.6084/m9.figshare.1247641.
Coverage was determined by mapping the quality checked reads to the nucleotide sequences corresponding to the viral ORFs using Bowtie 2 and default settings (Langmead et al., 2009).
Results and Discussion
In this study, we used RNA SMS to characterize the microbial communities associated with three mosquito species in northern California: Culex pipiens, Culiseta incidens, and Ochlerotatus sierrensis (previously Aedes sierrensis). Cx. pipiens (common house mosquito) is a known vector for West Nile virus and St. Louis encephalitis virus (Farajollahi et al., 2011), C. incidens (cool-weather mosquito) is not a major vector for West Nile virus or any other disease, and O. sierrensis (western treehole mosquito) is a major vector for dog heartworm (Ledesma and Harrington, 2011). Samples were collected from a variety of habitats including sylvatic/wild, rural, suburban, and urban (sensu Thongsripong et al., 2013). All three of these species have limited ranges (flight range <5 miles), making them good markers for the respective habitats in which they are found. Only female mosquitoes were used, as female mosquitoes, being hematophagous, are the primary vectors of most human and animal diseases.
As many clinically important mosquito-vectored viruses are RNA based (for example, certain Alphaviruses, Bunyaviridae, and Flaviviruses), we focused on the RNA metagenome of these mosquitoes. Nearly 250 million 100 bp paired-end reads were generated on the Illumina HiSeq platform, roughly evenly distributed among the seven libraries (Table 1). After quality control and contig assembly, the identity of the viral sequences associated with these samples was determined by querying the translated contigs against a custom database containing the entire NCBI non-redundant protein database, the PhAnToMe phage protein database (Aziz et al., 2012), the NCBI Viral RefSeq Database, and the Ae. aegypti, An. gambiae, and Cx. quinquefasciatus protein databases (Megy et al., 2012). The identity of the bacterial and fungal sequences associated with these samples was determined by first querying the non-translated contigs to the SILVA SSU and LSU Reference Databases (Quast et al., 2013). Any contigs with a closest match to either bacteria or fungi were then submitted to the RDP Classifier (Wang et al., 2007). Despite using Ribo-ZeroTM to deplete vertebrate rRNA, adequate bacterial and fungal RNA remained for microbial classification.
Viral Communities
Consistent with previous studies (Cook et al., 2013; Chandler et al., 2014; Coffey et al., 2014), viral sequences from the lineages Bunyaviridae, Rhabdoviridae, and Narnavirus were uncovered in these mosquitoes (Table 2). In addition, we also detected sequences related to an unassigned double-stranded RNA virus (dsRNA) which has been found to be associated with insects (Spear et al., 2010; Cook et al., 2013). No single viral taxon was found in all samples; however, members of the dsRNA group and the Rhabdoviridae were detected in three samples. Interestingly, the San Rafael Cx. pipiens sample and the San Francisco Cx. pipiens sample each contained representatives of the same three viral taxa – Bunyaviridae, Rhabdoviridae, and Narnavirus. Using the methods employed here, viral sequences were not detected in the Stinson Beach or Bolinas Cx. pipiens samples. If viruses are present in these samples, they fall below our detection limit (see Section “Materials and Methods”).
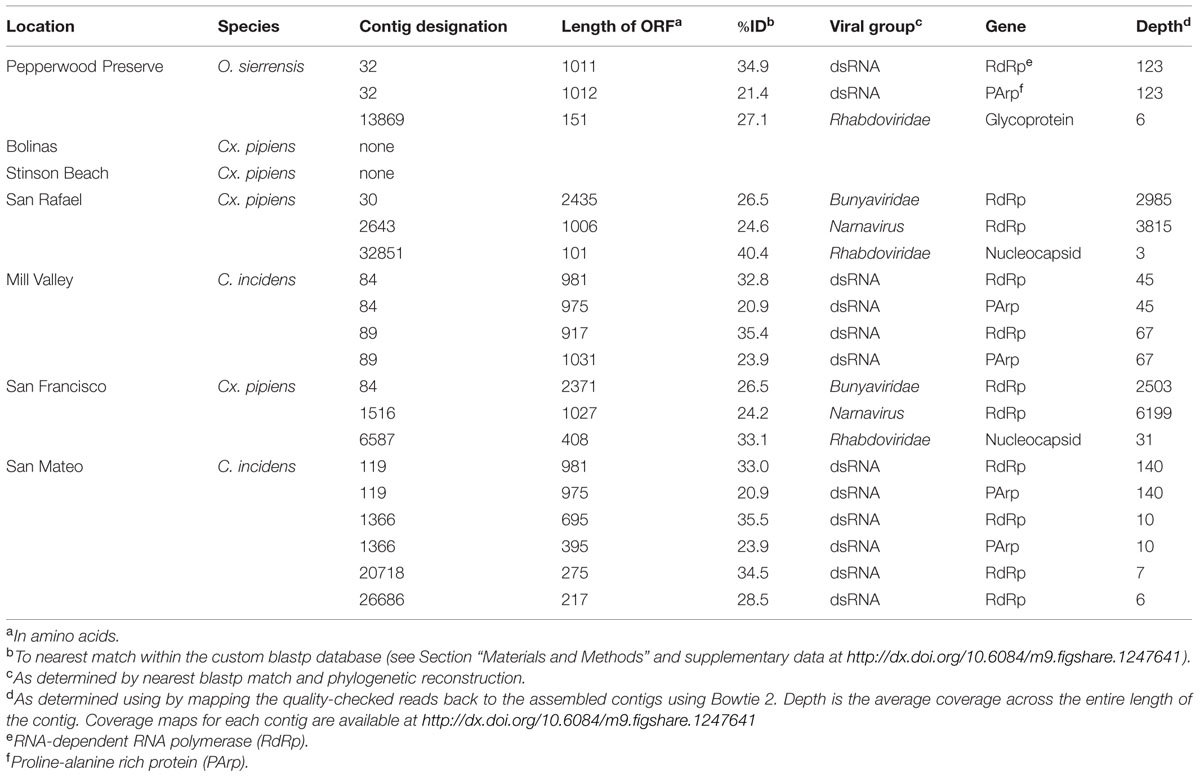
TABLE 2. Viral sequences identified in northern California mosquitoes.
Bunyaviridae
In two samples, Cx. pipiens from San Francisco and Cx. pipiens from San Rafael, we uncovered sequences related to the Bunyaviridae L segment. There are five recognized Bunyaviridae genera (Phlebovirus, Hantavirus, Nairovirus, Orthobunyavirus, and Tospovirus), and all except Hantavirus are known to be arthropod-vectored. The Bunyaviridae have single-stranded negative-sense genomes consisting of three segments. In the genus Phlebovirus (which our sequences are most closely related to, see next paragraph), these segments are designated L (encoding the RNA-dependent RNA polymerase), M (encoding the glycoprotein), and S (encoding the nucleocapsid and an ambisense non-structural gene, the NSs, which is necessary for virulence in vertebrates).
