 Beth M. Carpenter1†
Beth M. Carpenter1† Abby L. West2†
Abby L. West2† Hanan Gancz1
Hanan Gancz1 Stephanie L. Servetas1
Stephanie L. Servetas1 Oscar Q. Pich1Jeremy J. Gilbreath1Daniel R. Hallinger3
Oscar Q. Pich1Jeremy J. Gilbreath1Daniel R. Hallinger3 Mark H. Forsyth3
Mark H. Forsyth3 D. Scott Merrell1*
D. Scott Merrell1* Sarah L. J. Michel2*
Sarah L. J. Michel2*- 1Department of Microbiology and Immunology, Uniformed Services University of the Health Sciences, Bethesda, MD, USA
- 2Department of Pharmaceutical Sciences, School of Pharmacy, University of Maryland, Baltimore, Maryland, USA
- 3Department of Biology, The College of William and Mary, Williamsburg, VA, USA
Helicobacter pylori NikR (HpNikR) is a nickel dependent transcription factor that directly regulates a number of genes in this important gastric pathogen. One key gene that is regulated by HpNikR is ureA, which encodes for the urease enzyme. In vitro DNA binding studies of HpNikR with the ureA promoter (PureA) previously identified a recognition site that is required for high affinity protein/DNA binding. As a means to determine the in vivo significance of this recognition site and to identify the key DNA sequence determinants required for ureA transcription, herein, we have translated these in vitro results to analysis directly within H. pylori. Using a series of GFP reporter constructs in which the PureA DNA target was altered, in combination with mutant H. pylori strains deficient in key regulatory proteins, we confirmed the importance of the previously identified HpNikR recognition sequence for HpNikR-dependent ureA transcription. Moreover, we identified a second factor, the HpArsRS two-component system that was required for maximum transcription of ureA. While HpArsRS is known to regulate ureA in response to acid shock, it was previously thought to function independently of HpNikR and to have no role at neutral pH. However, our qPCR analysis of ureA expression in wildtype, ΔnikR and ΔarsS single mutants as well as a ΔarsS/nikR double mutant strain background showed reduced basal level expression of ureA when arsS was absent. Additionally, we determined that both HpNikR and HpArsRS were necessary for maximal expression of ureA under nickel, low pH and combined nickel and low pH stresses. In vitro studies of HpArsR-P with the PureA DNA target using florescence anisotropy confirmed a direct protein/DNA binding interaction. Together, these data support a model in which HpArsRS and HpNikR cooperatively interact to regulate ureA transcription under various environmental conditions. This is the first time that direct “cross-talk” between HpArsRS and HpNikR at neutral pH has been demonstrated.
Introduction
Helicobacter pylori is a microaerophilic, Gram negative pathogen that infects over half of the world's human population (Marshall and Warren, 1984; Loughlin, 2003). Colonization occurs in the highly acidic gastric mucosal layer as well as at the gastric epithelial surface of the human stomach. Prolonged H. pylori infection is associated with the development of gastritis, peptic ulcer disease, Mucosal-Associated Lymphoid Tissue (MALT) lymphoma and gastric adenocarcinoma (Marshall and Warren, 1984; Cover and Blaser, 1992; Sepulveda and Coelho, 2002; Loughlin, 2003; Kusters et al., 2006). Current therapies for H. pylori infection require antibiotic cocktails of two, three or four drugs that are often not well tolerated due to adverse side effects (Loughlin, 2003; Kusters et al., 2006). If H. pylori infection is left untreated, colonization will persist throughout a person's lifetime (Marshall and Warren, 1984; Blaser, 1990; Cover and Blaser, 1992; Dunn et al., 1997; Sepulveda and Coelho, 2002; Loughlin, 2003). The propensity for chronic infection by H. pylori, coupled with the large rate of infection, manifests as a significant disease burden worldwide (Kusters et al., 2006). Thus, there is a great need to develop novel targeted anti-Helicobacter agents that are well tolerated (Loughlin, 2003).
