 Tracy H. Hazen1,2*†
Tracy H. Hazen1,2*† Patricia C. Lafon2Nancy M. Garrett2
Patricia C. Lafon2Nancy M. Garrett2 Tiffany M. Lowe1†
Tiffany M. Lowe1† Daniel J. Silberger1†Lori A. Rowe3Michael Frace3Michele B. Parsons2
Daniel J. Silberger1†Lori A. Rowe3Michael Frace3Michele B. Parsons2 Cheryl A. Bopp2
Cheryl A. Bopp2 David A. Rasko4,5
David A. Rasko4,5 Patricia A. Sobecky1†
Patricia A. Sobecky1†- 1School of Biology, Georgia Institute of Technology, Atlanta, GA, USA
- 2Division of Foodborne, Waterborne, and Environmental Diseases, National Center for Emerging and Zoonotic Infectious Diseases, Centers for Disease Control and Prevention, Atlanta, GA, USA
- 3Division of Scientific Resources, National Center for Emerging and Zoonotic Infectious Diseases, Centers for Disease Control and Prevention, Atlanta, GA, USA
- 4Institute for Genome Sciences, University of Maryland School of Medicine, Baltimore, MD, USA
- 5Department of Microbiology and Immunology, University of Maryland, Baltimore, MD, USA
Vibrio parahaemolyticus is an aquatic halophilic bacterium that occupies estuarine and coastal marine environments, and is a leading cause of seafood-borne food poisoning cases. To investigate the environmental reservoir and potential gene flow that occurs among V. parahaemolyticus isolates, the virulence-associated gene content and genome diversity of a collection of 133 V. parahaemolyticus isolates were analyzed. Phylogenetic analysis of housekeeping genes, and pulsed-field gel electrophoresis, demonstrated that there is genetic similarity among V. parahaemolyticus clinical and environmental isolates. Whole-genome sequencing and comparative analysis of six representative V. parahaemolyticus isolates was used to identify genes that are unique to the clinical and environmental isolates examined. Comparative genomics demonstrated an O3:K6 environmental isolate, AF91, which was cultured from sediment collected in Florida in 2006, has significant genomic similarity to the post-1995 O3:K6 isolates. However, AF91 lacks the majority of the virulence-associated genes and genomic islands associated with these highly virulent post-1995 O3:K6 genomes. These findings demonstrate that although they do not contain most of the known virulence-associated regions, some V. parahaemolyticus environmental isolates exhibit significant genetic similarity to clinical isolates. This highlights the dynamic nature of the V. parahaemolyticus genome allowing them to transition between aquatic and host-pathogen states.
Introduction
Vibrio parahaemolyticus is halophilic aquatic bacterium that is ubiquitous in coastal marine and estuarine environments. The majority of isolates derived from environmental sources, that is water and sediments, are believed to be non-pathogenic (Depaola et al., 1990; Nair et al., 2007); however some V. parahaemolyticus isolates are capable of causing human illness, and are primarily associated with food-borne derived gastroenteritis and diarrhea. V. parahaemolyticus infection can also be associated with wound infections and sepsis (CDC, 2007, 2008; Tena et al., 2010). In 1996, an increase in diarrheal illness in India associated with V. parahaemolyticus infections were attributed to the emergence of a novel genetic variant in 1995 that had the O3:K6 serotype (Okuda et al., 1997). This novel disease-associated O3:K6 clone rapidly disseminated worldwide and is considered to be pandemic (Vuddhakul et al., 2000; Myers et al., 2003; Quilici et al., 2005; Ottaviani et al., 2008). Previously, isolates belonging to the post-1995 O3:K6 clone were identified with the serotypes O1:Kuk, O1:K25, and O4:K68, indicating the O3:K6 clone has undergone serogroup conversion in the years since the original clonal expansion (Nair et al., 2007; Chen et al., 2011). The disease-associated V. parahaemolyticus clinical isolates usually carry one or both of the thermostable direct hemolysins (tdh and trh) (Kaper et al., 1984; Nishibuchi et al., 1992; Nishibuchi and Kaper, 1995; Makino et al., 2003; Nair et al., 2007). In addition to the hemolysins, two type III secretion systems (T3SS) have been demonstrated to secrete effectors that induce cytotoxicity or enterotoxicity (Park et al., 2004; Lynch et al., 2005; Ono et al., 2006; Caburlotto et al., 2010; Broberg et al., 2011; Ham and Orth, 2012; Zhang and Orth, 2013). A previous study revealed a second version of T3SS2, T3SS2β, which was identified in clinical isolates that also possess the trh gene (Okada et al., 2009). Although the hemolysins and type III secretion have been identified as a major components of the V. parahaemolyticus virulence mechanism (Park et al., 2004; Burdette et al., 2008; Caburlotto et al., 2010; Ham and Orth, 2012; Zhang and Orth, 2013), disease-associated isolates have been identified that do not encode the thermostable direct hemolysins (Yu et al., 2006; Bhoopong et al., 2007; Meador et al., 2007), suggesting there may be additional, as yet, uncharacterized genes contributing to V. parahaemolyticus virulence mechanisms.
The genetic diversity of V. parahaemolyticus has been investigated using numerous molecular methods, including the identification of known virulence genes (Meador et al., 2007; Noriea et al., 2010; Jones et al., 2012), multi-locus sequence typing (MLST) (Chowdhury et al., 2004; González-Escalona et al., 2008; Gavilan et al., 2013; Turner et al., 2013), phylogenetic analysis of housekeeping genes (Thompson et al., 2005), microarray (Han et al., 2008), and pulsed-field gel electrophoresis (PFGE) (Parsons et al., 2007; Ludeke et al., 2014). MLST was used to identify two new clonal complexes in addition to a clonal complex of the post-1995 O3:K6 isolates (González-Escalona et al., 2008). The second clonal complex consisted of O4:K12 and O12:K12 isolates from the Pacific coast of the United States, and the third clonal complex was comprised primarily of isolates from oysters in the Gulf of Mexico (González-Escalona et al., 2008).
Genome sequencing and comparative analysis of the post-1995 V. parahaemolyticus O3:K6 isolate RIMD2210633 (Makino et al., 2003) revealed seven genomic islands, including four that are characteristic of post-1995 O3:K6 isolates (Hurley et al., 2006; Boyd et al., 2008; Chen et al., 2011). Genomic subtraction demonstrated that an 80-kb pathogenicity island (Vp-PAI) encoding T3SS2 was associated with the post-1995 O3:K6 pandemic isolates (Okura et al., 2005). The sequencing of additional V. parahaemolyticus genomes has confirmed that the emergence of the post-1995 O3:K6 pandemic isolates coincided with the acquisition of genomic islands as these regions were mostly absent from the genomes of pre-1995 O3:K6 isolates (Makino et al., 2003; Boyd et al., 2008; Chen et al., 2011).
The purpose of this study was to investigate the genetic diversity of V. parahaemolyticus isolates from human clinical (stool, blood, wound specimens, or unknown sample types) or environmental (sediment, water, oysters) sources using multiple molecular methods including a PCR assay of known V. parahaemolyticus virulence-associated genes, phylogenetic analysis of housekeeping genes, PFGE, and whole-genome sequencing. Investigation of the genomic diversity of two clinical isolates and four environmental isolates by whole-genome sequencing and comparative analysis identified genes that are shared or exclusive to the clinical or environmental isolate genomes sequenced. These methods highlight the genetic similarity among clinical and environmental isolates, and the different combinations of virulence-associated genes demonstrate the dynamic nature of the V. parahaemolyticus genome.
Materials and Methods
Bacterial Isolates and Media
V. parahaemolyticus clinical isolates included in this study were provided by the Centers for Disease Control and Prevention (Atlanta, GA). The V. parahaemolyticus environmental isolates were cultured from sediment, water, and oysters of Skidaway Island, GA, and Apalachicola Bay, FL in September 2006, and Skidaway Island, GA in September 2007 (Hazen et al., 2009). Additional environmental isolates were obtained from the rhizosphere sediment of a salt marsh in North Inlet, NC (Bagwell et al., 1998). The environmental isolates were cultured by plating environmental samples on thiosulfate citrate bile salts sucrose (TCBS) agar (Difco) and incubating them overnight at 30°C. Water samples were directly plated onto TCBS, while sediment, and oysters were homogenized with sterile water then plated onto TCBS agar. Presumptive V. parahaemolyticus colonies that were green on TCBS were confirmed by PCR by screening for the thermolabile hemolysin (tl) as previously developed (Bej et al., 1999), which is characteristic of V. parahaemolyticus (Meador et al., 2007). The culture collection strain, ATCC 17802, was used as a reference isolate for molecular characterizations of V. parahaemolyticus.
Serotyping
Serotypes were determined using V. parahaemolyticus Seiken typing antisera (Denka Seiken, Tokyo, Japan).
PCR Assay of Virulence-Associated Genes
The known V. parahaemolyticus virulence-associated genes were detected by PCR assay for all V. parahaemolyticus clinical and environmental isolates examined in this study using primers listed in Supplemental Table 5. All isolates that were positive for tl as described above were then PCR screened for previously-characterized virulence-associated genes. The thermostable direct hemolysins tdh and trh were detected as described (Bej et al., 1999; Meador et al., 2007). In addition, the ORF8 gene of the pandemic phage f237 was detected using primers that were previously developed (Myers et al., 2003). The presence of the T3SS1 and T3SS2 were determined by PCR assay for two effectors and one gene involved in translocation from each T3SS. The T3SS1 effectors vp1680 and vp1686 and the structural gene vp1670 was identified by PCR assay using previously developed (Vora et al., 2005; Meador et al., 2007) primers, and additional primers made in this study that are listed in Supplemental Table 5. The presence of T3SS2α and T3SS2β was determined by PCR assay for the effectors vpa1346 and vpa1362, and the export protein-encoding gene vpa1354 using primers listed in Supplemental Table 5.
