 Günther Schönrich
Günther Schönrich- Institute of Medical Virology, Helmut-Ruska-Haus, Charité-Universitätsmedizin Berlin, Berlin, Germany
Varicella zoster virus (VZV), a human alphaherpesvirus, causes varicella and subsequently establishes latency within sensory nerve ganglia. Later in life VZV can reactivate to cause herpes zoster. A reduced frequency of VZV-specific T cells is strongly associated with herpes zoster illustrating that these immune cells are central to control latency. Dendritic cells (DCs) are required for the generation of VZV-specific T cells. However, DCs can also be infected in vitro and in vivo allowing VZV to evade the antiviral immune response. Thus, DCs represent the immune systems’ Achilles heel. Uniquely among the human herpesviruses, VZV infects both DCs and T cells, and exploits both as Trojan horses. During primary infection VZV-infected DCs traffic to the draining lymph nodes and tonsils, where the virus is transferred to T cells. VZV-infected T cells subsequently spread infection throughout the body to give the typical varicella skin rash. The delicate interplay between VZV and DCs and its consequences for viral immune evasion and viral dissemination will be discussed in this article.
Introduction
In 1943 Helmut Ruska discovered that the virus particles isolated from varicella lesions and those isolated from zoster lesions had identical morphology (Ruska, 1943). The virus was subsequently named varicella zoster virus (VZV). Together with herpes simplex virus (HSV) type 1 and 2 it represents the human alphaherpesviruses (Arvin and Gilden, 2013). After transmission VZV replicates in epithelial cells of the upper respiratory tract. Subsequently, cell-associated viremia is observed which is followed by the appearance of skin infection, giving the typical varicella rash (commonly known as chickenpox) after an incubation period of 10–21 days.
After primary infection has been resolved VZV establishes latency in the dorsal ganglia of nerve cells that innervate the affected skin. It can reactivate, mostly in immunosuppressed individuals and in elderly adults, resulting in herpes zoster (commonly known as shingles) after anterograde transport to the corresponding dermatome. Skin blisters that occur during varicella and zoster harbor high numbers of infectious particles. The main route of transmission is through cell free virus particles that are carried by aerosols of infectious respiratory tract secretions or vesicular fluid. Pathological consequences of VZV include congenital varicella syndrome (Sauerbrei and Wutzler, 2000) and postherpetic neuralgia (PHN; Gnann and Whitley, 2002).
More than 30 years ago a live attenuated vaccine (vOka) was developed from the Oka parental strain (pOka; Takahashi et al., 1974). Intriguingly, vOka is a mixture of pOka genomes with distinct patterns of mutations (Gomi et al., 2002). In 1995 vOka was introduced in the United States and is now licensed for routine use in many countries worldwide. This resulted in significant reduction of the VZV-associated disease burden. Although complications are rare in childhood infection, adult infection, common in tropical countries, induces a more severe clinical course (Lee, 1998). The incidence and severity of herpes zoster increases with age. It was first postulated in 1965 that this is due to a decline in VZV-specific immunity (Hope-Simpson, 1965). It is now accepted that boosting VZV-reactive T cells in older adults using vOka is effective in preventing herpes zoster (Oxman et al., 2005; Gagliardi et al., 2012). Dendritic cells (DCs) are professional antigen-presenting cells that are key to a successful antiviral immune response. Upon sensing a pathogen DCs not only mount an innate response but also specifically instruct the adaptive immune system. This results in the activation of effector mechanisms that are tailor-made for the infecting pathogen (Medzhitov, 2007; Volz et al., 2012; Maisonneuve et al., 2014). Thus, DCs bridge the innate and adaptive arms of the immune system and orchestrate the different immune cells. In this review, we will discuss how VZV exploits DCs for its own purposes.
Tropism of VZV for DC Subsets
In the absence of inflammation (steady state) two main groups of DCs are observed in the skin and mucosa of the respiratory tract: conventional DCs (cDCs) and Langerhans cells (LCs; Pulendran, 2015). They migrate continuously via the lymphatics to draining lymph nodes. This migration is considerably increased during inflammation. cDCs subsets are derived from committed progenitor DCs in the bone marrow that traffic through the blood to peripheral tissue where they undergo a final differentiation step. In contrast, LCs are maintained independently of circulating progenitor DCs originating in the bone marrow. Migratory DCs form an intricate network that functions as a sentinel system in the peripheral tissue. Upon encounter with pathogens such as VZV they take up antigen and migrate through the lymphatics to the draining lymph nodes. They can localize in the T cell zone to present pathogen-derived antigen and activate specific T lymphocytes. Plasmacytoid DCs (pDCs), which produce large amounts of type I interferon (IFN) in response to viruses, fully differentiate in the bone marrow before they populate the lymphoid organs and become resident DCs.
Under inflammatory conditions during infection human monocytes can also develop in vivo into so-called inflammatory DCs (mo-DCs; Segura et al., 2013). Equivalent DCs can be generated in vitro by culturing CD14+ human monocytes in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin (IL)-4 and subsequently exposing them to maturation stimuli such as CD40L or tumor necrosis factor (TNF)-α (Sallusto and Lanzavecchia, 1994). These mo-DCs represent the most frequently used in vitro DC model and are permissive to VZV (Abendroth et al., 2001b; Morrow et al., 2003; Hu and Cohen, 2005; Gutzeit et al., 2010). VZV-infected immature mo-DCs do not undergo DNA fragmentation (Abendroth et al., 2001b) but can acquire other characteristics of cell death such as impaired membrane integrity and phosphatidylserine exposure (Gutzeit et al., 2010). The open reading frame (ORF) 47 encodes a serine/threonine protein kinase and promotes VZV replication in immature mo-DCs thereby enhancing spread of VZV to other cells (Hu and Cohen, 2005).