While it displays optimal growth at neutral pH, H. pylori is one of a select number of bacteria that can survive under highly acidic conditions; this makes it ideally suited to life in the gastric mucosa. The cytosolic pH of H. pylori ranges from 5.3 to 7.5, and the organism can endure periodic acid shocks of pH < 2 (Wen et al., 2003, 2009; van Vliet et al., 2004a). One key feature that enables H. pylori to survive under such harsh conditions is its ability to convert host urea into ammonia, which serves to buffer the cytoplasmic/periplasmic pH, as well as to neutralize the immediate environment upon excretion from the bacterial cell (Wen et al., 2003; van Vliet et al., 2004a,b; Sachs et al., 2005, 2006; Scott et al., 2007). The majority of ammonia is produced by the nickel dependent urease enzyme, which converts urea to ammonia and bicarbonate. Urease accounts for approximately 10% of the total protein content of H. pylori (van Vliet et al., 2004a; Carter et al., 2009), and expression of the operon of genes that encode urease is known to be subject to environmental regulation.
The nickel dependent metalloregulatory protein, HpNikR, is known to be a key regulator of urease expression (van Vliet et al., 2002; Contreras et al., 2003; Ernst et al., 2005; Dosanjh and Michel, 2006; Maier et al., 2007). HpNikR functions as a tetrameric metalloregulatory protein. Each HpNikR monomer is comprised of two domains, named the N-terminal and C-terminal domains (West et al., 2010, 2012; Benini et al., 2011). To form the functional tetrameric protein, all four C-terminal domains form a central tetrameric metal binding domain (MBD), which serves as the site of nickel coordination. The MBD is then flanked on either side by two N-terminal domains that fold to form two dimeric DNA binding domains (DBD). The DBDs adopt a classic ribbon-helix-helix fold, which is commonly found in DNA binding proteins (Figure 1A) (Chivers and Sauer, 1999, 2000, 2002; Chivers and Tahirov, 2005; Schreiter and Drennan, 2007; West et al., 2010).
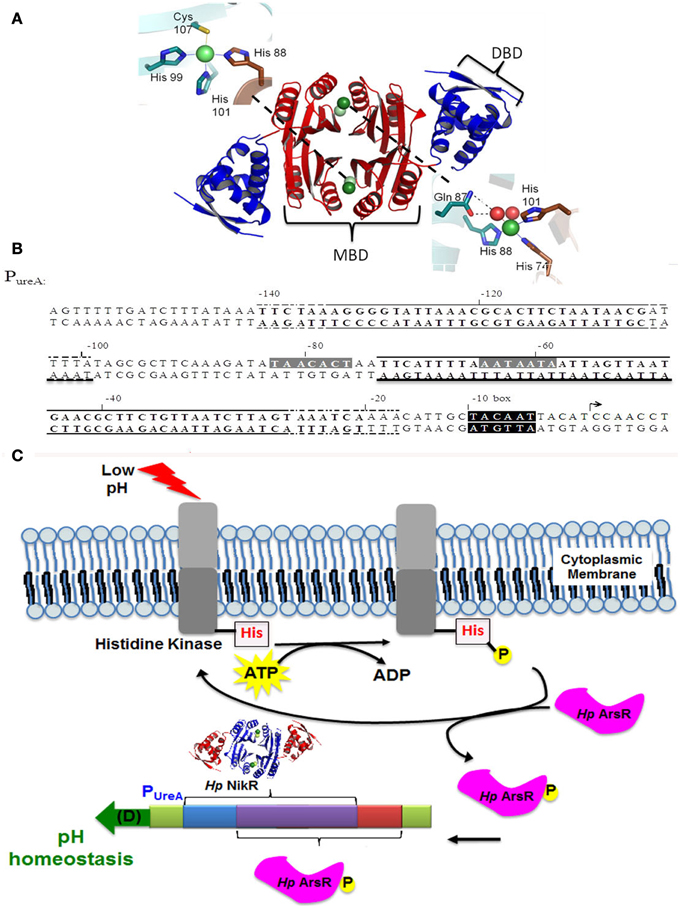
Figure 1. Structure of holo-HpNikR and architecture of the ureA promoter. (A) The structure of holo-HpNikR. Essential areas of the protein are highlighted as follows: the metal binding domain (MBD) in red, the DNA binding domain (DBD) in blue, 4-site (top left) and 5/6-site (bottom right). This image was constructed in pymol (accession number pdb 3LGH). (B) The recognition sites for HpNikR and HpArsR on the H. pylori ureA promoter. Highlighted in gray is the recognition site for HpNikR, while solid and dashed lines above the sequence designate the minimum and maximum protected regions from DNase protection assays for the two distinct binding sites of HpArsR as previously described (Pflock et al., 2005). (C) A cartoon demonstrating the operator overlap at the ureA promoter for HpNikR (pdb 3LGH) and HpArsR within the context of the biological role of HpArsR. The region colored purple represents the overlapping promoter sites.