Phylogenetic Analysis of Housekeeping Genes
The genetic similarity was investigated for 116 V. parahaemolyticus clinical and environmental isolates examined in this study by phylogenetic analysis of a concatenation of four housekeeping genes (recA, gyrB, pyrC, dtdS) using previously developed primers (González-Escalona et al., 2008). The genes were PCR amplified using NEB Phusion high-fidelity polymerase (NEB; Ipswich, MA) and purified by separation on a 0.7% Seakem LE agarose gel (Lonza; Allendale, NJ). The target amplicon was excised from the gel and the DNA was recovered using the Sigma GenElute gel extraction kit (Sigma Aldrich; St. Louis MO). Sequencing was performed with M13 primers at the Georgia Tech Genome Center on an ABI 3130 Genetic Analyzer (Applied Biosystems) using BigDye Terminator chemistry (Applied Biosystems). Sequences were assembled in BioEdit (v. 7.0.4.1) (Hall, 1999) and aligned using MEGA5 (Tamura et al., 2011), and all sequences for a particular gene were trimmed to the same length. The partial sequences of each housekeeping gene analyzed were concatenated in the same order for each V. parahaemolyticus isolate, generating a single representative sequence. A maximum-likelihood phylogeny with 100 bootstrap replicates was generated using RAxML v7.2.8 (Stamatakis, 2006) and visualized using FigTree v1.3.1 (http://tree.bio.ed.ac.uk/software/figtree/).
The genetic similarity was further investigated for 52 of the clinical and environmental isolates by analysis of three additional genes (dnaE, tnaA, pntA) for a total of seven genes. These three additional genes were PCR amplified and described above using previously developed primers (González-Escalona et al., 2008). Phylogenetic analysis of all seven (recA, gyrB, dnaE, pyrC, dtdS, tnaA, pntA) of the conserved genes for this subset of clinical and environmental isolates was performed as described above.
PFGE
Pulsed-field gel electrophoresis of 44 V. parahaemolyticus clinical and environmental isolates was performed according to the V. parahaemolyticus PulseNet USA standardized protocol (Parsons et al., 2007). Restriction endonuclease profiles were generated using the enzymes SfiI and NotI (Roche, Mannheim, Germany). Restricted plugs were run on a CHEF Mapper™ electrophoresis system (Bio-Rad Laboratories, Hercules, CA). Salmonella Braenderup H9812 restricted with 50 U of XbaI (Roche, Mannheim, Germany) was used as a control strain for gel normalization. PFGE patterns were analyzed with BioNumerics v. 5.1 (Applied-Maths, Kortrijk, Belgium) and dendrograms were generated using the Dice coefficient and unweighted pair group method with arithmetic averages (UPGMA) with a band position tolerance and optimization of 1.5% for cluster analysis.
Genome Sequencing and Assembly
Following the molecular characterization of the V. parahaemolyticus clinical and environmental isolates, we generated high-quality draft genome sequences of two clinical isolates (K1275, K1461) and four environmental isolates (AF91, SG176, J-C2-34, 22702) (Table 2). The clinical isolates analyzed have unique combinations of the known virulence-associated genes compared to the epidemic post-1995 O3:K6 isolates (Supplemental Table 1). The environmental isolates analyzed by genome sequencing were obtained from samples of three different states (NC, GA, FL) (Supplemental Table 1). The V. parahaemolyticus isolates analyzed by whole-genome sequencing were grown overnight in Luria Bertani (Difco) at 37°C with shaking (225 rpm). Genomic DNA was isolated from the overnight cultures using the Sigma GenElute genomic kit (Sigma Aldrich; St. Louis MO). The genome sequences of V. parahaemolyticus isolates K1461, K1275, SG176, J-C2-34, and AF91 were generated using the Roche 454-Titanium sequencing platform at the Centers for Disease Control and Prevention. The 454 reads were assembled into high-quality draft genomes at the Institute for Genome Sciences, using the Mira assembler (Chevreux et al., 1999), and the assemblies were filtered to contain contigs ≥500 bp.
The genome sequence of V. parahaemolyticus 22702 was generated using paired-end libraries with 300 bp inserts on the Illumina HiSeq2000 at the Institute for Genome Sciences, Genome Resource Center. The Illumina reads generated for 22702 were assembled into a high-quality draft genome using the Velvet assembly program (Zerbino and Birney, 2008) with kmer values determined using VelvetOptimiser v2.1.4 (http://bioinformatics.net.au/software.velvetoptimiser.shtml), and the assembly was filtered to contain contigs ≥500 bp.
Information regarding the genome assembly size, number of contigs, and the GenBank accession numbers for each of the genomes sequenced in this study are listed in Table 2.
Comparative Genomics
Phylogenomic analysis of the V. parahaemolyticus genomes sequenced in this study compared to previously sequenced V. parahaemolyticus genomes available in the public domain, was performed as previously described (Sahl et al., 2011). The genomes were aligned using Mugsy (Angiuoli and Salzberg, 2011), and the aligned regions were concatenated then used to construct a maximum-likelihood phylogeny with 100 bootstrap values using RAxML v7.2.8 (Stamatakis, 2006), and visualized using FigTree v1.3.1 (http://tree.bio.ed.ac.uk/software/figtree/).
BLAST score ratio (BSR) analysis was performed as previously described (Rasko et al., 2005) and used to identify the presence of virulence-associated genes in each of the genomes analyzed (Table 2). Briefly, the predicted amino acid sequences of virulence-associated genes and genomic regions (Hurley et al., 2006; Chen et al., 2011; Salomon et al., 2013) were compared using TBLASTN (Gertz et al., 2006) to all the V. parahaemolyticus genomes analyzed in this study. The protein-encoding genes that were considered present with significant similarity had BSR values ≥0.8.
Genetic similarity of chromosomes I and II of the O3:K6 isolate RIMD2210633 to genes in each of the genomes sequenced in this study was determined using BLASTN (Altschul et al., 1990) BSR analysis as previously described (Rasko et al., 2005). A circular display of the BLASTN BSR values was generated using Circos 0.65 (Krzywinski et al., 2009).
Large-scale BSR analysis (Hazen et al., 2013; Sahl et al., 2013, 2014) was used to identify the shared and unique features present in the six genomes sequenced in this study compared with eight previously sequenced genomes listed in Table 2. Each protein-encoding gene was considered present with high identity (BSR value ≥0.8), present but with sequence divergence (BSR value ≥0.4, ≤0.8), or absent (BSR value <0.4). A representative sequence of each predicted protein-encoding gene is included in Supplemental Data Set 1. The predicted function of protein-encoding genes was identified using the RAST annotation server (Overbeek et al., 2014).
Plasmid and Phage Analyses
The number of extrachromosomal elements (plasmids and prophage) was determined for a subset of the isolates using a modified acid phenol extraction method (Kieser, 1984; Sobecky et al., 1997). PCR amplification for sequencing was performed using NEB Phusion high-fidelity polymerase reaction mix with GC buffer and reaction and cycle conditions as recommended by the manufacturer (NEB; Ipswich, MA). Primers used to PCR amplify rstA are listed in Supplemental Table 5. The M13 sequence at the 5′ end of each rstA primer was used for sequencing. Sequencing was performed as described for the housekeeping genes. The sequences were aligned using MEGA5 (Tamura et al., 2011), and a maximum-likelihood phylogeny using the Kimura 2-parameter model (Kumar et al., 2004) and 1,000 bootstrap replications was constructed using MEGA5 (Tamura et al., 2011). Bootstrap values ≥50 are shown.
Nucleotide Sequence Accession Numbers
All individual gene sequences generated in this study are deposited in GenBank under the accession numbers FJ847518-FJ847829. The genome sequences are deposited in GenBank under the accession numbers JMMO00000000, JMMP00000000, JMMQ00000000, JMMR00000000, JMMS00000000, and JMMT00000000.
Results and Discussion
Identification of Virulence-Associated Genes in a Collection of Clinical and Environmental V. parahaemolyticus
As a measure of the virulence potential of V. parahaemolyticus clinical and environmental isolates analyzed, we detected common markers of virulence including: the ORF8 gene of the filamentous vibriophage, the hemolysins (tdh and trh), the type III secretion systems of chromosome I (T3SS1), and chromosome II (T3SS2α and T3SS2β) (Table 1). These genes have been previously identified in association with illness-associated V. parahaemolyticus, and the hemolysins and T3SS2 were characterized for their role in pathogenesis (Kaper et al., 1984; Nishibuchi and Kaper, 1995; Nasu et al., 2000; Park et al., 2004; Lynch et al., 2005; Ono et al., 2006; Nair et al., 2007; Broberg et al., 2011; Ham and Orth, 2012; Zhang and Orth, 2013). The ORF8 gene encoded by the filamentous vibriophage f237, which has previously been linked to the post-1995 O3:K6 pandemic clinical isolates (Nasu et al., 2000), was identified in 32% of the clinical isolates, and none of the environmental isolates in this study (Table 1). The ORF8 gene was identified in all post-1995 O3:K6 isolates analyzed, and only 13% of the non-O3:K6 clinical isolates (Table 1; Supplemental Table 1). The T3SS1 genes and tl were detected among all V. parahaemolyticus isolates, which is consistent with previous reports that these genes are universal among V. parahaemolyticus isolates (Vora et al., 2005) (Table 1, Supplemental Table 1). The T3SS2α genes were present in 93% (13/14) of the O3:K6 clinical isolates and 100% (4/4) of the O4:K8 clinical isolates, but only 25% (12/49) of the clinical isolates that had other serotypes (Table 1). T3SS2β was detected in 90% (9/10) of the O4:K12 isolates, and 46% (22/48) of the clinical isolates with other serotypes (Table 1). Included in this study were 10 clinical isolates with the O4:K12 serotype, and all but one (K4358) of these isolates were tdh+/trh+/T3SS2β+ (Table 1, Supplemental Table 1). Similar virulence-associated gene content (tdh+/trh+/T3SS2β+) was identified in clinical isolates of six other serotypes (O4:K53, O4:K63, O1:K56, O8:K21, O6:K18, O11:Kuk) (Supplemental Table 1). While T3SS2α was not identified in any of the environmental isolates, including the O3:K6 environmental isolate AF91, T3SS2β was present in two environmental isolates that were trh+ (Table 1, Supplemental Table 1). V. parahaemolyticus environmental isolates that possess T3SS2 have been demonstrated to adhere to eukaryotic cells and disrupt membrane tight junctions (Caburlotto et al., 2010). This study demonstrated that V. parahaemolyticus isolates residing in the environment that possessed some of the known virulence factors also had the potential to cause disease.