In the skin VZV affects both the dermis, where migratory cDCs reside, and the epidermis, where LCs are located (Nikkels et al., 1993). Both LCs and dermal cDCs isolated ex vivo are susceptible to VZV infection (Gutzeit et al., 2010). In VZV-infected human skin lesions the frequency and distribution of DC subsets changes most likely due to immigration and emigration similar to HSV-infected mouse skin (Eidsmo et al., 2009). The frequency of LCs is drastically reduced implying activation and migration of LCs to draining lymph nodes (Gutzeit et al., 2010; Huch et al., 2010). In contrast, an influx of other DCs such as inflammatory mo-DCs (Gutzeit et al., 2010) and pDCs (Gerlini et al., 2006; Huch et al., 2010) is observed. Skin biopsies from varicella lesions show occasional cDCs, pDCs, and LCs that express VZV proteins suggestive of VZV infection. Most likely, LC egress is induced by cytokines released during VZV infection. In accordance, inflammatory cytokines such as TNF-α or IL-1β are known to trigger LC migration (Cumberbatch et al., 1997). It is possible that migratory LCs represent the main route for transport of virus and viral antigen to the lymph nodes.
Role of DCs in VZV Dissemination
Varicella zoster virus-infected patients are extremely contagious. Primary VZV infection is by droplets of infectious saliva or respiratory tract secretions (Suzuki et al., 2004). Intriguingly, intraepithelial DCs in the lung extend cellular processes into the air lumen allowing continuous surveillance of the luminal surface of the alveoli and airways (Jahnsen et al., 2006) similar to gut DCs (Rescigno et al., 2001). Accordingly, these DCs are probably amongst the first cells to interact with VZV. The DC tropism and resultant migration permits VZV to shuttle to the tonsils and the regional lymph nodes. There, VZV-infected DCs establish intimate contact and infect T lymphocytes, as has been demonstrated in vitro (Abendroth et al., 2001b).
Confirming a role for T cells in shuttling virus to the skin VZV lesions appeared in human skin grafts of severe combined immunodeficiency mice (SCID) after injection of VZV-infected human T cells (Ku et al., 2004). Further evidence for the concept of a DC/T cells-axis during VZV dissemination comes from experiments with the closely related simian varicella virus (SVV; Messaoudi et al., 2009). Infected DC-like cells were detected in the lung of African green monkeys after intratracheal infection with recombinant enhanced green fluorescent protein (EGFP)-expressing SVV and transport the virus to the draining lymph nodes where it infects memory T cells (Ouwendijk et al., 2013). Other immune cells in the blood including DCs were also infected with SSV and may contribute to the hematogenous transfer of virus to the skin. The possible DC/T cells-axis during VZV dissemination is illustrated in Figure 1.
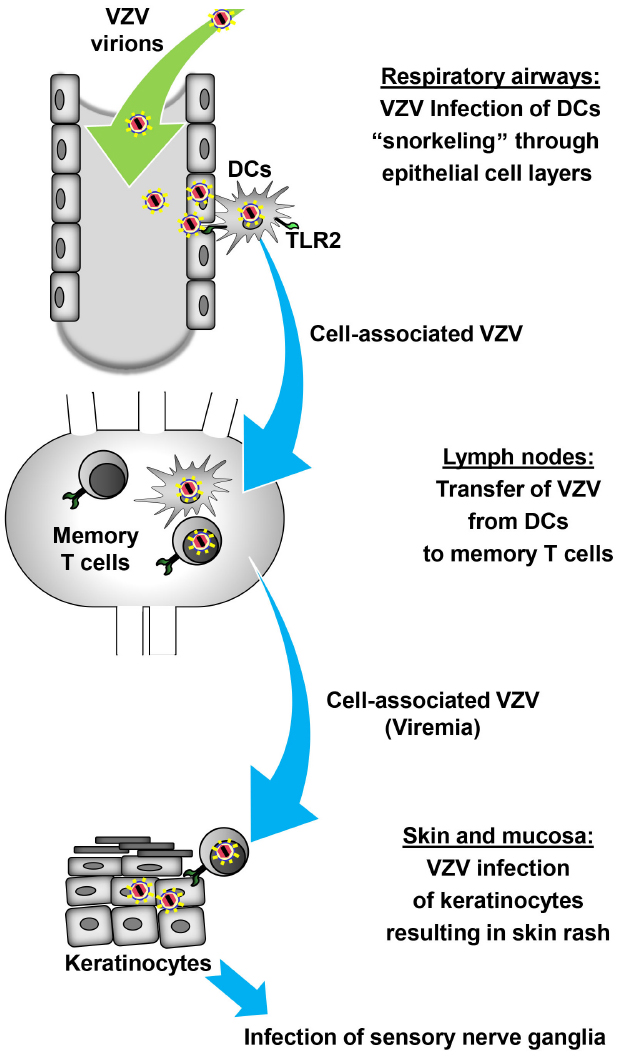
Figure 1. Model of DC-assisted viral dissemination during primary VZV infection. After inhalation of VZV-containing aerosols intraepithelial DCs that extend projections into the lumen of the airways, analogous to “snorkeling,” pick up VZV directly or from infected epithelial cells. Upon recognition of VZV through PRRs such as TLR2 intraepithelial DCs and possibly other cells infected with VZV start to migrate to the lung-draining lymph nodes in the first phase of cell-associated viral dissemination. At this site, DCs transfer VZV to memory T cells which subsequently carry VZV in the second phase of cell-associated viral dissemination to the skin. VZV-infected memory T cells infect keratinocytes either directly or indirectly through infection of other skin-resident cells which subsequently transfer the virus to keratinocytes. Finally, VZV replicating in the skin causes the typical rash followed by latent infection of sensory neurons.