HpNikR requires Ni(II) coordination to the MBD in order to recognize and bind to DNA via the DBD (Abraham et al., 2006; Benanti and Chivers, 2007; Dosanjh et al., 2007; Bahlawane et al., 2010). Crystallographic studies conducted by one of our laboratories have shown that Ni(II) coordinates to two distinct sites at the MBD of HpNikR: two Ni(II) ions coordinate at square planar sites that utilize a 3 His/1Cys ligand set and two Ni(II) ions coordinate to a square pyramidal/octahedral sites that utilize a 3 His/ 2–3 H2O ligand set (Figure 1A) (West et al., 2010, 2012). This heterogeneous nickel coordination controls the overall flexibility of the DBDs to favor high affinity DNA binding (West et al., 2012).
HpNikR is a global regulator of transcription in H. pylori (Contreras et al., 2003; van Vliet et al., 2004a; Ernst et al., 2005; Abraham et al., 2006). In addition to urease, HpNikR activates and represses the expression of at least forty genes in response to nickel availability (van Vliet et al., 2002; Contreras et al., 2003; Ernst et al., 2005, 2006; Abraham et al., 2006; Davis et al., 2006; Dosanjh and Michel, 2006; Benanti and Chivers, 2007, 2011; Dosanjh et al., 2007, 2009; Zambelli et al., 2008; Li and Zamble, 2009; Muller et al., 2011; Romagnoli et al., 2011; Evans and Michel, 2012; Jones et al., 2015). Genes which have been shown to be directly regulated by HpNikR encode for proteins that regulate nickel uptake (nixA, frpB4, fecA3, frpB2, ceuE), storage (hpn, hpn-like) and regulation (nikR), genes involved in iron uptake (exbB) and regulation (fur), genes involved in infection (hspA), and genes that encode for outer membrane proteins (exbB/tonB) (van Vliet et al., 2002; Contreras et al., 2003; Delany et al., 2005; Ernst et al., 2005, 2006; Abraham et al., 2006; Davis et al., 2006; Benanti and Chivers, 2007; Dosanjh et al., 2007, 2009; Jones et al., 2015). The direct binding of HpNikR to the promoter sequences of these genes has been shown via DNase footprinting, electrophoretic mobility shift assays (EMSA), fluorescence anisotropy (FA) and/or isothermal titration calorimetry (ITC) (Contreras et al., 2003; Delany et al., 2005; Ernst et al., 2005, 2006; Abraham et al., 2006; Davis et al., 2006; Benanti and Chivers, 2007, 2011; Dosanjh et al., 2007, 2009; Zambelli et al., 2008; Li and Zamble, 2009; Muller et al., 2011; Romagnoli et al., 2011; Jones et al., 2015). In addition, Krezel and co-workers recently reported a direct role for the RNA polymerase alpha-subunit C-terminal domain in promoting HpNikR/ureA binding (Borin et al., 2014). From those studies, a general model for HpNikR gene regulation has been formulated. In this model, HpNikR activates transcription by binding upstream of the RNA polymerase binding site, which aids in the recruitment of RNA polymerase (i.e., for ureA). In contrast, HpNikR represses transcription by a simple steric hindrance mechanism in which HpNikR blocks the interaction of RNA polymerase with the promoter by binding at promoter sequences overlapping the −10 or −35 hexameric boxes (e.g., nikR, nixA, fur, frpB4, exbB, fecA3) (van Vliet et al., 2002; Delany et al., 2005; Ernst et al., 2006; Wolfram et al., 2006; Danielli et al., 2009; Dosanjh et al., 2009). In addition to the genes for which direct protein/DNA binding has been established, approximately 30 additional genes have been predicted to be regulated by HpNikR (Contreras et al., 2003; Delany et al., 2005; Ernst et al., 2005; Abraham et al., 2006; Davis et al., 2006; Ernst et al., 2006; Benanti and Chivers, 2007; Dosanjh et al., 2007; Zambelli et al., 2008; Dosanjh et al., 2009; Li and Zamble, 2009; Benanti and Chivers, 2011; Muller et al., 2011; Romagnoli et al., 2011). Whether these additional genes are regulated directly or indirectly has yet to be established. These genes include components involved in motility (cheV, flaA, flaB) and stress response (hrcA, grpE, dnaK), as well as outer membrane proteins (omp11, omp31, omp32) (Contreras et al., 2003; Dosanjh et al., 2009).