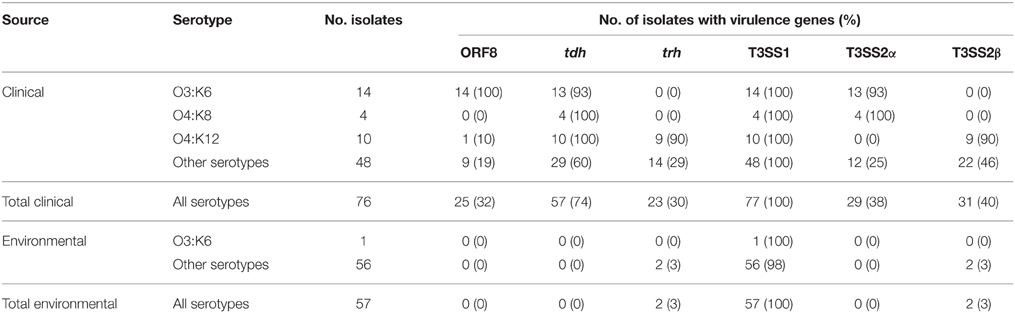
Table 1. Identification of V. parahaemolyticus virulence-associated genes in a collection of V. parahaemolyticus clinical and environmental isolates using PCR assays.
There were also clinical isolates that had an atypical combination of virulence genes, or were missing most of the known virulence-associated genes. Several clinical isolates contained only tdh (K0071, F5828, K4358, K4279), or both tdh and trh genes (K5067), but lacked detectable T3SS2 genes (Table 2). In addition, the clinical isolates K4763, K3528, and K4305 contained tdh and T3SS2β genes, which is unusual as the tdh gene is typically associated with T3SS2α (Sugiyama et al., 2008). It is possible these isolates may have contained both tdh and trh, similar to O4:K12, and they may have lost trh during infection or during laboratory passage. None of the isolates analyzed contained the trh gene and also the T3SS2α genes. Three clinical isolates (F8950, F8937, K4377) contained T3SS2α but lacked tdh and trh, and five clinical isolates (K0456, K4237, K4638, K5323G, K5330) contained T3SS2β but lacked tdh and trh (Supplemental Table 1). The presence of T3SS2 genes and the absence of hemolysins in clinical isolates has been previously described (Meador et al., 2007). There were 12 clinical isolates (K1275, K0851, K0850, F9974, F6658, F8132, F7979, F6179, K4434, F8190, K4981, K1000) that did not encode tdh, trh, or the T3SS2 genes (Supplemental Table 1). These isolates were obtained from blood (K1275), wound infections (F8132, K4434), or unknown clinical sample types (F6658, F6179, F8190, K4981, K1000). Although the V. parahaemolyticus clinical isolates that lacked the hemolysin and/or T3SS2 genes were obtained from clinical specimens, they may have been co-occurring in the host with other V. parahaemolyticus isolates that did encode the hemolysins or T3SS2 genes and were the primary cause of illness. A previous study demonstrated that multiple V. parahaemolyticus isolates were present in disease-associated samples; however, some of these isolates lacked the hemolysin genes (Bhoopong et al., 2007). Another possible explanation is that these isolates may have contained the hemolysin genes and T3SS2 genes and may have lost them following passage through a host or during passage in the laboratory. This was previously observed for the enteropathogenic Escherichia coli isolate E2348/69, which exhibited loss of the EPEC virulence plasmid in a subset of culturable isolates following passage through adults in a clinical trial (Levine et al., 1985). Also, it may be possible that some of these V. parahaemolyticus isolates have as yet uncharacterized virulence factors. Further research is necessary to determine whether these V. parahaemolyticus clinical isolates are capable of causing disease without the hemolysin genes and/or T3SS2 genes. These findings highlight the many combinations of virulence-associated genes in V. parahaemolyticus clinical isolates, demonstrating the dynamic nature of the virulence repertoires of V. parahaemolyticus isolates.
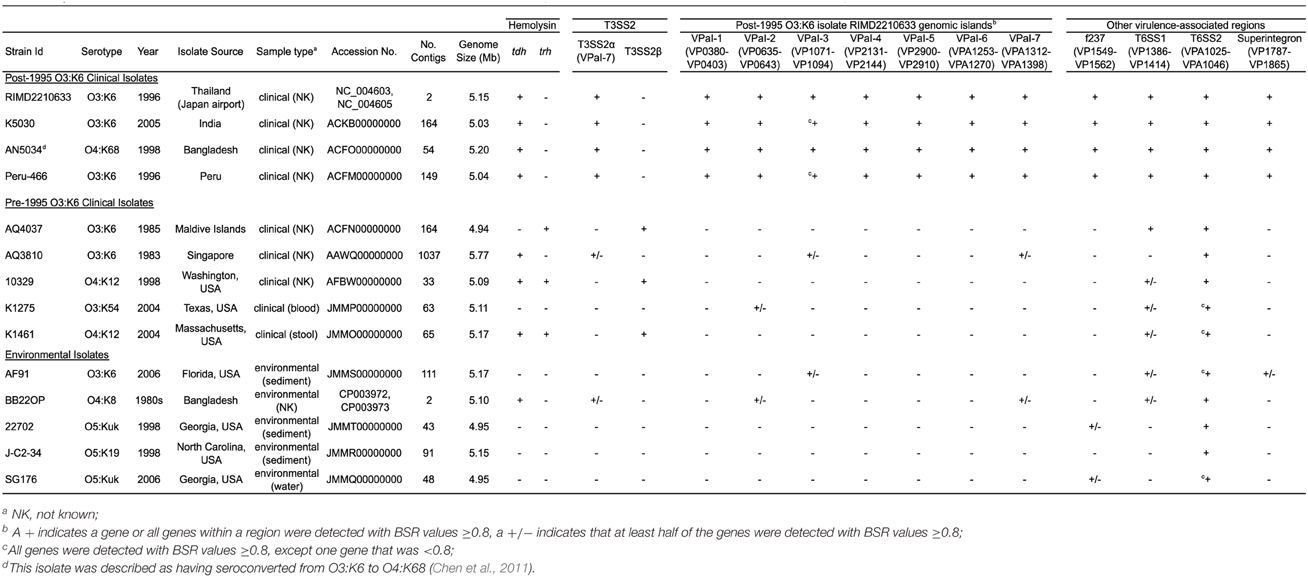
Table 2. Genome characteristics and in silico detection of virulence-associated genes in the sequenced V. parahaemolyticus genomes.
Molecular Analysis of the Genetic Similarity of Clinical and Environmental V. parahaemolyticus
Phylogenetic analysis of housekeeping genes was used to investigate the genetic similarity of V. parahaemolyticus clinical and environmental isolates representing diverse serotypes, isolation sources, and date of isolation (Figure 1, Supplemental Table 1). This approach has previously been used to investigate the evolutionary relationships of isolates within a single Vibrio species (Chowdhury et al., 2004; Boyd et al., 2008; González-Escalona et al., 2008; Turner et al., 2013), and among isolates belonging to multiple Vibrio species (Thompson et al., 2005, 2007, 2008; Sawabe et al., 2007, 2013; Lin et al., 2010). A phylogeny analyzing the genetic relatedness of 52 V. parahaemolyticus isolates (42 clinical, 10 environmental) was constructed using the partial nucleotide sequences of seven housekeeping genes (Figure 1). The phylogeny contained three distinct clades (colored boxes), which were primarily comprised of isolates with the O4:K12, O3:K6, and O4:K8 serotypes (Figure 1). Notably, all but two of the isolates (AF91 and BB22OP) that formed these three clades were derived from clinical sources (Figure 1). The other 18 isolates analyzed that were outside of these three clades included a mixture of clinical and environmental isolates that have diverse serotypes, and these isolates exhibited considerable phylogenetic diversity (Figure 1). Furthermore, this demonstrated that clinical isolates with serotypes other than O3:K6, O4:K12, and O4:K8 had genetic similarity to the environmental isolates analyzed in this study (Figure 1).