There are still open questions concerning this concept. Firstly, it has not been tested whether VZV-infected DCs still respond to chemotactic signals and are able to migrate in vivo, although the absence of LC in VZV skin lesions and the ability to infect primary LCs ex vivo strongly suggest this (Gutzeit et al., 2010; Huch et al., 2010). Secondly, virus cell-to-cell transmission is more than the sum of virus release and entry as these processes have to be coordinated and channeled to the site of cell-to-cell contact (Mothes et al., 2010). Thus far, the molecular details of how DCs transfer VZV to T cells have not been defined. It is possible but not proven that this process is similar to the DC-to-T cell trans infection observed during HIV pathogenesis (Rinaldo, 2013). This trans infection requires an intimate DC/T cell contact through membrane molecules and the formation of a virological synapse across which the virions are transferred. In addition, it is unclear which DC subset(s), epidermal and/or dermal DCs, transfers VZV to T cells in vivo. Finally, it has not been investigated whether mature DCs are more effective in transferring VZV to T cells as it is the case with HIV (Izquierdo-Useros et al., 2007).
VZV-associated Innate Signaling
Although DC and T cells can serve as Trojan horses, they also represent an enormous risk to the virus: DCs efficiently initiate the antiviral defense. Innate immune responses are triggered by virus-associated components through pattern recognition receptors (PRRs) such as toll-like receptors (TLRs; Vandevenne et al., 2010). Intact VZV virions activate innate signaling in monocytes and macrophages via TLR2 located on the cell membrane (Wang et al., 2005). However, the corresponding viral ligand has not yet been defined. Possibly, VZV-encoded glycoproteins in the viral envelope are involved similar to HSV (Leoni et al., 2012). A recent publication has shown that the VZV-encoded deoxyuridine triphosphate nucleotidohydrolase (dUTPase), a non-structural viral protein, induces TLR2-dependent secretion of inflammatory cytokines in DCs generated from CD34+ human stem cells (Ariza et al., 2014). Similar to other herpesviruses, the VZV-encoded dUTPase may be released by infected cells after induction of apoptosis or pyroptosis before it binds to TLR2 on the cell membrane. Alternatively, VZV-encoded dUTPase could be released through exosomes as demonstrated for Epstein-Barr virus, a gammaherpesvirus (Ariza et al., 2013).
Herpesviruses have developed diverse and numerous strategies to interfere with innate immune responses (Vandevenne et al., 2010). VZV prevents translocation of NF-κB subunits into the nucleus of mo-DCs (Sloan et al., 2012). This inhibition requires the E3 ubiquitin ligase domain of VZV ORF61 and is downstream of triggering receptors such as TLR3, TLR8, and TLR9. It is possible that further VZV proteins target NF-κB signaling. Moreover, it has been demonstrated that VZV ORF61 down-modulates the interferon regulatory factor (IRF) 3-mediated IFN-β pathway (Zhu et al., 2011). In accordance, infection of rhesus macaques with recombinant SVV lacking ORF61 resulted in an increased frequency of pDCs and increased expression of IFN-β as well as IL-6, which is dependent on NF-κB activation (Meyer et al., 2013). SVV ORF61 encodes a 54.1-kDa polypeptide that is homologous to VZV ORF61 and to other alpha herpesvirus proteins that are encoded in similar regions of the viral genome such as HSV-encoded infected cell protein 0 (ICP0). HSV ICP0 also inhibits the induction of IFN-stimulated genes with the help of its E3 ubiquitin ligase domain (Eidson et al., 2002; Lin et al., 2004). Moreover, several other VZV proteins interfere with type I IFN-mediated responses by targeting IRF3 activation including ORF62 (Sen et al., 2010) and ORF47 (Vandevenne et al., 2011). These viral proteins are most likely also functional in VZV-infected pDCs as they no longer produce type I IFN (Huch et al., 2010). In VZV skin accumulations of pDCs were observed (Gerlini et al., 2006; Huch et al., 2010). Similarly, after intrabronchial infection of rhesus macaques with SVV increased frequencies of pDCs are found that correlate with enhanced type I IFN production and a rapid decrease in viral load (Haberthur et al., 2014). These results implicate that pDCs are recruited to the site of VZV replication to support the antiviral host defense, however type I IFN release is inhibited in VZV-infected pDCs by not yet defined mechanisms. Upon stimulation pDCs are also able to present viral antigen and prime virus-specific T cells (Colonna et al., 2004). It is likely but not yet proven that VZV also interferes with T cell stimulation by pDCs.