Phylogenetic analysis indicates that the detected Bunyaviridae sequences are sister to the Phlebovirus genus, which includes the agent of Rift Valley fever, and relatives such as Gouléako virus (Figure 2). This is in congruence with several other mosquito-associated Bunyaviridae that have been found in Thailand (Chandler et al., 2014), France (Cook et al., 2013), and West Africa (Marklewitz et al., 2011). Although the two uncovered sequences are more closely related to each other than to any other taxa, these two sequences differ by approximately 6 amino acids substitutions and a 64 amino acid insertion/deletion at the terminal end (alignments available at http://dx.doi.org/10.6084/m9.figshare.1247641).
Attempts to find the M and S Bunyaviridae segments using specific blastp and vFam searches (Skewes-Cox et al., 2014) were unsuccessful (data not shown). While this is surprising given the depth of coverage of the L segment (Table 2), we note that a previous metagenomic shotgun sequencing study which found a Bunyaviridae L segment likewise did not find either the M or the S segments (Cook et al., 2013). The inability to recover these segments may reflect their greater degree of divergence than the L segment, or their reduced representation in the libraries. Targeted amplification of the missing segments by PCR could compensate for the latter, but may have limited success in the case of divergence. Previous work finds that some mosquito viruses lack the NSs gene, which is one of two genes, along with the nucleoprotein, present on the S segment (Marklewitz et al., 2011; Chandler et al., 2014). The absence of the NSs, which is found in all Phleboviruses and is necessary for virulence in vertebrates, and the presence of the nucleoprotein, which is required for replication, is suggestive of an arthropod-only lifecycle in these viruses (Marklewitz et al., 2011; Chandler et al., 2014).
The nucleic acid contigs of both Bunyaviridae detected here included stop codons on either side of the ORF suggesting that the entire ORF was found. However, the terminal hairpins that are present in all Bunyaviridae were not recovered, despite implementing PRICE (Ruby et al., 2013) to extend the fragments (data not shown). This suggests that the entire L segment was not present in our data.
Rhabdoviridae
In three of our samples, O. sierrensis from Pepperwood Preserve, Cx. pipiens from San Rafael, and Cx. pipiens from San Francisco, we detected sequences related to the family Rhabdoviridae. The Rhabdoviridae is a diverse family in the order Mononegavirales and consists of six established genera (Vesiculovirus, Lyssavirus, Ephemerovirus, Novirhabdovirus, Cytorhabdovirus, and Nucleorhabdovirus) along with more than 130 unassigned viruses, such as the Drosophila-associated Sigma viruses (Kuzmin et al., 2009; Longdon et al., 2010). Rhabdoviridae are the causative agents of numerous clinically and economically important diseases of humans, livestock, fish, and plants – some of which are arthropod-vectored (Kuzmin et al., 2009). The genome of the Rhabdoviridae consists of a single negative-sense RNA segment with, at minimum, five ORFs corresponding to the nucleoprotein, glycoprotein, phosphoprotein, matrix protein, and the RdRp.
Using our methods, we uncovered partial fragments of the Rhabdoviridae nucleoprotein (Cx. pipiens from San Rafael and Cx. pipiens from San Francisco) and the Rhabdoviridae glycoprotein (O. sierrensis from Pepperwood Preserve). Since the RdRp is the preferred gene for phylogenetic reconstruction (Bourhy et al., 2005; Longdon et al., 2010), we searched for it within these three libraries using specific blastp and vFam searches (Skewes-Cox et al., 2014), but did not recover the RdRp in our datasets. This is unsurprising since the depth of coverage for the three uncovered fragments was very low compared to many of the other uncovered viruses (Table 2).
Phylogenetic analysis was performed using MrBayes on concatenated alignments of the nucleoprotein and glycoprotein fragments against reference sequences for these regions (Figure 3). The resulting phylogeny successfully recapitulates the relationships between Rhabdoviridae lineages ((((Vesiculovirus, Ephemerovirus), Sigma viruses), Lyssavirus), (Cytorhabdovirus, Nucleorhabdovirus), Novirhabdovirus) found by other studies (Longdon et al., 2010; Coffey et al., 2014). Our mosquito-associated Rhabdoviridae form a clade with North Creek virus, isolated from Cx. sitiens in Australia (Coffey et al., 2014) and a unnamed virus isolated from Cx. tritaeniorhynchus in Japan (Kuwata et al., 2011). This mosquito-associated clade is sister to the Vesiculovirus, Ephemerovirus, and Sigma virus clades; basal to these is the mosquito-associated Moussa virus (Quan et al., 2010).
Double-Stranded RNA Viruses
In three libraries, we identified sequences that are related to dsRNA viruses previously identified in culicine mosquitoes from France (Cook et al., 2013). These viruses are related to, and have a similar genome structure as, viruses isolated from the plant-feeding insects Circulifer tenellus and Spissistilus festinus (Spear et al., 2010). The genomes of these viruses consist of a single segment with two ORFs, an RNA-dependent RNA polymerase (RdRp) and a proline–alanine rich protein (PArp) of unknown function. Phylogenetic analysis of the RdRp suggests that the viruses from plant-feeding insects belong to a novel genus of dsRNA viruses (Spear et al., 2010). Phylogenetic analysis using the PArp ORF has not been performed in any previous publication.
We identified sequences related to these dsRNA viruses in three of our samples: O. sierrensis from Pepperwood Preserve, C. incidens from Mill Valley, and C. incidens from San Mateo. Inspired by the discovery of these viral sequences, we searched a prior dataset from our laboratory [Ae. aegypti from Thailand (Chandler et al., 2014)] using the methods described above and identified another dsRNA virus. In several instances, a single contig spanned the entire RdRp and PArp ORFs (contig 32 in the O. sierrensis sample from Pepperwood Preserve, contigs 84 and 89 in the C. incidens sample from Mill Valley, and contig 119 in the C. incidens sample from San Mateo; Table 2). In the San Mateo sample, one low coverage contig contains partial sequences of both the RdRp and the PArp (contig 1366) and two additional low coverage contigs (contigs 20718 and 26686) contain fragments of the RdRp that partially overlap with, and are nearly identical to, the contig containing the partial sequences.
Phylogenetic analysis was performed using the complete RdRp ORFs and the partial ORF from the San Mateo sample (contig 1366). Viruses from three proposed novel genera, one Megabirnavirus, and Penicillium chrysogenum virus were included as comparison taxa (Spear et al., 2010; Ito et al., 2013). Our phylogeny reproduces the overall structure of previous work (Ito et al., 2013) and places all mosquito-associated viruses together (Figure 4). Interestingly, both the San Mateo and Mill Valley samples have two distinct dsRNA contigs. This suggests that multiple strains of this dsRNA virus are co-circulating within the northern California C. incidens population, a situation similar to that occurring in Circulifer tenellus in central California (Spear et al., 2013).
