 Achim Quaiser1*
Achim Quaiser1* Alexis Dufresne1
Alexis Dufresne1 Flore Ballaud1
Flore Ballaud1 Simon Roux2Yvan Zivanovic3Jonathan Colombet4
Simon Roux2Yvan Zivanovic3Jonathan Colombet4 Télesphore Sime-Ngando4
Télesphore Sime-Ngando4 André-Jean Francez1
André-Jean Francez1- 1UMR CNRS 6553 – ECOBIO, Université de Rennes 1, Rennes, France
- 2Department of Ecology and Evolutionary Biology, University of Arizona, Tucson, AZ, USA
- 3CNRS, UMR 8621, Université Paris Sud, Orsay, France
- 4CNRS, Laboratoire Microorganismes: Génome et Environnement – UMR 6023, Université Blaise Pascal, Clermont-Ferrand, France
Microviridae, a family of bacteria-infecting ssDNA viruses, is one of the still poorly characterized bacteriophage groups, even though it includes phage PhiX174, one of the main models in virology for genomic and capsid structure studies. Recent studies suggest that they are diverse and well represented in marine and freshwater virioplankton as well as in human microbiomes. However, their diversity, abundance, and ecological role are completely unknown in soil ecosystems. Here we present the comparative analysis of 17 completely assembled Microviridae genomes from 12 viromes of a Sphagnum-dominated peatland. Phylogenetic analysis of the conserved major capsid protein sequences revealed the affiliation to Gokushovirinae and Pichovirinae as well as to two newly defined subfamilies, the Aravirinae and Stokavirinae. Additionally, two new distinct prophages were identified in the genomes of Parabacteroides merdae and Parabacteroides distasonis representing a potential new subfamily of Microviridae. The differentiation of the subfamilies was confirmed by gene order and similarity analysis. Relative abundance analysis using the affiliation of the major capsid protein (VP1) revealed that Gokushovirinae, followed by Aravirinae, are the most abundant Microviridae in 11 out of 12 peat viromes. Sequences matching the Gokushovirinae and Aravirinae VP1 matching sequences, respectively, accounted for up to 4.19 and 0.65% of the total number of sequences in the corresponding virome, respectively. In this study we provide new genome information of Microviridae and pave the way toward quantitative estimations of Microviridae subfamilies.
Introduction
Viruses, in particular bacteriophages, have been identified in several ecosystems, and represent the most abundant biological entities on Earth. In marine ecosystems and freshwater lakes they are present at high concentrations of about 107 particles/mL on average (Wommack and Colwell, 2000). Viruses can control the microbial abundance and influence the composition of microbial communities by lysing their host organisms (Fuhrman and Schwalbach, 2003). In addition, they carry a highly diverse pool of genetic elements that might be exchanged with their hosts, thereby contributing to adaptation processes through the acquisition of new functions (Pedulla et al., 2003). Despite these potential influences the importance is almost solely illustrated in aquatic ecosystems (Wommack and Colwell, 2000; Suttle, 2007), and little is known about the diversity of viruses and in particular about ssDNA viruses in soil ecosystems. ssDNA viruses are divided into two families of bacteriophages (Inoviridae and Microviridae) and five eukaryotic viruses (King et al., 2012). Recent studies revealed that Microviridae seem to be ubiquitous. Through metagenomic approaches, Microviridae were identified in marine environments (Angly et al., 2006; Tucker et al., 2011; Labonté and Suttle, 2013a,b), freshwater habitats (López-Bueno et al., 2009; Roux et al., 2012a,b), human gut or feces (Roux et al., 2012b), stromatolites (Desnues et al., 2008), dragonflies (Rosario et al., 2012), sewage and sediments (Hopkins et al., 2014), and even as temperate phages integrated in the genomes of Bacteroidetes species (Krupovic and Forterre, 2011). These studies contribute significantly to the accumulation of Microviridae specific genomic information allowing more precise comparative genome analyses. These revealed that all Microviridae genomes possess several homologous genes including a well-conserved gene coding for the major capsid protein VP1, a roughly 500 amino acid long protein. This protein can be used as phylogenetic marker facilitating delineation of Microviridae clades or subfamilies (Wommack and Colwell, 2000; Desnues et al., 2008; Roux et al., 2012b; Labonté and Suttle, 2013b; Hopkins et al., 2014). All these studies applied whole-genome amplification methodology that preferentially amplifies small circular DNA templates such as ssDNA bacteriophages, which consequently introduces biases (Fuhrman and Schwalbach, 2003; Kim and Bae, 2011). We took advantage of this bias in that the ssDNA bacteriophages were likely preferentially enriched allowing for more targeted diversity analyses and exploration of new Microviridae genomes.
In this study, we analyzed 18 complete Microviridae genomes reconstructed through de novo assembly of reads from 12 viromes one microbial metagenome and obtained from peat soil and water samples collected in a Sphagnum-dominated peatland. Detailed phylogenetic analysis and genome comparisons revealed two new subfamilies. Combined with the available genomic information of Microviridae we provide new insights into the diversity, distribution and abundance of the Microviridae subfamilies.
Materials and Methods
Sampling and Accession to 12 Peat Viromes
Twelve samples were recovered from a Sphagnum-dominated peatland at “les Pradeaux mire” in the French Massif Central (3°55E; 45°32N) at an altitude of 1,250 m. Water and peat soils were both sampled in young states of peatland dynamics, called ‘Fen’ with Sphagnum fallax and Carex rostrata vegetation, and in older states of dynamics, called ‘Bog’ with S. magellanicum and S. capillifolium, Andromeda polyfolia, and Eriophorum vaginatum (Francez and Vasander, 1995). One peat sample (-5 cm to -15 cm depth) for each Fen and Bog was collected in June, August and October 2011 as well as three biological replicates of water that was extracted from Sphagnum-peat and filtered at 125 μm from both Fen and Bog (March 2012). Viruses were concentrated using PEGylation (Wommack and Colwell, 2000; Colombet et al., 2007; Suttle, 2007). Extracellular DNA was digested with DNAse RQ1 (Promega) at 37°C for 1 h. Viral DNA was extracted with the Nucleospin Extract II kit (Macherey–Nagel). Whole genome amplification (WGA) reactions were run in triplicate for each sample using Genomi-Phi following the instructions of the manufacturer (GE Healthcare) and subsequently pooled. Library construction and pyrosequencing was performed at the “Functional and Environmental Genomics” platform (OSUR, Rennes, France). After sequence quality and size trimming we obtained 481,402, and 618,487 reads from Fen and Bog, respectively, with an average length of 415 bp. Microviridae genomes reported in this publication have been deposited in the Sequence Read Archive under the study accession number KM589498–KM589516.