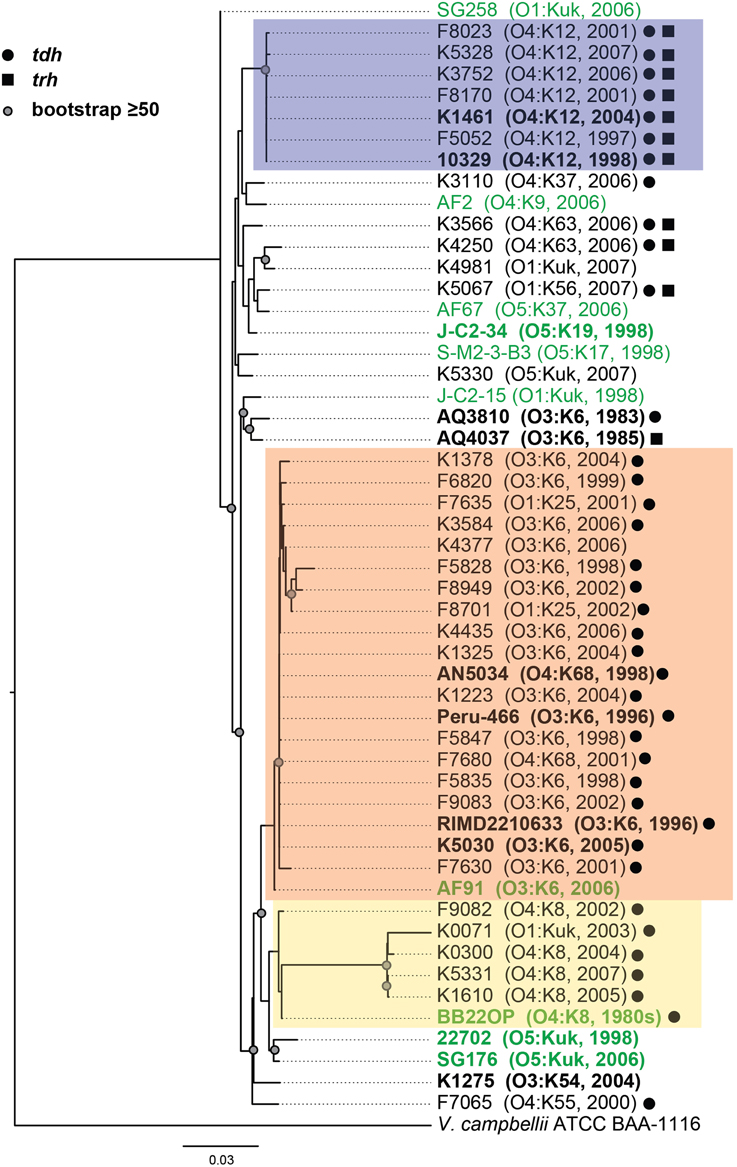
Figure 1. Maximum-likelihood phylogeny of V. parahaemolyticus clinical and environmental isolates analyzed in this study compared to V. parahaemolyticus isolates that have been previously characterized by complete or draft genome sequencing and are available in the public domain. The nucleotide sequences of seven conserved genes (recA, gyrB, pyrC, dtdS, tnaA, dnaE, and pntA) were concatenated for each V. parahaemolyticus isolate, and V. campbellii ATCC BAA-1116 was included as an outgroup. The phylogeny was constructed using RAxML (Stamatakis, 2006) with 100 bootstrap replications, and visualized using FigTree v1.3.1 (http://tree.bio.ed.ac.uk/software/figtree/). Only bootstrap values ≥50 are shown. The scale bar represents 0.03 nucleotide substitutions per site. The genomes that were sequenced in this study or in previous studies that are available in the public domain are indicated in bold and are listed in Table 2. The post-1995 V. parahaemolyticus O3:K6 isolates are indicated by an orange box, the O4:K12 isolates are indicated by a purple box, and the O4:K8 isolates are indicated in yellow. The V. parahaemolyticus isolates obtained from environmental sources are indicated in green, while the isolates from clinical sources are indicated in black. The presence of the virulence-associated thermostable direct hemolysins, tdh and trh, in each of the genomes is indicated by symbols.
To further investigate the genetic diversity observed for the clinical and environmental isolates that had serotypes other than O3:K6, O4:K12, and O4:K8, we analyzed partial sequences of two housekeeping genes of chromosome I (recA and gyrB), and two housekeeping genes of chromosome II (pyrC and dtdS) in a larger collection of 116 V. parahaemolyticus clinical and environmental isolates (Supplemental Figure 1). In a phylogenetic analysis of the concatenation of all four genes, there were four clinical isolates that formed a sub-clade with a long branch. To investigate whether the long branch of this sub-clade resulted from sequence divergence within a particular analyzed gene we generated individual phylogenies for each gene. Three of these genes (gyrB, pyrC, and dtdS) had similar topologies to the concatenated phylogeny, while the recA phylogeny demonstrated there was additional sequence divergence within recA for the four clinical isolates with the longer branch. Therefore, we analyzed the diversity of these four housekeeping genes by constructing a phylogeny for three of the genes (Supplemental Figure 1A), compared with a separate phylogeny of only recA sequences (Supplemental Figure 1B). Overall, phylogenetic analysis of the three genes (gyrB, pyrC, dtdS) indicated the clinical and environmental isolates analyzed have extensive genetic diversity (Supplemental Figure 1). Previous studies have demonstrated there is considerable genetic diversity of V. parahaemolyticus isolates from around the world (Chowdhury et al., 2004; González-Escalona et al., 2008).
The genetic relatedness of V. parahaemolyticus clinical and environmental isolates was also investigated using PFGE, which is a cost-effective method for routine identification of disease-associated bacteria, including V. parahaemolyticus (Parsons et al., 2007). PFGE was performed on a total of 37 clinical isolates and 7 environmental isolates (Supplemental Figure 2). Analysis of the SfiI and NotI patterns of these isolates demonstrated the presence of three main clades corresponding to those identified based on phylogenetic analysis of the housekeeping genes and the phylogenomic analysis (Figure 1, Supplemental Figure 1). A notable exception is that of the O3:K6 environmental isolate, AF91, which was not present in the O3:K6 clade by PFGE analysis as it was in the housekeeping gene phylogenies (Figure 1, Supplemental Figure 1). Similar to the housekeeping gene phylogenies, PFGE also demonstrated there is considerable genetic diversity among the clinical and environmental isolates analyzed that had serotypes other than those of the three main clades (O3:K6, O4:K12, and O4:K8) (Supplemental Figure 2). The PFGE pattern of the V. parahaemolyticus environmental isolate AF91 was different from the O3:K6 clinical isolates and the other environmental isolates examined. The NotI pattern of AF91 was similar to that of other V. parahaemolyticus isolates; however, the SfiI pattern had multiple large bands that ranged from approximately 485- to 693-kb. In addition, the SfiI pattern of this strain was missing four or more small bands that were present in the SfiI patterns of the other V. parahaemolyticus isolates. The presence of the larger bands in the SfiI pattern of AF91 suggested the absence of several SfiI restriction sites that may correlate with the absence of the genomic islands of the post-1995 O3:K6 isolates (Hurley et al., 2006).
Comparative Genomics of Clinical and Environmental V. parahaemolyticus
To investigate whether there are shared or exclusive genome features of V. parahaemolyticus clinical and environmental isolates, we generated high-quality draft genome sequences of six V. parahaemolyticus isolates (K1461, K1275, SG176, J-C2-34, AF91, and 22702) that had diverse isolation sources, serotypes, and virulence factor content (Supplemental Table 1). Phylogenomic analysis of the six V. parahaemolyticus genomes sequenced in this study compared to previously sequenced V. parahaemolyticus genomes (Table 2) demonstrated there is considerable genomic diversity among isolates from clinical and environmental sources (Figure 2). Three of the environmental isolate genomes (22702, SG176, and J-C2-34) grouped together in the whole-genome phylogeny, while the other two environmental isolate genomes (AF91, BB22OP (Jensen et al., 2013) were within a larger group that contained the clinical isolate genomes (Figure 2). The phylogenomic analysis further confirmed that the O3:K6 environmental isolate, AF91, was more related to the post-1995 O3:K6 genomes than to the pre-1995 O3:K6 isolate genomes that have been previously sequenced (Figure 2).
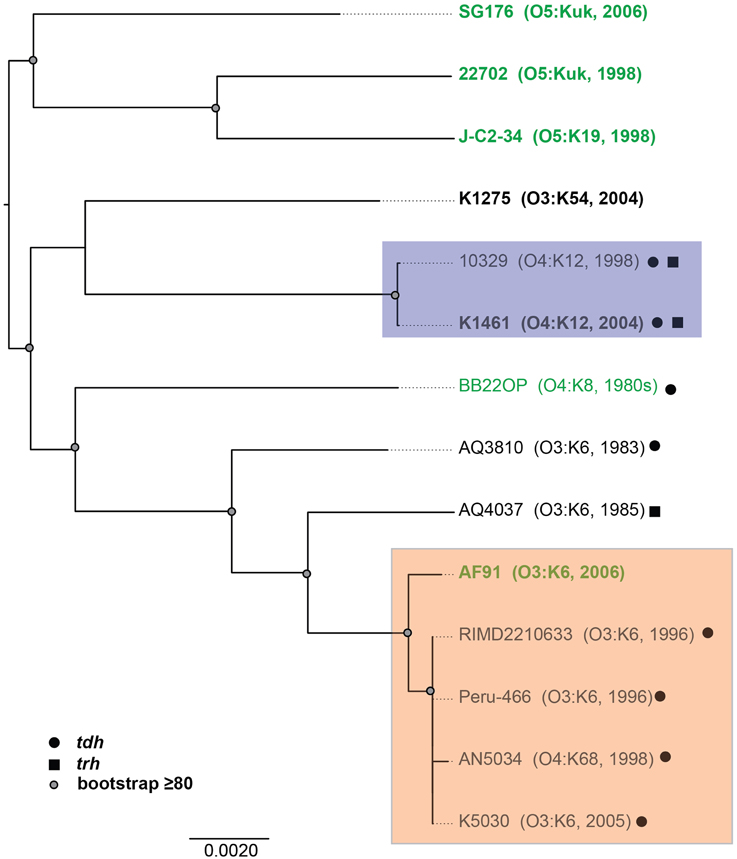
Figure 2. Phylogenomic analysis of the six V. parahaemolyticus clinical and environmental isolates sequenced in this study (indicated in bold) compared to eight V. parahaemolyticus genomes that have been previously sequenced (Makino et al., 2003; Boyd et al., 2008; Chen et al., 2011; Gonzalez-Escalona et al., 2011; Jensen et al., 2013) and are available in the public domain. The genomes were aligned using Mugsy (Angiuoli and Salzberg, 2011), and an approximately 4.0 Mb region of each genome that aligned was concatenated to generate a single sequence for each isolate as previously described (Sahl et al., 2011). A maximum-likelihood phylogeny with 100 bootstrap replicates was constructed using RAxML (Stamatakis, 2006) and visualized using FigTree v1.3.1 (http://tree.bio.ed.ac.uk/software/figtree/). Bootstrap values ≥80 are indicated by a circle. The genomes of V. parahaemolyticus isolates from environmental sources are indicated in green, while the isolates from clinical sources are indicated in black. The post-1995 O3:K6 isolates are identified by the orange box, and the O4:K12 isolates are indicated by a purple box. The presence of the virulence-associated thermostable direct hemolysins, tdh and trh, in each of the genomes is indicated by symbols.