Modulation of Adaptive Immune Responses by VZV
T cells not only transport VZV to the skin but also play a pivotal role in controlling VZV during primary infection (Malavige et al., 2008). Decreased VZV specific immunity in elder individuals is associated with increased risk for herpes zoster. This strongly suggests that T cells also control VZV reactivation (Hope-Simpson, 1965; Burke et al., 1982; Levin et al., 2003). At the time being it is unclear whether and how DCs contribute to control of latent VZV by memory T cells.
Varicella zoster virus interferes with T cell responses similar to most other herpesviruses (Smith and Khanna, 2013). For this purpose it targets DCs, which promote Th1-like adaptive immune responses by providing three major signals to T cells (Figure 2A). Signal 1 is delivered by antigen presentation through major histocompatibility complex (MHC) molecules whereas signal 2, also called costimulatory signal, is mediated by CD40, CD80, and CD86. Proinflammatory cytokines released by DCs, mainly IL-12 and type I IFN, represent a third signal that drives Th1-like adaptive immune responses. VZV clearly weakens signal 1–3 by targeting DCs (Figure 2B). After infection of mature mo-DCs VZV impairs signal 1 by downregulation of MHC class I (Abendroth et al., 2001a) as well as signal 2 by reducing surface density of CD80 and CD86 (Morrow et al., 2003). As a consequence of these phenotypic changes VZV-infected mo-DCs have a reduced capacity to stimulate allogeneic T lymphocytes. On the molecular level it has been found that VZV ORF66, a protein kinase, retains mature MCH class I complexes in the cis/medial Golgi (Abendroth et al., 2001a; Eisfeld et al., 2007). By which mechanism(s) VZV downregulates costimulatory molecules on mature mo-DCs is unknown. In addition, VZV-infected DCs also express diminished levels of CD83, an important maker of mature DCs, similar to mature DCs infected with HSV, or human cytomegalovirus (Kruse et al., 2000; Morrow et al., 2003; Senechal et al., 2004).
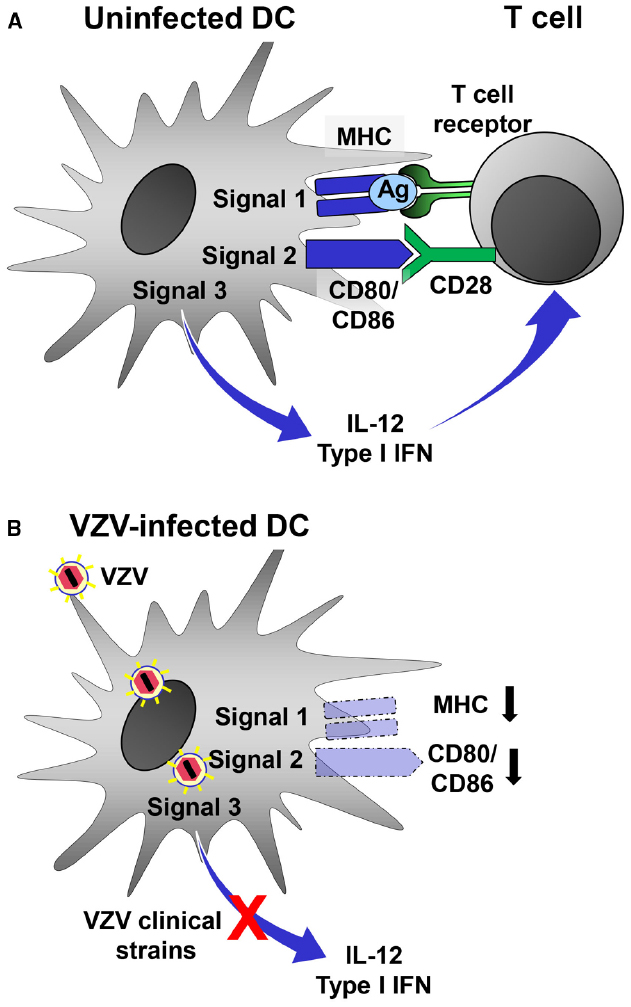
Figure 2. Evasion of DC function by VZV. (A) Stimulated uninfected DCs can induce efficient antiviral Th1-like adaptive immune responses by providing three signals to antiviral T cells. Virus-derived antigen (Ag) is presented by MHC molecules (signal 1) and recognized by the T cell-receptor (TCR) on T cells. Costimulatory molecules such as CD80 and CD86 interact with CD28 expressed by T cells (signal 2). DCs integrate stimuli received through a combination of different PRRs and release type I IFN and IL-12. (B) VZV-infected DCs are no longer able to deliver signal 1–3 to antiviral T cells. MHC as well as costimulatory molecules are downregulated. Moreover, VZV clinical strains but not the vaccine strain interfere with PRR-induced innate signaling resulting in the inability to release type I IFN and IL-12. Together, these functional changes prevent VZV-infected DCs to mount an effective antiviral Th1-like adaptive immune response.
