 Ramunas Stepanauskas
Ramunas StepanauskasOver the past decade, single cell genomics (SCG) made a swift transition from science fiction to a handy new tool in the biologist's toolset. The power of this technology lies in its ability to retrieve information-rich genomic blueprints from the most fundamental units of biological organization—individual cells. This is particularly significant in the case of bacteria, archaea, and protists, where single cells constitute complete organisms. Such unicellular individuals comprise the vast majority of biological diversity and biomass of our planet, yet only a small fraction of microbial diversity has been discovered and studied. Together with other modern research tools, SCG has been increasingly instrumental in deciphering the genomic composition, metabolic potential and evolutionary histories of the “microbial dark matter.” While cultivation-free recovery of discrete genomes was impossible in 2004, by 2009 it became a routine procedure that is accessible to the broad research community through open-access SCG core facilities (e.g., scgc.bigelow.org). This rapid development has enabled genomic studies of many previously unexplored branches of the tree of life (Marcy et al., 2007; Rinke et al., 2013) and findings of hitherto unrecognized biogeochemical processes and ecological patterns (Swan et al., 2011, 2013; Mason et al., 2012), paving the way for a new wave of discovery in microbiology and biotechnology.
An exciting feature that sets SCG apart from other cultivation-independent technologies is the retrieval of sequences of all the DNA molecules in a cell, in this way providing evidence for their physical co-occurrence (or absence) in the analyzed cell or consortia of multiple cells. Such molecules may include multiple chromosomes and plasmids of the host organism; genomes of organelles, symbionts, viruses, and other infecting agents and prey items; and naturally transformed DNA (Figure 1). The ability to collect this type of information offers a major, but still underutilized opportunity to microbiology. Infections, symbioses, phagotrophy, horizontal gene transfer, formation of consortia, and other interactions among unicellular, uncultured organisms and extracellular genetic elements can now be analyzed directly, in their natural environment. Such interactions are thought to be of paramount importance to the functioning of oceans, soils, macroorganismal (including human) biomes and other microbially-dominated ecosystems, although their in situ studies have been severely hampered by methodological difficulties.
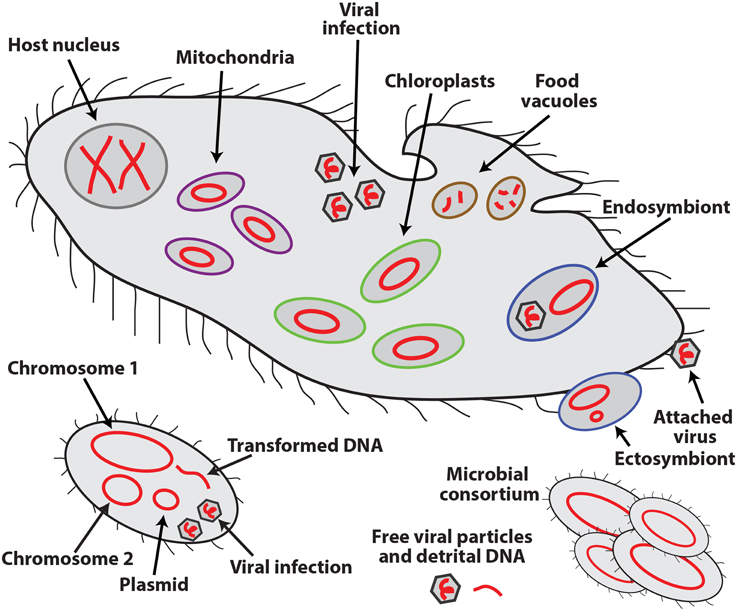
Figure 1. Schematic representation of the various types of DNA molecules (in red) and their occurrence inside and outside of eukaryotic (top) and bacterial (bottom) cells.
In one of the first applications of SCG on eukaryotes, multiple cells of the candidate phylum Picozoa (formerly Picobilliphyta) were found to contain fragments of bacterial and viral DNA, while no genes involved in photosynthesis were identified (Yoon et al., 2011). This provided the first evidence that Picozoa are phagotrophs, correcting prior suggestions of them being photosynthetic. The same SCG study also retrieved a complete genome of a novel nanovirus from an infected Picozoa cell. A phylogenetically broader screen of uncultured marine protists provided evidence for several novel symbiotic and phagotrophic interactions (Martinez-Garcia et al., 2012). More recently, SCG of planktonic bacteria from a model oxygen minimum zone revealed genomes and the spatiotemporal distribution of 69 novel phages infecting SUP05, an abundant but yet uncultured lineage of Gammaproteobacteria (Roux et al., 2014). Another SCG study, focused on surface ocean bacterioplankton, obtained genomes of the first known viruses of Thaumarchaeota, Marinimicrobia, Verrucomicrobia, and other ubiquitous, uncultured taxonomic groups of marine bacterioplankton (Labonte et al., in press). Using an innovative combination of SCG, metagenomics, and microarray hybridization, a virus infecting the candidate division Nanohaloarchaeota was identified (Martínez-García et al., 2014). It is likely that similar techniques will be increasingly utilized to untangle the complex inter-taxa interactions in “microbial jungles” of oceans, soils and other environments.