In the dsRNA plant-feeding insect viruses, the RdRp and PArp ORFs are not in frame, but are suspected to be transcribed as a fusion via a -1 ribosomal frameshift just prior to the stop codon of the 5′ (PArp) ORF. The site of this frameshift is predicted to occur at a G_GAA_AAC_stop motif in the virus of French culicine mosquitoes (Cook et al., 2013). In all of our contigs, the PArp and the RdRp were a single base out of frame. In three of our contigs (84 from Mill Valley C. incidens, 32 from Pepperwood Preserve Cx. pipiens, and 119 from San Mateo C. incidens), we identified a G_GAA_AAC_stop motif and in the remaining contigs (89 from Mill Valley C. incidens and 1366 from San Mateo C. incidens), we identified a G_GGA_AAC_stop motif. This suggests that the two dsRNA ORFs found here are transcribed as a fusion, although enabled by slightly different motifs.
Narnavirus
Two of our samples, Cx. pipiens from San Rafael and Cx. pipiens from San Francisco, included sequences that are related to the genus Narnavirus in the family Narnaviridae. Narnaviruses have single-stranded positive-sense genomes consisting of a single ORF that encodes an RdRp (King et al., 2012). The type strain Saccharomyces 20S virus infects the Ascomycota Saccharomyces cerevisiae, while similar sequences have been found associated with Basidiomycota fungi (Cook et al., 2013) and an oomycete plant pathogen (Cai et al., 2013). While the sister genus to Narnavirus is the mitochondrial-infecting Mitovirus, the RdRp of plant-infecting Ourmiavirus is more closely related to Narnavirus, as it was acquired via reassortment (Rastgou et al., 2009), and is therefore used as the outgroup in Figure 5.
Phylogenetic analysis finds that the Cx. pipiens Narnaviruses are more closely related to the other mosquito-associated Narnaviruses (Cook et al., 2013) than to any other taxa (Figure 5). Since most of the known Narnaviruses are suspected to infect Ascomycete or Basdiocymete fungi [the only exception being the oomycete-infecting Phytophthora infestans RNA virus (Cai et al., 2013)], we investigated the presence and identity of potential host fungal sequences in our samples (see Section “Fungal Communities”). We also used the methods described in Section “Sequence Processing and Taxonomy Assignment” to identify fungal sequences in the French mosquito dataset that contained two Narnavirus sequences (Cook et al., 2013). We did not find any oomycete SSU or LSU sequences in any of the three datasets (data not shown), thus ruling out that the mosquito-associated Narnaviruses are in fact associated with the presence of oomycetes.
All three mosquito samples positive for Narnaviruses also included sequences of Ascomycete fungi. Although no single fungal genus was common to all three mosquito samples, several [such as Cladosporium and Capnodiales (see Section “Fungal Communities”)] were shared between the two northern California samples. Considering that the mosquito-associated Narnavirus clade is sister to Narnaviruses of fungi, and that fungal sequences were also detected in the mosquitoes, the Narnaviruses from mosquitoes reported here and by Cook et al. (2013) may in fact be instances of virus-infected fungal infections of mosquitoes. Alternatively, these mosquito-associated Narnaviruses may represent direct infections of mosquitoes. However, Saccharomyces 20S RNA virus is vertically transmitted from mother to daughter cells and horizontally through mating (King et al., 2012), suggesting that host switching may be biologically improbable, particularly from a fungal host to a mosquito host. Taken together, our approach may have revealed an interesting scenario: since the method employed here simultaneously characterized both viruses and fungi within a given sample, it can identify patterns of multi-level nested host-parasite associations. In this case, we have identified putative fungal infecting viruses and the fungi they may be infecting, all in the same hosts. This illustrates the potential utility of our broad approach to microbiota characterization – one that could reveal microbial interactions that may ultimately lead to the development of infectious disease control measures.
Bacteriophages
No phage sequences were uncovered in this dataset, despite including the PhAnToMe phage database in our blastp search. Given that these mosquitoes were associated with a diversity of bacteria (see Section “Bacterial Communities”) and that the microbiota of other animals consists of abundant phage (De Paepe et al., 2014), it is reasonable to presume that phages are indeed associated with these mosquitoes as well. There exist two explanations for our inability to find phage associated with these samples. First, our dataset was RNA based and most phages are DNA based. Indeed, known phages of the two most widespread bacteria associated with these samples (Bacillus and Escherichia/Shigella; see Section “Bacterial Communities”) are double-stranded DNA based. Second, perhaps the phages infecting the bacteria within these mosquitoes are undescribed and therefore would not be represented in the databases.
Bacterial Communities
Contigs that could be confidently identified as bacteria using the RDP Classifier were found in all seven samples (Table 3). Since there are known limitations of using rRNA as a measurement of either microbial abundance or activity (Blazewicz et al., 2013), the quality-checked reads were not mapped back to these contigs to determine the number of reads used to build each contig. Therefore the data presented here (and of the fungi discussed below) should only be interpreted as representing the presence of that particular taxon and not that taxon’s abundance within the community.