Assembly, Annotation, and Comparative Genome Analysis
The peat viromes were assembled separately into contigs using the meta-assembler of CAMERA under standard conditions (Seshadri et al., 2007; King et al., 2012). The circularity of the genomes was checked manually. Only Microviridae genomes that showed overlapping reads at the start and end of the contigs, confirming their circular genomes were retained. Sequence analysis was performed using an integrated WEB-based annotation platform adapted to metagenomic sequence analysis (Genomic toolbox; Angly et al., 2006; Tucker et al., 2011; Labonté and Suttle, 2013a,b; Zivanovic et al., 2014; http://www-archbac.u-psud.fr/projects/aq_virome/aq_virome.html). Candidate coding sequences (CDSs) were defined with PRODIGAL (version 2.60; López-Bueno et al., 2009; Hyatt et al., 2010; Roux et al., 2012a,b). Semi-automated annotation was performed to identify genes by sequence similarity and coding probability using BLASTP (Altschul et al., 1997; Roux et al., 2012b) against the RefSeq NR protein database (GenBank), SWISSPROT (version 57.11), and COG databases (COG + KOG, seven eukaryotic genomes). Manual annotation was completed within the above-mentioned platform. Gene order comparison was performed using the platform tool GENOMAPPER (Desnues et al., 2008; Zivanovic et al., 2014) combined with multiple alignment and phylogenetic analysis. To allow the interactive visualization of genomic fragment comparisons, we used Artemis Comparison Tool ACTv.6 (Carver et al., 2012; Rosario et al., 2012).
The presence of Microviridae in peat metagenomes that were constructed from corresponding peat samples without virus enrichment (Quaiser et al., unpublished data) was analyzed by BLASTN and reads matching the virome-assembled Microviridae genomes were selected. The Microviridae contig from the metagenome was assembled manually using 21 reads. Since the reads showed nearly 100% identity to the Gokushovirinae genome Fen7875_21, it was used as a reference for recruitment of metagenome reads.
Phylogenetic Analysis
Sequences of full length major capsid protein and replication protein as well as the complete nucleic acid sequences of the Gokushovirinae genomes were aligned using MUSCLE (Edgar, 2004; Hopkins et al., 2014). Alignments were manually edited using ARB (Ludwig et al., 2004; Krupovic and Forterre, 2011). Gaps and ambiguously aligned positions were excluded from phylogenetic analysis. Maximum likelihood trees were reconstructed using TREEFINDER (Jobb et al., 2004) applying a JTT model (amino acid) and GTR3 (nucleic acid) of sequence evolution with a four category discrete approximation of a distribution plus invariant sites. The best-fit models were determined using TREEFINDER. Maximum likelihood bootstrap proportions were inferred using 1000 replicates.
Major Capsid Protein Structural Modeling
I-TASSER (Roy et al., 2010) was used to assess a first model of a VP1 protein from each of the two new clades (Bog5275_51 for Aravirinae, Fen51_42 for Stokavirinae). These initial models were processed through multiple steps of loops refinement with MODELER (Eswar et al., 2008) to improve their quality (PROSA-WEB; Wiederstein and Sippl, 2007). The final quality Z-scores of the obtained models was -5.15 and -4.59 for Bog5275_51 (Aravirinae) and Fen51_42 (Stokavirinae), respectively, which according to the ProSA-web server is in the range of X-ray based models, though slightly higher than the two reference VP1 models (-6.4 for φX174 F and -6.14 for SpV4 VP1). Visualization of the structural models and sequence conservation was performed with UCSF CHIMERA (Pettersen et al., 2004).
Relative Abundance of Microviridae
We compiled available full-length major capsid protein sequences from the complete Microviridae genomes to a total of 88 sequences and attributed the taxonomic affiliation according to the phylogenetic analysis in Figure 1. BLASTX analysis against the major capsid protein sequences was performed for the 12 Sphagnum-peat viromes and 69 viromes from public databases. To assure the reliability of matches a strict cut-off value of 1e-10 was applied. The number of matches was normalized to the total number of reads in the different viromes.
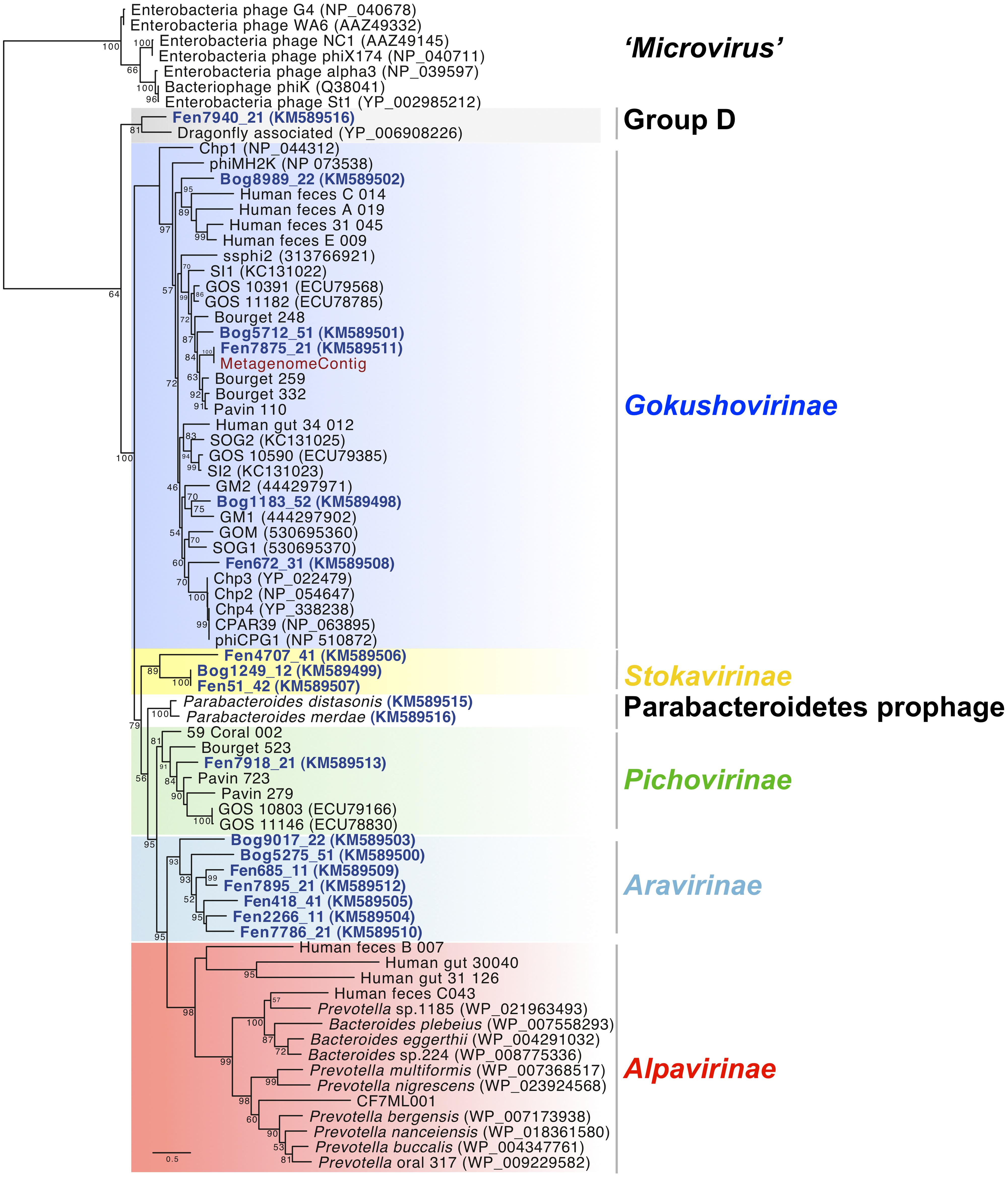
FIGURE 1. Maximum likelihood phylogenetic analysis of full-length major capsid protein sequences present in the Microviridae genomes from Sphagnum-dominated peat viromes. A total of 253 unambiguously aligned positions from 76 sequences were used in the analysis. Bootstrap values are indicated at the nodes. The scale bar indicates the number of substitutions per position for a unit branch length. Chlamydiaphages: Chp 1–4, CPAR39, phiCPG1; Bdellovibriophage: phiMH2K.