Central to viral immune evasion is the third signal which can be blocked only by VZV clinical strains but not by vOka, the VZV vaccine strain (Gutzeit et al., 2010). Efficient IL-12 secretion and Th1-polarization by DCs requires priming of DCs by synergistic signaling through multiple PRRs (Schulz et al., 2000; Napolitani et al., 2005; Roses et al., 2008) and is facilitated by innate T cells (Leslie et al., 2002; Fujii et al., 2007). In fact, TLRs control key functions of DCs and mediate signals that play a pivotal role in initiation of adaptive immune responses (Iwasaki and Medzhitov, 2004). In accordance, the VZV-associated block of IL-12 secretion was due to impaired signaling downstream of TLR2 (Gutzeit et al., 2010). TLR2 is triggered by VZV, most likely by a viral glycoprotein within the viral envelope and dUTPase released from VZV-infected cells (Wang et al., 2005; Ariza et al., 2013). Peripheral blood mononuclear cells from humans vaccinated with vOka produce high amounts of IL-12 after restimulation in vitro (Jenkins et al., 1998). This is in line with the notion that vOka instructs DCs to promote an efficient Th1-type of adaptive immune response. This instructive capability makes vOka a prime candidate vector for the development of novel vaccines. Its genome is large enough to accommodate even large DNA inserts encoding antigenic structures from other viral pathogens using bacterial artificial chromosome technology (Somboonthum et al., 2007; Matsuura et al., 2013).
Concluding Remarks
In this article we have discussed how VZV, one of the most successful human pathogens, exploits DCs to disseminate in the human organism, evade the antiviral immune responses and cause disease. In contrast, vOka, the only live attenuated vaccine available against human herpesviruses, does not cause significant disease. Although both wild type VZV and vOka target DCs only DCs infected with vOka can be instructed to become a Th1-promoting antigen-presenting cell. It will be important in the future to precisely define on the molecular level how DCs infected with wild type VZV differ from those infected with vOka. Moreover, it is necessary to pinpoint the viral proteins responsible. A better understanding of these differences could allow to design desperately needed vaccines against other human herpesviruses.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Deutsche Forschungsgemeinschaft (GraKo 1121). We thank DH Krüger for his kind support.
References
Abendroth, A., Lin, I., Slobedman, B., Ploegh, H., and Arvin, A. M. (2001a). Varicella-zoster virus retains major histocompatibility complex class I proteins in the Golgi compartment of infected cells. J. Virol. 75, 4878–4888. doi: 10.1128/JVI.75.10.4878-4888.2001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Abendroth, A., Morrow, G., Cunningham, A. L., and Slobedman, B. (2001b). Varicella-zoster virus infection of human dendritic cells and transmission to T cells: implications for virus dissemination in the host. J. Virol. 75, 6183–6192. doi: 10.1128/JVI.75.13.6183-6192.2001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ariza, M. E., Glaser, R., and Williams, M. V. (2014). Human herpesviruses-encoded dUTPases: a family of proteins that modulate dendritic cell function and innate immunity. Front. Microbiol. 5:504. doi: 10.3389/fmicb.2014.00504
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ariza, M. E., Rivailler, P., Glaser, R., Chen, M., and Williams, M. V. (2013). Epstein-Barr virus encoded dUTPase containing exosomes modulate innate and adaptive immune responses in human dendritic cells and peripheral blood mononuclear cells. PLoS ONE 8:e69827. doi: 10.1371/journal.pone.0069827
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Arvin, A. M., and Gilden, D. (2013). “Varicella-zoster virus,” in Fields’ Virology, eds D. Knipe and P. Howley (Philadelphia, PA: Lippincott Williams & Wilkins), 2015–2057.
Burke, B. L., Steele, R. W., Beard, O. W., Wood, J. S., Cain, T. D., and Marmer, D. J. (1982). Immune responses to varicella-zoster in the aged. Arch. Intern. Med. 142, 291–293. doi: 10.1001/archinte.1982.00340150091017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Colonna, M., Trinchieri, G., and Liu, Y. J. (2004). Plasmacytoid dendritic cells in immunity. Nat. Immunol. 5, 1219–1226. doi: 10.1038/ni1141
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cumberbatch, M., Dearman, R. J., and Kimber, I. (1997). Langerhans cells require signals from both tumour necrosis factor-alpha and interleukin-1 beta for migration. Immunology 92, 388–395. doi: 10.1046/j.1365-2567.1997.00360.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Eidsmo, L., Allan, R., Caminschi, I., van Rooijen, N., Heath, W. R., and Carbone, F. R. (2009). Differential migration of epidermal and dermal dendritic cells during skin infection. J. Biol. Chem. 182, 3165–3172. doi: 10.4049/jimmunol.0802950
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Eidson, K. M., Hobbs, W. E., Manning, B. J., Carlson, P., and DeLuca, N. A. (2002). Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral infection. J. Virol. 76, 2180–2191. doi: 10.1128/jvi.76.5.2180-2191.2002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Eisfeld, A. J., Yee, M. B., Erazo, A., Abendroth, A., and Kinchington, P. R. (2007). Downregulation of class I major histocompatibility complex surface expression by varicella-zoster virus involves open reading frame 66 protein kinase-dependent and -independent mechanisms. J. Virol. 81, 9034–9049. doi: 10.1128/JVI.00711-07