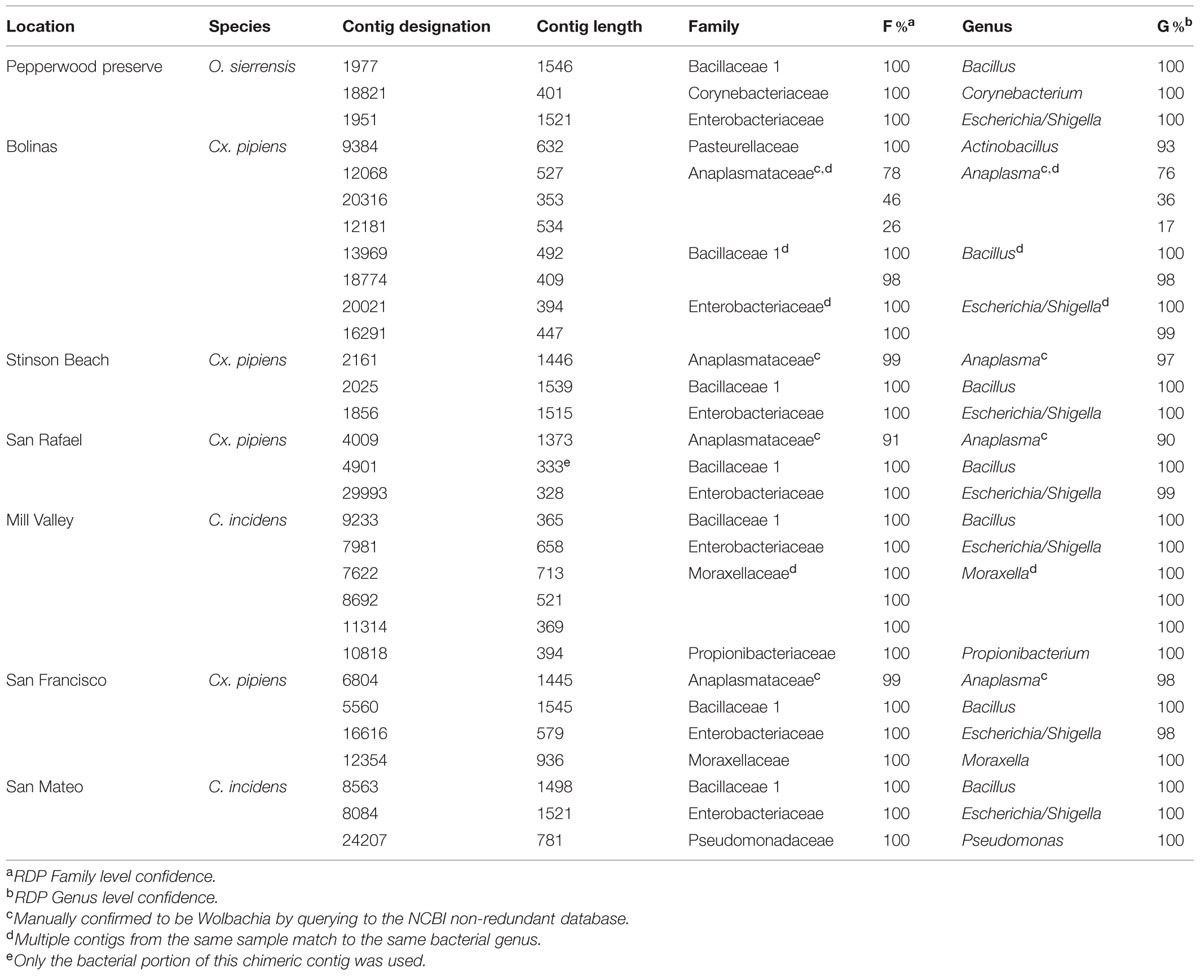
TABLE 3. Bacterial sequences associated with northern California mosquitoes.
Contigs (those that passed the initial 300 bp cutoff) ranged from 328 to 1546 bases in length, which means that some cover nearly the entire 16S gene and all cover at least one hyper-variable region. A single contig with a bacterial match (Bacillus in the Mill Valley C. incidens sample) was found to be chimeric (data not shown) and only the bacterial portion was used for classification. We identified the following bacterial genera in our samples: Actinobacillus, Anaplasma [Wolbachia (see below)], Bacillus, Corynebacterium, Escherichia/Shigella, Haemophilus, Moraxella, Propionibacterium, Pseudomonas and Wandonia. When a genus was found multiple times in the same sample (for example, Moraxella in the Mill Valley C. incidens sample), the contigs spanned different regions of the 16S gene (see blastn results at http://dx.doi.org/10.6084/m9.figshare.1247641).
Bacillus and Escherichia/Shigella were the most widespread genera, as they were found in all seven samples. Notably, a recent review by Minard et al. (2013) concludes that female mosquitoes are mostly colonized by Gammaproteobacteria (of which Escherichia/Shigella is a member) and that males are dominated by Firmicutes, such as Bacillus. The results of our study, in which only females were investigated, are therefore in partial agreement with this conclusion.
Anaplasma is a genus in the order Rickettsiales. Wolbachia is a notable and widespread arthropod-associated bacteria in this order (Hilgenboecker et al., 2008) and we therefore investigated the Anaplasma contigs further. Querying these contigs to the NCBI database finds them to be greater than 98% identical to Wolbachia pipiens (data not shown). The two samples containing Wolbachia were Cx. pipiens from San Rafael and Stinson beach. The third Cx. pipiens sample, from Bolinas, also had contigs identified as Anaplasma, although their confidence was below the 90% cutoff in RDP (Table 3). Querying these contigs to NCBI found they were greater than 99% identical to Wolbachia pipiens (data not shown, all contigs are available at http://dx.doi.org/10.6084/m9.figshare.1247641). Inspection of the contigs from C. incidens and O. sierrensis that were shorter than 300 bp or below the 90% confidence cutoff did not reveal any Anaplasma or Wolbachia-like sequences.
Wolbachia was first described in Cx. pipiens mosquitoes (Hertig and Wolbach, 1924). A survey of California mosquitoes (including Culex, C. incidens, and Ochlerotatus) for Wolbachia infections found that, out of 296 individuals and 14 species, only Cx. pipiens mosquitoes were infected (Rasgon and Scott, 2004). How Wolbachia, which is vertically transmitted, spreads into new taxa is not well understood, but its absence in certain species could be due to inhibition by other members of the mosquito microbiota, as has been shown in Anopheles mosquitoes (Hughes et al., 2014). While this study did not find any consistent microbiota differences between Wolbachia-infected and Wolbachia-free mosquitoes, more comprehensive studies may find such differences that might help account for natural Wolbachia distributions.
Of final note, two of the bacterial genera identified, Propionibacterium and Corynebacterium, are commonly associated with human skin (Grice and Segre, 2011). There are two potential explanations for their presence in these samples. One is that they were acquired from human hosts during a blood meal. Alternatively, these could be contaminants introduced during collection or processing for sequencing. Future studies will include a negative control to identify this possibility (Salter et al., 2014).
Fungal Communities
Contigs that could be confidently classified as fungi using the RDP Classifier were found in all samples except O. sierrensis from Pepperwood Preserve (Table 4). Manually querying the putative fungal contigs (identified by the initial SILVA database search) from this sample against the NCBI database confidently identified a fungus that was nonetheless below the RDP confidence cutoff (Table 4).