The promoters for the genes that are directly regulated by HpNikR share common sequences, but are not identical. These DNA targets share a partially palindromic sequence composed of two sets of seven base pairs (half-sites), separated by eleven bases (Delany et al., 2005; Dosanjh et al., 2007, 2009; Stoof et al., 2010; Evans and Michel, 2012). HpNikR binds to a subset of the promoters with high affinity (Kd ~ nM) and a subset of the promoters with low affinity (Kd ~ μM) (Dosanjh et al., 2009). The promoter sequences for which high affinity recognition is measured are from genes that encode for nickel regulated proteins (ureA, nixA, frpb4, fecA3), while the promoter sequences for which low affinity binding has been measured are from genes that encode for other proteins (fur, nikR, exbB) (Dosanjh et al., 2009). Thus, in vitro HpNikR preferentially recognizes genes that encode for nickel-regulated proteins. The DNA target sequences for which high affinity DNA binding is observed have greater conservation of sequence at the second half-site (Evans and Michel, 2012). Studies using the ureA promoter in which the DNA sequences have been systematically altered have identified key bases within the second half-site that are essential for high-affinity protein/DNA binding (Delany et al., 2005; Dosanjh et al., 2009; Evans and Michel, 2012). In addition, the intact partial-palindrome is required for high affinity DNA binding by HpNikR in vitro (Dosanjh et al., 2009). When either half of the partial palindrome was modified to all cytosines, the affinity of HpNikR for the ureA promoter decreased from 8.0 ± 1 nM to 1000 ± 94 nM for WT/C (CTTCAAAGATATAACACTAATTCATTTTACCCCCCCATTAGTTAATGA) and 4900 ± 780 nM for C/WT (CTTCAAAGATACCCCCCCAATTCATTTTAAATAATAATTAGTTAATGA) (Dosanjh et al., 2009; West et al., 2012). When both halves of the palindrome were modified, DNA binding was fully abrogated (Dosanjh et al., 2009).
The HpArsRS two-component system has also been shown to regulate a wide variety of genes in H. pylori. These genes include those that encode for proteins involved in acid resistance, (including urease), acetone metabolism (acetone carboxylase), resistance to oxidative stress (thioredoxin reductase), and quorum sensing (Pflock et al., 2005, 2006a,b; Wen et al., 2007; Goodwin et al., 2008; Loh et al., 2010). Within the regulatory pathway, HpArsS serves as a sensor protein that phosphorylates the HpArsR response regulator. Phosphorylation occurs via a two-step process: HpArsS autophosphorylates at histidine 214 and then transfers the phosphate to aspartic acid 52 on HpArsR to generate HpArsR-P (Schar et al., 2005; Pflock et al., 2006a; Joseph and Beier, 2007; Gupta et al., 2009; Muller et al., 2009). HpArsR appears essential for bacterial viability and binds to different promoter elements based on the phosporylation state. Binding at PureA requires phosphorylation and the binding site of HpArsR-P overlaps with the binding site recognized by HpNikR (Pflock et al., 2005) (Figures 1B,C). Given that HpArsR and HpNikR respond to different environmental conditions, acid and nickel, respectively, these two regulators are believed to function independently of one another (Pflock et al., 2005).