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fujii, S., Shimizu, K., Hemmi, H., and Steinman, R. M. (2007). Innate Valpha14+ natural killer T cells mature dendritic cells, leading to strong adaptive immunity. Immunol. Rev. 220, 183–198. doi: 10.1111/j.1600-065X.2007.00561.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gagliardi, A. M., Gomes Silva, B. N., Torloni, M. R., and Soares, B. G. (2012). Vaccines for preventing herpes zoster in older adults. Cochrane. Database. Syst. Rev. 10, CD008858. doi: 10.1002/14651858.CD008858.pub2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gerlini, G., Mariotti, G., Bianchi, B., and Pimpinelli, N. (2006). Massive recruitment of type I interferon producing plasmacytoid dendritic cells in varicella skin lesions. J. Invest. Dermatol. 126, 507–509. doi: 10.1038/sj.jid.5700052
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gnann, J. W. Jr., and Whitley, R. J. (2002). Clinical practice. Herpes zoster. N. Engl. J. Med. 347, 340–346. doi: 10.1056/NEJMcp013211
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gomi, Y., Sunamachi, H., Mori, Y., Nagaike, K., Takahashi, M., and Yamanishi, K. (2002). Comparison of the complete DNA sequences of the Oka varicella vaccine and its parental virus. J. Virol. 76, 11447–11459. doi: 10.1128/JVI.76.22.11447-11459.2002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gutzeit, C., Raftery, M. J., Peiser, M., Tischer, K. B., Ulrich, M., Eberhardt, M., et al. (2010). Identification of an important immunological difference between virulent varicella-zoster virus and its avirulent vaccine: viral disruption of dendritic cell instruction. J. Biol. Chem. 185, 488–497. doi: 10.4049/jimmunol.0902817
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Haberthur, K., Meyer, C., Arnold, N., Engelmann, F., Jeske, D. R., and Messaoudi, I. (2014). Intrabronchial infection of rhesus macaques with simian varicella virus results in a robust immune response in the lungs. J. Virol. 88, 12777–12792. doi: 10.1128/JVI.01814-14
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hope-Simpson, R. E. (1965). The nature of herpes zoster: a long-term study and a new hypothesis. Proc. R. Soc. Med. 58, 9–20.
Hu, H., and Cohen, J. I. (2005). Varicella-zoster virus open reading frame 47 (ORF47) protein is critical for virus replication in dendritic cells and for spread to other cells. Virology 337, 304–311. doi: 10.1016/j.virol.2005.04.024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huch, J. H., Cunningham, A. L., Arvin, A. M., Nasr, N., Santegoets, S. J., Slobedman, E., et al. (2010). Impact of varicella-zoster virus on dendritic cell subsets in human skin during natural infection. J. Virol. 84, 4060–4072. doi: 10.1128/JVI.01450-09
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Iwasaki, A., and Medzhitov, R. (2004). Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5, 987–995. doi: 10.1038/ni1112
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Izquierdo-Useros, N., Blanco, J., Erkizia, I., Fernandez-Figueras, M. T., Borras, F. E., Naranjo-Gomez, M., et al. (2007). Maturation of blood-derived dendritic cells enhances human immunodeficiency virus type 1 capture and transmission. J. Virol. 81, 7559–7570. doi: 10.1128/JVI.02572-06
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jahnsen, F. L., Strickland, D. H., Thomas, J. A., Tobagus, I. T., Napoli, S., Zosky, G. R., et al. (2006). Accelerated antigen sampling and transport by airway mucosal dendritic cells following inhalation of a bacterial stimulus. J. Biol. Chem. 177, 5861–5867. doi: 10.4049/jimmunol.177.9.5861
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jenkins, D. E., Redman, R. L., Lam, E. M., Liu, C., Lin, I., and Arvin, A. M. (1998). Interleukin (IL)-10, IL-12, and interferon-gamma production in primary and memory immune responses to varicella-zoster virus. J. Infect. Dis. 178, 940–948. doi: 10.1086/515702
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kruse, M., Rosorius, O., Kratzer, F., Stelz, G., Kuhnt, C., Schuler, G., et al. (2000). Mature dendritic cells infected with herpes simplex virus type 1 exhibit inhibited T-cell stimulatory capacity. J. Virol. 74, 7127–7136. doi: 10.1128/JVI.74.15.7127-7136.2000
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ku, C. C., Zerboni, L., Ito, H., Graham, B. S., Wallace, M., and Arvin, A. M. (2004). Varicella-zoster virus transfer to skin by T Cells and modulation of viral replication by epidermal cell interferon-alpha. J. Exp. Med. 200, 917–925. doi: 10.1084/jem.20040634
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lee, B. W. (1998). Review of varicella zoster seroepidemiology in India and Southeast Asia. Trop. Med. Int. Health 3, 886–890. doi: 10.1046/j.1365-3156.1998.00316.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Leoni, V., Gianni, T., Salvioli, S., and Campadelli-Fiume, G. (2012). Herpes simplex virus glycoproteins gH/gL and gB bind Toll-like receptor 2, and soluble gH/gL is sufficient to activate NF-kappaB. J. Virol. 86, 6555–6562. doi: 10.1128/JVI.00295-12
