 Matthew Ouellette
Matthew Ouellette Laura Jackson
Laura Jackson Scott Chimileski
Scott Chimileski R. Thane Papke
R. Thane Papke- Department of Molecular and Cell Biology, University of Connecticut, Storrs, CT, USA
Restriction-modification (RM) systems have evolved to protect the cell from invading DNAs and are composed of two enzymes: a DNA methyltransferase and a restriction endonuclease. Although RM systems are present in both archaeal and bacterial genomes, DNA methylation in archaea has not been well defined. In order to characterize the function of RM systems in archaeal species, we have made use of the model haloarchaeon Haloferax volcanii. A genomic DNA methylation analysis of H. volcanii strain H26 was performed using PacBio single molecule real-time (SMRT) sequencing. This analysis was also performed on a strain of H. volcanii in which an annotated DNA methyltransferase gene HVO_A0006 was deleted from the genome. Sequence analysis of H26 revealed two motifs which are modified in the genome: Cm4TAG and GCAm6BN6VTGC. Analysis of the ΔHVO_A0006 strain indicated that it exhibited reduced adenine methylation compared to the parental strain and altered the detected adenine motif. However, protein domain architecture analysis and amino acid alignments revealed that HVO_A0006 is homologous only to the N-terminal endonuclease region of Type IIG RM proteins and contains a PD-(D/E)XK nuclease motif, suggesting that HVO_A0006 is a PD-(D/E)XK nuclease family protein. Further bioinformatic analysis of the HVO_A0006 gene demonstrated that the gene is rare among the Halobacteria. It is surrounded by two transposition genes suggesting that HVO_A0006 is a fragment of a Type IIG RM gene, which has likely been acquired through gene transfer, and affects restriction-modification activity by interacting with another RM system component(s). Here, we present the first genome-wide characterization of DNA methylation in an archaeal species and examine the function of a DNA methyltransferase related gene HVO_A0006.
Introduction
Restriction-modification (RM) systems in bacteria and archaea provide individuals the ability to recognize self from non-self DNA and function as host defense mechanisms, protecting their genomes from foreign DNA invasion (Arber and Dussoix, 1962; Meselson et al., 1972; Vasu and Nagaraja, 2013). These systems are composed of a pair of enzymes: a restriction endonuclease and its cognate methyltransferase, which recognize identical short DNA sequences known as the recognition sequence. Endonucleases typically cleave dsDNA by hydrolyzing the phosphodiester bonds at one location after detection of an unmethylated recognition sequence (Loenen et al., 2014b). Methyltransferases catalyze a reaction that transfers a methyl group from a cofactor, S-adenosyl-L-methoinine (AdoMet), and modifies a specific nucleotide base (Wilson and Murray, 1991; Blumenthal and Cheng, 2002). Three separate types of methylated bases have been identified: N6 methyl-adenine (m6A), C5 methyl-cytosine (m5C), and M4 methyl-cytosine (m4C) (Wion and Casadesus, 2006). In nearly all cases, the addition of a methyl group to a base in the recognition sequence by the cognate methyltransferase prevents cleavage by the restriction endonuclease, thus protecting the host DNA from being digested (Kuhnlein and Arber, 1972). RM systems also play roles in recombination, where they mediate integration of horizontally transferred DNA into the host genome (Alm et al., 1999; Nobusato et al., 2000) and have been implicated in genetic isolation and subsequent speciation (Jeltsch, 2003).
RM systems are categorized into four types based on required cofactors and mechanism of activity. Type I RM systems function as pentamer complexes consisting of two restriction endonuclease (R) subunits, two methyltransferase (M) subunits, and a sequence specificity (S) subunit. The target DNA sequences are detected by the two tandem target recognition domains (TRDs) of the S subunit, and each TRD recognizes one half of the two-part target sites (Loenen et al., 2014a). When the complex encounters an unmethylated recognition sequence, the complex binds the DNA and cleaves it at an unpredictable distance from the binding site in an ATP-dependent manner (Murray, 2000). Type II RM systems, by contrast, have restriction endonucleases and methyltransferases that act independently. Type II restriction endonucleases recognize unmethylated sequences (Pingoud et al., 2014). The same recognition sequences are also targeted for modification by the equivalent type II methyltransferases (Murphy et al., 2013). There are also different subgroups of Type II systems, which function differently but primarily as independent RM proteins (Roberts et al., 2003). Type IIG RM proteins, for example, are fusion proteins containing three domains: an N-terminal restriction endonuclease, a central methyltransferase, and C-terminal site specificity (Roberts et al., 2003, 2014; Morgan et al., 2009; Shen et al., 2011). Type III RM systems are two-subunit enzymes consisting of a methyltransferase (mod) subunit and a restriction endonuclease (res) subunit (Bickle and Kruger, 1993; Rao et al., 2014). The recognition sequences of a Type III RM system are asymmetrical and only modified on one strand of DNA, with cleavage requiring two inversely-oriented, unmethylated recognition sequences (Meisel et al., 1992). Type IV systems consist of genes that target methylated recognition sequences (Roberts et al., 2003; Loenen and Raleigh, 2014).
Although RM systems and DNA methylation have been extensively studied in bacteria, few studies have examined these systems in archaea. Most research on archaeal RM systems has focused on the activity of restriction endonucleases and methyltransferases in hyperthermophilic organisms, such as those belonging to the genus Pyrococcus (Chinen et al., 2000; Ishikawa et al., 2005; Watanabe et al., 2006). Studies have also examined cytosine methylation in Sulfolobus acidocaldarius (Grogan, 2003), the structure of a type I S subunit in Methanococcus jannaschii (Kim et al., 2005), and the activity of a type II methyltransferase in a virus infecting Natrialba magadii (Baranyi et al., 2000). However, research on RM systems in other archaeal organisms, and the overall role of these systems, has been limited.
An organism that could prove useful as a model for archaeal RM systems and DNA methylation is Haloferax volcanii DS2 (Kuo et al., 1997; Allers and Ngo, 2003; Allers and Mevarech, 2005; Leigh et al., 2011), an archaeal species of the class Halobacteria first isolated from the Dead Sea (Mullakhanbhai and Larsen, 1975). H. volcanii is useful because it is easy to grow in the lab and has an advanced genetic system (Cline et al., 1989; Allers et al., 2004, 2010; Blaby et al., 2010). Also, the genome of wild-type strain DS2 has been fully-sequenced (Hartman et al., 2010).