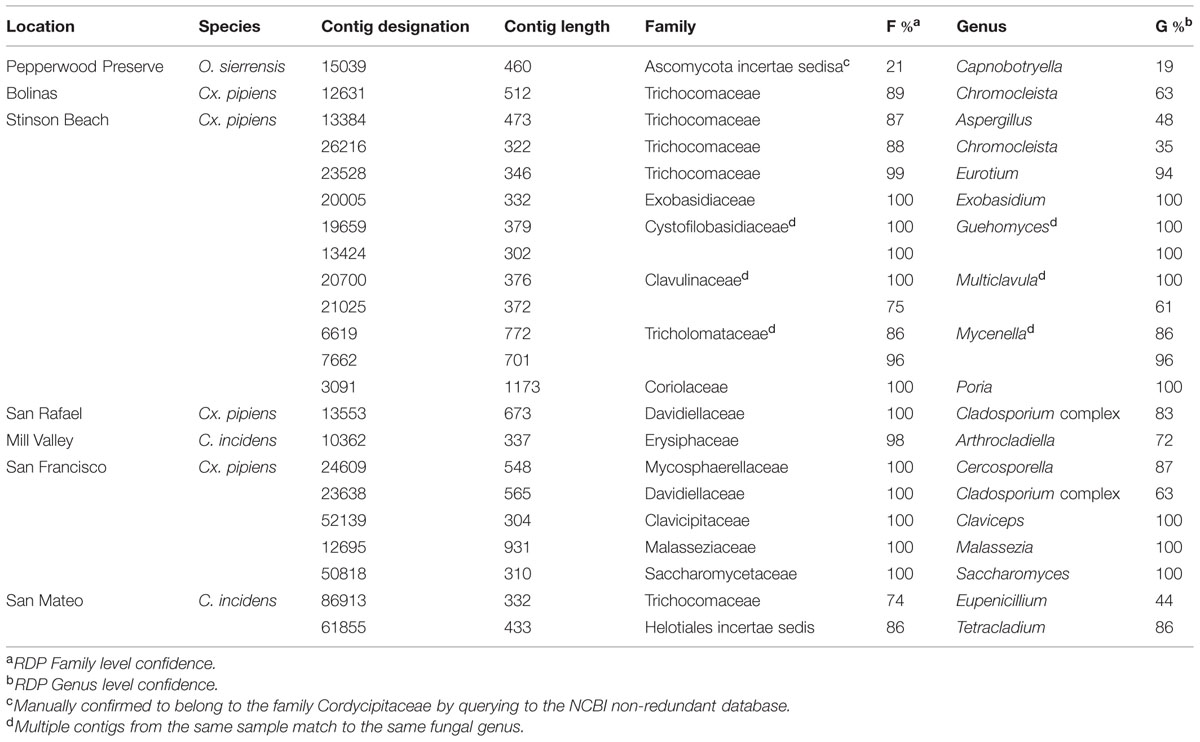
TABLE 4. Fungal sequences associated with northern California mosquitoes.
Fungal contigs ranged in length from 302 to 1173 bp. Fungi from 13 families were present in our data (Table 4). In contrast to the bacterial data, there is no single fungal genus that is present in all samples and only two fungi were found in multiple samples. Specifically, Chromocleista was found in Cx. pipiens from both Stinson Beach and Bolinas, and Cladosporium was found in Cx. pipiens from both San Rafael and San Francisco. All the remaining fungal taxa identified were unique to a single sample. More in-depth sequencing may reveal widespread fungi that exist at numbers within the host too low for our methods to detect.
Some of the fungi identified within these samples may not exist in an intimate relationship with mosquitoes. For example, several taxa within the Basidiomycota have a characteristic multicellular stage, such as the Mycenella gilled mushrooms identified in the Cx. pipiens sample from Stinson Beach. Most likely, dispersing spores of these fungi contaminated water sources used by adult mosquitoes as breeding sites. Similarly, the fungal taxa Malassezia, identified in Cx. pipiens from San Francisco, is a common human commensal and opportunistic pathogen (Gupta et al., 2004) that likely contaminated the sample through human contact, as with the bacteria discussed above.
Much previous work has focused on using entomopathogenic fungal taxa for mosquito control (Scholte et al., 2004; de Faria and Wraight, 2007). Interestingly, several of the fungi identified belong to families that include many known entomopathogenic fungi including Clavicipitaceae (found in the Cx. pipiens sample from San Francisco) and Cordycipitaceae (found in the O. sierrensis sample from Pepperwood Preserve). For future mosquito collections, histologic examination would be necessary to identify visible signs of disease in positive mosquitoes.
This study represents, to our knowledge, the first to use culture-independent techniques to characterize fungal communities in natural populations of mosquitoes. As with other animal hosts [e.g., humans (Huffnagle and Noverr, 2013) and Drosophila (Broderick and Lemaitre, 2012)], fungi associated with mosquitoes are relatively understudied compared to bacteria. One study of laboratory-raised Ae. aegypti identified Saccharomyces, Penicillium, and Aspergillus using methods similar to those employed here (Bishop-Lilly et al., 2010). While the current study did find these, or closely related, taxa, more work is needed to fully determine how the fungal communities of laboratory-raised and wild-caught mosquitoes compare.
Mosquito Identification
Five of the seven mosquitoes were identified morphologically or by PCR amplification and Sanger sequencing of the COI gene prior to SMS. For the two specimens that were not identified beforehand, contigs matching mosquito COI and mosquito LSU and SSU were queried against the NCBI database and indicated that these mosquitoes were Cx. pipiens and O. sierrensis (data not shown, all contigs available at http://dx.doi.org/10.6084/m9.figshare.1247641).
Conclusion
In this study, we used SMS to identify viral, bacterial, and fungal sequences associated with naturally collected mosquitoes. Notably, in all three cases of mosquito-associated viruses (i.e., the Bunyaviridae, Rhabdoviridae, and dsRNA viruses) the viruses described here were more closely related to other mosquito-associated viruses than to any other viruses. This is despite the fact that the mosquitoes belonged to a variety of taxa (e.g., Ae. aegypti, C. incidens, O. sierrensis, and several species of Culex) and were collected from a variety of locations (e.g., northern California, Thailand, France, western Africa, and Australia). Furthermore, it should be noted that two of the viral groups discussed here (Bunyaviridae and Rhabdoviridae) have been extensively studied due to their importance in human and animal health and are therefore well characterized in terms of host breadth. Taken together, a pattern of family level host-specificity (i.e., to the family Culicidae) is emerging. We look forward to future research to sample the viral taxa more extensively and help answer the question of host-specificity and host-switching in the viral tree of life.
With respect to the bacterial and fungal components of the mosquito microbiota, two opposing patterns emerged: several bacterial taxa were widespread regardless of mosquito species or habitat, whereas fungal communities were distinct between individuals even within host species or habitat. These patterns may be an artifact of insufficient sample size and sequencing depth. Much of our sequencing effort was needlessly expended on host rRNA, as over a quarter of the total data can be attributed to the mosquito ribosomal large subunit or small subunit. However, recent techniques have been developed to selectively remove sample-specific rRNA from environmental (Stewart et al., 2010) and mosquito (Kukutla et al., 2013) samples. The use of these techniques will greatly increase the power to detect bacteria, fungi, and viruses in field-caught mosquitoes. Furthermore, many more samples could be combined into a single experiment to allow for the inclusion of multiple replicates per population, host species, or habitat type. Additionally, the physical environment of the mosquitoes, such as larval breeding sites and attractants used in collection, can be sampled. In this way, the complex interactions between host, environment, and all the microbes inhabiting both, can be determined.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the California Academy of Sciences’ Student Science Fellows high school enrichment program for support, especially Roberta Brett, Sean Edgerton, and Ryan Hulett. This program was generously supported by the Gordon and Betty Moore Foundation, with additional support provided by Mary Austin and Brewster Kahle. V. Piper Kimball and team members of the Marin/Sonoma Mosquito and Vector Control District executed mosquito collections at Stinson Beach, Bolinas, and San Rafael. Durrell Kapan assisted in other mosquito collections. Panpim Thongsripong, Barbara Arrighi, and Anna Sellas provided technical assistance. We thank Jonathan Eisen, Jack Dumbacher, and Joseph DeRisi for use of their servers.
