 Günther Schönrich
Günther Schönrich- Institute of Medical Virology, Helmut-Ruska-Haus, Charité–Universitätsmedizin Berlin, Berlin, Germany
Viral hemorrhagic fever caused by hantaviruses is an emerging infectious disease for which suitable treatments are not available. In order to improve this situation a better understanding of hantaviral pathogenesis is urgently required. Hantaviruses infect endothelial cell layers in vitro without causing any cytopathogenic effect and without increasing permeability. This implies that the mechanisms underlying vascular hyperpermeability in hantavirus-associated disease are more complex and that immune mechanisms play an important role. In this review we highlight the latest developments in hantavirus-induced immunopathogenesis. A possible contribution of neutrophils has been neglected so far. For this reason, we place special emphasis on the pathogenic role of neutrophils in disrupting the endothelial barrier.
Introduction
Viral hemorrhagic fever (VHF) is caused by viruses belonging to different virus families, one of which is the Bunyaviridae (Schmaljohn and Nichol, 2007). Structurally, hantaviruses have an envelope derived from the host cell membrane. Their genome consists of three negative-strand RNA segments encoding a nucleoprotein (N), two glycoproteins (Gn and Gc), and a RNA-dependent RNA polymerase (Schmaljohn and Nichol, 2007). According to the geographic location of the natural reservoir hosts and the disease syndrome induced, hantaviruses are divided into Old World and New World hantavirus species.
Humans become infected with hantaviruses after inhalation of aerosols derived from excreta of persistently infected but asymptomatic natural reservoir hosts, in general rodents. Depending on the hantavirus species involved the severity of hantavirus-induced disease varies with case fatality rates from less than 1% to up to more than 40% (Jonsson et al., 2010; Krüger et al., 2015). Old World hantavirus species such as Hantaan virus (HTNV) are associated with hemorrhagic fever with renal syndrome (HFRS). After an incubation period of approximately 3 weeks HFRS starts with a febrile phase and further unspecific symptoms. Subsequently, hypotension and oliguria is observed that may finally result in fatal shock. Patients recover after a polyuric phase that starts in the second week of illness. A mild form of HFRS, also termed nephropathia epidemica, with a case fatality rate of less than 1% is endemic in Europe and is in large part due to infection with Puumala virus (PUUV; Mustonen et al., 2013). In contrast, infection with New World hantavirus species such as Sin Nombre virus (SNV) can result in hantavirus cardio-pulmonary syndrome (HCPS; Nichol et al., 1993). In the course of HCPS patients develop pulmonary edema and cardiac failure whereas in HFRS kidney failure is the prominent clinical feature. Andes virus (ANDV) is the most lethal New World hantavirus species with case fatality rates of up to more than 40%. It is the only hantavirus species for which human-to-human-transmission has been reported (Martinez-Valdebenito et al., 2014). Some hantavirus species such as Prospect Hill virus (PHV) are non-pathogenic whereas others such as Tula virus (TULV) cause only sporadically disease (Klempa et al., 2003; Zelena et al., 2013). In China 20,000 to 50,000 HFRS cases are reported annually, which represents 90% of HFRS cases worldwide (Fang et al., 2015).
It is now increasingly apparent that the paradigm of two distinct syndromes induced by Old Word and New World hantaviruses needs to be reconsidered (Krautkrämer et al., 2013; Clement et al., 2014). For example, cardiopulmonary dysfunction can dominate the clinical picture after infection with Old World hantavirus species (Clement et al., 1994; Rasmuson et al., 2011a,b; Gizzi et al., 2013). Vice versa, kidney function is also impaired in patients suffering from infection with New World hantavirus species (Pergam et al., 2009; MacNeil et al., 2011).
As with other VHF dysregulation of the endothelial cell (EC) barrier resulting in capillary leakage is the key finding in hantavirus-induced disease (Duchin et al., 1994). The extent of vascular dysfunction determines the severity of the clinical course. So far no preventive or therapeutic strategies for hantavirus-induced disease have been approved by the Food and Drug Administration (Schmaljohn, 2009; Krüger et al., 2011). However, an experimental HCPS DNA vaccine has been successfully tested in non-human primates (Kwilas et al., 2014). Moreover, the HCPS DNA vaccine elicits production of neutralizing human IgG (immunoglobulin G) in trans-chromosomal bovines which could be used for passive immunoprophylaxis in humans (Hooper et al., 2014). In this review we will focus on concepts explaining how hantavirus-induced immune responses interfere with the endothelial barrier function and briefly mention also non-immunological mechanisms.
Non-immunological Mechanisms
Hantaviruses infect and replicate in EC cultures without causing any cytopathic effect or increasing permeability (Pensiero et al., 1992; Temonen et al., 1993; Khaiboullina et al., 2000; Sundstrom et al., 2001). However, in the presence of vascular endothelial growth factor (VEGF) replication of HTNV or ANDV in human umbilical EC downregulates vascular endothelial (VE)-cadherin, a major component of adherens junctions, thereby disrupting the endothelial barrier (Gavrilovskaya et al., 2008; Gorbunova et al., 2010; Li et al., 2012). Recently, VE-cadherin degradation was observed even in the absence of exogenous VEGF after ANDV infection of primary human pulmonary microvascular EC (Shrivastava-Ranjan et al., 2010). This was not confirmed in another experimental setup using in vitro capillary blood vessels (Taylor et al., 2013). In this system it was found that infection with HTNV or ANDV results in activation of the kallikrein–kinin system and liberation of bradykinin, a potent inducer of vascular permeability (Taylor et al., 2013). In accordance, a bradykinin receptor antagonist improved the clinical outcome in a case of PUUV infection (Antonen et al., 2013). Finally, glomerular EC infected with PUUV show disruption of cell-to-cell contacts (Krautkrämer et al., 2011).
Hantaviral Immunopathogenesis
Both innate and adaptive as well as humoral and cellular immune mechanisms contribute to hantavirus-associated disease. Human dendritic cells (DC) are highly mobile and bridge innate and adaptive immunity. DC reside at the pathogen-host interface in peripheral tissue including the respiratory mucosa and alveoli of the lung. They can push their dendritic projections into the airway lumen thereby “snorkeling” through the epithelial-tight junctions (Jahnsen et al., 2006). Thus, DC may become infected with hantavirus in the lung shortly after inhalation of viral particles. In accordance, human DC are susceptible to infection with HTNV and ANDV in vitro (Raftery et al., 2002; Marsac et al., 2011). Moreover, monocytes infected with HTNV develop into DC-like cells (Markotic et al., 2007; Schönrich et al., 2008). DC might act as a Trojan horse helping the pathogens to disseminate within the human organism and finally infect EC in various organs. Alternatively, DC may become infected later when they get in contact with the already infected human EC barrier. In striking contrast to most other DC-tropic viruses both Old World and New World hantavirus species induce DC maturation in vitro (Raftery et al., 2002; Marsac et al., 2011). This implies that in humans hantavirus-infected DC migrate to the draining lymph nodes and induce a vigorous adaptive immune response.
In accordance, histopathological analysis of tissue collected from fatal human HCPS cases has revealed strong mononuclear cell infiltrates especially in lung tissue (Nolte et al., 1995; Zaki et al., 1995). Similarly, endobronchial mucosal biopsies and bronchoalveolar lavage fluid from HFRS patients revealed activated CD8+ T cells and strong upregulation of vascular cell adhesion molecule 1 (VCAM-1) at the site of infection (Rasmuson et al., 2011b). Animal models of HCPS based on non-human primates and Syrian hamsters confirmed that an excessive and aberrant tissue-specific host response correlates with increased vascular hyperpermeability (Safronetz et al., 2015). For unknown reasons, however, T cell depletion neither influenced the viral load nor the clinical course of HCPS in Syrian hamsters. Intriguingly, most of the host genes that are linked to hantavirus disease severity are associated with abnormal immune responses or even autoimmune diseases (Charbonnel et al., 2014). In line with this view elevated levels of autoantibodies to nuclear antigen are found in hantavirus-infected patients (Raftery et al., 2014).