Previous research indicated that H. volcanii DS2 uses DNA methylation to identify its own DNA from foreign sources. A study on DNA extracted from H. volcanii demonstrated that it is resistant to digestion from restriction endonucleases, such as XbaI, which recognize motifs containing CTAG (Charlebois et al., 1987). These results indicated that H. volcanii DNA was methylated at CTAG tetranucleotide regions; it has since been hypothesized that a putative Type II CTAG methyltransferase HVO_0794 is responsible for this methylation (Hartman et al., 2010). Another study demonstrated that transformation efficiency in H. volcanii is greater when using unmethylated DNA from a dam− E. coli strain (Holmes et al., 1991). The difference in transformation was hypothesized to be the result of cleavage from the putative type IV Mrr restriction endonuclease HVO_0682 (Hartman et al., 2010). This hypothesis was confirmed in another study in which the HVO_0682 gene was deleted, resulting in higher transformation efficiency when adding methylated DNA (Allers et al., 2010). Overall, this evidence supports the hypothesis that archaeal organisms such as the archaeon H. volcanii use RM systems to identify and defend against foreign DNA.
Another potential role for DNA methylation in archaea was uncovered in a recent study on extracellular DNA (eDNA) metabolism in H. volcanii (Chimileski et al., 2014). H. volcanii was provided with eDNA from different species as a growth substrate and was able to grow using its own DNA as a phosphorus source, but not with herring sperm DNA or methylated E. coli DNA. However, H. volcanii was able to grow on unmethylated E. coli DNA isolated from a dam−/dcm− mutant strain (i.e., without methyltransferase genes). Therefore, methylation may also be used as a means for cells to recognize self and non-self when exploiting eDNA for nutritional purposes and possibly for natural transformation. Determining the methylation patterns of haloarchaeal DNA may shed light on the phenomenon of discriminatory eDNA metabolism.
Recently, a new DNA sequencing technique has been developed which can detect methylated bases, known as single molecule, real-time (SMRT) sequencing. This process determines the sequence of DNA as a new strand is synthesized in real time, and the kinetic signals of incorporated bases can also be measured (Flusberg et al., 2010). Unique kinetic signals produced at modified bases during strand synthesis are used to detect methylation patterns of sequenced DNA. This process has been used to sequence the genomic methylation patterns, or methylomes, of several different bacterial species (Fang et al., 2012; Murray et al., 2012; Lluch-Senar et al., 2013; Furuta et al., 2014; Krebes et al., 2014). In this study, we used SMRT sequencing to characterize the methylomes of H. volcanii H26, a laboratory strain derived from wild-type strain DS2, and a derivative strain in which the gene HVO_A0006 was deleted. HVO_A0006 is annotated in the H. volcanii genome as an adenine methyltransferase and is located on the native replicon pHV4 (Hartman et al., 2010). It is predicted to encode a protein that is 219 amino acids in length and with a molecular weight of 24.794 kDa. HVO_A0006 was selected for further characterization because, despite being annotated as a methyltransferase, it is not recognized as an RM protein by the RM database REBASE (Roberts et al., 2014).
Materials and Methods
Strains and Growth Conditions
Descriptions of all strains and plasmids used in this study are provided in Table 1. E. coli strains were grown in Lysogeny Broth (LB; Ampicillin was added at 100 μg mL−1 when necessary). H. volcanii strains were grown in Hv-YPC (rich medium) or Hv-CA (selective rich medium) developed by Allers et al. (2004) per instructions in the Halohandbook (Dyall-Smith, 2008). Uracil (50 μg mL−1), tryptophan (50 μg mL−1), and 5-fluoroorotic acid (at 50 μg mL−1) were added to the media as needed for ΔpyrE2 and ΔtrpA strains. All H. volcanii cultures were incubated at 42°C and shaken at 200 rpm unless otherwise specified.

Table 1. Strains and plasmids used in this study.
Deletion of HVO_A0006 Gene
The annotated adenine methyltransferase gene HVO_A0006 (accession number ADE01899) was selected for deletion in H. volcanii strain H53. The gene deletion strategy used in this study was modified from the methodology previously developed (Blaby et al., 2010) and uses the In-Fusion HD Cloning Kit (Clontech). The primer sequences used in this study are listed in Table 2. H. volcanii strain H53 was then transformed with pΔHVO_A0006 using the PEG-mediated transformation of haloarchaea protocol from Cline et al. (1989), Bitan-Banin et al. (2003), Allers et al. (2004), and Blaby et al. (2010). The transformed cells were then plated on Hv-CA agar media without uracil and incubated for 5–7 days at 42°C. Colony PCR was performed using forward and reverse M13 primers to screen for pop-ins. The positive colonies from the pop-in screen were plated on Hv-CA plates with 50 μg mL−1 5-FOA and 50 μg mL−1 uracil to obtain pop-outs. Final pop-outs were confirmed through PCR (see primers in Table 2) as visualized through gel electrophoresis (Figure 1).

Table 2. Oligonucleotide primers used in this study.
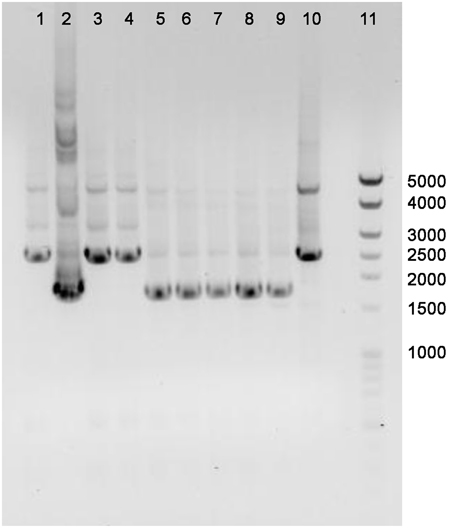
Figure 1. PCR confirmation of HVO_A0006 deletion strain. The template DNA amplified was from H. volcanii DS2 (Lane 1), pΔHVO_A0006 (lane 2; as a positive control), unsuccessful pop-outs (lanes 3, 4, and 10) and successful pop-out colonies (lanes 5–9). A Mid-Range DNA ladder (Fisher Scientific) is shown in Lane 11.
DNA Preparation for PacBio Sequencing
To extract DNA from H. volcanii strains H26 and ΔHVO_A0006 for PacBio SMRT sequencing, 5 mL of cell cultures in log phase were pelleted and resuspended in 5 mL DNA buffer (10 mM Tris-HCl, pH 8.0) to lyse the cells. Lysates were treated with 100 μg mL−1 RNase A and incubated overnight at 42°C to degrade RNA. Proteinase K 10 mg mL−1 was added to the lysates to a final concentration of 50 μg mL−1 and incubated at 37°C for 1 h to degrade protein, followed by ethanol precipitation. Three rounds of phenol/chloroform extraction were then performed to purify the DNA (until no interphase was visible). The top aqueous layer from each tube was removed and a final ethanol precipitation was performed as described above. The 260/280 ratio and 260/230 ratio for each DNA sample were measured for purity (H26: 260/280 = 1.83, 260/230 = 2.28; ΔHVO_A0006: 260/280 = 1.78, 260/230 = 2.27).