In silico identification of the known V. parahaemolyticus virulence-associated genes and genomic islands (Hurley et al., 2006; Boyd et al., 2008) in the clinical and environmental genomes sequenced demonstrated that these regions were primarily identified in the post-1995 O3:K6 genomes (Table 2). However, some but not all of the genes in a few of these regions (VPaI-2, VPaI-3, and VPaI-7 encoding T3SS2α) were identified in some of the other clinical or the environmental isolate genomes (Table 2). This finding is similar to previous studies that demonstrated the T6SS gene cluster of chromosome I (T6SS1) is more frequently associated with V. parahaemolyticus clinical isolates than environmental isolates (Yu et al., 2012). The genes of T6SS1 were identified in nearly all the clinical isolate genomes, except the pre-1995 O3:K6 isolate AQ3810, and they were not identified in the genomes of the environmental isolates except for AF91 and BB22OP, which encode genes with similarity to those of T6SS1 (Table 2). However, AF91 and BB22OP have the serotypes O3:K6 and O4:K8, respectively, which are serotypes that have been linked to cases of human illness (Okuda et al., 1997; Matsumoto et al., 2000; Chowdhury et al., 2013; Ma et al., 2014). Further investigation is necessary to determine whether these environmental isolates may be more likely to cause disease than other environmental isolates that do not possess the T6SS1 genes.
Comparison of the V. parahaemolyticus genome content was analyzed using large-scale BLAST score ratio (LS-BSR) (Sahl et al., 2014) and further demonstrated the extent of the overall genome similarity among the clinical and environmental isolate genomes analyzed (Table 3). There were a total of 7782 genes identified in the 14 genomes analyzed in this study, 3494 of these genes were present with significant similarity (LS-BSR value ≥0.8) in all of the genomes analyzed (Table 3). Of the total genes identified there were 755 that were present in one or more of the clinical isolate genomes with significant similarity (LS-BSR value ≥0.8) that were not identified (LS-BSR value <0.4) in any of the environmental genomes sequenced (Table 3, Supplemental Table 2). Among these were genes encoding T3SS proteins, which likely belong to T3SS2 since these genes were not identified in any of the environmental isolates sequenced (Table 2). Also included among these genes were a multidrug resistance efflux pump and a putative RTX toxin (Supplemental Table 2). There were a similar number of genes (838) that were identified in one or more of the environmental isolate genomes that were not identified in any of the clinical isolate genomes (Table 3, Supplemental Table 2).
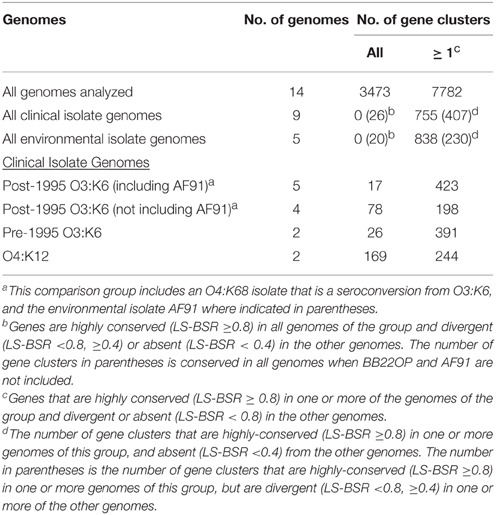
Table 3. LS-BSR analysis of the genomic similarity of select V. parahaemolyticus clinical and environmental isolates.
There were no genes identified in all of the clinical isolate genomes that were not present in one or more of the environmental isolate genomes, or vice versa (Table 3). This is likely due to significant genetic similarities between clinical and environmental isolate genomes such as the O3:K6 environmental isolate AF91 and the O3:K6 clinical isolates (Figures 1, 2). The inability to identify genes that are exclusive to all clinical isolate genomes also can likely be attributed to the inclusion of the environmental isolate BB22OP, which encodes known virulence-associated genes such as tdh (Jensen et al., 2013). However, upon exclusion of the AF91 and BB22OP genomes from the analysis, there were 26 genes that were highly-conserved (LS-BSR values ≥0.8) in all clinical isolate genomes that were divergent (LS-BSR values <0.8, ≥0.4) or absent (LS-BSR values < 0.4) from the three remaining environmental isolate genomes (22702, J-C2-34, SG176), and 20 that were highly-conserved (LS-BSR values ≥0.8) in the three environmental isolate genomes that were divergent (LS-BSR values <0.8, ≥0.4) or absent (LS-BSR values <0.4) from the clinical isolate genomes (Table 3). The small number of genes that were universal to clinical or environmental isolates could also be a result of the genetic diversity or misclassification of the clinical and environmental isolates (Figures 1, 2).
The number of genes that were exclusive (LS-BSR values ≥0.8, and <0.4 in all other genomes) to the six V. parahaemolyticus genomes sequenced in this study ranged from 20 to 173 (Supplemental Table 3). The fewest number of exclusive genes (20) was identified in the genome of the O4:K12 isolate K1461, which can be attributed to the significant genomic similarity of this isolate to the previously sequenced genome of the O4:K12 isolate 10329 (Gonzalez-Escalona et al., 2011) (Figure 2). The genome of the V. parahaemolyticus clinical isolate that was tdh−/trh−/T3SS2−, K1275, encoded 150 genes that were exclusive to this isolate (Supplemental Table 3). Among these unique genes were many hypothetical proteins and other genes that lacked similarity to any previously characterized genes, which suggests there is extensive genomic diversity that has yet to be characterized from V. parahaemolyticus isolates (Supplemental Table 3).
Many of the genes identified as exclusive to a particular genome were hypothetical or were similar to genes of mobile genetic elements including plasmids and phage (Supplemental Table 3), highlighting the contribution of mobile elements such as plasmids and phage to the diversification of V. parahaemolyticus. In addition to being present in the post-1995 O3:K6 genomes, the protein-encoding genes of the filamentous vibriophage f237 of RIMD2210633 were identified in the genomes of two environmental isolate genomes (22702, SG176) (Table 2). Sequence analysis of partial nucleotide sequences of the vibriophage replication protein-encoding gene, rstA obtained from V. parahaemolyticus clinical and environmental isolates revealed these sequences had 95–100% nucleotide identity to the rstA of the filamentous phage f237 (Nasu et al., 2000). Phylogenetic analysis of the partial nucleotide sequences of rstA, demonstrated there is no discernible pattern of genetic similarity of the filamentous vibriophage based on serotype, isolation source, or geographical location, with the exception of the O4:K12 isolates and the post-1995 O3:K6 isolates (Supplemental Figure 3).
In addition to the identification of genetic similarity of filamentous vibriophage from clinical and environmental isolates, there was an approximately 90-kb phage-like element identified in the genome sequences of the O4:K12 clinical isolate K1461, and the environmental isolate and J-C2-34. Analysis of the plasmid content of K1461 and J-C2-34 using a modified acid-phenol extraction method (Kieser, 1984; Sobecky et al., 1997) demonstrated that both of these isolates contain a single large extrachromosomal element that is approximately 90-kb, which is likely the prophage identified in the genomes of these isolates. Sequence characterization of these prophage demonstrated they encode numerous phage-like genes with a conserved organization that exhibited 80–100% nucleotide identity to each other (Supplemental Figure 4). This finding provides additional evidence of the horizontal transfer of similar prophage-like elements among V. parahaemolyticus clinical and environmental isolates. These phage-like elements also exhibited divergent similarity (BSR values ≥0.4, <0.8) to genes encoded by a previously sequenced plasmid, p0908, from V. fluvialis (Hazen et al., 2007) and the bacteriophage P1 (Lobocka et al., 2004), suggesting they belong to a phage family that has relatives in other enteric bacteria.
Genomic Similarity of the O3:K6 Environmental Isolate, AF91, to Pre- and Post-1995 O3:K6 Isolates
Phylogenomic analysis demonstrated that the 2006 O3:K6 environmental isolate, AF91, exhibited greater genomic similarity to the post-1995 O3:K6 isolate genomes (Makino et al., 2003; Chen et al., 2011) than to the two pre-1995 O3:K6 isolate genomes (Boyd et al., 2008; Chen et al., 2011) (Figure 2). A PCR-based assay and in silico analysis of the AF91 genome demonstrated that AF91 does not encode the hemolysins or T3SS2 that are typically found in V. parahaemolyticus clinical isolates (Table 2, Supplemental Table 1). The AF91 genome was also missing most of the genomic islands of the post-1995 O3:K6 isolate RIMD2210633 (Table 2, Figure 3). However, AF91 did contain genes with similarity to those encoded by the genomic island VPaI-3 (Table 2), which was previously identified in post-1995 O3:K6 isolates and related isolates (AN5034) that have undergone seroconversion (Boyd et al., 2008; Chen et al., 2011) (Table 2). AF91 also encoded genes with significant similarity to genes of T6SS1 (Table 2, Figure 3), which has primarily been identified in V. parahaemolyticus clinical isolates (Yu et al., 2012). Their findings demonstrated that the T6SS1 genes exhibited bacteriolytic activity against other bacteria when grown in conditions similar to the marine environment, suggesting T6SS1 provides a fitness advantage that allows the disease-associated isolates to be competitive and persist in marine environments (Salomon et al., 2013). T6SS1 may have contributed to the emergence and spread of the post-1995 O3:K6 isolates (Okuda et al., 1997; Matsumoto et al., 2000).