Activation of Endothelial Cells
Immunohistological studies of kidney biopsies derived from HFRS patients revealed that EC become activated during PUUV infection and increase expression of chemokines and adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1), E-Selectin, and VCAM-1 (Temonen et al., 1996). The latter are important for regulating the interaction of EC with immune cells (Razakandrainibe et al., 2013). It is questionable whether hantavirus directly upregulate adhesion molecules on EC (Sundstrom et al., 2001; Geimonen et al., 2002; Yu et al., 2014). It has been established, however, that immune cells stimulated during hantavirus infection release tumor necrosis factor alpha (TNF-α), a strong inducer of adhesion molecules in EC (Pober, 2002). The chemokines that are upregulated during hantavirus infection include interleukin (IL)-8 (Klingstrom et al., 2008; Sadeghi et al., 2011; Libraty et al., 2012; Kyriakidis and Papa, 2013), a key neutrophil-recruiting chemokine and activator (Amulic et al., 2012). Intriguingly, in some studies IL-8 levels were positively correlated with severe acute disease suggesting that it is part of an important pathogenic link (Libraty et al., 2012; Kyriakidis and Papa, 2013). Moreover, expression of HLA (human leucocyte antigen) class I molecules is increased on EC (Kraus et al., 2004). These include HLA-E (Bjorkstrom et al., 2011) which serves as a ligand for the activating NK (natural killer) cell receptor NKG2C. Thus, hantavirus-infected EC can interact with a variety of immune effector cells such as HLA class I-restricted CD8+ T cells, HLA-E stimulated NK cells and neutrophils.
Cytotoxic Immune Cells
Cytotoxic activity of activated immune cells may eliminate hantavirus-infected EC thereby causing vascular leakage. A SNV-specific CD8+ T cell line lysed HLA-matched SNV-infected EC thereby increasing vascular permeability (Hayasaka et al., 2007). Moreover, involvement of T cells is also supported by genetic susceptibility studies (Terajima and Ennis, 2011). In accordance, researchers have recently detected enhanced endothelial repair activity in HFRS patients (Krautkrämer et al., 2014). A role for cytotoxic immune mechanisms is further supported by increased serum levels of perforin and granzyme B (Klingstrom et al., 2006) as well as cell-free DNA (Outinen et al., 2012a; Raftery et al., 2014) in HFRS patients. However, histopathological examination of tissue from fatal HCPS cases did not reveal necrosis or any overtly visible lesions that can account for the vascular leakage in HCPS patients (Lukes, 1954; Nolte et al., 1995; Zaki et al., 1995). This may be due to difficulties in visualizing small but functionally relevant morphological correlates of endothelial damage. Moreover, it is possible that apoptotic EC are immediately phagocytosed by macrophages or neutrophils.
There is evidence that hantavirus-infected EC are protected, at least to some degree, from attack by cytotoxic T cells and NK cells (Gupta et al., 2013). However, uninfected EC are susceptible to cytotoxic attack and might be prone to bystander killing. For example, a subset of NK cells is activated through increased HLA-E expression on hantavirus-infected EC and may subsequently attack uninfected EC (Braun et al., 2014). This bystander NK attack could be facilitated by the fact that uninfected cells express less inhibitory HLA class I molecules on the cell surface than hantavirus-infected EC (Kraus et al., 2004; Lalwani et al., 2013).
Neutrophils
Metchnikoff (1887) first postulated that polymorphonuclear cells release substances that damage EC function. Nevertheless, neutrophils have been overlooked in models of hantavirus-induced disease although they represent the most abundant type of immune cell. In fact, neutrophil-rich infiltrates were reported in HCPS patients (Zaki et al., 1995). Moreover, increased numbers of neutrophils with band cell morphology are observed in the blood during hantavirus-associated disease (Hjelle et al., 1995; Zaki et al., 1995). This neutrophil subtype represents most likely a typical left-shift response that is usually found after bacterial challenge and regulates T cell responses (Pillay et al., 2012; Nauseef and Borregaard, 2014). Recent research indicates that neutrophils can contribute generally to hantavirus-induced immunopathogenesis. Upon interaction with activated EC neutrophils undergo NETosis (Gupta et al., 2010; Saffarzadeh et al., 2012), a recently discovered form of programmed neutrophil cell death (Brinkmann et al., 2004). It is characterized by the generation and release of neutrophil extracellular traps (NETs). NETs are a fibrillary network composed of a double-stranded DNA backbone and coated with histones as well as granule molecules such as myeloperoxidase, elastase and cathepsin G. NETosis in close proximity to EC is harmful and results in increased vascular permeability (Gupta et al., 2010; Villanueva et al., 2011; Saffarzadeh et al., 2012).
Neutrophils express β2 integrins, i.e., β2αL (CD18/CD11a), β2αM (CD18/CD11b) and β2αX (CD18/CD11c; Langereis, 2013). A recent study has demonstrated that hantaviruses strongly activate neutrophils through β2 integrin signaling resulting in NETosis (Raftery et al., 2014). In addition, activated platelets recruit neutrophils rapidly to the site of inflamed EC during VHF. Subsequently, platelet-leukocyte aggregation is mediated by the interaction of platelet proteins with β2 integrins on neutrophils (Zapata et al., 2014). Several infection models have demonstrated that platelet-neutrophil interactions through β2 integrins result also in NETosis (Clark et al., 2007; Caudrillier et al., 2012; McDonald et al., 2012; Jenne et al., 2013). Thus, β2 integrins may act as a master switch of NETosis during VHF (Figure 1A).
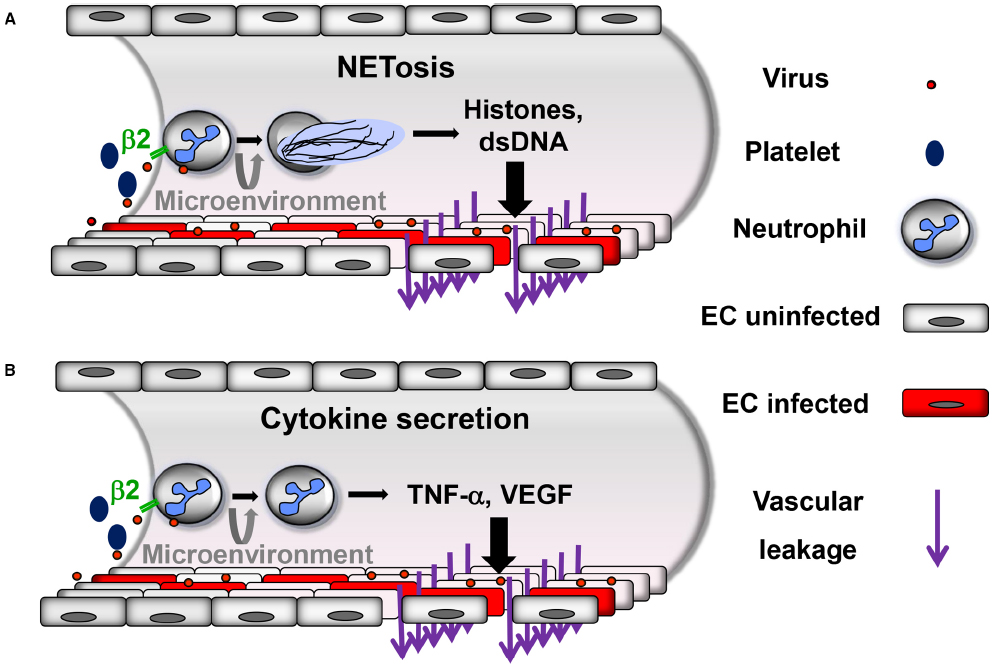
Figure 1. Proposed neutrophil-mediated mechanisms contributing to vascular leakage during hantavirus infection. Neutrophils are activated through virus-induced β2 integrin signaling. Activated platelets also stimulate neutrophils through β2 integrins. Depending on the β2 integrin ligands involved and possibly further microenvironmental stimuli neutrophils can be activated in a different way resulting in (A) NETosis or (B) secretion of inflammatory cytokines such as TNF-α or VEGF. In both cases increased vascular leakage is generated, although most likely by distinct mechanisms.