PacBio SMRT Sequencing
The methylation patterns of DNA extracted from H. volcanii H26 and the ΔHVO_A0006 strain were sequenced using PacBio SMRT sequencing. The prepared samples were processed by the Keck Sequencing facility of the Yale School of Medicine for analysis using PacBio SMRT sequencing. The SMRT method is described in detail in the PacBio manual “Detecting DNA Base Modifications: SMRT Analysis of Microbial Methylomes” (http://www.pacb.com/pdf/TN_Detecting_DNA_Base_Modifications.pdf). Using an estimated input DNA size range of ~4000 kb, 500–800 bp libraries were prepared for each strain and were run in one SMRT cell, yielding ~60x coverage for H26 and ~80x coverage for ΔHVO_A0006. Analysis of the methylated bases and motifs in each strain was performed using the “RS_Modification_and_Motif_Analysis.1” program in SMRT Portal under default parameter settings, with the H. volcanii DS2 genome used as a reference (Hartman et al., 2010). The “motifs.gff” output files for both strains are available in Supplementary Data (Supplementary Data Sheet 1 for H26 and Supplementary Data Sheet 2 for ΔHVO_A0006).
Protein Domain Architecture Analysis and Multiple Alignment
Homologs of the HVO_A0006 protein were identified through a blastp search (Altschul et al., 1990) of the non-redundant protein database (based on an E-value threshold for inferring homology of 1e-4). Structural Classification of Proteins (SCOP) database superfamilies (Gough and Chothia, 2002) and sequence features shown in domain architecture diagrams were detected with InterProScan (Quevillon et al., 2005). Multiple alignments were generated with Clustal Omega (Sievers et al., 2011). Secondary structure predictions for the multiple alignment were made using PRALINE (Heringa, 1999) and PSIPRED (Jones, 1999). PD-(D/E)XK motifs were identified with the PD-EXK web server (Laganeckas et al., 2011).
Results
Characterization of DNA Methylation in Haloferax volcanii H26
In order to characterize the methylome of H. volcanii, the genome of laboratory strain H26 (Table 1) was sequenced via SMRT sequencing. The data from the SMRT sequencing analysis for H26 is summarized in Table 3. SMRT sequencing of H. volcanii H26 identified two modification motifs for H26: one in which cytosine was methylated (m4C) and another in which adenine was methylated (m6A). The m4C motif was identified as Cm4TAG and the m6A sequence motif was identified as GCAm6 BN6VTGC. These two motifs are also the same as those predicted for H. volcanii DS2, the parental strain of H26, in REBASE (Roberts et al., 2014). The CTAG motif was identified 1342 times in the H26 genome, and methylation was detected at 374 of these motifs (28% methylation). The GCABN6VTGC motif occurred 410 times in the H26 genome, and 316 of the copies were detected to have methylated bases (77%).
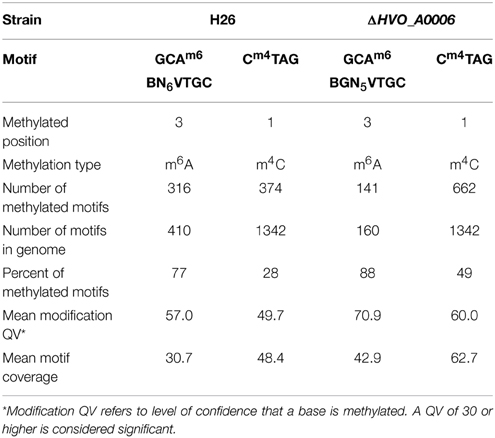
Table 3. DNA methylation patterns detected for H. volcanii H26 and ΔHVO_A0006.
Deletion of HVO_A0006 Increases Specificity of m6A Motif and Reduces m6A Methylation
In order to determine the effect of HVO_A0006 on genome wide methylation, the chromosome of strain H53 with the HVO_A0006 gene deleted (ΔHVO_A0006) was sequenced via SMRT sequencing. The data from the SMRT sequencing analysis is summarized in Table 3 for ΔHVO_A0006. In ΔHVO_A0006, the Cm4TAG motif was identified like in H26, although the sequencing coverage was much higher and more methylated motifs were detected. However, the m6A motif differed significantly for the knockout strain. First, the methylated m6A motif in the ΔHVO_A0006 strain was more specific than in its H26 counterpart, with one of the unspecified nucleotides in the H26 sequence (N) being identified as a guanine (G) in the ΔHVO_A0006 strain. The resulting methylation motif from the ΔHVO_A0006 strain is GCAm6 BGN5VTGC. Thus, the m6A sequence became more specific with the deletion of HVO_A0006. Secondly, the total number of detected motifs, and the number of detected methylated m6A motifs in comparison to the reference genome also decreased when the HVO_A0006 gene was deleted. In ΔHVO_A0006, the total number of detected GCABGN5VTGC motifs was 160, 61% less than H26, and the number of methylated GCAm6 BGN5VTGC sequences was 141, a decrease of 55% from H26. This decrease was due to GCABHN5VTGC sequences not being included in the motif total for ΔHVO_A0006, since those sites were not methylated in that strain. This should also explain why the percentage of methylated m6A motifs is higher in ΔHVO_A0006 (88%) compared to H26 (77%) even though ΔHVO_A0006 has fewer methylated m6A motifs. Differences were also observed in the number of methylated m4C sites detected, with ΔHVO_A0006 exhibiting an increase in the number of methylated Cm4TAG sequences compared to H26. However, this change was likely the result of a sequence coverage artifact, since the motif coverage for CTAG was higher in ΔHVO_A0006 (62.7) than in H26 (48.4). It is also important to note that SMRT sequencing only reports coverage of a motif above a certain modification quality value (QV) score. For both the adenine and cytosine motifs, the knockout strain QV was higher overall with an average value of 60.9 as compared to 49.7 for the H26 strain. It is highly plausible that the cytosine motifs were equally methylated but some of the methylated bases did not reach the QV threshold and were therefore not counted. Therefore, the discrepancy in methylated m4C sites is unlikely to be caused by the deletion of the HVO_A0006 gene. Overall, the results are conclusive that deletion of HVO_A0006 reduces m6A methylation in H. volcanii.