Most of the studies that have investigated binding of HpNikR to target promoters have been conducted in vitro. However, it is clear that the cytoplasm of the bacterial cell is a much more complex environment than the one modeled in an in vitro experiment. There is one in vivo study of HpNikR mediated nickel response in which quantitative real-time PCR was used to measure HpNikR regulation of target genes (Muller et al., 2011). In this study, a complex relationship between Ni(II) availability and activation or repression of a series of genes was determined, suggesting that HpNikR activity in vivo may be more complex than observed in vitro. The goal of the studies described herein was to determine if the DNA sequences required for recognition of PureA by HpNikR are the same in vitro and in vivo. To this end, we constructed transcriptional reporters in which the wildtype ureA promoter or mutant versions of the promoter were fused to the gene encoding GFP. Reporter assays were then used to monitor ureA promoter activity directly in H. pylori. The wildtype PureA sequence exhibited high levels of GFP expression that increased with increasing concentrations of nickel. Unexpectedly, mutation of the half-sites did not prevent basal level urease expression, but negated the Ni(II) dependence of ureA expression. These results suggested that another factor rescues PureA transcription when the DNA target sequence for HpNikR is compromised. Herein, we show that the HpArsRS acid response regulatory system, which has previously been shown to regulate ureA expression in response to low pH, also affects ureA transcription at neutral pH. This is the first time that HpArsR has been shown to regulate urease expression in conjunction with HpNikR, and we propose that there is a cooperative interaction between these two regulators to control urease expression in H pylori.
Materials and Methods
Bacterial Strains and Growth
Primer sequences are listed in Table 1 and bacterial strains and plasmids used in this study are listed in Table 2. The H. pylori strains used in this study are all derivatives of G27 (Covacci et al., 1993; Baltrus et al., 2009). H. pylori strains were maintained as frozen stocks at −80°C in brain heart infusion broth (BD Biosciences) supplemented with 10% fetal bovine serum (Gibco) and 20% glycerol (EMD chemicals, Inc.). Bacterial strains were cultured essentially as previously described (Carpenter et al., 2007). Briefly, strains were grown on horse blood agar (HBA) plates that contained 4% Columbia agar base (Neogen Corporation), 5% defibrinated horse blood (HemoStat Laboratories, Dixon, CA), 0.2% β-cyclodextrin (Sigma), 10 μg/ml vancomycin (Amresco), 5 μg/ml cefsulodin (Sigma), 2.5 U/ml polymyxin B (Sigma), 5 μg/ml trimethoprim (Sigma), and 8 μg/ml amphotericin B (Amresco). Liquid cultures of H. pylori were grown in brucella broth (Neogen Corporation) supplemented with 10% fetal bovine serum and 10 μg/ml vancomycin at 37°C with shaking at 100 rpm. As noted in Table 2, where appropriate, cultures and plates were supplemented with 8 μg/ml chloramphenicol (Cm) (EMD Chemicals, Inc.) and/or 25 μg/ml kanamycin (Kan) (Gibco). In addition, where detailed below, some HBA plates contained 5% sucrose (Suc) (Sigma). Both liquid and plate cultures were grown under microaerobic conditions (5% O2, 10% CO2, and 85% N2) generated with an Anoxomat gas evacuation and replacement system (Spiral Biotech) in gas evacuation jars. Exponential phase cultures were grown in liquid culture for 20 h, while stationary phase cultures were grown for 44 h.
Table 1. List of oligonucleotides used in this study.
Table 2. List of strains used in this study.
Construction of a ΔnikR::cat H. pylori Strain
The HpnikR mutant strain was constructed using a strategy that resulted in replacement of the HpnikR sequence with the cat gene, which encodes for chloramphenicol resistance. Briefly, a 968 bp fragment containing the region directly upstream of nikR was amplified with primers U1338-F and U1338-R, the latter of which was engineered to contain SmaI and XhoI restriction sites. This fragment was then fused via splicing by overlap extension (SOE) PCR to a 1098 bp fragment containing the region immediately downstream of nikR, which was amplified with primers D1338-F and D1338-R, the former of which contains SmaI and XhoI restriction sites. The SOE product was cloned into pGEM-T Easy to create pDSM923. Sequence analysis showed that only the SmaI site was preserved in the SOE fusion product. The cat gene (Carpenter et al., 2007), which had first been cloned into pGEM-T Easy as pDSM278, was liberated via restriction digestion with EcoR1 New England Biolabs (NEB), Klenow (NEB) treated, and then ligated with the SmaI (NEB) digested and calf intestine phosphatase (NEB) treated, pDSM923; the resulting nikR deletion construct was named pDSM924. pDSM924 was subsequently transformed into G27, and transformants were selected for on HBA plates containing 8 μg/mL Cm. Resulting colonies were screened for differences in size in the nikR region via PCR with the U1338-F and D1338-R primers. For those colonies showing the expected change in size, the PCR product was next sequenced to verify deletion of nikR. One such colony showing a deletion insertion of nikR was named DSM975.