Results and Discussion
Assembly, Identification, and Analysis of Complete Microviridae-Like Genomes
To get insights into the genome structure and the distribution of Microviridae, reads from 12 peat viromes were assembled separately into contigs using the meta-assembler of CAMERA (Seshadri et al., 2007). In total 840 contigs longer than 3 kbp were obtained. Of these, 107 contigs showed high sequence similarity to the major capsid protein VP1 of Microviridae identified with TBLASTX analysis and by GENE RELATIONS and SYNTENY ANALYSIS tools from the Genomics toolbox (see Material and Methods). Only contigs corresponding to complete circular genomes were retained. In total, we obtained 17 new complete bacteriophage genomes affiliated to Microviridae. Sequencing coverage ranged from 8.31 to 94.63 times (Supplementary Table S1). One additional genome was assembled from reads of a microbial metagenome constructed from a peat sample without viral enrichment (Quaiser, unpublished data). Interestingly, several genomes assembled independently from different samples exhibited very high levels of similarity, thus validating the assembly process of viral genomes from metagenomic reads used in this study and confirming that the obtained assemblies were not chimeric and represented real Microviridae genomes.
Diversity of Peat Microviridae Accessed by Phylogenetic Analysis of the Conserved Major Capsid Protein VP1
To assess the diversity of the new Microviridae-affiliated genomes, phylogenetic analysis was performed using the translated sequence of the gene coding for the major capsid protein VP1 (Figure 1). Eight different clades were formed using representative sequences from already identified Microviridae subfamilies and VP1 sequences from the 18 peat Microviridae genomes.
Five Microviridae genomes recovered from the viral fraction of the peat samples as well as the genome assembled from a peat metagenome clustered within the subfamily of Gokushovirinae. Two of these genomes derived from Fen and three from Bog samples. Peat Gokushovirinae were clearly separated from cultured ssDNA bacteriophage representatives (Chlamydiaphages: Chp 1–4, phiCPG1, CPAR39; Bdellovibriophage: phiMJ2K) and clustered with recently assembled Gokushovirinae genomes from planktonic microbial communities (GM1; Labonté and Suttle, 2013a), freshwater lake (Bourget_248, Bourget_259; Roux et al., 2012a), and human feces (Roux et al., 2012b).
One new clade was formed by three genomes from three different viromes (Fen4707_41, Bog1249_12, Fen51_42) representing a putative novel subfamily. We propose to name it Stokavirinae (Stoka: small in Sanskrit). A second new clade, which we propose to name Aravirinae (Ara: little in Sanskrit), consisted of seven new genomes assembled from five different peat viromes (Bog9017_22, Bog5275_51, Fen685_11, Fen7895_21, Fen418_41, Fen2266_11, Fen7786_21). Only one genome, Fen7918_21, was affiliated with the recently defined subfamily of Pichovirinae that, to date, consists of seven assembled genomes isolated from freshwater and marine environments (Roux et al., 2012b). Genome Fen7940_21 was affiliated with the dragonfly associated microvirus representing the second member of this clade of ssDNA microviruses (Rosario et al., 2012) and thereby confirming its distinction into another potentially novel subfamily (Group D). Alpavirinae that were identified as integrated prophages in Bacteroidetes genomes associated with human gut and oral microbiota (Krupovic and Forterre, 2011; Roux et al., 2012b) as well as sequences affiliated to the genus Microvirus (proposed subfamily Microvirinae) could not be identified in any of the peat viral genomes. Phylogenetic analysis of VP1 including recently amplified sequences from Gokushovirinae changed slightly the tree topology (Supplementary Figure S1; Labonté and Suttle, 2013a). In this case Bog9017_22 was clearly separated from Aravirinae and Fen7940_21 was apart from dragonfly associated microvirus. This is probably due to the shorter alignment imposed by the PCR-generated sequences that lower the resolution of the phylogenetic analysis.
Taxonomic affiliation was additionally determined using sequences of the replication protein (VP4), which is shared by all the Microviridae genomes (Supplementary Figure S2). While the replication protein is shorter and less conserved than the major capsid protein, phylogenetic analysis revealed similar clades with the sole exception of Bog9017_22, which moved from Aravirinae (major capsid protein phylogeny) to Pichovirinae in replication protein phylogeny. While this could hint for a chimeric assembly of the genome, it is noteworthy that the replication protein sequence is shorter and more variable, and therefore provides a less reliable phylogenetic signal than the VP1 sequence (Supplementary Figure S2; Roux et al., 2012b).
Structure of the Aravirinae and Stokavirinae Major Capsid Protein
Structural modeling of the major capsid protein from the two new Microviridae subfamilies indicated that they harbor a conserved eight-stranded β-barrel core (“viral jelly- roll”) and a loop extension (Figure 2A) similar to Alpavirinae and Pichovirinae which is known to form mushroom-like protrusions in SpV4 (Roux et al., 2012b). These protrusions, found in every type of Microviridae except for the Microvirinae, are thought to bind to host receptors (Roux et al., 2012b). Based on the seven different Aravirinae, we analyzed the level of residue conservation along the major capsid protein sequences. As expected, the protrusion loop is highly variable (region 1 on Figure 2B). Moreover we identified two additional variable regions backing the separation into a new subfamily (Figure 2B). Interestingly, when mapping the protein model on the virion structure, these two additional variable regions appear to be situated on the virion surface. We speculate that these regions may form additional outer structures involved in protein–protein interaction.