HVO_A0006 is a Fragment Homologous to Larger, Multi-domain RM Proteins
A blastp search indicated that HVO_A0006 is a rare protein among the sequenced Halobacteria. The only significant hit within this group was an annotated adenine specific DNA methyltransferase domain protein belonging to Halorubrum sp. AJ67 (Table 4). Out of the top 10 significant hits only a few belonged to archaeal species and the rest were bacterial (Table 4). The blastp analysis also found homologs experimentally characterized as methyltransferases from Borrelia burgdorferi and Helicobacter pylori (Rego et al., 2011; Krebes et al., 2014). However, those homologs are much larger than HVO_A0006, spanning between 700 and 1000 amino acids (Figure 2). A reciprocal blastp of those hits against the H. volcanii DS2 and Halorubrum sp. AJ67 genomes revealed there are additional homologs. One is H. volcanii gene HVO_A0237 (accession number ADE02204), also annotated as an adenine methyltransferase, listed in REBASE as HvoDSORF237P, and located on pHV4 231 genes downstream from HVO_A0006. Another identified homolog is a small annotated adenine methyltransferase in Halorubrum sp. AJ67.
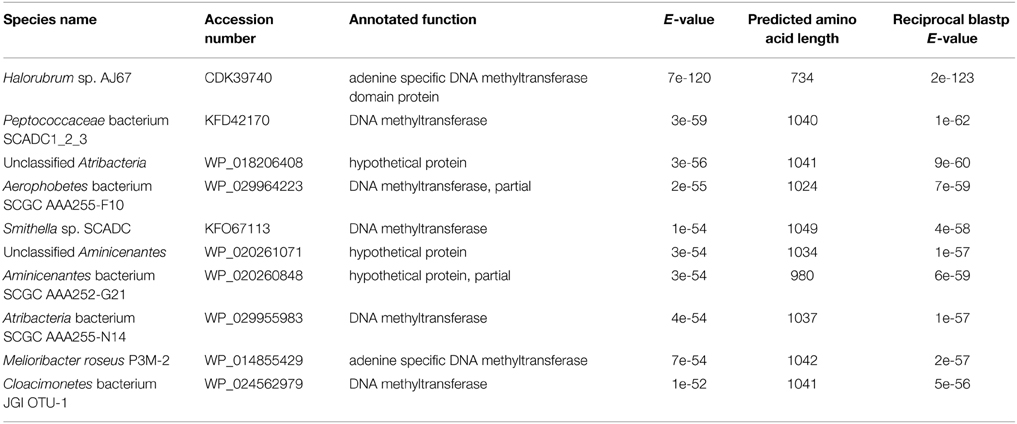
Table 4. Top 10 blastp hits for H. volcanii HVO_A0006.

Figure 2. Domain architecture and multiple alignment of Type IIG homologs. (A) The position of predicted domains present within HVO_A0006 (accession number ADE01899) homologs is shown, including experimentally characterized DNA methyltransferases from B. burgdorferi and H. pylori and several annotated methyltransferases from archaeal species. The common regions of homology between all proteins are shown in gray. Detected s-adenosyl-L-methionine-dependent methyltransferase superfamily domains (SSF53335) are shown in light blue, along with N6 adenine specific DNA methlytransferase signatures (IPR002296; dark blue). The conserved structural core (αβββαβ) of a PD-(D/E)XK nuclease domain is shown in purple. All sequences shown contained a PD-(D/E)XK motif with a confidence score of 1.0 (Laganeckas et al., 2011). (B) Multiple alignment of HVO_A0006 homologs. Predicted DNA methlytransferase signatures (blue) and PD-(D/E)XK signatures (purple) from part A are highlighted. Conserved secondary structure predictions are shown above: as red boxes for α-helices and yellow arrows for β-sheets. Components of the conserved αβββαβ core of the predicted PD-(D/E)XK nuclease domain shown in part A are outlined in black. Amino acid shading represents Clustal sequence similarity. All sequences other than HVO_A0006 are truncated (see Figure S1 for entire alignment).
In REBASE, homologs of HVO_A0006 are predicted to belong to the Type IIG subgroup. A multiple sequence alignment of HVO_A0006 (Figure 2) and several of its homologs indicate that the shared sequence identity occurs only in the N-terminal, putative endonuclease region. The homologs of HVO_A0006 contain known methyltransferase features in their central regions, including the s-adenosyl-L-methionine-dependent methyltransferse superfamily domain (SSF53335) within the SCOP database and predicted N6 adenine specific DNA methyltransferase signatures (IPR002296; Figure 2A), but these do not overlap with HVO_A0006 in the alignment. The N-terminal region of HVO_A0237 contains this methyltransferase domain, but it aligns with the central region of the other sequences, not with HVO_A0006. The smaller annotated adenine methyltransferase in Halorubrum sp. AJ67 also did not align with HVO_A0006, or with the larger annotated methyltransferase in the same Halorubrum strain, but instead aligned with the C-terminal region of the other homologs in Figure 2 (see also Figure S1). Overall, these results indicate that HVO_A0006 is not an adenine methyltransferase, as it was annotated, but is instead a fragment homologous to the N terminus of Type IIG RM fusion proteins.
HVO_A0006 contains a Conserved PD-(D/E)XK Nuclease Motif
Analysis of the HVO_A0006 sequence for endonuclease signatures revealed that it contains a PD-(D/E)XK nuclease motif (PD60-X14-E75AK77), a Mg2+-dependent catalytic motif common to many restriction endonucleases (Kosinski et al., 2005). The active sites of this motif (D60, E75, and K77) are conserved in all homologs in the multiple sequence alignment which align with HVO_A0006 (Figure 2B). Analysis of these sequences in the alignment using the PD-EXK web server (Laganeckas et al., 2011) predicted each sequence belonged to a PD-(D/E)XK family with a probability of 1.0. Secondary structural analysis also demonstrated that the region containing the PD-(D/E)XK motif in the aligned sequences all share an αβββαβ core structure common to this domain family (Venclovas et al., 1994; Kinch et al., 2005). Overall, this evidence indicates that HVO_A0006 is a PD-(D/E)XK nuclease family protein.
HVO_A0006 and HVO_A0237 are Flanked by Predicted Integrase and Transposase Genes
The gene neighborhoods of HVO_A0006 and HVO_A0237 were examined. Analysis of the surrounding genomic region of HVO_A0006 (Figure 3A) revealed that the gene is flanked by three transposition genes: a XerC/D-like integrase and two putative transposases. A putative transposase was also found directly upstream of HVO_A0237 (Figure 3B). This predicted protein (HVO_A0238) is the same length and 100% identical to the ISH5 transposase adjacent to HVO_A0006: HVO_A0007 (Figure 3).