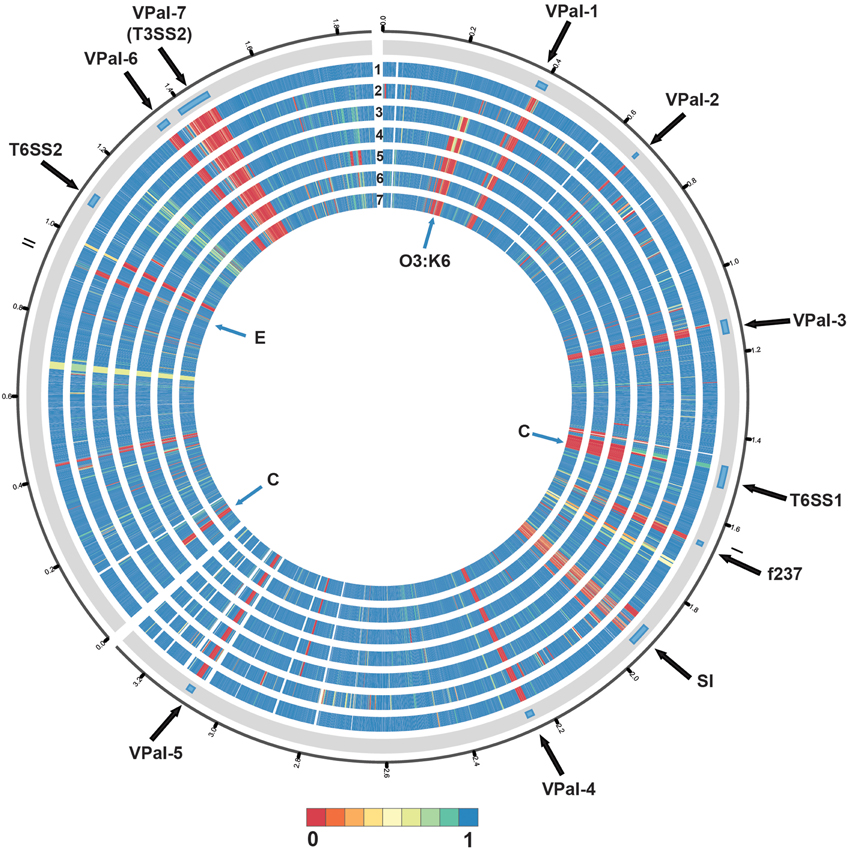
Figure 3. Circular display of the BLASTN BSR values demonstrating the genetic similarity of each gene of RIMD2210633 in the genome of the pre-1995 O3:K6 isolate AQ4037 (Chen et al., 2011), and the genomes sequenced in this study. The previously described putative pathogenicity islands (Hurley et al., 2006) and other regions of interest (Chen et al., 2011) in the genome of RIMD2210633 (Makino et al., 2003) are indicated in the outermost track. The genomes in tracks labeled 1–7 are as follows: (1) AF91, (2) AQ4037, (3) K1275, (4) K1461, (5) SG176, (6) J-C2-34, and (7) 22702. The location of the O3:K6 O-antigen lipopolysaccharide region is indicated by “O3:K6.” An “E” designates the location of RIMD2210633 genes that are present with greater similarity in the environmental isolate genomes than in the clinical isolate genomes. A “C” designates the location of RIMD2201633 genes that are present in the clinical isolate genomes analyzed and not in the environmental isolate genomes, with the exception of AF91. The circular display of the BSR values was generated using Circos (Krzywinski et al., 2009). Blue indicates genes were present with significant similarity, yellow indicates the genes were present but divergent, and red indicates genes were absent.
Comparative analysis of the O3:K6 genomes using LS-BSR demonstrated there were only 17 gene clusters identified in all post-1995 O3:K6 isolates, including the O3:K6 environmental isolate AF91 (Table 3) that were divergent (LS-BSR value <0.8, ≥0.4) or absent (LS-BSR value <0.4) from the other genomes. However, there were 78 gene clusters present with significant similarity in all of the post-1995 O3:K6 isolate genomes when AF91 was not included (Table 3). This finding demonstrates that although AF91 has the O3:K6 serotype and was isolated a decade after the emergence and spread of the O3:K6 pandemic clone (Okuda et al., 1997; Vuddhakul et al., 2000; Myers et al., 2003; Quilici et al., 2005; Ottaviani et al., 2008), the genome of AF91 exhibits genetic differences compared to other post-1995 O3:K6 isolate genomes. Not surprisingly, many of these genes were encoded within the genomic regions that were previously described as unique to the post-1995 O3:K6 genomes (Hurley et al., 2006; Chen et al., 2011) (Supplemental Table 4). In addition to the 78 genes that are divergent or missing (LS-BSR value <0.8) from the AF91 genome compared to other post-1995 O3:K6 genomes, there were 173 genes that were identified by LS-BSR as being unique to the AF91 genome compared to the other genomes analyzed (Table 3). The genes that were unique to AF91 included integrases and transposases, putative transcriptional regulators, a GGDEF domain-containing protein, and many genes encoding proteins with unknown functions (Supplemental Table 3).
This study describes an environmental O3:K6 isolate that exhibits significant genetic similarity to the post-1995 O3:K6 isolates, yet does not encode most of the known virulence-associated genes of these isolates. Due to the genomic similarity of this O3:K6 environmental isolate to the post-1995 O3:K6 isolates, this isolate may have originally carried the missing genomic islands, and after entering the environment may have begun to transition to an environmental niche by losing the virulence-associated genomic regions. For example, genomic islands of the uropathogenic Escherichia coli isolate 536, a strain isolated from a urinary tract infection, were demonstrated to be unstable (Middendorf et al., 2004; Soto et al., 2006). Interestingly, two of the E. coli 536 genomic islands, including one island that encoded a hemolysin, were lost in response to altered environmental conditions such as lower temperature and higher cell density (Middendorf et al., 2004). Further experiments would be necessary to determine whether AF91 would have a fitness advantage over other V. parahaemolyticus environmental isolates, and whether the absence of the other post-1995 O3:K6 genomic islands and virulence-associated regions gives it an additional advantage for surviving in the environment. Another possibility is that AF91 may represent an intermediate isolate that was involved in the emergence of the post-1995 O3:K6 isolates, and AF91 may have persisted without acquiring the post-1995 O3:K6 genomic islands due to a fitness advantage for surviving in the environment. Further experiments are needed to determine if AF91 would have a similar pathogenic potential as other post-1995 O3:K6 isolates, following the acquisition of the post-1995 O3:K6 genomic islands and other virulence-associated regions.
Previous research investigating the disease-causing potential of V. parahaemolyticus in the environment has typically examined the presence of prevalent clinical serotypes and known virulence-associated genes. However, we have demonstrated that investigating the genetic diversity of environmental isolates that do not carry the known virulence-associated genes can yield insight into the emergence of human disease-associated V. parahaemolyticus. Sequencing and characterization of V. parahaemolyticus AF91, an O3:K6 environmental isolate, demonstrated that environmental isolates that do not carry the known virulence-associated genes can have significant genetic similarity to disease-associated V. parahaemolyticus clinical isolates, including the pandemic post-1995 O3:K6 isolates. Additional genome sequencing of V. parahaemolyticus clinical and environmental isolates that have diverse serotypes and unique combinations of known virulence-associated genes and genomic regions would yield further insight into the ability of V. parahaemolyticus isolates to transition from an environmental niche and to emerge as pathogens.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank C. Lovell for kindly providing V. parahaemolyticus environmental isolates from NC. We thank S. Chen for laboratory assistance. This project was supported by a GT/CDC seed grant no. 1241326 to PS and CB. Instrumentation for sequencing of PCR amplicons at Georgia Tech was supported by NSF grant DBI-0304606.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2015.00204/abstract
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Angiuoli, S. V., and Salzberg, S. L. (2011). Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics 27, 334–342. doi: 10.1093/bioinformatics/btq665
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bagwell, C. E., Piceno, Y. M., Ashburne-Lucas, A., and Lovell, C. R. (1998). Physiological diversity of the rhizosphere diazotroph assemblages of selected salt marsh grasses. Appl. Environ. Microbiol. 64, 4276–4282.
Bej, A. K., Patterson, D. P., Brasher, C. W., Vickery, M. C., Jones, D. D., and Kaysner, C. A. (1999). Detection of total and hemolysin-producing Vibrio parahaemolyticus in shellfish using multiplex PCR amplification of tl, tdh, and trh. J. Microbiol. Methods 36, 215–225. doi: 10.1016/S0167-7012(99)00037-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bhoopong, P., Palittapongarnpim, P., Pomwised, R., Kiatkittipong, A., Kamruzzaman, M., Nakaguchi, Y., et al. (2007). Variability of properties of Vibrio parahaemolyticus strains isolated from individual patients. J. Clin. Microbiol. 45, 1544–1550. doi: 10.1128/JCM.02371-06
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Boyd, E. F., Cohen, A. L., Naughton, L. M., Ussery, D. W., Binnewies, T. T., Stine, O. C., et al. (2008). Molecular analysis of the emergence of pandemic Vibrio parahaemolyticus. BMC Microbiol. 8:110. doi: 10.1186/1471-2180-8-110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Broberg, C. A., Calder, T. J., and Orth, K. (2011). Vibrio parahaemolyticus cell biology and pathogenicity determinants. Microbes Infect. 13, 992–1001. doi: 10.1016/j.micinf.2011.06.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Burdette, D. L., Yarbrough, M. L., Orvedahl, A., Gilpin, C. J., and Orth, K. (2008). Vibrio parahaemolyticus orchestrates a multifaceted host cell infection by induction of autophagy, cell rounding, and then cell lysis. Proc. Natl. Acad. Sci. U.S.A. 105, 12497–12502. doi: 10.1073/pnas.0802773105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Caburlotto, G., Lleó, M. M., Hilton, T., Huq, A., Colwell, R. R., and Kaper, J. B. (2010). Effect on human cells of environmental Vibrio parahaemolyticus strains carrying type III secretion system 2. Infect. Immun. 78, 3280–3287. doi: 10.1128/IAI.00050-10
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
CDC. (2007). Preliminary FoodNet data on the incidence of infection with pathogens transmitted commonly through food—10 states, 2006. Morb. Mortal. Wkly. Rep. 55, 854–856.
CDC. (2008). Surveillance for waterborne disease and outbreaks associated with recreational water use and other aquatic facility-associated health events—United States, 2005–2006. Morb. Mortal. Wkly. Rep. 57, 1–29.
Chen, Y., Stine, O. C., Badger, J. H., Gil, A. I., Nair, G. B., Nishibuchi, M., et al. (2011). Comparative genomic analysis of Vibrio parahaemolyticus: serotype conversion and virulence. BMC Genomics 12:294. doi: 10.1186/1471-2164-12-294
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chevreux, B., Wetter, T., and Suhai, S. (1999). Genome sequence assembly using trace signals and additional sequence information. Comp. Sci. Biol. 99, 45–56.
Chowdhury, G., Ghosh, S., Pazhani, G. P., Paul, B. K., Maji, D., Mukhopadhyay, A. K., et al. (2013). Isolation and characterization of pandemic and nonpandemic strains of Vibrio parahaemolyticus from an outbreak of diarrhea in North 24 Parganas, West Bengal, India. Foodborne Pathog. Dis. 10, 338–342. doi: 10.1089/fpd.2012.1340
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chowdhury, N. R., Stine, O. C., Glenn Morris, J., and Nair, G. B. (2004). Assessment of evolution of pandemic Vibrio parahaemolyticus by multilocus sequence typing. J. Bacteriol. 42, 1280–1282.