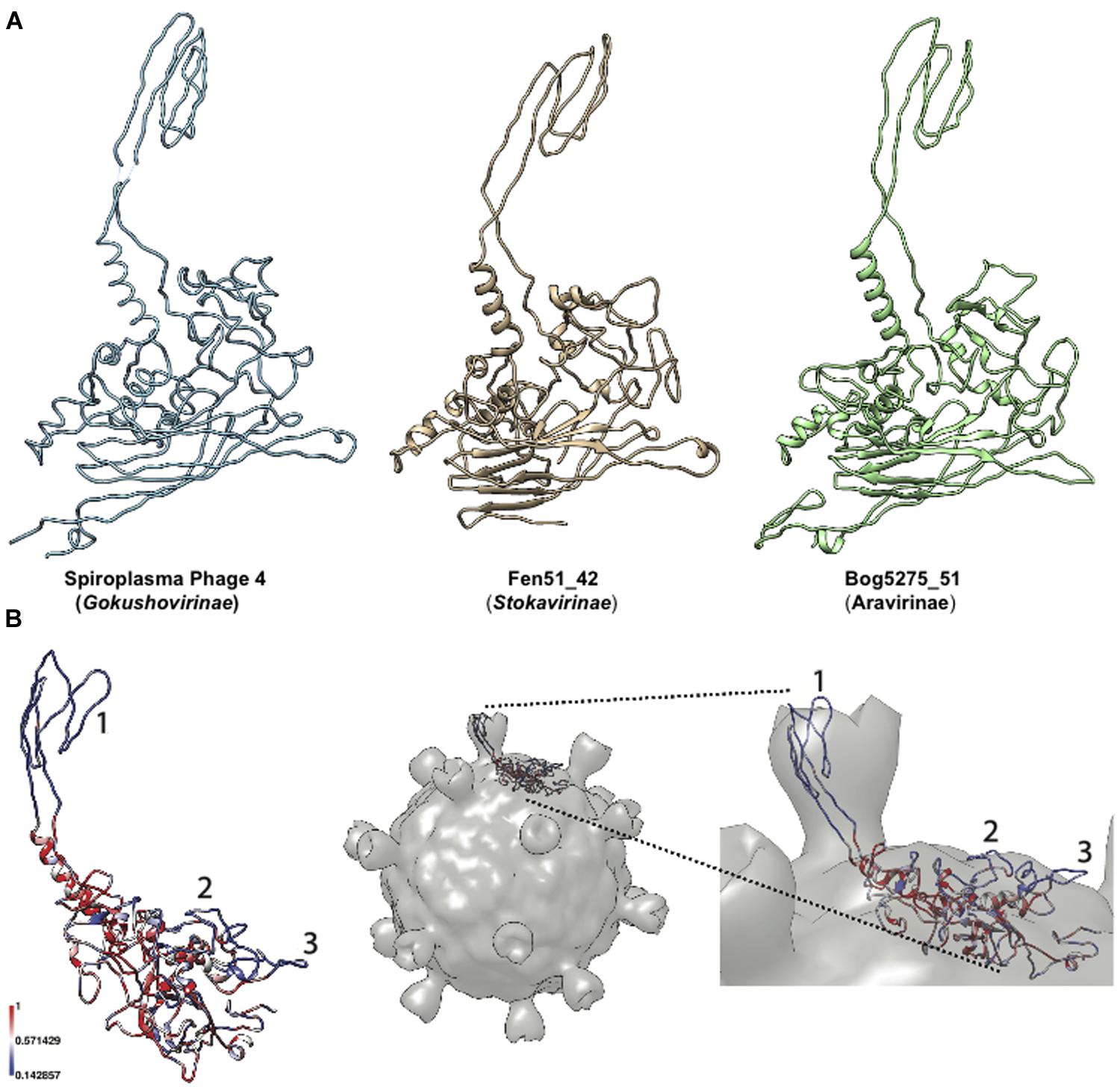
FIGURE 2. Modeling of major capsid proteins. (A) Three-dimensional models of Microviridae major capsid protein. Different subfamilies are depicted with, from left to right, the reference model from Spiroplasma Phage 4 (Pdb Id: 1KVP), representatives from Stokavirinae and Aravirinae. (B) Hypervariable regions of Aravirinae major capsid protein. The sequence conservation across the seven Aravirinae sequences was mapped on the three-dimensional model obtained for the Bog5275_51 genome. The three hypervariable regions were numbered (left panel). This model was mapped on the capsid structure of Spiroplasma phage 4 to estimate the position of these hypervariable regions relative to the whole virion (right panel).
Identification of Microviridae Prophages in Bacterial Genomes
Blast searches with the major capsid protein sequences from the assembled genomes showed similarities to VP1 proteins encoded in the genomes of the bacteria Parabacteroides distasonis and Parabacteroides merdae (Sakamoto and Benno, 2006). A detailed inspection of the genomic environment of the Parabacteroidetes VP1 gene showed that genes coding for homologs of VP2 and VP4 were located next to the VP1 gene in a 5 kbp region. This indicates the presence of a prophage affiliated to Microviridae in both Parabacteroidetes species. In order to compare their genomic organization, these identified prophage regions were cut from the two genomes and considered as circular genomes. For comparative analysis the genome start was arbitrarily fixed at VP1. Synteny analysis confirmed that the two prophages shared a common ancestor showing the same gene order (VP1-ORF1-ORF2-ORF3-VP2-VP4 for Parabacteroides distasonis and VP1-ORF1-VP2-VP4 for Parabacteroides merdae; Figure 3). The gene coding for an uncharacterized protein (ORF1) located downstream of the VP1 coding gene was specific to the two prophages and did not match with genes from other Microviridae genomes or from the NCBI non-redundant database. This strengthens the hypothesis that these prophages represent a distinct subfamily of Microviridae as suggested by the phylogenetic analysis of the major capsid protein sequences (Figure 1).
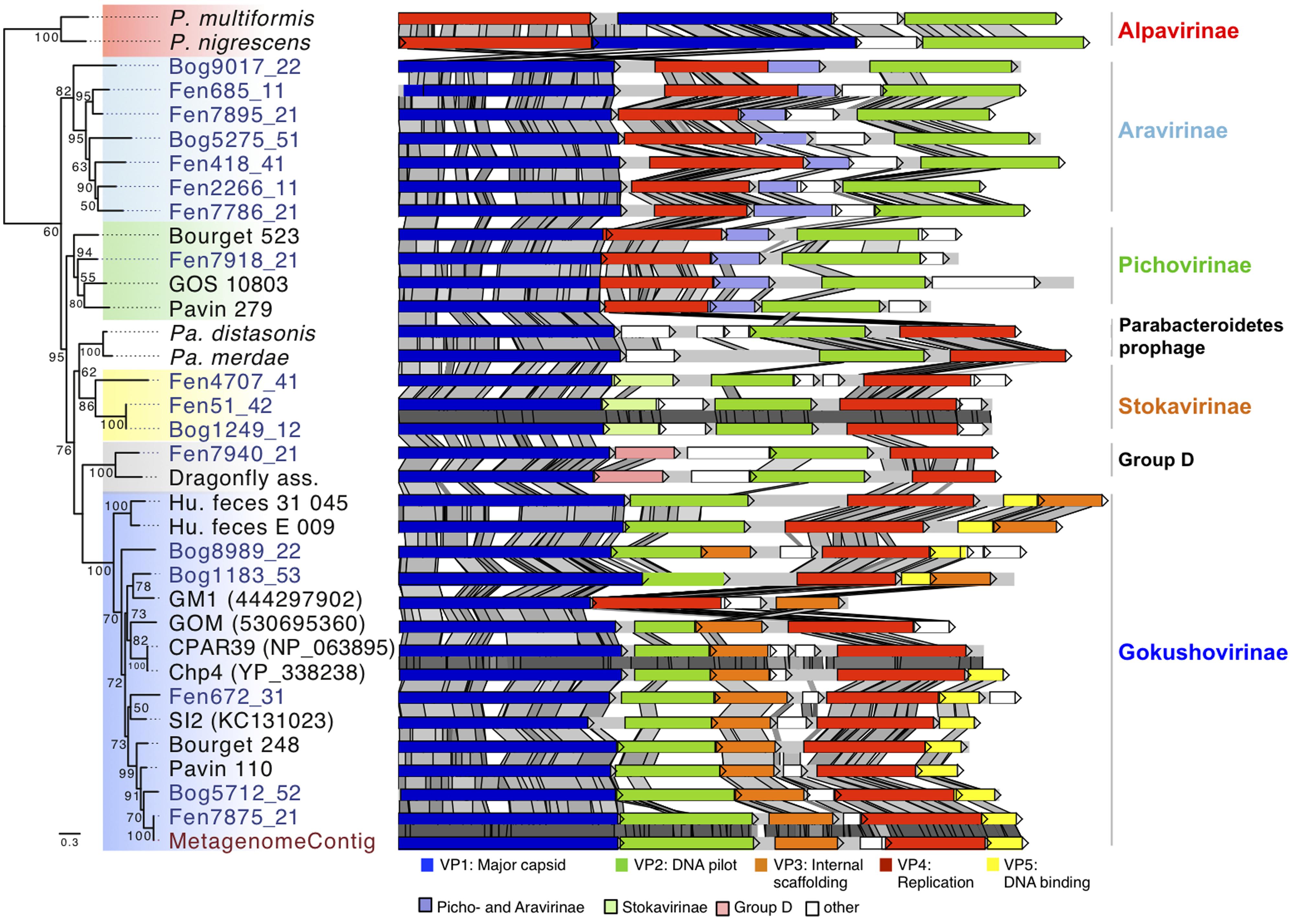
FIGURE 3. Major capsid protein phylogeny and genome structure of major subfamilies of Microviridae bacteriophages. The affiliation of the peat genomes to Microviridae is shown by major capsid protein phylogeny (Maximum likelihood, 252aa positions, 1000 iterations, JTT+G model). Bootstrap values above 50% are indicated at the nodes. The genome structures of the assembled viral peat genomes (blue) were compared to other Microviridae genomes. Pairwise comparison (TBLASTX) was visualized with ACT (Carver et al., 2005). Gray shading indicates the level of similarities. Homologous genes specific for some subfamilies are color-coded as indicated in the legend. P. multiformis, Prevotella multiformis (WP_007368517); P. nigrescens, Prevotella nigrescens (WP_023924568); Pa. distasonis, Parabacteroides distasonis (ZP_17317332); Pa. merdae, Parabacteroides merdae (WP_022322420). Dragonfly, Dragonfly associated phage (YP_006908226); GOS_10803 (ECU79166); GM1 (444297902); GOM (530695360); CPAR39 (NP_063895); Chp4 (YP_338238); SI2 (KC131023); Metagenome: genome assembled from the metagenome.
Comparative Genome Analysis