Footnotes
- ^ https://github.com/jts/sga/tree/master/src
- ^ http://mafft.cbrc.jp/alignment/software/algorithms/algorithms.html
Supplementary Material
The supplemental data associated with this work is available at: http://dx.doi.org/10.6084/m9.figshare.1247641. This includes alignments, tree files, blast outputs, and other intermediate files.
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Amann, R. I., Ludwig, W., and Schleifer, K. H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143–169.
Ashelford, K. E., Chuzhanova, N. A., Fry, J. C., Jones, A. J., and Weightman, A. J. (2006). New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Appl. Environ. Microbiol. 72, 5734–5741. doi: 10.1128/AEM.00556-06
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aziz, R. K., Devoid, S., Disz, T., Edwards, R. A., Henry, C. S., Olsen, G. J.,et al. (2012). SEED servers: high-performance access to the SEED genomes, annotations, and metabolic models. PLoS ONE 7:e48053. doi: 10.1371/journal.pone.0048053
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bishop-Lilly, K. A., Turell, M. J., Willner, K. M., Butani, A., Nolan, N. M. E., Lentz, S. M.,et al. (2010). Arbovirus detection in insect vectors by rapid, high-throughput pyrosequencing. PLoS Negl. Trop. Dis. 4:e878. doi: 10.1371/journal.pntd.0000878
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Blazewicz, S. J., Barnard, R. L., Daly, R. A., and Firestone, M. K. (2013). Evaluating rRNA as an indicator of microbial activity in environmental communities: limitations and uses. ISME J. 7, 2061–2068. doi: 10.1038/ismej.2013.102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bohart, R. M., and Washino, R. K. (1978). Mosquitoes of California, 3rd Edn. Berkeley, CA: University of California, Division of Agricultural Sciences.
Bourhy, H., Cowley, J. A., Larrous, F., Holmes, E. C., and Walker, P. J. (2005). Phylogenetic relationships among rhabdoviruses inferred using the L polymerase gene. J. Gen. Virol. 86, 2849–2858. doi: 10.1099/vir.0.81128-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Broderick, N. A., and Lemaitre, B. (2012). Gut-associated microbes of Drosophila melanogaster. Gut Microbes 3, 307–321. doi: 10.4161/gmic.19896
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cai, G., Krychiw, J. F., Myers, K., Fry, W. E., and Hillman, B. I. (2013). A new virus from the plant pathogenic oomycete Phytophthora infestans with an 8 kb dsRNA genome: the sixth member of a proposed new virus genus. Virology 435, 341–349. doi: 10.1016/j.virol.2012.10.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chandler, J. A., Lang, J. M., Bhatnagar, S., Eisen, J. A., and Kopp, A. (2011). Bacterial communities of diverse Drosophila species: ecological context of a host-microbe model system. PLoS Genet. 7:e1002272. doi: 10.1371/journal.pgen.1002272
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chandler, J. A., Thongsripong, P., Green, A., Kittayapong, P., Wilcox, B. A., Schroth, G. P.,et al. (2014). Metagenomic shotgun sequencing of a Bunyavirus in wild-caught Aedes aegypti from Thailand informs the evolutionary and genomic history of the Phleboviruses. Virology 464–465, 312–319. doi: 10.1016/j.virol.2014.06.036
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cirimotich, C. M., Ramirez, J. L., and Dimopoulos, G. (2011). Native microbiota shape insect vector competence for human pathogens. Cell Host Microbe 10, 307–310. doi: 10.1016/j.chom.2011.09.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Coffey, L. L., Page, B. L., Greninger, A. L., Herring, B. L., Russell, R. C., Doggett, S. L.,et al. (2014). Enhanced arbovirus surveillance with deep sequencing: identification of novel rhabdoviruses and bunyaviruses in Australian mosquitoes. Virology 448, 146–158. doi: 10.1016/j.virol.2013.09.026
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cook, S., Chung, B. Y.-W., Bass, D., Moureau, G., Tang, S., McAlister, E.,et al. (2013). Novel virus discovery and genome reconstruction from field RNA samples reveals highly divergent viruses in dipteran hosts. PLoS ONE 8:e80720. doi: 10.1371/journal.pone.0080720
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Coon, K. L., Vogel, K. J., Brown, M. R., and Strand, M. R. (2014). Mosquitoes rely on their gut microbiota for development. Mol. Ecol. 23, 2727–2739. doi: 10.1111/mec.12771
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
de Faria, M. R., and Wraight, S. P. (2007). Mycoinsecticides and mycoacaricides: a comprehensive list with worldwide coverage and international classification of formulation types. Biol. Control 43, 237–256. doi: 10.1016/j.biocontrol.2007.08.001
De Paepe, M., Leclerc, M., Tinsley, C. R., and Petit, M.-A. (2014). Bacteriophages: an underestimated role in human and animal health? Front. Cell. Infect. Microbiol. 4:39. doi: 10.3389/fcimb.2014.00039
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Donachie, S. P., Foster, J. S., and Brown, M. V. (2007). Culture clash: challenging the dogma of microbial diversity. ISME J. 1, 97–99. doi: 10.1038/ismej.2007.22
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Engel, P., Martinson, V. G., and Moran, N. A. (2012). Functional diversity within the simple gut microbiota of the honey bee. Proc. Natl. Acad. Sci. U.S.A. 109, 11002–11007. doi: 10.1073/pnas.1202970109
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Farajollahi, A., Fonseca, D. M., Kramer, L. D., and Marm Kilpatrick, A. (2011). “Bird biting” mosquitoes and human disease: a review of the role of Culex pipiens complex mosquitoes in epidemiology. Infect. Genet. Evol. 11, 1577–1585. doi: 10.1016/j.meegid.2011.08.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Foster, J. A., Bunge, J., Gilbert, J. A., and Moore, J. H. (2012). Measuring the microbiome: perspectives on advances in DNA-based techniques for exploring microbial life. Brief. Bioinform. 13, 420–429. doi: 10.1093/bib/bbr080
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fukuda, M. M., Klein, T. A., Kochel, T., Quandelacy, T. M., Smith, B. L., Villinski, J.,et al. (2011). Malaria and other vector-borne infection surveillance in the U.S. Department of Defense Armed Forces Health Surveillance Center-Global Emerging Infections Surveillance program: review of 2009 accomplishments. BMC Public Health 11:S9. doi: 10.1186/1471-2458-11-S2-S9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ghyselinck, J., Pfeiffer, S., Heylen, K., Sessitsch, A., and De Vos, P. (2013). The effect of primer choice and short read sequences on the outcome of 16S rRNA gene based diversity studies. PLoS ONE 8:e71360. doi: 10.1371/journal.pone.0071360
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Glaser, R. L., and Meola, M. A. (2010). The native Wolbachia endosymbionts of Drosophila melanogaster and Culex quinquefasciatus increase host resistance to West Nile virus infection. PLoS ONE 5:e11977. doi: 10.1371/journal.pone.0011977
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Grice, E. A., and Segre, J. A. (2011). The skin microbiome. Nat. Rev. Microbiol. 9, 244–253. doi: 10.1038/nrmicro2537
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gupta, A. K., Batra, R., Bluhm, R., Boekhout, T., and Dawson, T. L. (2004). Skin diseases associated with Malassezia species. J. Am. Acad. Dermatol. 51, 785–798. doi: 10.1016/j.jaad.2003.12.034
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Haas, B. J., Gevers, D., Earl, A. M., Feldgarden, M., Ward, D. V., Giannoukos, G.,et al. (2011). Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21, 494–504. doi: 10.1101/gr.112730.110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hall-Mendelin, S., Allcock, R., Kresoje, N., van den Hurk, A. F., and Warrilow, D. (2013). Detection of arboviruses and other micro-organisms in experimentally infected mosquitoes using massively parallel sequencing. PLoS ONE 8:e58026. doi: 10.1371/journal.pone.0058026
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Handelsman, J., Rondon, M. R., Brady, S. F., Clardy, J., and Goodman, R. M. (1998). Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem. Biol. 5, R245–R249. doi: 10.1016/S1074-5521(98)90108-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hertig, M., and Wolbach, S. B. (1924). Studies on Rickettsia-like micro-organisms in insects. J. Med. Res. 44, 329–374.7.