In accordance with hantavirus-induced NETosis, high levels of extracellular histones are found in sera from hantavirus-infected patients (Raftery et al., 2014; Vaheri et al., 2014). Histones are known to cause microvascular injury and mediate death in sepsis (Xu et al., 2009). Moreover, thrombocytopenia, prolonged prothrombin time and fibrin deposition are hallmarks of hantavirus-induced disease (Laine et al., 2010) and are observed upon histone injection into mice (Fuchs et al., 2011). In fact, extracellular nucleosomes derived from neutrophils induce formation of thrombosis in microvessels which is regarded as an innate host defense mechanism (Massberg et al., 2010). Importantly, depletion of neutrophils prevents pneumonia and vascular hyperpermeability in the SCID (severe combined immunodeficiency) mouse model of hantavirus infection (Koma et al., 2014). The observation that methylprednisolone treatment is not beneficial for HCPS patients is also in accordance with hantavirus-induced NETosis playing an important pathogenic role (Vial et al., 2013) as corticosteroids do not suppress NET formation (Lapponi et al., 2013).
Neutrophils may also contribute to microvascular plasma protein leakage by mechanisms other than NETosis (Figure 1B). Depending on the β2 integrin ligand involved in signaling and further as yet unknown microenvironmental stimuli neutrophils may be activated without undergoing NETosis. After adhering to activated endothelium and crawling along EC neutrophils start to transmigrate and release TNF-α which strongly increases vascular permeability (Finsterbusch et al., 2014). Subsequent binding of TNF-α to its receptor on EC induces endocytosis and degradation of VE-cadherin (Schulte et al., 2011). Similarly, stimulated neutrophils also secrete VEGF (Taichman et al., 1997), an important mediator of VE-cadherin degradation in hantavirus-infected EC (Gavrilovskaya et al., 2008; Gorbunova et al., 2010; Shrivastava-Ranjan et al., 2010; Li et al., 2012). In accordance, high VEGF serum levels are found during HFRS and HCPS (Shrivastava-Ranjan et al., 2010; Gavrilovskaya et al., 2012; Ma et al., 2012). Taken together, neutrophils represent a long-sought missing piece in the puzzle of hantaviral immunopathogenesis.
Complement System
There is compelling evidence that the severity of HFRS symptoms correlates with the degree of complement activation (Paakkala et al., 2000; Sane et al., 2012). The complement system functions as an important inducer of vascular leakage alongside the kinin and the coagulation system (Bossi et al., 2011). During acute HFRS complement is activated by pentraxin-related protein 3 (PTX3), which represents a humoral pattern recognition receptor (Outinen et al., 2012b). Intriguingly, PTX3 is stored in neutrophil granules and released upon outside-in signals through integrins (Jaillon et al., 2007; Razvina et al., 2014). The soluble complement components C3a and C5a generated during complement activation by antibodies and PTX3 not only induce cytoskeletal rearrangements in EC but also IL-8 secretion (Monsinjon et al., 2003). Consequently, PTX3 attracts more neutrophils to the endothelial barrier aggravating vascular inflammation.
Inflammatory Cytokines
High levels of proinflammatory cytokines are detected in sera from hantavirus-infected patients especially TNF-α (Linderholm et al., 1996; Mori et al., 1999; Borges et al., 2008; Klingstrom et al., 2008; Sadeghi et al., 2011; Saksida et al., 2011; Libraty et al., 2012; Kyriakidis and Papa, 2013). TNF-α is released by activated antiviral immune cells such as neutrophils, NK cells and CD8+ T cells as well as hantavirus-infected DC and macrophages (Raftery et al., 2002; Marsac et al., 2011; Shin et al., 2012).
TNF-α represents a double-edged sword. On one side it may help to control hantaviral dissemination by purging virus from infected cells through non-cytolytic mechanisms (Khaiboullina et al., 2000; Guidotti and Chisari, 2001). On the other side, if it is administered exogenously in quantities that are found during hantavirus infection, vascular leakage and respiratory distress are induced (Tracey and Cerami, 1994; Wimer, 1998). Local release of TNF-α at the EC interface could increase vascular permeability by direct and indirect mechanisms. Firstly, TNF-α not only upregulates adhesion molecules such as ICAM-1, a natural ligand for β2 integrin, but also IL-8. This cytokine both recruits and activates neutrophils, and furthermore induces NETs (Brinkmann et al., 2004). Secondly, TNF-α can directly increase vascular permeability by inducing cytoskeletal rearrangements resulting in redistribution of human microvascular endothelial tight junctions (Blum et al., 1997; Ozaki et al., 1999). Hantaviruses may further enhance this direct TNF-α effect as HTNV-infected EC show prolonged hyperpermeability after exposure to TNF-α in comparison to uninfected control cells (Niikura et al., 2004). The pivotal role of TNF-α in hantaviral immunopathogenesis may explain the relatively poor activity of ribavirin in HCPS; it blocks ANDV replication and suppresses release of some inflammatory mediators but not TNF-α (Khaiboullina et al., 2013).
A high-producing TNF-α genotype (polymorphism at position –308) was linked to more severe HFRS in Finish patients although not independently of the HLA-B8-DR3 haplotype (Kanerva et al., 1998; Makela et al., 2002). This high-producing TNF-α genotype was also more frequently found in HCPS patients than in seropositive individuals without HCPS (Borges et al., 2010). Another study in Belgium showed a link between a low-producing TNF-α genotype (polymorphism at position –238) with more severe HFRS (Maes et al., 2006). This discrepancy may be reconciled by assuming that TNF-α release at the hantavirus-infected EC barrier must be tightly controlled. If there is not enough TNF-α the virus may replicate and disseminate more vigorously especially as hantavirus N protein can interfere with signaling through the TNF receptor (Taylor et al., 2009; Ontiveros et al., 2010). This likely increases vascular permeability due to non-immunological effects of viral particles on subcellular structures. On the other hand, too much local TNF-α allows better control of the virus but at the same time may increase immune-mediated damage.
Concluding Remarks
Humoral as well as cellular mechanisms of the adaptive and innate immune system contribute to hantavirus-induced disruption of the endothelial barrier (Figure 2). Intriguingly, neutrophils which so far have not been regarded as a player in hantavirus-induced immunopathogenesis seem to be important. NETs as well as neutrophil-derived factors such as VEGF, PTX3, and TNF-α can cause vascular dysfunction. Further studies are needed to reveal whether strategies aiming at neutrophil function can prevent hantavirus-induced immunopathogenesis. Furthermore, it is possible that NETs and other neutrophil-derived mediators of vascular hyperpermeability play a role in VHF caused by members of other virus families.