To get further insights into the diversity and evolution of the new Microviridae genomes the genome structures and gene sequence conservation levels were compared (Figure 3). The characteristic genes encoding the major capsid protein (VP1), replication protein (VP4), and DNA pilot protein (VP2) were identified in all Microviridae genomes. All analyzed Gokushovirinae possessed one additional gene coding for an internal scaffolding protein (VP3) and in most cases a DNA binding protein (VP5). Both genes were absent in all other subfamilies of Microviridae. The sequence similarity of VP3 and VP5 among the peat Gokushovirinae ranged from 31 to 100 and 26 to 100% identity, respectively. The gene order in Gokushovirinae was relatively well conserved, but we could distinguish two clades. In one clade the gene order consists of VP1, VP2, VP3, VP4, and VP5 (i.e., Fen672_31) while in the other clade the organization was VP1, VP2, VP4, VP5, and VP3 (Bog1183_53, Human_feces_E_009, and Human_feces_31_045). However, this distinction was not supported by phylogenetic analysis of the major capsid protein or the replication protein.
The two genomes, representative for the Group D (Dragonfly associated and Fen7940_21), contain five genes. Compared to other groups, they possess two additional genes organized in the same order between VP1 and VP2. The gene situated downstream of VP1 was conserved with up to 50% similarity but only over 41% of their amino acid sequences (Figure 3, Group D, pink). Since no other matches to this gene were found, neither in the NCBI non-redundant protein database nor in other peat Microviridae genomes, this gene was a unique feature for this clade confirming the distinction from other Microviridae clades. The second protein encoded upstream of VP2 appeared to be unique to each genome as they exhibited no similarity between each other or with proteins found in the NCBI nr database.
The three Stokavirinae genomes showed a similar organization. The protein encoded downstream of the major capsid protein gene was homologous within the Stokavirinae (Figure 3, light green) but without similarity to Parabacteroidetes prophage proteins or to the NCBI NR database. Stokavirinae possess a characteristic additional ORF downstream of VP4 that is absent in other subfamilies confirming that they present a distinct clade. Interestingly, the two genomes Bog1249_12 and Fen51_42 showed 99% nucleic acid identity while the third Fen4704_41 was more distant. Both were assembled independently not only from different viromes but also from bog and fen samples recovered at different dates. This indicates that the viral community was at least partially shared among different peat samples.
Whole Genome Phylogeny and Comparative Genome Analysis of Gokushovirinae
In order to determine the precise affiliation of the Gokushovirinae peat genomes, whole genome phylogeny was performed for this subfamily (Figure 4). The phylogenetic tree obtained reveals three groups that could not be identified based on analysis of the sole VP1 sequences: Chlamydiaphage Gokushovirinae, Group 2 and Group 3. As reported recently (Labonté and Suttle, 2013a) all cultured Gokushovirinae deriving from Chlamydia species, with the exception of Chp1 that appeared more distantly related, clustered together and their genome sequences were strongly conserved (Chp2, Chp3, Chp4, CPAR39, and phiCPG1). Group 2 contained phiMH2K (Bdellovibrio) and SpV4 (Spiroplasma) as well as recently assembled genomes from marine seawater (SOG1, GOM, ssphi2, SOG2, SI2; Labonté and Suttle, 2013a) and marine sediments (GM1; Yoshida et al., 2013). Group 3 contained the five peat Gokushovirinae and two assembled marine genomes SI1 (Labonté and Suttle, 2013a) and GM2 (Yoshida et al., 2013). Four peat Gokushovirinae exhibited the typical gene ordering, but in Bog1183_53 the VP3 gene was found downstream of the gene encoding VP5. Identities among the analyzed genomes ranged from 44.1 to 100% identical positions (Supplementary Table S2). Interestingly, the metagenome derived genome showed 100% similarity to the Fen7875_21 genome.