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Leslie, D. S., Vincent, M. S., Spada, F. M., Das, H., Sugita, M., Morita, C. T., et al. (2002). CD1-mediated gamma/delta T cell maturation of dendritic cells. J. Exp. Med. 196, 1575–1584. doi: 10.1084/jem.20021515
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Levin, M. J., Smith, J. G., Kaufhold, R. M., Barber, D., Hayward, A. R., Chan, C. Y., et al. (2003). Decline in varicella-zoster virus (VZV)-specific cell-mediated immunity with increasing age and boosting with a high-dose VZV vaccine. J. Infect. Dis. 188, 1336–1344. doi: 10.1086/379048
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lin, R., Noyce, R. S., Collins, S. E., Everett, R. D., and Mossman, K. L. (2004). The herpes simplex virus ICP0 RING finger domain inhibits. J. Virol. 78, 1675–1684. doi: 10.1128/JVI.78.4.1675-1684.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maisonneuve, C., Bertholet, S., Philpott, D. J., and De Gregorio, E. (2014). Unleashing the potential of NOD- and Toll-like agonists as vaccine adjuvants. Proc. Natl. Acad. Sci. U.S.A. 111, 12294–12299. doi: 10.1073/pnas.1400478111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Malavige, G. N., Jones, L., Kamaladasa, S. D., Wijewickrama, A., Seneviratne, S. L., Black, A. P., et al. (2008). Viral load, clinical disease severity and cellular immune responses in primary varicella zoster virus infection in Sri Lanka. PLoS ONE 3:e3789. doi: 10.1371/journal.pone.0003789
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Matsuura, M., Somboonthum, P., Murakami, K., Ota, M., Shoji, M., Kawabata, K., et al. (2013). Novel polyvalent live vaccine against varicella-zoster and mumps virus infections. Microbiol. Immunol. 57, 704–714. doi: 10.1111/1348-0421.12087
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Medzhitov, R. (2007). Recognition of microorganisms and activation of the immune response. Nature 449, 819–826. doi: 10.1038/nature06246
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Messaoudi, I., Barron, A., Wellish, M., Engelmann, F., Legasse, A., Planer, S., et al. (2009). Simian varicella virus infection of rhesus macaques recapitulates essential features of varicella zoster virus infection in humans. PLoS Pathog. 5:e1000657. doi: 10.1371/journal.ppat.1000657
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meyer, C., Kerns, A., Haberthur, K., Dewane, J., Walker, J., Gray, W., et al. (2013). Attenuation of the adaptive immune response in rhesus macaques infected with simian varicella virus lacking open reading frame 61. J. Virol. 87, 2151–2163. doi: 10.1128/JVI.02369-12
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Morrow, G., Slobedman, B., Cunningham, A. L., and Abendroth, A. (2003). Varicella-zoster virus productively infects mature dendritic cells and alters their immune function. J. Virol. 77, 4950–4959. doi: 10.1128/JVI.77.8.4950-4959.2003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mothes, W., Sherer, N. M., Jin, J., and Zhong, P. (2010). Virus cell-to-cell transmission. J. Virol. 84, 8360–8368. doi: 10.1128/JVI.00443-10
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Napolitani, G., Rinaldi, A., Bertoni, F., Sallusto, F., and Lanzavecchia, A. (2005). Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 6, 769–776. doi: 10.1038/ni1223
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nikkels, A. F., Debrus, S., Sadzot-Delvaux, C., Piette, J., Delvenne, P., Rentier, B., et al. (1993). Comparative immunohistochemical study of herpes simplex and varicella-zoster infections. Virchows Arch. A Pathol. Anat. Histopathol. 422, 121–126. doi: 10.1007/BF01607163
Ouwendijk, W. J., Mahalingam, R., de Swart, R. L., Haagmans, B. L., van Amerongen, G., Getu, S., et al. (2013). T-Cell tropism of simian varicella virus during primary infection. PLoS Pathog. 9:e1003368. doi: 10.1371/journal.ppat.1003368
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Oxman, M. N., Levin, M. J., Johnson, G. R., Schmader, K. E., Straus, S. E., Gelb, L. D., et al. (2005). A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N. Engl. J. Med. 352, 2271–2284. doi: 10.1056/NEJMoa051016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pulendran, B. (2015). The varieties of immunological experience: of pathogens, stress, and dendritic cells. Annu. Rev. Immunol. 33, 563–606. doi: 10.1146/annurev-immunol-020711-075049
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rescigno, M., Urbano, M., Valzasina, B., Francolini, M., Rotta, G., Bonasio, R., et al. (2001). Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2, 361–367. doi: 10.1038/86373
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rinaldo, C. R. (2013). HIV-1 trans infection of CD4+ T cells by professional antigen presenting cells. Scientifica 2013, 30. doi: 10.1155/2013/164203
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roses, R. E., Xu, S., Xu, M., Koldovsky, U., Koski, G., and Czerniecki, B. J. (2008). Differential production of IL-23 and IL-12 by myeloid-derived dendritic cells in response to TLR agonists. J. Biol. Chem. 181, 5120–5127. doi: 10.4049/jimmunol.181.7.5120
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ruska, H. (1943). Über das virus der varizellen und des zoster. Klin. Wochenschr. 22, 703–704. doi: 10.1007/BF01768631
Sallusto, F., and Lanzavecchia, A. (1994). Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 179, 1109–1118. doi: 10.1084/jem.179.4.1109