Construction of a Markerless ΔarsS H. pylori Strain
The arsS mutant strain was created using pDSM922, which is a suicide vector that contains a counter selectable marker (generous gift of Liz Marcus and David Scott, UCLA School of Medicine). Briefly, the plasmid contains a kan-sacB counter selectable cassette, previously described by Copass et al. (1997), that is flanked by the 600 and 400 bp immediately upstream and downstream, respectively, of HP0165 (arsS). These regions were originally amplified using strain 43,504 as the template (Marshall and Goodwin, 1987). pDSM922 was naturally transformed into G27, and transformants were selected on HBA plates containing 25 μg/ml kanamycin. The resulting transformants were patched on HBA plates containing 5% sucrose to ensure sucrose sensitivity, and deletion insertion of arsS was then confirmed by PCR amplification of the region with HP0165_up_F and HP0165_down_R primers followed by sequencing with the same primers. One such ΔarsS mutant was named DSM1069, which then served as the background strain to create the markerless mutant.
To create the markerless deletion strain, the region immediately upstream and downstream of arsS were fused together via SOE PCR; the upstream region was amplified with the HP0165_Up_F and HP0165_Up_R primers, the downstream region was amplified with the HP0165_Down_F and HP0165_Down_R primers, and the products from these reactions were gel purified and fused together via SOE PCR using primers (HP0165_Up_F and HP0165_Down_R). The resulting product was gel purified, and naturally transformed into DSM1069. Transformants were selected on HBA plates containing 5% sucrose and then patched onto HBA plates containing 25 μg/ml kanamycin to ensure kanamycin sensitivity; double crossover homologous recombination resulted in the replacement of the kan-sacB cassette to create a markerless deletion of the arsS gene. Proper integration was confirmed by PCR and sequencing with the HP0165_Up_F and HP0165_Down_R primers. The resulting strain was named DSM983.
Construction of a ΔarsS/nikR::cat H. pylori Strain
The ΔarsS/nikR::cat mutant strain was created by naturally transforming pDSM924, the nikR::cat deletion construct, into H. pylori strain DSM983. Transformants were then screened on HBA plates supplemented with 8 μg/mL Cm to ensure chloramphenicol resistance. Proper integration was confirmed first by PCR with the U_1338_F and D_1338_R primers. For those colonies showing the expected change in size for the nikR gene, the PCR product was sequenced to verify deletion of nikR. To ensure that the ΔarsS markerless deletion was still intact for this strain, PCR with the HP0165_Up_F and HP0165_Down_R primers followed by sequencing of the PCR product was performed. The resulting strain was named DSM1071.
Construction of ureA::GFP Transcriptional Fusions
Transcriptional fusions to the promoterless gfpmut3 allele carried on pTM117 (Carpenter et al., 2007) were constructed to monitor ureA expression. Briefly, the wildtype ureA promoter was PCR amplified from G27 using the UreA_F_Prom_KpnI and UreA_R_Prom_XbaI primers; these primers incorporate KpnI and XbaI restriction sites, respectively. The resulting PCR fragment was subcloned into pGEM-T Easy (Promega) to create pDSM462. The ureA promoter was then removed via digestion with KpnI (NEB) and XbaI (NEB) and ligated into the appropriately digested pTM117 vector to create pDSM463. The proper fusion was confirmed by PCR amplification with the UreA_F_Prom_KpnI and UreA_R_Prom_XbaI primers and by sequencing with the same primers. pDSM463 was then naturally transformed into G27, DSM975, DSM983 and DSM1071, and transformants were selected on HBA plates containing 25 μg/ml Kan. The individual strains transformed with pDSM463 are described in Table 2.