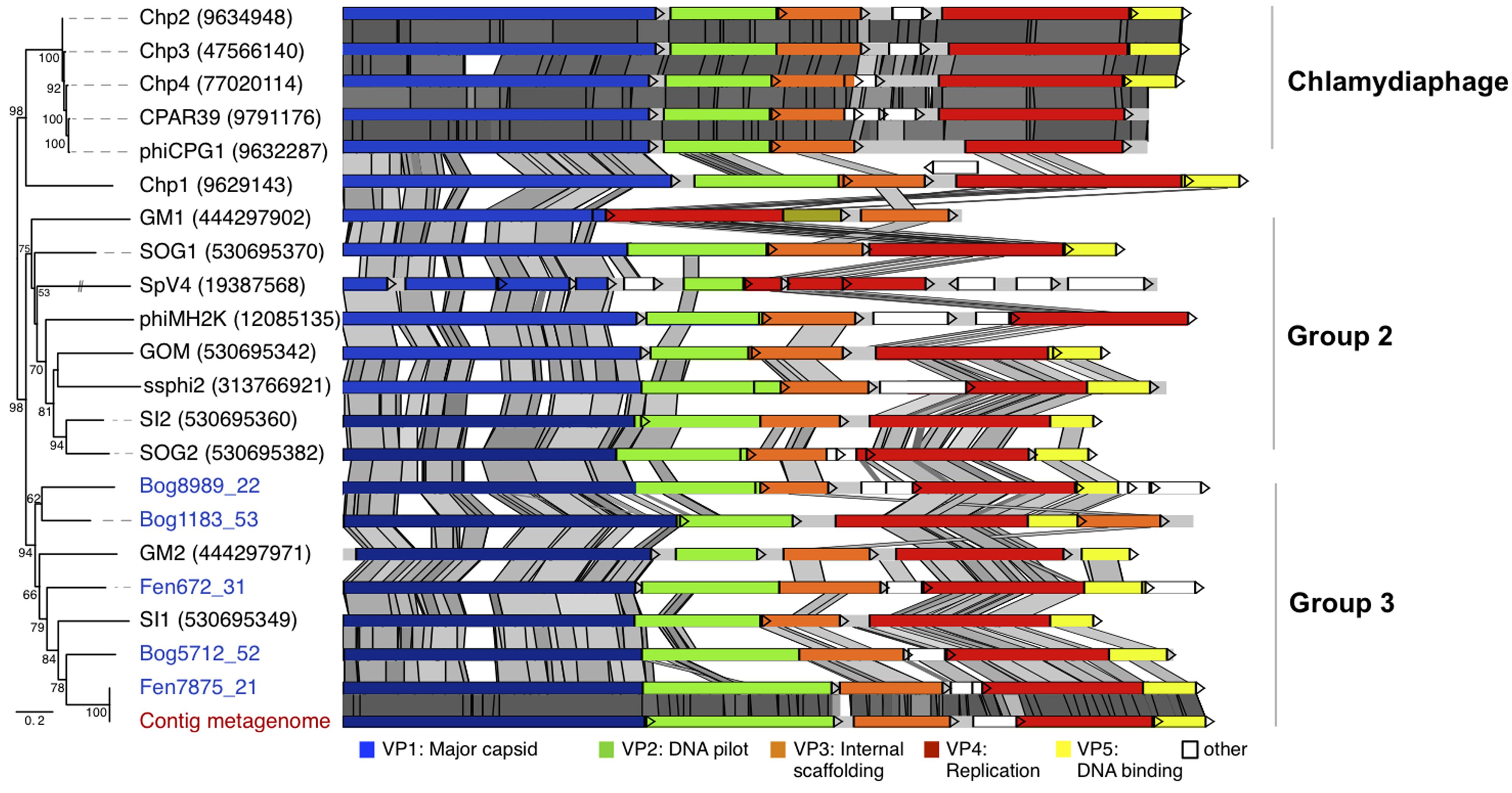
FIGURE 4. Whole genome phylogeny and conserved genome structure of peat Gokushovirinae. The affiliation of the peat genomes to Gokushovirinae was shown by whole genome phylogeny (Maximum likelihood, 2052 positions, 1000 iterations, GTR3 model). Maximum likelihood bootstrap values above 50% are indicated at the nodes. The genome structure of the assembled viral peat genomes (virome blue, metagenome red) was compared to Gokushovirinae. Pairwise comparisons (TBLASTX) were visualized by ACT (Carver et al., 2005). Gray shading indicates the level of similarities.
Relative Abundance of Microviridae Subfamilies in Peatland and Other Habitats Based on Matches to the Major Capsid Protein Sequences
In an attempt to estimate the distribution and abundance of Microviridae subfamilies the relative abundance of the major capsid protein coding genes was determined in the 12 peat viromes and in 69 additional viromes from 12 types of ecosystems (Figure 5). Quantitative estimations of viruses based on the detection of particular signature sequences is currently the best representative proxy, but should be considered with a grain of salt as WGA leads to overrepresentation of small ssDNA viruses (Kim and Bae, 2011), Nevertheless, to date WGA is the best way to recover sufficient viral genomic DNA for sequencing. Accordingly, all published viromes and the viromes included in this analysis were generated using this technique. Based on the phylogenetic analyses a database containing 88 major capsid protein sequences representing the different subfamilies of Microviridae was constructed. Each virome was searched with BLASTX against the major capsid protein sequence database and best matches were counted using strict count conditions (e-value 10-10). To take into account virome size variation, relative proportions of Microviridae subfamilies were obtained through the normalization of the matches by the total number of sequences in each virome. Taxonomic affiliation was determined according to the eight different subfamilies established in phylogenetic analyses (Figure 1). Alpavirinae represented the major subfamily of Microviridae in all human feces and human saliva viromes reaching up to 18.78% in the virome Human feces A (Kim et al., 2011; Figure 5A) while they were absent in all peat viromes as well as in most other analyzed viromes. Gokushovirinae were the most abundant subfamily in most peat viromes ranging from 0.11% in sample vBog_Oct11 to 4.19% in sample vBog_Mar12_B (Figure 5B). Gokushovirinae also represented the majority of the Microviridae retrieved in Shimokita and Ogasaware marine sediment viromes (Yoshida et al., 2013) representing 6.00 and 7.47%, respectively, and even reaching 21.66% of the Coral A5 virome (Soffer et al., 2014; Figure 5A).