Figure 3. Diagram of the genomic neighborhood for H. volcanii HVO_A0006 (A) and HVO_A0237 (B). Genes are depicted along with two upstream and downstream flanking genes, with annotated functional predictions below. Gene sizes and intergenic spaces are shown in nucleotides. Both regions shown are found on replicon pHV4.
Discussion
Previous research on DNA methylation has focused primarily on eukaryotic and bacterial organisms. The few studies which have examined DNA methylation and RM systems in archaeal organisms have concentrated on a few restriction endonucleases and methyltransferases, with no emphasis on overall genomic methylation (Baranyi et al., 2000; Chinen et al., 2000; Grogan, 2003; Ishikawa et al., 2005; Kim et al., 2005; Watanabe et al., 2006). In this study, we characterized genome-wide DNA methylation patterns of an archaeal organism. We used H. volcanii due to its developed genetic system and ease of growth, which allowed us to examine the effect of deleting one of the seven annotated methyltransferases in the H. volcanii genome.
The SMRT sequencing analysis detected two motifs, which are modified throughout the H. volcanii H26 genome: a Cm4TAG motif and a GCAm6BN6VTGC motif. The presence of methylated CTAG motifs is consistent with observations from previous restriction digest experiments, and the putative Type II CTAG methyltransferase HVO_0794 may be responsible for modifying these motifs (Charlebois et al., 1987). The GCAm6BN6VTGC motif resembles the type of sequence targeted by Type I RM systems, which typically consist of two partial sequences separated by a gap of unspecified nucleotides (Loenen et al., 2014a). The only Type I RM system predicted in H. volcanii, according to the RM database REBASE, is the operon HVO_2269-2271 named rmeRMS (Roberts et al., 2014). Therefore, we predict that the Type I complex encoded by the rmeRMS operon is responsible for at least some of the modifications detected by the SMRT sequencing. Further studies would need to be performed to confirm the role of these putative RM systems in methylating the identified motifs, as well as determining the role of the other annotated methyltransferase genes in the H. volcanii genome.
The gene HVO_A0006 was selected for investigation via gene knockout in this study because in genomic analysis it was annotated as an adenosine specific methyltransferase (Hartman et al., 2010). The results indicate that the HVO_A0006 gene has an effect on m6A methylation in H. volcanii according to the motif analysis produced by SMRT sequencing. This is evidenced by the observed change in adenine motif recognition between the H26 strain, which was GCAm6 BN6VTGC, and the HVO_A0006 knockout, which was GCAm6BGN5VTGC. Our conclusion is also supported by the reduction in the total amount of motifs recognized, with the knockout strain exhibiting a 61% decrease in the total number of recognized motifs compared to the parental strain and a 55% reduction in the number of methylated motifs. These data appear to support the genome annotation of HVO_A0006 as an adenine methyltransferase. However, the amino acid alignment and domain architecture analysis of the protein and its homologs reveals that HVO_A0006 is only homologous to the N-terminal region of characterized and predicted Type IIG RM fusion proteins, which typically contain a restriction endonuclease domain. Analysis of the conserved region in HVO_A0006 and its homologs revealed the presence of a PD-(D/E)XK nuclease motif with conserved active sites and secondary structure. While methyltransferase domains were detected in homologs of HVO_A0006, they do not correspond to the region of shared sequence identity with HVO_A0006. Therefore, HVO_A0006 likely functions as a nuclease, not a complete methyltransferase as its annotation suggests.
We hypothesize that HVO_A0006 influences adenine methylation patterns through interaction with another RM protein or system in H. volcanii. One possible candidate for interaction is the Type I RM system encoded by rmeRMS. In Type I RM systems, the M subunits are known to interact with each other to coordinate restriction and modification activity in the Type I complex. This interaction primarily occurs via N-terminal helical regions in the M subunits, which coordinate to detect adenine methylation when the complex binds to the target sequence and mediate methylation if the site is hemimethylated (maintenance methylation), or cleavage if the site is unmethylated (Kelleher et al., 1991). Mutation of this N-terminal region alters the activity of the Type I complex to de novo methylation (modification of unmethylated target sites) rather than maintenance methylation, indicating that changes to molecular communication in the Type I complex can alter RM activity (Kelleher et al., 1991). In HVO_A0006, we detect a helical region in the C-terminal region of the protein. This region is likely part of the helical connector domain that links the restriction endonuclease and methyltransferase domains in Type IIG RM proteins (Shen et al., 2011). The Type IIG RM systems are hypothesized to be evolutionarily related to the Type I systems, and this helical connector domain is predicted to be homologous to the N-terminal helical region of the Type I M subunits, with a similar function of molecular communication between the methyltransferase and restriction endonuclease domains (Nakonieczna et al., 2009). Therefore, it is possible that HVO_A0006 uses its partial helical domain to interact with the Type I RmeRMS complex and acts as an R subunit, thus altering the restriction and modification activity of the complex and resulting in the different methylation patterns seen in the parental strain of H. volcanii compared to the HVO_A0006 knockout. However, since the RmeRMS system already has an R subunit encoded in the operon, any possible interaction between HVO_A0006 and the Type I complex would be complicated by competition with the native subunit.
Another possible candidate protein for interaction with HVO_A0006 is HVO_A0237 which are both located on the native plasmid pHV4. The multiple alignment analysis (Figure 2) indicates that HVO_A0237 is also a homolog of a Type IIG system, but it is missing an N-terminal restriction endonuclease domain. This absent region might be supplied by HVO_A0006, which interacts with HVO_A0237 via the helical connector region to form a complete Type IIG RM enzyme, and forms a split protein complex, rather than a fused one as is typical of these systems. Previous analysis has demonstrated that the activity of a Type IIG protein is coordinated by the interaction between the restriction endonuclease domain and the other two regions of the enzyme (Shen et al., 2011). Therefore, deletion of the HVO_A0006 gene might compromise the integrity of the required Type IIG protein-protein interactions and thus prevent methylation by the remaining methyltransferase encoded by HVO_A0237, resulting in the change in adenine methylation observed in the deletion mutant. Complicating this proposed mechanism is that motifs modified by Type IIG systems do not typically resemble Type I motifs. For example, the motif modified by the H. pylori homolog is GGWTAAm6 (Krebes et al., 2014). However, some Type IIG proteins in Campylobacter jejuni have been reported to use S subunits similar to those found in Type I S RM systems (Furuta et al., 2010). Further studies will be needed to characterize the interactions between HVO_A0006 and other RM systems.