Depaola, A., Hopkins, L. H., Peeler, J. T., Wentz, B., and McPhearson, R. M. (1990). Incidence of Vibrio parahaemolyticus in U.S. coastal waters and oysters. Appl. Environ. Microbiol. 56, 2299–2302.
Gavilan, R. G., Zamudio, M. L., and Martinez-Urtaza, J. (2013). Molecular epidemiology and genetic variation of pathogenic Vibrio parahaemolyticus in Peru. PLoS Negl. Trop. Dis. 7:e2210. doi: 10.1371/journal.pntd.0002210
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gertz, E. M., Yu, Y. K., Agarwala, R., Schaffer, A. A., and Altschul, S. F. (2006). Composition-based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. BMC Biol. 4:41. doi: 10.1186/1741-7007-4-41
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
González-Escalona, N., Martinez-Urtaza, J., Romero, J., Espejo, R. T., Jaykus, L., and Depaola, A. (2008). Determination of molecular phylogenetics of Vibrio parahaemolyticus strains by multilocus sequence typing. J. Bacteriol. 190, 2831–2840. doi: 10.1128/JB.01808-07
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gonzalez-Escalona, N., Strain, E. A., De Jesus, A. J., Jones, J. L., and Depaola, A. (2011). Genome sequence of the clinical O4:K12 serotype Vibrio parahaemolyticus strain 10329. J. Bacteriol. 193, 3405–3406. doi: 10.1128/JB.05044-11
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and anlysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98.
Ham, H., and Orth, K. (2012). The role of type III secretion system 2 in Vibrio parahaemolyticus pathogenicity. J. Microbiol. 50, 719–725. doi: 10.1007/s12275-012-2550-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Han, H., Wong, H. C., Kan, B., Guo, Z., Zeng, X., Yin, S., et al. (2008). Genome plasticity of Vibrio parahaemolyticus: microevolution of the ‘pandemic group’. BMC Genomics 9:570. doi: 10.1186/1471-2164-9-570
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hazen, T. H., Kennedy, K. D., Chen, S., Yi, S. V., and Sobecky, P. A. (2009). Inactivation of mismatch repair increases the diversity of Vibrio parahaemolyticus. Environ. Microbiol. 11, 1254–1266. doi: 10.1111/j.1462-2920.2008.01853.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hazen, T. H., Sahl, J. W., Fraser, C. M., Donnenberg, M. S., Scheutz, F., and Rasko, D. A. (2013). Refining the pathovar paradigm via phylogenomics of the attaching and effacing Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 110, 12810–12815. doi: 10.1073/pnas.1306836110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hazen, T. H., Wu, D., Eisen, J. A., and Sobecky, P. A. (2007). Sequence characterization and comparative analysis of three plasmids isolated from environmental Vibrio spp. Appl. Environ. Microbiol. 73, 7703–7710. doi: 10.1128/AEM.01577-07
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hurley, C. C., Quirke, A. M., Reen, F. J., and Boyd, E. F. (2006). Four genomic islands that mark post-1995 pandemic Vibrio parahaemolyticus isolates. BMC Genom. 7:104. doi: 10.1186/1471-2164-7-104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jensen, R. V., Depasquale, S. M., Harbolick, E. A., Hong, T., Kernell, A. L., Kruchko, D. H., et al. (2013). Complete Genome Sequence of Prepandemic Vibrio parahaemolyticus BB22OP. Genome Announc. 1:e00002-12. doi: 10.1128/genomeA.00002-12
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jones, J. L., Ludeke, C. H., Bowers, J. C., Garrett, N., Fischer, M., Parsons, M. B., et al. (2012). Biochemical, serological, and virulence characterization of clinical and oyster Vibrio parahaemolyticus isolates. J. Clin. Microbiol. 50, 2343–2352. doi: 10.1128/JCM.00196-12
Kaper, J. B., Campen, R. K., Seidler, R. J., Baldini, M. M., and Falkow, S. (1984). Cloning of the thermostable direct or Kanagawa phenomenon-associated hemolysin of Vibrio parahaemolyticus. Infect. Immun. 45, 290–292.
Kieser, T. (1984). Factors affecting the isolation of CCC DNA from Streptomyces lividans and Escherichia coli. Plasmid 12, 19–36. doi: 10.1016/0147-619X(84)90063-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kumar, S., Tamura, K., and Nei, M. (2004). MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Briefings Bioinf. 5, 150–163. doi: 10.1093/bib/5.2.150
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Levine, M. M., Nataro, J. P., Karch, H., Baldini, M. M., Kaper, J. B., Black, R. E., et al. (1985). The diarrheal response of humans to some classic serotypes of enteropathogenic Escherichia coli is dependent on a plasmid encoding an enteroadhesiveness factor. J. Infect. Dis. 152, 550–559. doi: 10.1093/infdis/152.3.550
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lin, B., Wang, Z., Malanoski, A. P., O'grady, E. A., Wimpee, C. F., Vuddhakul, V., et al. (2010). Comparative genomic analyses identify the Vibrio harveyi genome sequenced strains BAA-1116 and HY01 as Vibrio campbellii. Environ. Microbiol. Rep. 2, 81–89. doi: 10.1111/j.1758-2229.2009.00100.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lobocka, M. B., Rose, D. J., Plunkett, G. III., Rusin, M., Samojedny, A., Lehnherr, H., et al. (2004). Genome of bacteriophage P1. J. Bacteriol. 186, 7032–7068. doi: 10.1128/JB.186.21.7032-7068.2004
Ludeke, C. H., Fischer, M., Lafon, P., Cooper, K., and Jones, J. L. (2014). Suitability of the molecular subtyping methods intergenic spacer region, direct genome restriction analysis, and pulsed-field gel electrophoresis for clinical and environmental Vibrio parahaemolyticus isolates. Foodborne Pathog. Dis. 11, 520–528. doi: 10.1089/fpd.2013.1728
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lynch, T., Livingstone, S., Buenaventura, E., Lutter, E., Fedwick, J., Buret, A. G., et al. (2005). Vibrio parahaemolyticus disruption of epithelial cell tight junctions occurs independently of toxin production. Infect. Immun. 73, 1275–1283. doi: 10.1128/IAI.73.3.1275-1283.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ma, C., Deng, X., Ke, C., He, D., Liang, Z., Li, W., et al. (2014). Epidemiology and etiology characteristics of foodborne outbreaks caused by Vibrio parahaemolyticus during 2008-2010 in Guangdong province, China. Foodborne Pathog. Dis. 11, 21–29. doi: 10.1089/fpd.2013.1522
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Makino, K., Oshima, K., Kurokawa, K., Yokoyama, K., Uda, T., Tagomori, K., et al. (2003). Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V. cholerae. Lancet 361, 743–749. doi: 10.1016/S0140-6736(03)12659-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Matsumoto, C., Okuda, J., Ishibashi, M., Iwanaga, M., Garg, P., Rammamurthy, T., et al. (2000). Pandemic spread of an O3:K6 clone of Vibrio parahaemolyticus and emergence of related strains evidenced by arbitrarily primed PCR and toxRS sequence analyses. J. Clin. Microbiol. 38, 578–585.
Meador, C. E., Parsons, M. B., Bopp, C. A., Gerner-Smidt, P., Painter, J. A., and Vora, G. J. (2007). Virulence gene- and pandemic group-specific marker profiling of clinical Vibrio parahaemolyticus isolates. J. Clin. Microbiol. 45, 1133–1139. doi: 10.1128/JCM.00042-07
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Middendorf, B., Hochhut, B., Leipold, K., Dobrindt, U., Blum-Oehler, G., and Hacker, J. (2004). Instability of pathogenicity islands in uropathogenic Escherichia coli 536. J. Bacteriol. 186, 3086–3096. doi: 10.1128/JB.186.10.3086-3096.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Myers, M. L., Panicker, G., and Bej, A. K. (2003). PCR detection of a newly emerged pandemic Vibrio parahaemolyticus O3:K6 pathogen in pure cultures and seeded waters from the Gulf of Mexico. Appl. Environ. Microbiol. 69, 2194–2200. doi: 10.1128/AEM.69.4.2194-2200.2003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nair, G. B., Ramamurthy, T., Bhattacharya, S. K., Dutta, B., Takeda, Y., and Sack, D. A. (2007). Global dissemination of Vibrio parahaemolyticus serotype O3:K6 and its serovariants. Clin. Microbiol. Rev. 20, 39–48. doi: 10.1128/CMR.00025-06
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nasu, H., Iida, T., Sugahara, T., Yamaichi, Y., Park, K. S., Yokoyama, K., et al. (2000). A filamentous phage associated with recent pandemic Vibrio parahaemolyticus O3:K6 strains. J. Clin. Microbiol. 38, 2156–2161.
Nishibuchi, M., Fasano, A., Russell, R. G., and Kaper, J. B. (1992). Enterotoxigenicity of Vibrio parahaemolyticus with and without genes encoding thermostable direct hemolysin. Infect. Immun. 60, 3539–3545.
Nishibuchi, M., and Kaper, J. B. (1995). Thermostable direct hemolysin gene of Vibrio parahaemolyticus: a virulence gene acquired by a marine bacterium. Infect. Immun. 63, 2093–2099.
Noriea, N. F. III., Johnson, C. N., Griffitt, K. J., and Grimes, D. J. (2010). Distribution of type III secretion systems in Vibrio parahaemolyticus from the northern Gulf of Mexico. J. Appl. Microbiol. 109, 953–962. doi: 10.1111/j.1365-2672.2010.04722.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Okada, N., Iida, T., Park, K. S., Goto, N., Yasunaga, T., Hiyoshi, H., et al. (2009). Identification and characterization of a novel type III secretion system in trh-positive Vibrio parahaemolyticus strain TH3996 reveal genetic lineage and diversity of pathogenic machinery beyond the species level. Infect. Immun. 77, 904–913. doi: 10.1128/IAI.01184-08
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Okuda, J., Ishibashi, M., Hayakawa, E., Nishino, T., Takeda, Y., Mukhopadhyay, A. K., et al. (1997). Emergence of a unique O3:K6 clone of Vibrio parahaemolyticus in Calcutta, India, and isolation of strains from the same clonal group from southeast Asian travelers arriving in Japan. J. Clin. Microbiol. 35, 3150–3155.