SCG is also becoming progressively instrumental in studies of microbial horizontal gene exchange (sexual interactions) and other microevolutionary processes in the environment. Microbial genomics came a long way since the first sequencing of a complete prokaryote genome, Haemophilus influenzae Rd, in 1995 (Fleischmann et al., 1995). As of January 2015, over 30,000 genomes of bacteria and archaea have been deposited in public databases, at an accelerating rate. While this is a truly revolutionary achievement for microbiology, it is important to acknowledge that we are still only scratching the surface of the full genetic complexity and the underlying evolutionary processes in many environmental microbial communities. To put current sequencing efforts in perspective, roughly 30,000 genomes of bacteria and archaea are present in each 30 μL of ocean water or 30 μg of a fertile soil (the same 30 μL water/30 μg soil also contain ~300,000 viruses, a few eukaryotes and a large amount of detrital DNA). The total number of microbial cells on the planet is in the order of 1030 (Whitman et al., 1998), which collectively encode at least 1030 Mbp of genetic information. An important fundamental question in microbiology is the extent and underlying mechanisms of genome variation among cells that share highly similar small subunit rRNA genes, i.e., belong to the same phylotype. Already the earliest comparative genomics studies revealed massive differences in gene content among members of the same, operationally defined bacterial species (Welch et al., 2002). Subsequent studies confirmed that substantial genome content variation among individuals, indicative of extensive horizontal gene exchange, is the rule rather than the exception in natural microbial populations (Ochman et al., 2000; Papke et al., 2007; Shapiro et al., 2012), with likely major implications to their resilience and adaptability to new conditions, such as climate change or exposure to antibiotics. From a technical perspective, the limited clonality of many natural microbial populations makes it difficult to assemble discrete genomes from metagenomic reads, and to unambiguously interpret such assemblies (Rusch et al., 2007; Hess et al., 2011). Our current understanding remains rudimentary when it comes to the extent of genetic variability within most microbial populations, specific evolutionary and ecological processes that govern this variability, and rates of these evolutionary processes. By eliminating the need for arbitrary taxonomic binning of omics data, SCG is well-suited to enable a breakthrough in studies of microbial microevolution, and first publications in this area have already provided valuable insights (Engel et al., 2014; Kashtan et al., 2014). These pilot studies suggest that each cell in a natural microbial population may have a unique subset of mutations, horizontally acquired and recombined genes, mobile genetic elements, and other genetic features. Instead of viewing this natural complexity as a nuisance, SCG enables “forensic DNA investigations” of individual microbial cells in the environment, opening a new window into their life histories.
In order to obtain statistically representative samples of members of natural microbial assemblages and their intra- and inter-species interactions, the scalability of SCG will be of key importance. In the past 5 years, the scale of SCG projects grew from single genomes (Marcy et al., 2007; Woyke et al., 2009) to 10 s and 100 s of genomes (Rinke et al., 2013; Swan et al., 2013; Kashtan et al., 2014), and further technology improvements are well-underway. Increasingly sophisticated research applications will also drive improvements in SCG data quality, such as better genome recovery and reduced frequency of assembly errors. Like any technology, SCG is at its best when combined with other research tools, in order to most effectively address transformative science questions. There is little doubt that SCG will be increasingly utilized in diverse microbial studies, complementing cultivation-based, community omics, biogeochemical, and other research approaches.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank David Emerson for insightful comments and stimulating discussions. Funding has been provided by the U.S. National Science Foundation grants OCE-1232982 and OCE-1136488.
References
Engel, P., Stepanauskas, R., and Moran, N. A. (2014). Hidden diversity in honey bee gut symbionts detected by single-cell genomics. PLoS Genet. 10:e1004596. doi: 10.1371/journal.pgen.1004596
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fleischmann, R. D., Adams, M. D., White, O., Clayton, R. A., Kirkness, E. F., Kerlavage, A. R., et al. (1995). Whole-genome random sequencing and assembly of Haemophilus influenzae RD. Science 269, 496–512. doi: 10.1126/science.7542800
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hess, M., Sczyrba, A., Egan, R., Kim, T. W., Chokhawala, H., Schroth, G., et al. (2011). Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331, 463–467. doi: 10.1126/science.1200387
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kashtan, N., Roggensack, S. E., Rodrigue, S., Thompson, J. W., Biller, S. J., Coe, A., et al. (2014). Single cell genomics reveals hundreds of coexisting subpopulations in wild Prochlorococcus. Science 344, 416–420. doi: 10.1126/science.1248575
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Labonte, J. M., Swan, B. K., Poulos, B., Luo, H., Koren, S., Hallam, S. J., et al. (in press). Single cell genomics-based analysis of virus-host interactions in marine surface bacterioplankton. ISME J.