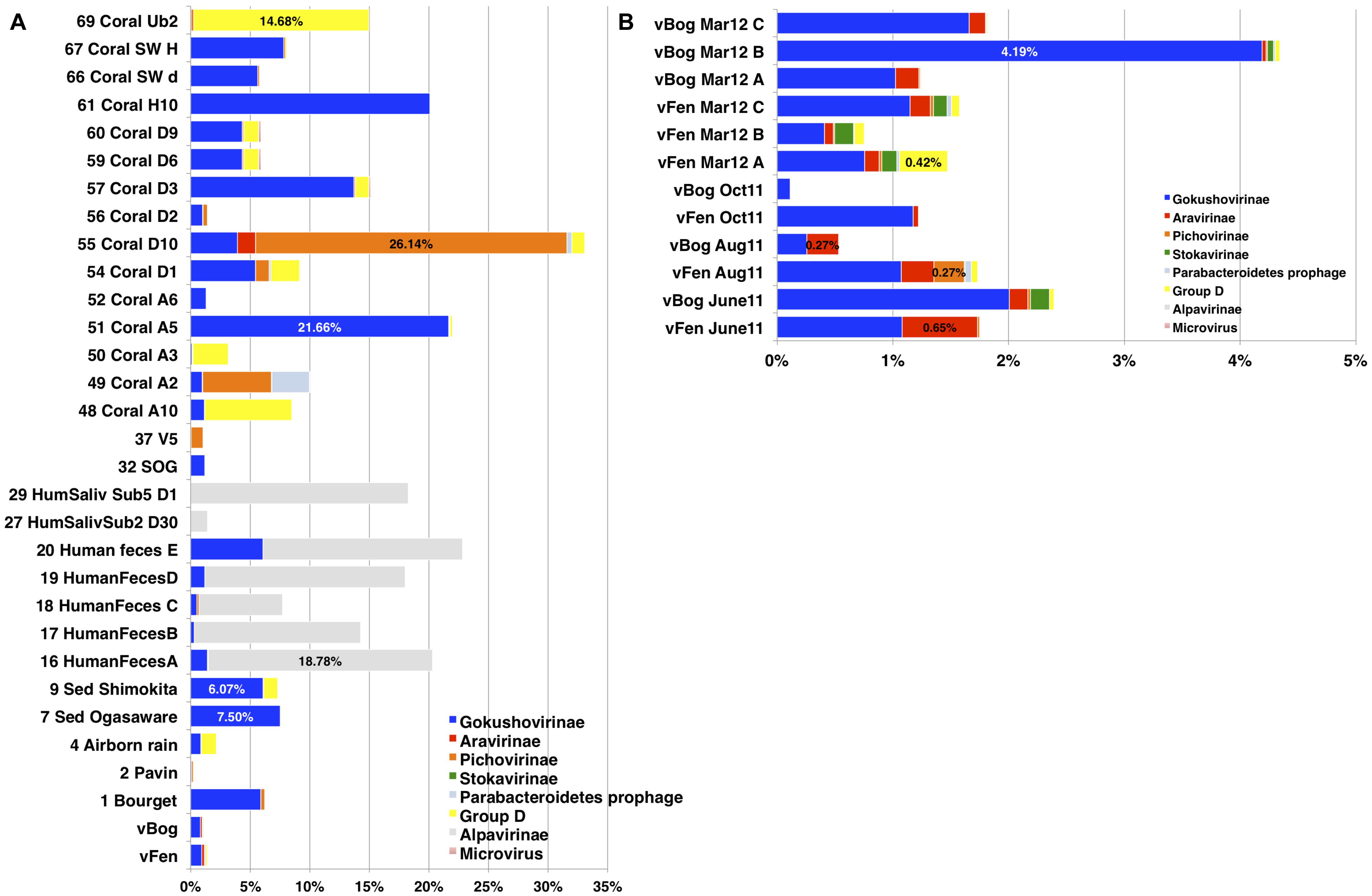
FIGURE 5. Relative proportions of Microviridae subfamilies based on their content of major capsid protein sequences. (A) VP1 match counts in 69 viromes from public databases; (B) in the 12 peat viromes. Matches to major capsid proteins were normalized to the number of total sequences in the viromes. Best matches were determined by BLASTX with an e-value 10-10 cut off.
With major capsid protein matches accounting for 0.27% of the total virome sequences, Aravirinae were the most abundant Microviridae subfamily in vBog_Aug11. They represented the second most abundant subfamily in vFen_June11, vFen_Aug11, vFen_Mar12_C, vBog_Mar12_A, and vBog_Mar12_B. Stokavirinae were the second most abundant in vBog_June11, vFen_Mar12_B, and vBog_Mar12_B. Stokavirinae and Aravirinae were not restricted to peatland. They were present in other viromes but seemed to be much less frequent as only 10 and 270 matches were detected, respectively, from a total of 282,508 major capsid protein matches identified in the 69 non-peat viromes. Matches to Parabacteroidetes prophages were low, ranging from 0 in vBog_June11 to 59 in vFen_Aug11, which corresponds to 0.055% of total viral sequences. Group D was the second most abundant subfamily in the virome vFen_Mars12_A with major capsid protein sequences accounting for 0.42%. As for other viromes, Group D affiliated major capsid protein sequences represented up to 14.68% in the virome Coral Ub2 (Soffer et al., 2014). Pichovirinae were present in all peat viromes with the exception of vBog Oct11 reaching up to 0.27% in vFen_Aug11. In the virome Lake Bourget (Roux et al., 2012b), where Pichovirinae were first detected, they accounted for 0.31%. The highest proportion was found in Coral D10 (Soffer et al., 2014), where the major capsid protein sequences of Pichovirinae accounted for 26.14% (Figure 5A) indicating that the viral community at the time of sampling consisted in the large majority of Pichovirinae.
The relative abundances of Microviridae subfamilies based on the analysis of the available viromes showed considerable variations, even between viral communities sampled from the same environment. For now, it is only possible to identify broad trends in the habitat preferences of Microviridae subfamilies. Further data and the identification of the corresponding hosts should help to determine the ecological and functional significance of the genome variations between Microviridae subfamilies.
Conclusion
While bacteriophages are already considered as important biological actors in seawater, freshwater, and human gut ecosystems, this study sheds light onto the specific diversity of Microviridae in peatland. Namely, we identified two completely new Microviridae subfamilies from Sphagnum-peat habitats that are distinct by phylogenetic analysis and by their genome structure. By combining whole genome phylogeny, major capsid protein sequence phylogeny and genome structure analysis; we gained new detailed insights into the evolution and genomic diversity of Microviridae subfamilies. Notably, we highlighted new hypervariable regions in the major capsid protein sequence as well as the co-existence of different genome organizations. Diversity and relative abundance analysis using the major capsid protein as a marker gene revealed that Gokushovirinae have the largest environmental distribution and contributed the most to the pool of Microviridae-affiliated reads in a majority of viromes. Moreover, these new genomes allowed for the detection of a second group of Microviridae prophages, along with the Alpavirinae (Krupovic and Forterre, 2011), in two genomes of Bacteroidetes species that likely represent a novel Microviridae subfamily. These new Microviridae genomes significantly augment the currently available genome information facilitating subsequent comparative and diversity analyses.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the ‘Environmental and Functional Genomics Platform’ (Biogenouest Genomics) for sequencing and the molecular ecology facility (UMR 6553 ECOBIO) for support. We thank Mart Krupovic for assistance and advice on capsid modeling. We thank Daniel Revillini for manuscript editing. This work was supported by funding from the INSU-CNRS EC2CO program to A-JF. (“VIRTOU”) and grants from the University of Rennes/CNRS and from the “Observatoire des Sciences de l’Univers de Rennes” (OSUR) to AQ.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.00375/abstract
References
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Angly, F. E., Felts, B., Breitbart, M., Salamon, P., Edwards, R. A., Carlson, C., et al. (2006). The marine viromes of four oceanic regions. PLoS Biol. 4:e368. doi: 10.1371/journal.pbio.0040368
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Carver, T., Harris, S. R., Berriman, M., Parkhill, J., and McQuillan, J. A. (2012). Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469. doi: 10.1093/bioinformatics/btr703
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Carver, T. J., Rutherford, K. M., Berriman, M., Rajandream, M. A., Barrell, B. G., and Parkhill, J. (2005). ACT: the Artemis comparison tool. Bioinformatics 21, 3422–3423. doi: 10.1093/bioinformatics/bti553
Colombet, J., Robin, A., Lavie, L., Bettarel, Y., Cauchie, H. M., and Sime-Ngando, T. (2007). Virioplankton “pegylation”: use of PEG (polyethylene glycol) to concentrate and purify viruses in pelagic ecosystems. J. Microbiol. Methods 71, 212–219. doi: 10.1016/j.mimet.2007.08.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Desnues, C., Rodriguez-Brito, B., Rayhawk, S., Kelley, S., Tran, T., Haynes, M., et al. (2008). Biodiversity and biogeography of phages in modern stromatolites and thrombolites. Nature 452, 340–343. doi: 10.1038/nature06735
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Eswar, N., Eramian, D., Webb, B., Shen, M.-Y., and Sali, A. (2008). Protein structure modeling with MODELLER. Methods Mol. Biol. 426, 145–159. doi: 10.1007/978-1-60327-058-8_8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Francez, A.-J., and Vasander, H. (1995). Peat accumulation and peat decomposition after human disturbance in French and finnish mires. Acta Oecol. 16, 599–608.