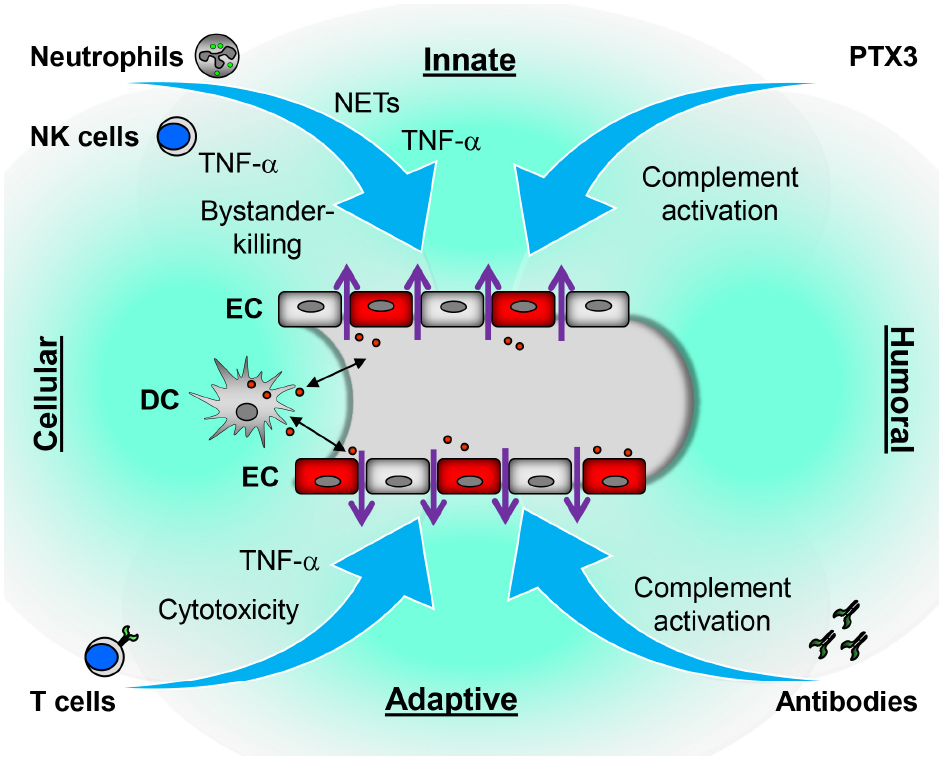
Figure 2. Proposed immune mechanisms contributing to hantavirus-induced disruption of the endothelial barrier. Both cellular and humoral components of innate and adaptive immune responses could contribute to vascular leakage of hantavirus-infected vessels. In response to hantavirus-infected EC, neutrophils generate NETs or may secrete inflammatory cytokines such as TNF-α which directly or indirectly increase vascular permeability. The humoral pattern recognition receptor PTX3 and antibodies activate complement. Activated complement components induce cytoskeletal rearrangements in EC further increasing dysfunction of the EC barrier. NK cells may kill bystander EC or also secrete TNF-α. DC may carry the virus from lung tissue to EC of the microvasculature in various organs or become infected after interaction with virus-infected EC. As hantavirus-infected DC mature they migrate to draining lymph nodes to initiate a vigorous CD8+ T cell response. The latter could contribute to vascular leakage by direct killing of hantavirus-infected EC or, more likely, by releasing TNF-α.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
This work was supported by Deutsche Forschungsgemeinschaft (GraKo 1121).
References
Amulic, B., Cazalet, C., Hayes, G. L., Metzler, K. D., and Zychlinsky, A. (2012). Neutrophil function: from mechanisms to disease. Annu. Rev. Immunol. 30, 459–489. doi: 10.1146/annurev-immunol-020711-074942
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Antonen, J., Leppanen, I., Tenhunen, J., Arvola, P., Makela, S., Vaheri, A., et al. (2013). A severe case of Puumala hantavirus infection successfully treated with bradykinin receptor antagonist icatibant. Scand. J. Infect. Dis. 45, 494–496. doi: 10.3109/00365548.2012.755268
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bjorkstrom, N. K., Lindgren, T., Stoltz, M., Fauriat, C., Braun, M., Evander, M., et al. (2011). Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J. Exp. Med. 208, 13–21. doi: 10.1084/jem.20100762
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Blum, M. S., Toninelli, E., Anderson, J. M., Balda, M. S., Zhou, J., O’Donnell, L., et al. (1997). Cytoskeletal rearrangement mediates human microvascular endothelial tight junction modulation by cytokines. Am. J. Physiol. 273, H286–H294.
Borges, A. A., Campos, G. M., Moreli, M. L., Moro Souza, R. L., Saggioro, F. P., Figueiredo, G. G., et al. (2008). Role of mixed Th1 and Th2 serum cytokines on pathogenesis and prognosis of hantavirus pulmonary syndrome. Microbes Infect. 10, 1150–1157. doi: 10.1016/j.micinf.2008.06.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Borges, A. A., Donadi, E. A., Campos, G. M., Moreli, M. L., de Sousa, R. L., Saggioro, F. P., et al. (2010). Association of –308G/A polymorphism in the tumor necrosis factor-α gene promoter with susceptibility to development of hantavirus cardiopulmonary syndrome in the Ribeirao Preto region, Brazil. Arch. Virol. 155, 971–975. doi: 10.1007/s00705-010-0655-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bossi, F., Peerschke, E. I., Ghebrehiwet, B., and Tedesco, F. (2011). Cross-talk between the complement and the kinin system in vascular permeability. Immunol. Lett. 140, 7–13. doi: 10.1016/j.imlet.2011.06.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Braun, M., Bjorkstrom, N. K., Gupta, S., Sundstrom, K., Ahlm, C., Klingstrom, J., et al. (2014). NK cell activation in human hantavirus infection explained by virus-induced IL-15/IL15Rα expression. PLoS Pathog. 10:e1004521. doi: 10.1371/journal.ppat.1004521
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brinkmann, V., Reichard, U., Goosmann, C., Fauler, B., Uhlemann, Y., Weiss, D. S., et al. (2004). Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535. doi: 10.1126/science.1092385
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Caudrillier, A., Kessenbrock, K., Gilliss, B. M., Nguyen, J. X., Marques, M. B., Monestier, M., et al. (2012). Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Invest. 122, 2661–2671. doi: 10.1172/JCI61303
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Charbonnel, N., Pages, M., Sironen, T., Henttonen, H., Vapalahti, O., Mustonen, J., et al. (2014). Immunogenetic factors affecting susceptibility of humans and rodents to hantaviruses and the clinical course of hantaviral disease in humans. Viruses 6, 2214–2241. doi: 10.3390/v6052214
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Clark, S. R., Ma, A. C., Tavener, S. A., McDonald, B., Goodarzi, Z., Kelly, M. M., et al. (2007). Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 13, 463–469. doi: 10.1038/nm1565
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Clement, J., Colson, P., and McKenna, P. (1994). Hantavirus pulmonary syndrome in New England and Europe. N. Engl. J. Med. 331, 545–546. doi: 10.1056/NEJM199408253310813
Clement, J., Maes, P., and Van, R. M. (2014). Hemorrhagic fever with renal syndrome in the new, and Hantavirus Pulmonary Syndrome in the old world: paradi(se)gm lost or regained? Virus Res. 187, 55–58. doi: 10.1016/j.virusres.2013.12.036
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duchin, J. S., Koster, F. T., Peters, C. J., Simpson, G. L., Tempest, B., Zaki, S. R., et al. (1994). Hantavirus pulmonary syndrome: a clinical description of 17 patients with a newly recognized disease. The Hantavirus Study Group. N. Engl. J. Med. 330, 949–955. doi: 10.1056/NEJM199404073301401
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fang, L. Z., Zhao, L., Wen, H. L., Zhang, Z. T., Liu, J. W., He, S. T., et al. (2015). Reservoir host expansion of hantavirus, china. Emerg. Infect. Dis. 21, 170–171. doi: 10.3201/eid2101.140960
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Finsterbusch, M., Voisin, M. B., Beyrau, M., Williams, T. J., and Nourshargh, S. (2014). Neutrophils recruited by chemoattractants in vivo induce microvascular plasma protein leakage through secretion of TNF. J. Exp. Med. 211, 1307–1314. doi: 10.1084/jem.20132413
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fuchs, T. A., Bhandari, A. A., and Wagner, D. D. (2011). Histones induce rapid and profound thrombocytopenia in mice. Blood 118, 3708–3714. doi: 10.1182/blood-2011-01-332676
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gavrilovskaya, I., Gorbunova, E., Koster, F., and Mackow, E. (2012). Elevated VEGF levels in pulmonary edema fluid and PBMCs from patients with acute Hantavirus Pulmonary Syndrome. Adv. Virol. 2012, 674360. doi: 10.1155/2012/674360
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gavrilovskaya, I. N., Gorbunova, E. E., Mackow, N. A., and Mackow, E. R. (2008). Hantaviruses direct endothelial cell permeability by sensitizing cells to the vascular permeability factor, VEGF, while angiopoietin-1 and sphingosine 1-phosphate inhibit hantavirus-directed permeability. J. Virol. 82, 5797–5806. doi: 10.1128/JVI.02397-07