Based on a multiple alignment and the presence of integrase and transposition genes upstream and downstream of the gene, we propose that HVO_A0006 is likely the result of a gene transfer event of a Type IIG RM in which a gene fragment (the N-terminal restriction endonuclease region) of the whole gene was acquired. The other regions of the Type IIG RM gene were likely also acquired or were the result of internal genomic rearrangements from a once intact gene in H. volcanii, since HVO_A0237 is missing an N-terminal restriction endonuclease region, but is homologous to the same Type IIG RM genes as HVO_A0006. A similar event also appears to have occurred with a Type IIG RM gene in Halorubrum sp. AJ67, with the C-terminal site specificity region fragmenting from the restriction endonuclease and methyltransferase regions. This is consistent with previous findings of gene transfer that have resulted in gene fragmentation (Kobayashi et al., 1999; Chan et al., 2009). These RM systems are highly mobile, and gene transfer events of RM domains and subunits likely results in the formation of new fusion proteins, when the domains are transferred next to each other, as well as new independent RM proteins via fragmentation.
Using SMRT sequencing, this study was able to characterize the methylome of the archaeal organism H. volcanii. We were also able to demonstrate that an annotated methyltransferase gene, HVO_A0006, affects adenine methylation patterns in the H. volcanii genome, although it is likely a PD-(D/E)XK nuclease family protein that interacts with another RM protein or system. Future SMRT sequencing studies utilizing a methyltransferase null mutant could help provide more precise characterization of methyltransferase genes and provide a more detailed picture of DNA methylation in H. volcanii and the role of RM systems in the organism.
Conflict of Interest Statement
The Review Editor Antonio Ventosa declares that, despite having published with author Thane Papke, the review process was handled objectively and no conflict of interest exists. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Thorsten Allers from the University of Nottingham, UK for the Haloferax volcanii strains and plasmids. This work was supported by the National Science foundation (award numbers DEB-0910290 and DEB-0830024) and the NASA Astrobiology: Exobiology and Evolutionary Biology Program grant number NNX12AD70G.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2015.00251/abstract
References
Allers, T., Barak, S., Liddell, S., Wardell, K., and Mevarech, M. (2010). Improved strains and plasmid vectors for conditional overexpression of His-tagged proteins in Haloferax volcanii. Appl. Environ. Microbiol. 76, 1759–1769. doi: 10.1128/AEM.02670-09
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Allers, T., and Mevarech, M. (2005). Archaeal genetics - the third way. Nat. Rev. Genet. 6, 58–73. doi: 10.1038/nrg1504
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Allers, T., and Ngo, H. P. (2003). Genetic analysis of homologous recombination in Archaea: Haloferax volcanii as a model organism. Biochem. Soc. Trans. 31, 706–709. doi: 10.1042/BST0310706
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Allers, T., Ngo, H. P., Mevarech, M., and Lloyd, R. G. (2004). Development of additional selectable markers for the halophilic archaeon Haloferax volcanii based on the leuB and trpA genes. Appl. Environ. Microbiol. 70, 943–953. doi: 10.1128/AEM.70.2.943-953.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Alm, R. A., Ling, L. S., Moir, D. T., King, B. L., Brown, E. D., Doig, P. C., et al. (1999). Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397, 176–180. doi: 10.1038/16495
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Arber, W., and Dussoix, D. (1962). Host specificity of DNA produced by Escherichia coli. I. Host controlled modification of bacteriophage lambda. J. Mol. Biol. 5, 18–36. doi: 10.1016/S0022-2836(62)80058-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baranyi, U., Klein, R., Lubitz, W., Kruger, D. H., and Witte, A. (2000). The archaeal halophilic virus-encoded Dam-like methyltransferase M. phiCh1-I methylates adenine residues and complements dam mutants in the low salt environment of Escherichia coli. Mol. Microbiol. 35, 1168–1179. doi: 10.1046/j.1365-2958.2000.01786.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bitan-Banin, G., Ortenberg, R., and Mevarech, M. (2003). Development of a gene knockout system for the halophilic archaeon Haloferax volcanii by use of the pyrE gene. J. Bacteriol. 185, 772–778. doi: 10.1128/JB.185.3.772-778.2003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Blaby, I. K., Phillips, G., Blaby-Haas, C. E., Gulig, K. S., El Yacoubi, B., and De Crecy-Lagard, V. (2010). Towards a systems approach in the genetic analysis of archaea: accelerating mutant construction and phenotypic analysis in Haloferax volcanii. Archaea 2010:426239. doi: 10.1155/2010/426239
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Blumenthal, R. M., and Cheng, X. (2002). “Restriction-modification systems,” in Modern Microbial Genetics, 2nd Edn., eds U. N. Streips and R. E. Yasbin (New York, NY: John Wiley & Sons, Inc.), 177–217.