Fuhrman, J. A., and Schwalbach, M. (2003). Viral influence on aquatic bacterial communities. Biol. Bull. 204, 192–195. doi: 10.2307/1543557
Hopkins, M., Kailasan, S., Cohen, A., Roux, S., Tucker, K. P., Shevenell, A., et al. (2014). Diversity of environmental single-stranded DNA phages revealed by PCR amplification of the partial major capsid protein. ISME J. 8, 2093–2103. doi: 10.1038/ismej.2014.43
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hyatt, D., Chen, G.-L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jobb, G., von Haeseler, A., and Strimmer, K. (2004). TREEFINDER: a powerful graphical analysis environment for molecular phylogenetics. BMC Evol. Biol. 4:18. doi: 10.1186/1471-2148-4-18
Kim, K.-H., and Bae, J.-W. (2011). Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl. Environ. Microbiol. 77, 7663–7668. doi: 10.1128/AEM.00289-11
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, M.-S., Park, E.-J., Roh, S. W., and Bae, J.-W. (2011). Diversity and abundance of single-stranded DNA viruses in human feces. Appl. Environ. Microbiol. 77, 8062–8070. doi: 10.1128/AEM.06331-11
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
King, A., Adams, M. J., Lefkowitz, E. J., and Carstens, E. B. (2012). Virus Taxonomy: Ninth Report of the International Commitee on Taxonomy of Viruses. London: Elsevier/Academic Press.
Krupovic, M., and Forterre, P. (2011). Microviridae goes temperate: microvirus-related proviruses reside in the genomes of Bacteroidetes. PLoS ONE 6:e19893. doi: 10.1371/journal.pone.0019893
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Labonté, J. M., and Suttle, C. A. (2013a). Metagenomic and whole-genome analysis reveals new lineages of gokushoviruses and biogeographic separation in the sea. Front. Microbiol. 4:404. doi: 10.3389/fmicb.2013.00404
Labonté, J. M., and Suttle, C. A. (2013b). Previously unknown and highly divergent ssDNA viruses populate the oceans. ISME J. 7, 2169–2177. doi: 10.1038/ismej.2013.110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
López-Bueno, A., Tamames, J., Velázquez, D., Moya, A., Quesada, A., and Alcamí, A. (2009). High diversity of the viral community from an Antarctic lake. Science 326, 858–861. doi: 10.1126/science.1179287
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar, et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pedulla, M. L., Ford, M. E., Houtz, J. M., Karthikeyan, T., Wadsworth, C., Lewis, J. A., et al. (2003). Origins of highly mosaic mycobacteriophage genomes. Cell 113, 171–182. doi: 10.1016/S0092-8674(03)00233-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Rosario, K., Dayaram, A., Marinov, M., Ware, J., Kraberger, S., Stainton, D., et al. (2012). Diverse circular ssDNA viruses discovered in dragonflies (Odonata: Epiprocta). J. Gen. Virol. 93, 2668–2681. doi: 10.1099/vir.0.045948-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roux, S., Enault, F., Robin, A., Ravet, V., Personnic, S., Theil, S., et al. (2012a). Assessing the diversity and specificity of two freshwater viral communities through metagenomics. PLoS ONE 7:e33641. doi: 10.1371/journal.pone.0033641
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roux, S., Krupovic, M., Poulet, A., Debroas, D., and Enault, F. (2012b). Evolution and diversity of the Microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLoS ONE 7:e40418. doi: 10.1371/journal.pone.0040418
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roy, A., Kucukural, A., and Zhang, Y. (2010). I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738. doi: 10.1038/nprot.2010.5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sakamoto, M., and Benno, Y. (2006). Reclassification of Bacteroides distasonis, Bacteroides goldsteinii and Bacteroides merdae as Parabacteroides distasonis gen. nov., comb. nov., Parabacteroides goldsteinii comb. nov. and Parabacteroides merdae comb. nov. Int. J. Syst. Evol. Microbiol. 56, 1599–1605. doi: 10.1099/ijs.0.64192-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Seshadri, R., Kravitz, S. A., Smarr, L., Gilna, P., and Frazier, M. (2007). CAMERA: a community resource for metagenomics. PLoS Biol. 5:e75. doi: 10.1371/journal.pbio.0050075
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Soffer, N., Brandt, M. E., Corrla, A. M., Smith, T. B., and Thurber, R. V. (2014). Potential role of viruses in white plague coral disease. ISME J. 8, 271–283. doi: 10.1038/ismej.2013.137
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Suttle, C. A. (2007). Marine viruses–major players in the global ecosystem. Nat. Rev. Microbiol. 5, 801–812. doi: 10.1038/nrmicro1750
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tucker, K. P., Parsons, R., Symonds, E. M., and Breitbart, M. (2011). Diversity and distribution of single-stranded DNA phages in the North Atlantic Ocean. ISME J. 5, 822–830. doi: 10.1038/ismej.2010.188
Wiederstein, M., and Sippl, M. J. (2007). ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35, W407–W410. doi: 10.1093/nar/gkm290
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wommack, K. E., and Colwell, R. R. (2000). Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 64, 69–114. doi: 10.1128/MMBR.64.1.69-114.2000
Yoshida, M., Takaki, Y., Eitoku, M., Nunoura, T., and Takai, K. (2013). Metagenomic analysis of viral communities in (hado)pelagic sediments. PLoS ONE 8:e57271. doi: 10.1371/journal.pone.0057271
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zivanovic, Y., Confalonieri, F., Ponchon, L., Lurz, R., Chami, M., Flayhan, A., et al. (2014). Insights into bacteriophage T5 structure from analysis of its morphogenesis genes and protein components. J. Virol. 88, 1162–1174. doi: 10.1128/JVI.02262-13
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: virus ecology, viral metagenomics, virus diversity, ssDNA phage, Microviridae, Gokushovirinae, Aravirinae, Stokavirinae
Citation: Quaiser A, Dufresne A, Ballaud F, Roux S, Zivanovic Y, Colombet J, Sime-Ngando T and Francez A-J (2015) Diversity and comparative genomics of Microviridae in Sphagnum-dominated peatlands. Front. Microbiol. 6:375. doi: 10.3389/fmicb.2015.00375
Received: 09 January 2015; Paper pending published: 25 March 2015;
Accepted: 12 April 2015; Published online: 28 April 2015
Edited by:
Frank T. Robb, University of Maryland, USAReviewed by:
Awdhesh Kalia, University of Texas MD Anderson Cancer Center, USASvetlana N. Dedysh, University of Texas MD Anderson Cancer Center, USA
Copyright © 2015 Quaiser, Dufresne, Ballaud, Roux, Zivanovic, Colombet, Sime-Ngando and Francez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Achim Quaiser, UMR CNRS 6553 – ECOBIO, Université de Rennes 1, Bâtiment 14A, Campus de Beaulieu, 35042 Rennes Cedex, France,YWNoaW0ucXVhaXNlckB1bml2LXJlbm5lczEuZnI=