Marcy, Y., Ouverney, C., Bik, E. M., Lösekann, T., Ivanova, N., Martin, H. G., et al. (2007). Dissecting biological “dark matter” with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc. Natl. Acad. Sci. U.S.A. 104, 11889–11894. doi: 10.1073/pnas.0704662104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Martinez-Garcia, M., Brazel, D., Poulton, N. J., Swan, B. K., Gomez, M. L., Masland, D., et al. (2012). Unveiling in situ interactions between marine protists and bacteria through single cell sequencing. ISME J. 6, 703–707. doi: 10.1038/ismej.2011.126
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Martínez-García, M., Santos, F., Moreno-Paz, M., Parro, V., and Antón, J. (2014). Unveiling viral-host interactions within the ‘microbial dark matter’. Nat. Commun. 5, 4542. doi: 10.1038/ncomms5542
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mason, O. U., Hazen, T. C., Borglin, S., Chain, P. S. G., Dubinsky, E. A., Fortney, J. L., et al. (2012). Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill. ISME J. 6, 1715–1727. doi: 10.1038/ismej.2012.59
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ochman, H., Lawrence, J. G., and Grolsman, E. A. (2000). Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304. doi: 10.1038/35012500
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Papke, R. T., Zhaxybayeva, O., Feil, E. J., Sommerfeld, K., Muise, D., and Doolittle, W. F. (2007). Searching for species in haloarchaea. Proc. Natl. Acad. Sci. U.S.A. 104, 14092–14097. doi: 10.1073/pnas.0706358104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rinke, C., Schwientek, P., Sczyrba, A., Ivanova, N. N., Anderson, I. J., Cheng, J. F., et al. (2013). Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437. doi: 10.1038/nature12352
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roux, S., Hawley, A. K., Torres Beltran, M., Scofield, M., Schwientek, P., Stepanauskas, R., et al. (2014). Ecology and evolution of viruses infecting uncultivated SUP05 bacteria as revealed by single-cell- and meta- genomics. eLife 3:e03125. doi: 10.7554/eLife.03125
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rusch, D. B., Halpern, A. L., Sutton, G., Heidelberg, K. B., Williamson, S., Yooseph, S., et al. (2007). The sorcerer II global ocean sampling expedition: Northwest Atlantic through Eastern Tropical Pacific. PLoS Biol. 5:e77. doi: 10.1371/journal.pbio.0050077
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shapiro, B. J., Friedman, J., Cordero, O. X., Preheim, S. P., Timberlake, S. C., Szabo, G., et al. (2012). Population genomics of early events in the ecological differentiation of bacteria. Science 335, 48–51. doi: 10.1126/science.1218198
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Swan, B. K., Martinez-Garcia, M., Preston, C. M., Sczyrba, A., Woyke, T., Lamy, D., et al. (2011). Potential for chemolithoautotrophy among ubiquitous bacteria lineages in the dark ocean. Science 333, 1296–1300. doi: 10.1126/science.1203690
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Swan, B. K., Tupper, B., Sczyrba, A., Lauro, F. M., Martinez-Garcia, M., GonŸalez, J. M., et al. (2013). Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. Proc. Natl. Acad. Sci. U.S.A. 110, 11463–11468. doi: 10.1073/pnas.1304246110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Welch, R. A., Burland, V., Plunkett, G., Redford, P., Roesch, P., Rasko, D., et al. (2002). Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 99, 17020–17024. doi: 10.1073/pnas.252529799
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Whitman, W. B., Coleman, D. C., and Wiebe, W. J. (1998). Prokaryotes: the unseen majority. Proc. Natl. Acad. Sci. U.S.A. 95, 6578–6583. doi: 10.1073/pnas.95.12.6578
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Woyke, T., Xie, G., Copeland, A., Gonzalez, J. M., Han, C., Kiss, H., et al. (2009). Assembling the marine metagenome, one cell at a time. PLoS ONE 4:e5299. doi: 10.1371/journal.pone.0005299
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yoon, H. S., Price, D. C., Stepanauskas, R., Rajah, V. D., Sieracki, M. E., Wilson, W. H., et al. (2011). Single-cell genomics reveals organismal interactions in uncultivated marine protists. Science 332, 714–717. doi: 10.1126/science.1203163
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: single cell genomics, microbial interactions, horizontal gene exchange, microevolution, microbial dark matter
Citation: Stepanauskas R (2015) Wiretapping into microbial interactions by single cell genomics. Front. Microbiol. 6:258. doi: 10.3389/fmicb.2015.00258
Received: 12 January 2015; Paper pending published: 16 February 2015;
Accepted: 16 March 2015; Published: 08 April 2015.
Edited by:
Manuel Martinez Garcia, University of Alicante, SpainCopyright © 2015 Stepanauskas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ramunas Stepanauskas,cnN0ZXBhbmF1c2thc0BiaWdlbG93Lm9yZw==