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Geimonen, E., Neff, S., Raymond, T., Kocer, S. S., Gavrilovskaya, I. N., and Mackow, E. R. (2002). Pathogenic and nonpathogenic hantaviruses differentially regulate endothelial cell responses. Proc. Natl. Acad. Sci. U.S.A. 99, 13837–13842. doi: 10.1073/pnas.192298899
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gizzi, M., Delaere, B., Weynand, B., Clement, J., Maes, P., Vergote, V., et al. (2013). Another case of “European hantavirus pulmonary syndrome” with severe lung, prior to kidney, involvement, and diagnosed by viral inclusions in lung macrophages. Eur. J. Clin. Microbiol. Infect. Dis. 32, 1341–1345. doi: 10.1007/s10096-013-1885-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gorbunova, E., Gavrilovskaya, I. N., and Mackow, E. R. (2010). Pathogenic hantaviruses Andes virus and Hantaan virus induce adherens junction disassembly by directing vascular endothelial cadherin internalization in human endothelial cells. J. Virol. 84, 7405–7411. doi: 10.1128/JVI.00576-10
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Guidotti, L. G., and Chisari, F. V. (2001). Noncytolytic control of viral infections by the innate and adaptive immune response. Annu. Rev. Immunol. 19, 65–91. doi: 10.1146/annurev.immunol.19.1.65
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gupta, A. K., Joshi, M. B., Philippova, M., Erne, P., Hasler, P., Hahn, S., et al. (2010). Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 584, 3193–3197. doi: 10.1016/j.febslet.2010.06.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gupta, S., Braun, M., Tischler, N. D., Stoltz, M., Sundstrom, K. B., Bjorkstrom, N. K., et al. (2013). Hantavirus-infection confers resistance to cytotoxic lymphocyte-mediated apoptosis. PLoS. Pathog. 9:e1003272. doi: 10.1371/journal.ppat.1003272
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hayasaka, D., Maeda, K., Ennis, F. A., and Terajima, M. (2007). Increased permeability of human endothelial cell line EA.hy926 induced by hantavirus-specific cytotoxic T lymphocytes. Virus Res. 123, 120–127. doi: 10.1016/j.virusres.2006.08.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hjelle, B., Jenison, S. A., Goade, D. E., Green, W. B., Feddersen, R. M., and Scott, A. A. (1995). Hantaviruses: clinical, microbiologic, and epidemiologic aspects. Crit. Rev. Clin. Lab. Sci. 32, 469–508. doi: 10.3109/10408369509082592
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hooper, J. W., Brocato, R. L., Kwilas, S. A., Hammerbeck, C. D., Josleyn, M. D., Royals, M., et al. (2014). DNA vaccine-derived human IgG produced in transchromosomal bovines protect in lethal models of hantavirus pulmonary syndrome. Sci. Transl. Med. 6, 264ra162. doi: 10.1126/scitranslmed.3010082
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jahnsen, F. L., Strickland, D. H., Thomas, J. A., Tobagus, I. T., Napoli, S., Zosky, G. R., et al. (2006). Accelerated antigen sampling and transport by airway mucosal dendritic cells following inhalation of a bacterial stimulus. J. Immunol. 177, 5861–5867. doi: 10.4049/jimmunol.177.9.5861
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jaillon, S., Peri, G., Delneste, Y., Fremaux, I., Doni, A., Moalli, F., et al. (2007). The humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J. Exp. Med. 204, 793–804. doi: 10.1084/jem.20061301
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jenne, C. N., Wong, C. H., Zemp, F. J., McDonald, B., Rahman, M. M., Forsyth, P. A., et al. (2013). Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe 13, 169–180. doi: 10.1016/j.chom.2013.01.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jonsson, C. B., Figueiredo, L. T., and Vapalahti, O. (2010). A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol. Rev. 23, 412–441. doi: 10.1128/CMR.00062-09
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kanerva, M., Vaheri, A., Mustonen, J., and Partanen, J. (1998). High-producer allele of tumour necrosis factor-alpha is part of the susceptibility MHC haplotype in severe Puumala virus-induced nephropathia epidemica. Scand. J. Infect. Dis. 30, 532–534. doi: 10.1080/00365549850161629
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Khaiboullina, S. F., Netski, D. M., Krumpe, P., and St Jeor, S. C. (2000). Effects of tumor necrosis factor alpha on sin nombre virus infection in vitro. J. Virol. 74, 11966–11971. doi: 10.1128/JVI.74.24.11966-11971.2000
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Khaiboullina, S. F., Rizvanov, A. A., Lombardi, V. C., Morzunov, S. P., Reis, H. J., Palotas, A., et al. (2013). Andes-virus-induced cytokine storm is partially suppressed by ribavirin. Antivir. Ther. 18, 575–584. doi: 10.3851/IMP2524
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Klempa, B., Meisel, H., Rath, S., Bartel, J., Ulrich, R., and Kruger, D. H. (2003). Occurrence of renal and pulmonary syndrome in a region of northeast Germany where Tula hantavirus circulates. J. Clin. Microbiol. 41, 4894–4897. doi: 10.1128/JCM.41.10.4894-4897.2003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Klingstrom, J., Hardestam, J., Stoltz, M., Zuber, B., Lundkvist, A., Linder, S., et al. (2006). Loss of cell membrane integrity in Puumala hantavirus-infected patients correlates with levels of epithelial cell apoptosis and perforin. J. Virol. 80, 8279–8282. doi: 10.1128/JVI.00742-06
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Klingstrom, J., Lindgren, T., and Ahlm, C. (2008). Sex-dependent differences in plasma cytokine responses to hantavirus infection. Clin. Vaccine Immunol. 15, 885–887. doi: 10.1128/CVI.00035-08
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Koma, T., Yoshimatsu, K., Nagata, N., Sato, Y., Shimizu, K., Yasuda, S. P., et al. (2014). Neutrophil depletion suppresses pulmonary vascular hyperpermeability and occurrence of pulmonary edema caused by hantavirus infection in C.B-17 SCID mice. J. Virol. 88, 7178–7188. doi: 10.1128/JVI.00254-14
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kraus, A. A., Raftery, M. J., Giese, T., Ulrich, R., Zawatzky, R., Hippenstiel, S., et al. (2004). Differential antiviral response of endothelial cells after infection with pathogenic and nonpathogenic hantaviruses. J. Virol. 78, 6143–6150. doi: 10.1128/JVI.78.12.6143-6150.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Krautkrämer, E., Grouls, S., Hettwer, D., Rafat, N., Tonshoff, B., and Zeier, M. (2014). Mobilization of circulating endothelial progenitor cells correlates with the clinical course of hantavirus disease. J. Virol. 88, 483–489. doi: 10.1128/JVI.02063-13