Chan, C. X., Beiko, R. G., Darling, A. E., and Ragan, M. A. (2009). Lateral transfer of genes and gene fragments in prokaryotes. Genome Biol. Evol. 1, 429–438. doi: 10.1093/gbe/evp044
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Charlebois, R. L., Lam, W. L., Cline, S. W., and Doolittle, W. F. (1987). Characterization of pHV2 from Halobacterium volcanii and its use in demonstrating transformation of an archaebacterium. Proc. Natl. Acad. Sci. U.S.A. 84, 8530–8534. doi: 10.1073/pnas.84.23.8530
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chimileski, S., Dolas, K., Naor, A., Gophna, U., and Papke, R. T. (2014). Extracellular DNA metabolism in Haloferax volcanii. Front. Microbiol. 5:57. doi: 10.3389/fmicb.2014.00057
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chinen, A., Uchiyama, I., and Kobayashi, I. (2000). Comparison between Pyrococcus horikoshii and Pyrococcus abyssi genome sequences reveals linkage of restriction-modification genes with large genome polymorphisms. Gene 259, 109–121. doi: 10.1016/S0378-1119(00)00459-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cline, S. W., Lam, W. L., Charlebois, R. L., Schalkwyk, L. C., and Doolittle, W. F. (1989). Transformation methods for halophilic archaebacteria. Can. J. Microbiol. 35, 148–152. doi: 10.1139/m89-022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dyall-Smith, M. (ed.). (2008). The Halohandbook: Protocols for Haloarchaeal Genetics. Available online at: http://www.haloarchaea.com/resources/halohandbook/index.html
Fang, G., Munera, D., Friedman, D. I., Mandlik, A., Chao, M. C., Banerjee, O., et al. (2012). Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat. Biotechnol. 30, 1232–1239. doi: 10.1038/nbt.2432
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Flusberg, B. A., Webster, D. R., Lee, J. H., Travers, K. J., Olivares, E. C., Clark, T. A., et al. (2010). Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 7, 461–465. doi: 10.1038/nmeth.1459
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Furuta, Y., Abe, K., and Kobayashi, I. (2010). Genome comparison and context analysis reveals putative mobile forms of restriction-modification systems and related rearrangements. Nucleic Acids Res. 38, 2428–2443. doi: 10.1093/nar/gkp1226
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Furuta, Y., Namba-Fukuyo, H., Shibata, T. F., Nishiyama, T., Shigenobu, S., Suzuki, Y., et al. (2014). Methylome diversification through changes in DNA methyltransferase sequence specificity. PLoS Genet. 10:e1004272. doi: 10.1371/journal.pgen.1004272
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gough, J., and Chothia, C. (2002). SUPERFAMILY: HMMs representing all proteins of known structure. SCOP sequence searches, alignments and genome assignments. Nucleic Acids Res. 30, 268–272. doi: 10.1093/nar/30.1.268
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Grogan, D. W. (2003). Cytosine methylation by the SuaI restriction-modification system: implications for genetic fidelity in a hyperthermophilic archaeon. J. Bacteriol. 185, 4657–4661. doi: 10.1128/JB.185.15.4657-4661.2003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hartman, A. L., Norais, C., Badger, J. H., Delmas, S., Haldenby, S., Madupu, R., et al. (2010). The complete genome sequence of Haloferax volcanii DS2, a model archaeon. PLoS ONE 5:e9605. doi: 10.1371/journal.pone.0009605
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Heringa, J. (1999). Two strategies for sequence comparison: profle-preprocessed and secondary structure-induced multiple alignment. Comput. Chem. 23, 341–364. doi: 10.1016/S0097-8485(99)00012-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Holmes, M. L., Nuttall, S. D., and Dyall-Smith, M. L. (1991). Construction and use of halobacterial shuttle vectors and further studies on Haloferax DNA gyrase. J. Bacteriol. 173, 3807–3813.
Ishikawa, K., Watanabe, M., Kuroita, T., Uchiyama, I., Bujnicki, J. M., Kawakami, B., et al. (2005). Discovery of a novel restriction endonuclease by genome comparison and application of a wheat-germ-based cell-free translation assay: PabI (5'-GTA/C) from the hyperthermophilic archaeon Pyrococcus abyssi. Nucleic Acids Res. 33:e112. doi: 10.1093/nar/gni113
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jeltsch, A. (2003). Maintenance of species identity and controlling speciation of bacteria: a new function for restriction/modification systems? Gene 317, 13–16. doi: 10.1016/S0378-1119(03)00652-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jones, D. T. (1999). Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202. doi: 10.1006/jmbi.1999.3091
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kelleher, J. E., Daniel, A. S., and Murray, N. E. (1991). Mutations that confer de novo activity upon a maintenance methyltransferase J. Mol. Biol. 221, 431–440. doi: 10.1016/0022-2836(91)80064-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, J. S., Degiovanni, A., Jancarik, J., Adams, P. D., Yokota, H., Kim, R., et al. (2005). Crystal structure of DNA sequence specificity subunit of a type I restriction-modification enzyme and its functional implications. Proc. Natl. Acad. Sci. U.S.A. 102, 3248–3253. doi: 10.1073/pnas.0409851102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kinch, L. N., Ginalski, K., Rychlewski, L., and Grishin, N. V. (2005). Identification of novel restriction endonuclease-like fold families among hypothetical proteins. Nucleic Acids Res. 33, 3598–3605. doi: 10.1093/nar/gki676
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kobayashi, I., Nobusato, A., Kobayashi-Takahashi, N., and Uchiyama, I. (1999). Shaping the genome–restriction-modification systems as mobile genetic elements. Curr. Opin. Genet. Dev. 9, 649–656. doi: 10.1016/S0959-437X(99)00026-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kosinski, J., Feder, M., and Bujnicki, J. M. (2005). The PD-(D/E)XK superfamily revisited: identification of new members among proteins involved in DNA metabolism and functional predictions for domains of (hitherto) unknown function. BMC Bioinformatics 6:172. doi: 10.1186/1471-2105-6-172
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Krebes, J., Morgan, R. D., Bunk, B., Sproer, C., Luong, K., Parusel, R., et al. (2014). The complex methylome of the human gastric pathogen Helicobacter pylori. Nucleic Acids Res. 42, 2415–2432. doi: 10.1093/nar/gkt1201
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kuhnlein, U., and Arber, W. (1972). Host specificity of DNA produced by Escherichia coli. XV. The role of nucleotide methylation in in vitro B-specific modification. J. Mol. Biol. 63, 9–19. doi: 10.1016/0022-2836(72)90518-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kuo, Y., Thompson, D. K., Jean, A., Charlebois, R. L., and Daniels, C. (1997). Characterization of two heat shock genes from Haloferax volcanii: a model system for transcription regulation in the Archaea. J. Bacteriol. 179, 6318–6324.