Hilgenboecker, K., Hammerstein, P., Schlattmann, P., Telschow, A., and Werren, J. H. (2008). How many species are infected with Wolbachia?–a statistical analysis of current data. FEMS Microbiol. Lett. 281, 215–220. doi: 10.1111/j.1574-6968.2008.01110.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huffnagle, G. B., and Noverr, M. C. (2013). The emerging world of the fungal microbiome. Trends Microbiol. 21, 334–341. doi: 10.1016/j.tim.2013.04.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hughes, G. L., Dodson, B. L., Johnson, R. M., Murdock, C. C., Tsujimoto, H., Suzuki, Y.,et al. (2014). Native microbiome impedes vertical transmission of Wolbachia in Anopheles mosquitoes. Proc. Natl. Acad. Sci. U.S.A. 111, 12498–12503. doi: 10.1073/pnas.1408888111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ito, T., Suzaki, K., and Nakano, M. (2013). Genetic characterization of novel putative rhabdovirus and dsRNA virus from Japanese persimmon. J. Gen. Virol. 94, 1917–1921. doi: 10.1099/vir.0.054445-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kembel, S. W., Wu, M., Eisen, J. A., and Green, J. L. (2012). Incorporating 16S gene copy number information improves estimates of microbial diversity and abundance. PLoS Comput. Biol. 8:e1002743. doi: 10.1371/journal.pcbi.1002743
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
King, A. M. Q., Adams, M. J., Lefkowitz, E., and Carstens, E. B. (2012). Virus Taxonomy. London: Elsevier.
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M.,et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1. doi: 10.1093/nar/gks808
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kukutla, P., Steritz, M., and Xu, J. (2013). Depletion of ribosomal RNA for mosquito gut metagenomic RNA-seq. J. Vis. Exp. e50093. doi: 10.3791/50093
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kuwata, R., Isawa, H., Hoshino, K., Tsuda, Y., Yanase, T., Sasaki, T.,et al. (2011). RNA splicing in a new rhabdovirus from Culex mosquitoes. J. Virol. 85, 6185–6196. doi: 10.1128/JVI.00040-11
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kuzmin, I. V., Novella, I. S., Dietzgen, R. G., Padhi, A., and Rupprecht, C. E. (2009). The rhabdoviruses: biodiversity, phylogenetics, and evolution. Infect. Genet. Evol. 9, 541–553. doi: 10.1016/j.meegid.2009.02.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. doi: 10.1186/gb-2009-10-3-r25
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lassmann, T., Hayashizaki, Y., and Daub, C. O. (2009). TagDust–a program to eliminate artifacts from next generation sequencing data. Bioinformatics 25, 2839–2840. doi: 10.1093/bioinformatics/btp527
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ledesma, N., and Harrington, L. (2011). Mosquito vectors of dog heartworm in the United States: vector status and factors influencing transmission efficiency. Top. Companion Anim. Med. 26, 178–185. doi: 10.1053/j.tcam.2011.09.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Longdon, B., Obbard, D. J., and Jiggins, F. M. (2010). Sigma viruses from three species of Drosophila form a major new clade in the rhabdovirus phylogeny. Proc. Biol. Soc. 277, 35–44. doi: 10.1098/rspb.2009.1472
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ma, M., Huang, Y., Gong, Z., Zhuang, L., Li, C., Yang, H.,et al. (2011). Discovery of DNA viruses in wild-caught mosquitoes using small RNA high throughput sequencing. PLoS ONE 6:e24758. doi: 10.1371/journal.pone.0024758
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marklewitz, M., Handrick, S., Grasse, W., Kurth, A., Lukashev, A., Drosten, C.,et al. (2011). Gouléako virus isolated from West African mosquitoes constitutes a proposed novel genus in the family Bunyaviridae▽. J. Virol. 85, 9227–9234. doi: 10.1128/JVI.00230-11
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McFall-Ngai, M., Hadfield, M. G., Bosch, T. C. G., Carey, H. V., Domazet-Lošo, T., Douglas, A. E.,et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. U.S.A. 110, 3229–3236. doi: 10.1073/pnas.1218525110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Megy, K., Emrich, S. J., Lawson, D., Campbell, D., Dialynas, E., Hughes, D. S. T.,et al. (2012). VectorBase: improvements to a bioinformatics resource for invertebrate vector genomics. Nucleic Acids Res. 40, D729–D734. doi: 10.1093/nar/gkr1089
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Minard, G., Mavingui, P., and Moro, C. V. (2013). Diversity and function of bacterial microbiota in the mosquito holobiont. Parasit. Vectors 6, 146. doi: 10.1186/1756-3305-6-146
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Morens, D. M., Folkers, G. K., and Fauci, A. S. (2004). The challenge of emerging and re-emerging infectious diseases. Nature 430, 242–249. doi: 10.1038/nature02759
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Muegge, B. D., Kuczynski, J., Knights, D., Clemente, J. C., González, A., Fontana, L.,et al. (2011). Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332, 970–974. doi: 10.1126/science.1198719
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ng, T. F. F., Willner, D. L., Lim, Y. W., Schmieder, R., Chau, B., Nilsson, C.,et al. (2011). Broad surveys of DNA viral diversity obtained through viral metagenomics of mosquitoes. PLoS ONE 6:e20579. doi: 10.1371/journal.pone.0020579
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Peng, Y., Leung, H. C. M., Yiu, S. M., and Chin, F. Y. L. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428. doi: 10.1093/bioinformatics/bts174
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C.,et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. doi: 10.1038/nature08821
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Quan, P.-L., Junglen, S., Tashmukhamedova, A., Conlan, S., Hutchison, S. K., Kurth, A.,et al. (2010). Moussa virus: a new member of the Rhabdoviridae family isolated from Culex decens mosquitoes in Côte d’Ivoire. Virus Res. 147, 17–24. doi: 10.1016/j.virusres.2009.09.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P.,et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rambaut, A. (2012). FigTree v1.4.0. Available at: http://tree.bio.ed.ac.uk/software/figtree