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Krautkrämer, E., Grouls, S., Stein, N., Reiser, J., and Zeier, M. (2011). Pathogenic old world hantaviruses infect renal glomerular and tubular cells and induce disassembling of cell-to-cell contacts. J. Virol. 85, 9811–9823. doi: 10.1128/JVI.00568-11
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Krautkrämer, E., Zeier, M., and Plyusnin, A. (2013). Hantavirus infection: an emerging infectious disease causing acute renal failure. Kidney Int. 83, 23–27. doi: 10.1038/ki.2012.360
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Krüger, D. H., Figueiredo, L. T., Song, J. W., and Klempa, B. (2015). Hantaviruses-Globally emerging pathogens. J. Clin. Virol. 64, 128–136. doi: 10.1016/j.jcv.2014.08.033
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Krüger, D. H., Schönrich, G., and Klempa, B. (2011). Human pathogenic hantaviruses and prevention of infection. Hum. Vaccin. 7, 685–693. doi: 10.4161/hv.7.6.15197
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kwilas, S., Kishimori, J. M., Josleyn, M., Jerke, K., Ballantyne, J., Royals, M., et al. (2014). A hantavirus pulmonary syndrome (HPS) DNA vaccine delivered using a spring-powered jet injector elicits a potent neutralizing antibody response in rabbits and nonhuman primates. Curr. Gene Ther. 14, 200–210. doi: 10.2174/1566523214666140522122633
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kyriakidis, I., and Papa, A. (2013). Serum TNF-α, sTNFR1, IL-6, IL-8 and IL-10 levels in hemorrhagic fever with renal syndrome. Virus Res. 175, 91–94. doi: 10.1016/j.virusres.2013.03.020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Laine, O., Makela, S., Mustonen, J., Huhtala, H., Szanto, T., Vaheri, A., et al. (2010). Enhanced thrombin formation and fibrinolysis during acute Puumala hantavirus infection. Thromb. Res. 126, 154–158. doi: 10.1016/j.thromres.2010.05.025
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lalwani, P., Raftery, M. J., Kobak, L., Rang, A., Giese, T., Matthaei, M., et al. (2013). Hantaviral mechanisms driving HLA class I antigen presentation require both RIG-I and TRIF. Eur. J. Immunol. 43, 2566–2576. doi: 10.1002/eji.201243066
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Langereis, J. D. (2013). Neutrophil integrin affinity regulation in adhesion, migration, and bacterial clearance. Cell Adh. Migr. 7, 476–481. doi: 10.4161/cam.27293
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lapponi, M. J., Carestia, A., Landoni, V. I., Rivadeneyra, L., Etulain, J., Negrotto, S., et al. (2013). Regulation of neutrophil extracellular trap formation by anti-inflammatory drugs. J. Pharmacol. Exp. Ther. 345, 430–437. doi: 10.1124/jpet.112.202879
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, Y., Wang, W., Wang, J. P., Pan, L., Zhang, Y., Yu, H. T., et al. (2012). Elevated vascular endothelial growth factor levels induce hyperpermeability of endothelial cells in hantavirus infection. J. Int. Med. Res. 40, 1812–1821. doi: 10.1177/030006051204000519
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Libraty, D. H., Makela, S., Vlk, J., Hurme, M., Vaheri, A., Ennis, F. A., et al. (2012). The degree of leukocytosis and urine GATA-3 mRNA levels are risk factors for severe acute kidney injury in Puumala virus nephropathia epidemica. PLoS ONE 7:e35402. doi: 10.1371/journal.pone.0035402
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Linderholm, M., Ahlm, C., Settergren, B., Waage, A., and Tarnvik, A. (1996). Elevated plasma levels of tumor necrosis factor (TNF)-α, soluble TNF receptors, interleukin (IL)-6, and IL-10 in patients with hemorrhagic fever with renal syndrome. J. Infect. Dis. 173, 38–43. doi: 10.1093/infdis/173.1.38
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lukes, R. J. (1954). The pathology of thirty-nine fatal cases of epidemic hemorrhagic fever. Am. J. Med. 16, 639–650. doi: 10.1016/0002-9343(54)90270-3
Ma, Y., Liu, B., Yuan, B., Wang, J., Yu, H., Zhang, Y., et al. (2012). Sustained high level of serum VEGF at convalescent stage contributes to the renal recovery after HTNV infection in patients with hemorrhagic fever with renal syndrome. Clin. Dev. Immunol. 2012, 812386. doi: 10.1155/2012/812386
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
MacNeil, A., Ksiazek, T. G., and Rollin, P. E. (2011). Hantavirus pulmonary syndrome, United States, 1993–2009. Emerg. Infect. Dis. 17, 1195–1201. doi: 10.3201/eid1707.101306
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maes, P., Clement, J., Groeneveld, P. H., Colson, P., Huizinga, T. W., and Van Ranst, M. (2006). Tumor necrosis factor-α genetic predisposing factors can influence clinical severity in nephropathia epidemica. Viral Immunol. 19, 558–564. doi: 10.1089/vim.2006.19.558
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Makela, S., Mustonen, J., Ala-Houhala, I., Hurme, M., Partanen, J., Vapalahti, O., et al. (2002). Human leukocyte antigen-B8-DR3 is a more important risk factor for severe Puumala hantavirus infection than the tumor necrosis factor-α(–308) G/A polymorphism. J. Infect. Dis. 186, 843–846. doi: 10.1086/342413
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Markotic, A., Hensley, L., Daddario, K., Spik, K., Anderson, K., and Schmaljohn, C. (2007). Pathogenic hantaviruses elicit different immunoreactions in THP-1 cells and primary monocytes and induce differentiation of human monocytes to dendritic-like cells. Coll. Antropol. 31, 1159–1167.
Marsac, D., Garcia, S., Fournet, A., Aguirre, A., Pino, K., Ferres, M., et al. (2011). Infection of human monocyte-derived dendritic cells by ANDES Hantavirus enhances pro-inflammatory state, the secretion of active MMP-9 and indirectly enhances endothelial permeability. Virol. J. 8, 223. doi: 10.1186/1743-422X-8-223
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Martinez-Valdebenito, C., Calvo, M., Vial, C., Mansilla, R., Marco, C., Palma, R. E., et al. (2014). Person-to-person household and nosocomial transmission of andes hantavirus, Southern Chile, 2011. Emerg. Infect. Dis. 20, 1629–1636. doi: 10.3201/eid2010.140353
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Massberg, S., Grahl, L., von Bruehl, M. L., Manukyan, D., Pfeiler, S., Goosmann, C., et al. (2010). Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 16, 887–896. doi: 10.1038/nm.2184
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McDonald, B., Urrutia, R., Yipp, B. G., Jenne, C. N., and Kubes, P. (2012). Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 12, 324–333. doi: 10.1016/j.chom.2012.06.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Metchnikoff, E. (1887). Sur la lutte des cellules de l’organisme contre l’invasion de microbes. Ann. Inst. Pasteur. 1, 321–326.