Laganeckas, M., Margelevicius, M., and Venclovas, C. (2011). Identification of new homologs of PD-(D/E)XK nucleases by support vector machines trained on data derived from profile-profile alignments. Nucleic Acids Res. 39, 1187–1196. doi: 10.1093/nar/gkq958
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Leigh, J. A., Albers, S. V., Atomi, H., and Allers, T. (2011). Model organisms for genetics in the domain Archaea: methanogens, halophiles, Thermococcales and Sulfolobales. FEMS Microbiol. Rev. 35, 577–608. doi: 10.1111/j.1574-6976.2011.00265.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lluch-Senar, M., Luong, K., Llorens-Rico, V., Delgado, J., Fang, G., Spittle, K., et al. (2013). Comprehensive methylome characterization of Mycoplasma genitalium and Mycoplasma pneumoniae at single-base resolution. PLoS Genet. 9:e1003191. doi: 10.1371/journal.pgen.1003191
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Loenen, W. A., Dryden, D. T., Raleigh, E. A., and Wilson, G. G. (2014a). Type I restriction enzymes and their relatives. Nucleic Acids Res. 42, 20–44. doi: 10.1093/nar/gkt847
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Loenen, W. A., Dryden, D. T., Raleigh, E. A., Wilson, G. G., and Murray, N. E. (2014b). Highlights of the DNA cutters: a short history of the restriction enzymes. Nucleic Acids Res. 42, 3–19. doi: 10.1093/nar/gkt990
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Loenen, W. A., and Raleigh, E. A. (2014). The other face of restriction: modification-dependent enzymes. Nucleic Acids Res. 42, 56–69. doi: 10.1093/nar/gkt747
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meisel, A., Bickle, T. A., Kruger, D. H., and Schroeder, C. (1992). Type III restriction enzymes need two inversely oriented recognition sites for DNA cleavage. Nature 355, 467–469. doi: 10.1038/355467a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meselson, M., Yuan, R., and Heywood, J. (1972). Restriction and modification of DNA. Annu. Rev. Biochem. 41, 447–466. doi: 10.1146/annurev.bi.41.070172.002311
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Morgan, R. D., Dwinell, E. A., Bhatia, T. K., Lang, E. M., and Luyten, Y. A. (2009). The MmeI family: type II restriction-modification enzymes that employ single-strand modification for host protection. Nucleic Acids Res. 37, 5208–5221. doi: 10.1093/nar/gkp534
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mullakhanbhai, M. F., and Larsen, H. (1975). Halobacterium volcanii spec. nov., a Dead Sea halobacterium with a moderate salt requirement. Arch. Microbiol. 104, 207–214. doi: 10.1007/BF00447326
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Murphy, J., Mahony, J., Ainsworth, S., Nauta, A., and Van Sinderen, D. (2013). Bacteriophage orphan DNA methyltransferases: insights from their bacterial origin, function, and occurrence. Appl. Environ. Microbiol. 79, 7547–7555. doi: 10.1128/AEM.02229-13
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Murray, I. A., Clark, T. A., Morgan, R. D., Boitano, M., Anton, B. P., Luong, K., et al. (2012). The methylomes of six bacteria. Nucleic Acids Res. 40, 11450–11462. doi: 10.1093/nar/gks891
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Murray, N. E. (2000). Type I restriction systems: sophisticated molecular machines (a legacy of Bertani and Weigle). Microbiol. Mol. Biol. Rev. 64, 412–434. doi: 10.1128/MMBR.64.2.412-434.2000
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakonieczna, J., Kaczorowski, T., Obarska-Kosinska, A., and Bujnicki, J. M. (2009). Functional analysis of MmeI from methanol utilizer Methylophilus methylotrophus, a subtype IIC restriction-modification enzyme related to type I enzymes. Appl. Environ. Microbiol. 75, 212–223. doi: 10.1128/AEM.01322-08
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nobusato, A., Uchiyama, I., Ohashi, S., and Kobayashi, I. (2000). Insertion with long target duplication: a mechanism for gene mobility suggested from comparison of two related bacterial genomes. Gene 259, 99–108. doi: 10.1016/S0378-1119(00)00456-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pingoud, A., Wilson, G. G., and Wende, W. (2014). Type II restriction endonucleases–a historical perspective and more. Nucleic Acids Res. 42, 7489–7527. doi: 10.1093/nar/gku447
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Quevillon, E., Silventoinen, V., Pillai, S., Harte, N., Mulder, N., Apweiler, R., et al. (2005). InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120. doi: 10.1093/nar/gki442
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rao, D. N., Dryden, D. T., and Bheemanaik, S. (2014). Type III restriction-modification enzymes: a historical perspective. Nucleic Acids Res. 42, 45–55. doi: 10.1093/nar/gkt616
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rego, R. O., Bestor, A., and Rosa, P. A. (2011). Defining the plasmid-borne restriction-modification systems of the Lyme disease spirochete Borrelia burgdorferi. J. Bacteriol. 193, 1161–1171. doi: 10.1128/JB.01176-10
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roberts, R. J., Belfort, M., Bestor, T., Bhagwat, A. S., Bickle, T. A., Bitinaite, J., et al. (2003). A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 31, 1805–1812. doi: 10.1093/nar/gkg274
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roberts, R. J., Vincze, T., Posfai, J., and Macelis, D. (2014). REBASE-a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 43, D298–D299. doi: 10.1093/nar/gku1046
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shen, B. W., Xu, D., Chan, S. H., Zheng, Y., Zhu, Z., Xu, S. Y., et al. (2011). Characterization and crystal structure of the type IIG restriction endonuclease RM.BpuSI. Nucleic Acids Res. 39, 8223–8236. doi: 10.1093/nar/gkr543
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vasu, K., and Nagaraja, V. (2013). Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev. 77, 53–72. doi: 10.1128/MMBR.00044-12
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Venclovas, C., Timinskas, A., and Siksnys, V. (1994). Five-stranded β-sheet sandwiched with two α-helices: a structural link between restriction endonucleases EcoRI and EcoRV. Proteins 20, 279–282. doi: 10.1002/prot.340200308
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Watanabe, M., Yuzawa, H., Handa, N., and Kobayashi, I. (2006). Hyperthermophilic DNA methyltransferase M.PabI from the archaeon Pyrococcus abyssi. Appl. Environ. Microbiol. 72, 5367–5375. doi: 10.1128/AEM.00433-06
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wilson, G. G., and Murray, N. E. (1991). Restriction and modification systems. Annu. Rev. Genet. 25, 585–627. doi: 10.1146/annurev.ge.25.120191.003101
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wion, D., and Casadesus, J. (2006). N6-methyl-adenine: an epigenetic signal for DNA-protein interactions. Nat. Rev. Microbiol. 4, 183–192. doi: 10.1038/nrmicro1350
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: DNA methylation, restriction modification system, haloarchaea, Haloferax volcanii, Halobacteria, methylome, PD-(D/E)XK nuclease, methylation
Citation: Ouellette M, Jackson L, Chimileski S and Papke RT (2015) Genome-wide DNA methylation analysis of Haloferax volcanii H26 and identification of DNA methyltransferase related PD-(D/E)XK nuclease family protein HVO_A0006. Front. Microbiol. 6:251. doi: 10.3389/fmicb.2015.00251
Received: 15 November 2014; Accepted: 13 March 2015;
Published: 08 April 2015.
Edited by:
Jesse Dillon, California State University, Long Beach, USAReviewed by:
Antonio Ventosa, University of Sevilla, SpainIchizo Kobyashi, University of Tokyo, Japan
Copyright © 2015 Ouellette, Jackson, Chimileski and Papke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: R. Thane Papke, Department Molecular and Cell Biology, University of Connecticut, 91 N. Eagleville Rd. Unit 3125, Storrs, CT 06268, USAdGhhbmVAdWNvbm4uZWR1
†These authors have contributed equally to this work.