Mutant ureA promoter constructs were each created using SOE and primer pairs that incorporated the desired mutation during the process of amplification. Specifically, the C/WT mutant promoter was created using the primer pairs F1_ureA_prom_switch and UreA_R_Prom_XbaI, and UreA_F_Prom_KpnI and R1_ureA_prom_switch. The WT/C mutant promoter was created using the primer pairs F2_ureA_prom_switch and UreA_R_Prom_XbaI, and UreA_F_Prom_KpnI and R2_ureA_prom_switch. Finally, the C/C ureA promoter mutant was created using the primer pairs F3_ureA_prom_switch and UreA_R_Prom_XbaI, and UreA_F_Prom_KpnI and R3_ureA_prom_switch. Each of the mutant ureA promoters were subcloned into pGEM-T Easy (Promega), removed by digestion with KpnI (NEB) and XbaI (NEB), and ligated into the appropriately digested pTM117 vector. The constructed plasmids are pDSM697 (C/C), pDSM698 (C/WT), and pDSM796 (WT/C). All fusions were confirmed by PCR amplification with UreA_F_Prom_KpnI and UreA_R_Prom_XbaI primers and by sequencing with the same primers. Each of the resulting plasmids containing the individual ureA promoter mutations as well as a promoterless GFP fusion vector (pDSM199) were naturally transformed into G27, DSM975, DSM983, and DSM1071, and transformants were selected on HBA plates containing 8 μg/ml Cm and 25 μg/ml Kan (DSM 975 and DSM 1071), or 25 μg/ml Kan alone (G27 and DSM 983).
GFP Reporter Assays
The ability of the ureA transcriptional fusions to drive expression of GFP was assessed visually utilizing an Olympus BX61 fluorescent microscope, as well as using flow cytometry as previously described (Carpenter et al., 2007). Briefly, strains containing the ureA promoter fusions were grown overnight in liquid cultures containing varying NiSO4 concentrations (0, 0.5, 1.0, 10 μM) (Sigma). Following overnight growth, 0.5–1.5 ml of each culture was pelleted and resuspended in 1–2 ml of sterile 1× phosphate-buffered saline depending on the density of the culture. Bacterial clumps and culture debris were subsequently removed by passing the resuspended culture through a 1.2-μm Acrodisc PSF syringe filter (Pall). Flow cytometry analysis for the ureA fusion constructs was performed using either a Beckman Coulter Epics XL-MCL flow cytometer with a laser setting of 750 V or a BD SLR II flow cytometer. 20,000 events were collected for each assay. WinList 3D, version 6.0 (Verity Software House) and FlowJo, version X (FLOWJO, LLC) were used to analyze the flow cytometry data. These experiments were performed 3–5 times for each strain-reporter plasmid combination.
RNA Isolation, cDNA Synthesis and RT-PCR
In addition to the GFP reporter assays, RT-PCR was utilized to measure ureA expression under normal, 10 μM nickel supplemented, low pH (pH 5.0), and combined nickel supplementation and low pH conditions. Bacterial liquid cultures of DSM1 (WT G27), DSM975 (ΔnikR), DSM983 (ΔarsS), and DSM1071 (ΔarsS/nikR) were grown for 18 h, and then each culture was divided into four equal portions. The first portion was utilized for RNA isolation and represents the normal media sample. The remaining portions were pelleted and resuspended in one of the following supplemented liquid culture medias: 10 μM NiSO4(Sigma-Aldrich), pH 5.0 (achieved through the addition of HCl to the media), or pH 5.0 with 10 μM NiSO4. These portions of each culture were then maintained for another 90 min prior to RNA isolation. RNA isolation was performed as previously described (Thompson et al., 2003). The integrity of the RNA was determined through visualization on agarose gels. Next, cDNA was generated as previously described (Gilbreath et al., 2012) using the Quantitect reverse transcriptase kit (Qiagen) according to the manufacturer's protocol. Control reactions for each sample were also performed without the addition of reverse transcriptase (noRT) enzyme. Following cDNA synthesis, quantitative real-time PCR (qPCR) for ureA as well as the 16S internal reference gene was performed using the primers listed in Table 1. qPCR was conducted similar to the methods used by Gilbreath et al. (2012). Briefly, qPCR reaction mixtures composed of 1x Roto-Gene SYBR green RT-PCR master mix, 3 pmol each of forward and reverse primer pair, and 1 μL of either cDNA or noRT reaction for use as template were combined and brought to a total volume of 20 μL with water. The following 2-step cycling conditions were used: 5 min at 95°C (initial activation) followed by 35 cycles of 5 s at 95°C (denaturation) and 10 s at 50°C (annealing/extension); SYBR green fluorescence was measured at each extension step. Relative gene expression was calculated using the 2−ΔΔCT method. Post-run melt curve analysis were performed to ensure specificity of amplification. Four biologically independent replicates of these experiments were conducted.