Monsinjon, T., Gasque, P., Chan, P., Ischenko, A., Brady, J. J., and Fontaine, M. C. (2003). Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J. 17, 1003–1014. doi: 10.1096/fj.02-0737com
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mori, M., Rothman, A. L., Kurane, I., Montoya, J. M., Nolte, K. B., Norman, J. E., et al. (1999). High levels of cytokine-producing cells in the lung tissues of patients with fatal hantavirus pulmonary syndrome. J. Infect. Dis. 179, 295–302. doi: 10.1086/314597
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mustonen, J., Makela, S., Outinen, T., Laine, O., Jylhava, J., Arstila, P. T., et al. (2013). The pathogenesis of nephropathia epidemica: new knowledge and unanswered questions. Antiviral Res. 100, 589–604. doi: 10.1016/j.antiviral.2013.10.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nauseef, W. M., and Borregaard, N. (2014). Neutrophils at work. Nat. Immunol. 15, 602–611. doi: 10.1038/ni.2921
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nichol, S. T., Spiropoulou, C. F., Morzunov, S., Rollin, P. E., Ksiazek, T. G., Feldmann, H., et al. (1993). Genetic identification of a hantavirus associated with an outbreak of acute respiratory illness. Science 262, 914–917. doi: 10.1126/science.8235615
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Niikura, M., Maeda, A., Ikegami, T., Saijo, M., Kurane, I., and Morikawa, S. (2004). Modification of endothelial cell functions by Hantaan virus infection: prolonged hyper-permeability induced by TNF-α of hantaan virus-infected endothelial cell monolayers. Arch. Virol. 149, 1279–1292. doi: 10.1007/s00705-004-0306-y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nolte, K. B., Feddersen, R. M., Foucar, K., Zaki, S. R., Koster, F. T., Madar, D., et al. (1995). Hantavirus pulmonary syndrome in the United States: a pathological description of a disease caused by a new agent. Hum. Pathol. 26, 110–120. doi: 10.1016/0046-8177(95)90123-X
Ontiveros, S. J., Li, Q., and Jonsson, C. B. (2010). Modulation of apoptosis and immune signaling pathways by the Hantaan virus nucleocapsid protein. Virology 401, 165–178. doi: 10.1016/j.virol.2010.02.018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Outinen, T. K., Kuparinen, T., Jylhava, J., Leppanen, S., Mustonen, J., Makela, S., et al. (2012a). Plasma cell-free DNA levels are elevated in acute Puumala hantavirus infection. PLoS ONE 7:e31455. doi: 10.1371/journal.pone.0031455
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Outinen, T. K., Makela, S., Huhtala, H., Hurme, M., Meri, S., Porsti, I., et al. (2012b). High pentraxin-3 plasma levels associate with thrombocytopenia in acute Puumala hantavirus-induced nephropathia epidemica. Eur. J. Clin. Microbiol. Infect. Dis. 31, 957–963. doi: 10.1007/s10096-011-1392-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ozaki, H., Ishii, K., Horiuchi, H., Arai, H., Kawamoto, T., Okawa, K., et al. (1999). Cutting edge: combined treatment of TNF-α and IFN-γ causes redistribution of junctional adhesion molecule in human endothelial cells. J. Immunol. 163, 553–557.
Paakkala, A., Mustonen, J., Viander, M., Huhtala, H., and Pasternack, A. (2000). Complement activation in nephropathia epidemica caused by Puumala hantavirus. Clin. Nephrol. 53, 424–431.
Pensiero, M. N., Sharefkin, J. B., Dieffenbach, C. W., and Hay, J. (1992). Hantaan virus infection of human endothelial cells. J. Virol. 66, 5929–5936.
Pergam, S. A., Schmidt, D. W., Nofchissey, R. A., Hunt, W. C., Harford, A. H., and Goade, D. E. (2009). Potential renal sequelae in survivors of hantavirus cardiopulmonary syndrome. Am. J. Trop. Med. Hyg. 80, 279–285.
Pillay, J., Kamp, V. M., van, H. E., Visser, T., Tak, T., Lammers, J. W., et al. (2012). A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J. Clin. Invest. 122, 327–336. doi: 10.1172/JCI57990
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pober, J. S. (2002). Endothelial activation: intracellular signaling pathways. Arthritis Res. 4(Suppl. 3), S109–S116. doi: 10.1186/ar576
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Raftery, M. J., Kraus, A. A., Ulrich, R., Krüger, D. H., and Schönrich, G. (2002). Hantavirus infection of dendritic cells. J. Virol. 76, 10724–10733. doi: 10.1128/JVI.76.21.10724-10733.2002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Raftery, M. J., Lalwani, P., Krautkrämer, E., Peters, T., Scharffetter-Kochanek, K., Krüger, R., et al. (2014). β2 integrin mediates hantavirus-induced release of neutrophil extracellular traps. J. Exp. Med. 211, 1485–1497. doi: 10.1084/jem.20131092
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rasmuson, J., Andersson, C., Norrman, E., Haney, M., Evander, M., and Ahlm, C. (2011a). Time to revise the paradigm of hantavirus syndromes? Hantavirus pulmonary syndrome caused by European hantavirus. Eur. J. Clin. Microbiol. Infect. Dis. 30, 685–690. doi: 10.1007/s10096-010-1141-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rasmuson, J., Pourazar, J., Linderholm, M., Sandstrom, T., Blomberg, A., and Ahlm, C. (2011b). Presence of activated airway T lymphocytes in human puumala hantavirus disease. Chest 140, 715–722. doi: 10.1378/chest.10-2791
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Razakandrainibe, R., Combes, V., Grau, G. E., and Jambou, R. (2013). Crossing the wall: the opening of endothelial cell junctions during infectious diseases. Int. J. Biochem. Cell Biol. 45, 1165–1173. doi: 10.1016/j.biocel.2013.03.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Razvina, O., Jiang, S., Matsubara, K., Ohashi, R., Hasegawa, G., Aoyama, T., et al. (2014). Differential expression of pentraxin 3 in neutrophils. Exp. Mol. Pathol. 98, 33–40. doi: 10.1016/j.yexmp.2014.11.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sadeghi, M., Eckerle, I., Daniel, V., Burkhardt, U., Opelz, G., and Schnitzler, P. (2011). Cytokine expression during early and late phase of acute Puumala hantavirus infection. BMC Immunol. 12:65. doi: 10.1186/1471-2172-12-65
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Saffarzadeh, M., Juenemann, C., Queisser, M. A., Lochnit, G., Barreto, G., Galuska, S. P., et al. (2012). Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE 7:e32366. doi: 10.1371/journal.pone.0032366
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Safronetz, D., Feldmann, H., and de, W. E. (2015). Birth and pathogenesis of rogue respiratory viruses. Annu. Rev. Pathol. 10, 449–471. doi: 10.1146/annurev-pathol-012414-040501
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Saksida, A., Wraber, B., and Avsic-Zupanc, T. (2011). Serum levels of inflammatory and regulatory cytokines in patients with hemorrhagic fever with renal syndrome. BMC Infect. Dis. 11:142. doi: 10.1186/1471-2334-11-142
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sane, J., Laine, O., Makela, S., Paakkala, A., Jarva, H., Mustonen, J., et al. (2012). Complement activation in Puumala hantavirus infection correlates with disease severity. Ann. Med. 44, 468–475. doi: 10.3109/07853890.2011.573500
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schmaljohn, C. (2009). Vaccines for hantaviruses. Vaccine 27(Suppl. 4), D61–D64. doi: 10.1016/j.vaccine.2009.07.096
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schmaljohn, C. S., and Nichol, S. T. (2007). “Bunyaviridae,” in Fields Virology, eds D. M. Knipe and P. M. Howley (Philadelphia, PA: Lippincott Williams & Wilkins), 1741–1789.