Rambaut, A., Suchard, M. A., Xie, D., and Drummond, A. J. (2013). Tracer v1.5.0. Available at: http://beast.bio.ed.ac.uk/Tracer
Rasgon, J. L., and Scott, T. W. (2004). An initial survey for Wolbachia (Rickettsiales: Rickettsiaceae) infections in selected California mosquitoes (Diptera: Culicidae). J. Med. Entomol. 41, 255–257. doi: 10.1603/0022-2585-41.2.255
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rastgou, M., Habibi, M. K., Izadpanah, K., Masenga, V., Milne, R. G., Wolf, Y. I.,et al. (2009). Molecular characterization of the plant virus genus Ourmiavirus and evidence of inter-kingdom reassortment of viral genome segments as its possible route of origin. J. Gen. Virol. 90, 2525–2535. doi: 10.1099/vir.0.013086-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ruby, J. G., Bellare, P., and DeRisi, J. L. (2013). PRICE: software for the targeted assembly of components of (Meta) genomic sequence data. G3(Bethesda) 3, 865–880. doi: 10.1534/g3.113.005967
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Runckel, C., Flenniken, M. L., Engel, J. C., Ruby, J. G., Ganem, D., Andino, R.,et al. (2011). Temporal analysis of the honey bee microbiome reveals four novel viruses and seasonal prevalence of known viruses, Nosema, and Crithidia. PLoS ONE 6:e20656. doi: 10.1371/journal.pone.0020656
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F.,et al. (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12:87. doi: 10.1186/s12915-014-0087-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schmidt, T. M., DeLong, E. F., and Pace, N. R. (1991). Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J. Bacteriol. 173, 4371–4378.
Scholte, E.-J., Knols, B. G. J., Samson, R. A., and Takken, W. (2004). Entomopathogenic fungi for mosquito control: a review. J. Insect Sci. 4, 19. doi: 10.1093/jis/4.1.19
Skewes-Cox, P., Sharpton, T. J., Pollard, K. S., and DeRisi, J. L. (2014). Profile hidden Markov models for the detection of viruses within metagenomic sequence data. PLoS ONE 9:e105067. doi: 10.1371/journal.pone.0105067
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Spear, A., Sisterson, M. S., Yokomi, R., and Stenger, D. C. (2010). Plant-feeding insects harbor double-stranded RNA viruses encoding a novel proline-alanine rich protein and a polymerase distantly related to that of fungal viruses. Virology 404, 304–311. doi: 10.1016/j.virol.2010.05.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Spear, A., Yokomi, R., French, R., and Stenger, D. C. (2013). Occurrence, sequence polymorphism and population structure of Circulifer tenellus virus 1 in a field population of the beet leafhopper. Virus Res. 176, 307–311. doi: 10.1016/j.virusres.2013.06.017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stewart, F. J., Ottesen, E. A., and DeLong, E. F. (2010). Development and quantitative analyses of a universal rRNA-subtraction protocol for microbial metatranscriptomics. ISME J. 4, 896–907. doi: 10.1038/ismej.2010.18
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thongsripong, P., Green, A., Kittayapong, P., Kapan, D., Wilcox, B., and Bennett, S. (2013). Mosquito vector diversity across habitats in central Thailand endemic for dengue and other arthropod-borne diseases. PLoS Negl. Trop. Dis. 7:e2507. doi: 10.1371/journal.pntd.0002507
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tritt, A., Eisen, J. A., Facciotti, M. T., and Darling, A. E. (2012). An integrated pipeline for de novo assembly of microbial genomes. PLoS ONE 7:e42304. doi: 10.1371/journal.pone.0042304
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Venter, J. C., Remington, K., Heidelberg, J. F., Halpern, A. L., Rusch, D., Eisen, J. A.,et al. (2004). Environmental genome shotgun sequencing of the Sargasso Sea. Science 304, 66–74. doi: 10.1126/science.1093857
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Walker, T., Johnson, P. H., Moreira, L. A., Iturbe-Ormaetxe, I., Frentiu, F. D., McMeniman, C. J.,et al. (2011). The wMel Wolbachia strain blocks dengue and invades caged Aedes aegypti populations. Nature 476, 450–453. doi: 10.1038/nature10355
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, S., Ghosh, A. K., Bongio, N., Stebbings, K. A., Lampe, D. J., and Jacobs-Lorena, M. (2012). Fighting malaria with engineered symbiotic bacteria from vector mosquitoes. Proc. Natl. Acad. Sci. U.S.A. 109, 12734–12739. doi: 10.1073/pnas.1204158109
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Warnecke, F., Luginbühl, P., Ivanova, N., Ghassemian, M., Richardson, T. H., Stege, J. T.,et al. (2007). Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450, 560–565. doi: 10.1038/nature06269
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weiss, B., and Aksoy, S. (2011). Microbiome influences on insect host vector competence. Trends Parasitol. 27, 514–522. doi: 10.1016/j.pt.2011.05.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: metagenomics, shotgun sequencing, microbiota, Culex pipiens Culiseta, Ochlerotatus, Bunyaviridae, Rhabdoviridae
Citation: Chandler JA, Liu RM and Bennett SN (2015) RNA shotgun metagenomic sequencing of northern California (USA) mosquitoes uncovers viruses, bacteria, and fungi. Front. Microbiol. 6:185. doi: 10.3389/fmicb.2015.00185
Received: 03 December 2014; Accepted: 19 February 2015;
Published online: 24 March 2015.
Edited by:
Katrine L. Whiteson, University of California, Irvine, USAReviewed by:
Thawornchai Limjindaporn, Mahidol University, ThailandLark L. Coffey, University of California, Davis, USA
Copyright © 2015 Chandler, Liu and Bennett. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James Angus Chandler and Shannon N. Bennett, Department of Microbiology, California Academy of Sciences, 55 Music Concourse Drive, Golden Gate Park, San Francisco, CA 94118, USAamNoYW5kbGVyQGNhbGFjYWRlbXkub3Jn;c2Jlbm5ldHRAY2FsYWNhZGVteS5vcmc=