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sauerbrei, A., and Wutzler, P. (2000). The congenital varicella syndrome. J. Perinatol. 20, 548–554. doi: 10.1038/sj.jp.7200457
Schulz, O., Edwards, A. D., Schito, M., Aliberti, J., Manickasingham, S., Sher, A., et al. (2000). CD40 triggering of heterodimeric IL-12 p70 production by dendritic cells in vivo requires a microbial priming signal. Immunity 13, 453–462. doi: 10.1016/S1074-7613(00)00045-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Segura, E., Touzot, M., Bohineust, A., Cappuccio, A., Chiocchia, G., Hosmalin, A., et al. (2013). Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity 38, 336–348. doi: 10.1016/j.immuni.2012.10.018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sen, N., Sommer, M., Che, X., White, K., Ruyechan, W. T., and Arvin, A. M. (2010). Varicella-zoster virus immediate-early protein 62 blocks interferon regulatory factor 3 (IRF3) phosphorylation at key serine residues: a novel mechanism of IRF3 inhibition among herpesviruses. J. Virol. 84, 9240–9253. doi: 10.1128/JVI.01147-10
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Senechal, B., Boruchov, A. M., Reagan, J. L., Hart, D. N., and Young, J. W. (2004). Infection of mature monocyte-derived dendritic cells with human cytomegalovirus inhibits stimulation of T-cell proliferation via the release of soluble CD83. Blood 103, 4207–4215. doi: 10.1182/blood-2003-12-4350
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sloan, E., Henriquez, R., Kinchington, P. R., Slobedman, B., and Abendroth, A. (2012). Varicella-zoster virus inhibition of the NF-kappaB pathway during infection of human dendritic cells: role for open reading frame 61 as a modulator of NF-kappaB activity. J. Virol. 86, 1193–1202. doi: 10.1128/JVI.06400-11
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Smith, C., and Khanna, R. (2013). Immune regulation of human herpesviruses and its implications for human transplantation. Am. J. Transplant. 13(Suppl. 3), 9–23. doi: 10.1111/ajt.12005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Somboonthum, P., Yoshii, H., Okamoto, S., Koike, M., Gomi, Y., Uchiyama, Y., et al. (2007). Generation of a recombinant Oka varicella vaccine expressing mumps virus hemagglutinin-neuraminidase protein as a polyvalent live vaccine. Vaccine 25, 8741–8755. doi: 10.1016/j.vaccine.2007.10.039
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Suzuki, K., Yoshikawa, T., Tomitaka, A., Matsunaga, K., and Asano, Y. (2004). Detection of aerosolized varicella-zoster virus DNA in patients with localized herpes zoster. J. Infect. Dis. 189, 1009–1012. doi: 10.1086/382029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Takahashi, M., Otsuka, T., Okuno, Y., Asano, Y., and Yazaki, T. (1974). Live vaccine used to prevent the spread of varicella in children in hospital. Lancet 2, 1288–1290. doi: 10.1016/S0140-6736(74)90144-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vandevenne, P., Lebrun, M., El Mjiyad, N., Ote, I., Di Valentin, E., Habraken, Y., et al. (2011). The varicella-zoster virus ORF47 kinase interferes with host innate immune response by inhibiting the activation of IRF3. PLoS ONE 6:e16870. doi: 10.1371/journal.pone.0016870
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vandevenne, P., Sadzot-Delvaux, C., and Piette, J. (2010). Innate immune response and viral interference strategies developed by human herpesviruses. Biochem. Pharmacol. 80, 1955–1972. doi: 10.1016/j.bcp.2010.07.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Volz, T., Kaesler, S., and Biedermann, T. (2012). Innate immune sensing 2.0—from linear activation pathways to fine tuned and regulated innate immune networks. Exp. Dermatol. 21, 61–69. doi: 10.1111/j.1600-0625.2011.01393.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, J. P., Kurt-Jones, E. A., Shin, O. S., Manchak, M. D., Levin, M. J., and Finberg, R. W. (2005). Varicella-zoster virus activates inflammatory cytokines in human monocytes and macrophages via Toll-like receptor 2. J. Virol. 79, 12658–12666. doi: 10.1128/JVI.79.20.12658-12666.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhu, H., Zheng, C., Xing, J., Wang, S., Li, S., Lin, R., et al. (2011). Varicella-zoster virus immediate-early protein ORF61 abrogates the IRF3-mediated innate immune response through degradation of activated IRF3. J. Virol. 85, 11079–11089. doi: 10.1128/JVI.05098-11
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: dendritic cells, viral pathogenesis, viral immune evasion, varicella zoster virus, herpesviruses
Citation: Schönrich G and Raftery MJ (2015) Dendritic cells as Achilles’ heel and Trojan horse during varicella zoster virus infection. Front. Microbiol. 6:417. doi: 10.3389/fmicb.2015.00417
Received: 23 February 2015; Paper pending published: 22 March 2015;
Accepted: 20 April 2015; Published: 08 May 2015.
Edited by:
Laura Hertel, Children’s Hospital Oakland Research Institute, USAReviewed by:
Jennifer F. Moffat, State University of New York Upstate Medical University, USALeigh Zerboni, Stanford University School of Medicine, USA
Copyright © 2015 Schönrich and Raftery. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Günther Schönrich, Institute of Medical Virology, Helmut-Ruska-Haus, Charité-Universitätsmedizin Berlin, Charitéplatz 1, 10117 Berlin, Germany,Z3VlbnRoZXIuc2Nob2VucmljaEBjaGFyaXRlLmRl