Schönrich, G., Rang, A., Lutteke, N., Raftery, M. J., Charbonnel, N., and Ulrich, R. G. (2008). Hantavirus-induced immunity in rodent reservoirs and humans. Immunol. Rev. 225, 163–189. doi: 10.1111/j.1600-065X.2008.00694.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schulte, D., Kuppers, V., Dartsch, N., Broermann, A., Li, H., Zarbock, A., et al. (2011). Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J. 30, 4157–4170. doi: 10.1038/emboj.2011.304
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shin, O. S., Yanagihara, R., and Song, J. W. (2012). Distinct innate immune responses in human macrophages and endothelial cells infected with shrew-borne hantaviruses. Virology 434, 43–49. doi: 10.1016/j.virol.2012.08.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shrivastava-Ranjan, P., Rollin, P. E., and Spiropoulou, C. F. (2010). Andes virus disrupts the endothelial cell barrier by induction of vascular endothelial growth factor and downregulation of VE-cadherin. J. Virol. 84, 11227–11234. doi: 10.1128/JVI.01405-10
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sundstrom, J. B., McMullan, L. K., Spiropoulou, C. F., Hooper, W. C., Ansari, A. A., Peters, C. J., et al. (2001). Hantavirus infection induces the expression of RANTES and IP-10 without causing increased permeability in human lung microvascular endothelial cells. J. Virol. 75, 6070–6085. doi: 10.1128/JVI.75.13.6070-6085.2001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Taichman, N. S., Young, S., Cruchley, A. T., Taylor, P., and Paleolog, E. (1997). Human neutrophils secrete vascular endothelial growth factor. J. Leukoc. Biol. 62, 397–400.
Taylor, S. L., Frias-Staheli, N., Garcia-Sastre, A., and Schmaljohn, C. S. (2009). Hantaan virus nucleocapsid protein binds to importin α proteins and inhibits tumor necrosis factor alpha-induced activation of nuclear factor kappa B. J. Virol. 83, 1271–1279. doi: 10.1128/JVI.00986-08
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Taylor, S. L., Wahl-Jensen, V., Copeland, A. M., Jahrling, P. B., and Schmaljohn, C. S. (2013). Endothelial cell permeability during hantavirus infection involves factor XII-dependent increased activation of the kallikrein–kinin system. PLoS. Pathog. 9:e1003470. doi: 10.1371/journal.ppat.1003470
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Temonen, M., Mustonen, J., Helin, H., Pasternack, A., Vaheri, A., and Holthofer, H. (1996). Cytokines, adhesion molecules, and cellular infiltration in nephropathia epidemica kidneys: an immunohistochemical study. Clin. Immunol. Immunopathol. 78, 47–55. doi: 10.1006/clin.1996.0007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Temonen, M., Vapalahti, O., Holthofer, H., Brummer-Korvenkontio, M., Vaheri, A., and Lankinen, H. (1993). Susceptibility of human cells to Puumala virus infection. J. Gen. Virol. 74(Pt 3), 515–518. doi: 10.1099/0022-1317-74-3-515
Terajima, M., and Ennis, F. A. (2011). T cells and pathogenesis of hantavirus cardiopulmonary syndrome and hemorrhagic fever with renal syndrome. Viruses 3, 1059–1073. doi: 10.3390/v3071059
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tracey, K. J., and Cerami, A. (1994). Tumor necrosis factor: a pleiotropic cytokine and therapeutic target. Annu. Rev. Med. 45, 491–503. doi: 10.1146/annurev.med.45.1.491
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vaheri, A., Strandin, T., Jaaskelainen, A. J., Vapalahti, O., Jarva, H., Lokki, M. L., et al. (2014). Pathophysiology of a severe case of Puumala hantavirus infection successfully treated with bradykinin receptor antagonist icatibant. Antiviral Res. 111C, 23–25. doi: 10.1016/j.antiviral.2014.08.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vial, P. A., Valdivieso, F., Ferres, M., Riquelme, R., Rioseco, M. L., Calvo, M., et al. (2013). High-dose intravenous methylprednisolone for hantavirus cardiopulmonary syndrome in Chile: a double-blind, randomized controlled clinical trial. Clin. Infect. Dis. 57, 943–951. doi: 10.1093/cid/cit394
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Villanueva, E., Yalavarthi, S., Berthier, C. C., Hodgin, J. B., Khandpur, R., Lin, A. M., et al. (2011). Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 187, 538–552. doi: 10.4049/jimmunol.1100450
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wimer, B. M. (1998). Implications of the analogy between recombinant cytokine toxicities and manifestations of hantavirus infections. Cancer Biother. Radiopharm. 13, 193–207. doi: 10.1089/cbr.1998.13.193
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, J., Zhang, X., Pelayo, R., Monestier, M., Ammollo, C. T., Semeraro, F., et al. (2009). Extracellular histones are major mediators of death in sepsis. Nat. Med. 15, 1318–1321. doi: 10.1038/nm.2053
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yu, H., Jiang, W., Du, H., Xing, Y., Bai, G., Zhang, Y., et al. (2014). Involvement of the Akt/NF-κB pathways in the HTNV-mediated increase of IL-6, CCL5, ICAM-1, and VCAM-1 in HUVECs. PLoS ONE 9:e93810. doi: 10.1371/journal.pone.0093810
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zaki, S. R., Greer, P. W., Coffield, L. M., Goldsmith, C. S., Nolte, K. B., Foucar, K., et al. (1995). Hantavirus pulmonary syndrome. Pathogenesis of an emerging infectious disease. Am. J. Pathol. 146, 552–579.
Zapata, J. C., Cox, D., and Salvato, M. S. (2014). The role of platelets in the pathogenesis of viral hemorrhagic fevers. PLoS Negl. Trop. Dis. 8:e2858. doi: 10.1371/journal.pntd.0002858
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zelena, H., Mrazek, J., and Kuhn, T. (2013). Tula hantavirus infection in immunocompromised host, Czech Republic. Emerg. Infect. Dis. 19, 1873–1875. doi: 10.3201/eid1911.130421
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: viral hemorrhagic fever, hantaviruses, immunopathogenesis, neutrophils, neutrophil extracellular traps, vascular hyperpermeability
Citation: Schönrich G, Krüger DH and Raftery MJ (2015) Hantavirus-induced disruption of the endothelial barrier: neutrophils are on the payroll. Front. Microbiol. 6:222. doi: 10.3389/fmicb.2015.00222
Received: 06 December 2014; Accepted: 05 March 2015;
Published: 25 March 2015.
Edited by:
Marco Goeijenbier, Erasmus University Medical Center, NetherlandsReviewed by:
Sofia Kouidou, Aristotle University of Thessaloniki, GreeceMarcelo Lopez-Lastra, Pontificia Universidad Católica de Chile, Chile
Copyright © 2015 Schönrich, Krüger and Raftery. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Günther Schönrich, Institute of Medical Virology, Helmut-Ruska-Haus, Charité–Universitätsmedizin Berlin, Charitéplatz 1, 10117 Berlin, GermanyZ3VlbnRoZXIuc2Nob2VucmljaEBjaGFyaXRlLmRl