Okura, M., Osawa, R., Arakawa, E., Terajima, J., and Watanabe, H. (2005). Identification of Vibrio parahaemolyticus pandemic group-specific DNA sequence by genomic subtraction. J. Clin. Microbiol. 43, 3533–3536. doi: 10.1128/JCM.43.7.3533-3536.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ono, T., Park, K. S., Ueta, M., Iida, T., and Honda, T. (2006). Identification of proteins secreted via Vibrio parahaemolyticus type III secretion system 1. Infect. Immun. 74, 1032–1042. doi: 10.1128/IAI.74.2.1032-1042.2006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ottaviani, D., Leoni, F., Rocchegiani, E., Santarelli, S., Canonico, C., Masini, L., et al. (2008). First clinical report of pandemic Vibrio parahaemolyticus O3:K6 infection in Italy. J. Clin. Microbiol. 46, 2144–2145. doi: 10.1128/JCM.00683-08
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Park, K. S., Ono, T., Rokuda, M., Jang, M. H., Okada, K., Iida, T., et al. (2004). Functional characterization of two type III secretion systems of Vibrio parahaemolyticus. Infect. Immun. 72, 6659–6665. doi: 10.1128/IAI.72.11.6659-6665.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Parsons, M. B., Cooper, K. L., Kubota, K. A., Puhr, N., Simington, S., Calimlim, P. S., et al. (2007). PulseNet USA standardized pulsed-field gel electrophoresis protocol for subtyping of Vibrio parahaemolyticus. Foodborne Pathog. Dis. 4, 285–292. doi: 10.1089/fpd.2007.0089
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Quilici, M. L., Robert-Pillot, A., Picart, J., and Fournier, J. M. (2005). Pandemic Vibrio parahaemolyticus O3:K6 spread, France. Emerg. Infec. Dis. 11, 1148–1149. doi: 10.3201/eid1107.041008
Rasko, D. A., Myers, G. S., and Ravel, J. (2005). Visualization of comparative genomic analyses by BLAST score ratio. BMC Bioinformatics 6:2. doi: 10.1186/1471-2105-6-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sahl, J. W., Caporaso, J. G., Rasko, D. A., and Keim, P. (2014). The large-scale blast score ratio (LS-BSR) pipeline: a method to rapidly compare genetic content between bacterial genomes. PeerJ 2:e332. doi: 10.7717/peerj.332
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sahl, J. W., Gillece, J. D., Schupp, J. M., Waddell, V. G., Driebe, E. M., Engelthaler, D. M., et al. (2013). Evolution of a pathogen: a comparative genomics analysis identifies a genetic pathway to pathogenesis in Acinetobacter. PLoS ONE 8:e54287. doi: 10.1371/journal.pone.0054287
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sahl, J. W., Steinsland, H., Redman, J. C., Angiuoli, S. V., Nataro, J. P., Sommerfelt, H., et al. (2011). A comparative genomic analysis of diverse clonal types of enterotoxigenic Escherichia coli reveals pathovar-specific conservation. Infect. Immun. 79, 950–960. doi: 10.1128/IAI.00932-10
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Salomon, D., Gonzalez, H., Updegraff, B. L., and Orth, K. (2013). Vibrio parahaemolyticus type VI secretion system 1 is activated in marine conditions to target bacteria, and is differentially regulated from system 2. PLoS ONE 8:e61086. doi: 10.1371/journal.pone.0061086
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sawabe, T., Kita-Tsukamoto, K., and Thompson, F. L. (2007). Inferring the evolutionary history of Vibrios by means of multilocus sequence analysis. J. Bacteriol. 189, 7932–7936. doi: 10.1128/JB.00693-07
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sawabe, T., Ogura, Y., Matsumura, Y., Feng, G., Amin, A. R., Mino, S., et al. (2013). Updating the Vibrio clades defined by multilocus sequence phylogeny: proposal of eight new clades, and the description of Vibrio tritonius sp. nov. Front. Microbiol. 4:414. doi: 10.3389/fmicb.2013.00414
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sobecky, P. A., Mincer, T. J., Chang, M. C., and Helinski, D. R. (1997). Plasmids isolated from marine sediment microbial communities contain replication and incompatibility regions unrelated to those of known plasmid groups. Appl. Environ. Microbiol. 63, 888–895.
Soto, S. M., Jimenez De Anta, M. T., and Vila, J. (2006). Quinolones induce partial or total loss of pathogenicity islands in uropathogenic Escherichia coli by SOS-dependent or -independent pathways, respectively. Antimicrob. Agents Chemother. 50, 649–653. doi: 10.1128/AAC.50.2.649-653.2006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sugiyama, T., Iida, T., Izutsu, K., Park, K. S., and Honda, T. (2008). Precise region and the character of the pathogenicity island in clinical Vibrio parahaemolyticus strains. J. Bacteriol. 190, 1835–1837. doi: 10.1128/JB.01293-07
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tena, D., Arias, M., Alvarez, B. T., Mauleon, C., Jimenez, M. P., and Bisquert, J. (2010). Fulminant necrotizing fasciitis due to Vibrio parahaemolyticus. J. Med. Microbiol. 59, 235–238. doi: 10.1099/jmm.0.014654-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thompson, C. C., Thompson, F. L., and Vicente, A. C. (2008). Identification of Vibrio cholerae and Vibrio mimicus by multilocus sequence analysis (MLSA). Int. J. Syst. Evol. Microbiol. 58, 617–621. doi: 10.1099/ijs.0.65461-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thompson, F. L., Gevers, D., Thompson, C. C., Dawyndt, P., Naser, S., Hoste, B., et al. (2005). Phylogeny and molecular identification of Vibrios on the basis of multilocus sequence analysis. Appl. Environ. Microbiol. 71, 5107–5115. doi: 10.1128/AEM.71.9.5107-5115.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thompson, F. L., Gomez-Gil, B., Vasconcelos, A. T., and Sawabe, T. (2007). Multilocus sequence analysis reveals that Vibrio harveyi and V. campbellii are distinct species. Appl. Environ. Microbiol. 73, 4279–4285. doi: 10.1128/AEM.00020-07
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Turner, J. W., Paranjpye, R. N., Landis, E. D., Biryukov, S. V., Gonzalez-Escalona, N., Nilsson, W. B., et al. (2013). Population structure of clinical and environmental Vibrio parahaemolyticus from the Pacific Northwest coast of the United States. PLoS ONE 8:e55726. doi: 10.1371/journal.pone.0055726
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vora, G. J., Meador, C. E., Bird, M. M., Bopp, C. A., Andreadis, J. D., and Stenger, D. A. (2005). Microarray-based detection of genetic heterogeneity, antimicrobial resistance, and the viable but nonculturable state in human pathogenic Vibrio spp. Proc. Natl. Acad. Sci. U.S.A. 102, 19109–19114. doi: 10.1073/pnas.0505033102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vuddhakul, V., Chowdhury, A., Laohaprertthisan, V., Pungrasamee, P., Patararungrong, N., Thianmontri, P., et al. (2000). Isolation of a pandemic O3:K6 clone of a Vibrio parahaemolyticus strain from environmental and clinical sources in Thailand. Appl. Environ. Microbiol. 66, 2685–2689. doi: 10.1128/AEM.66.6.2685-2689.2000
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yu, P. A., Puhr, N. D., Bopp, C. A., Gerner-Smidt, P., and Painter, J. A. (2006). “Vibrio parahaemolyticus hemolysins associated with decreased hospitalization from shellfish-borne illness,” in Fifth International Conference on Emerging Infectious Diseases. (Atlanta, GA).
Yu, Y., Yang, H., Li, J., Zhang, P., Wu, B., Zhu, B., et al. (2012). Putative type VI secretion systems of Vibrio parahaemolyticus contribute to adhesion to cultured cell monolayers. Arch. Microbiol. 194, 827–835. doi: 10.1007/s00203-012-0816-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, L., and Orth, K. (2013). Virulence determinants for Vibrio parahaemolyticus infection. Curr. Opin. Microbiol. 16, 70–77. doi: 10.1016/j.mib.2013.02.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: genomics, Vibrio parahaemolyticus, environment, phylogenomics, O3:K6
Citation: Hazen TH, Lafon PC, Garrett NM, Lowe TM, Silberger DJ, Rowe LA, Frace M, Parsons MB, Bopp CA, Rasko DA and Sobecky PA (2015) Insights into the environmental reservoir of pathogenic Vibrio parahaemolyticus using comparative genomics. Front. Microbiol. 6:204. doi: 10.3389/fmicb.2015.00204
Received: 08 December 2014; Accepted: 26 February 2015;
Published: 24 March 2015.
Edited by:
Iddya Karunasagar, Food Safety Consultant, IndiaReviewed by:
Sheng Chen, Hong Kong Polytechnic University, Hong KongYukiko Hara-Kudo, National Institute of Health Sciences, Japan
Angelo Depaola, United States Food and Drug Administration, USA
Copyright © 2015 Hazen, Lafon, Garrett, Lowe, Silberger, Rowe, Frace, Parsons, Bopp, Rasko and Sobecky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tracy H. Hazen, School of Medicine, Institute for Genome Sciences, University of Maryland, 801 W. Baltimore Street, Baltimore, MD 21201, USAdGhhemVuQHNvbS51bWFyeWxhbmQuZWR1
†Present Address: Tracy H. Hazen, Institute for Genome Sciences, University of Maryland School of Medicine, Baltimore, MD, USA
Tiffany M. Lowe, Department of Plant Pathology, University of Wisconsin, Madison, Madison, WI, USA
Daniel J. Silberger, Department of Pathology, University of Alabama at Birmingham, Birmingham, AL, USA
Patricia A. Sobecky, Department of Biological Sciences, University of Alabama, Tuscaloosa, AL, USA