H. pylori HpArsR Cloning, Expression and Purification
PCR primers were designed to amplify the arsR gene from H. pylori J99 with BamH1 and HindIII restriction sites included. The arsR gene was ligated into a pQE3 vector (Qiagen), which includes an N-terminus hexa-histidine coding sequence. The identity of the cloned arsR gene was confirmed by DNA sequencing. For protein expression, arsR-pQE3 was transformed into E. coli M15-pREP4 cells, and grown in LB medium containing ampicillin (100 μg/mL) and kanamycin (20 μg/mL). When the culture reached an A600 of 0.6, protein expression was induced for 4 h with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Cells were collected by centrifugation at 12,000 × g for 10 min. All buffers utilized in the HpArsR studies contained 5 mM tris(2-carboxyethyl) phosphine hydrochloride (TCEP) to prevent oligomerization of the protein. Cell pellets were resuspended in 20 mM Tris-HCl, 500 mM NaCl, 5 mM TCEP, 1 mM phenylmethylsulfonyl fluoride (PMSF) pH 7.5 and then lysed by sonication on ice. After sonication, the preparation was centrifuged at 31,000 × g for 20 min. The supernatant (~ 100 ml) was collected and applied to a 30-ml metal affinity chromatography column (His-Bind, Novagen) charged with Ni(II). The column was washed with a 50 mM imidazole gradient to remove proteins bound non-specifically to the column, and the protein of interest, including the hexa-histidine tag, was eluted with 250 mM imidazole over 45 ml. Five milliliter fractions corresponding to the elution were collected, and the purity of the proteins was visually assessed using 15% SDS-PAGE gels stained with coomasie. Fractions determined to be >95% pure were pooled, and buffer exchanged into 50 mM Tris-HCl, 5 mM MgCl2, 50 mM KCl, 5 mM TCEP, pH 7.5 and concentrated to a volume of 6 ml using Amicon Ultra-15 centrifugal filters [5-kDa molecular weight cut-off (MWCO) membrane] for use in DNA binding assays.
In vitro Phosphorylation of HpArsR
HpArsR was phosphorylated (HpArsR-P) in vitro by incubating the protein in phosphorylation buffer (50 mM Tris-HCl, 5 mM MgCl2, 50 mM KCl, 5 mM TCEP, pH 7.5) with 50 mM acetylphosphate for 60 min at 25°C (McCleary and Stock, 1994; Dietz et al., 2002). Phosphorylation yields were not independently measured. Post-phosphorylation, the KCl concentration was adjusted to 100 mM to be consistent with past salt concentrations used in the studies of HpNikR DNA recognition (Dosanjh et al., 2007, 2009; West et al., 2012).
Oligonucleotide Probes
HPLC-purified oligonucleotide probes were purchased from Integrated DNA Technologies (Coralville, IA) and were either labeled with fluorescein (F) or unlabeled as indicated in Table 1. Upon receipt, the probes were resuspended in DNase-free water and quantified. To obtain double stranded probes, each oligonucleotide probe was mixed with a probe with the complementary sequence such that there was a 1.25:1 ratio of unlabeled to labeled oligonucleotide probe in annealing buffer (10 mM Tris, 10 mM NaCl at pH 8.0). The annealing reaction mixtures were placed in a water bath set to a temperature 10°C higher than the melting temperatures (Tms) of the component oligonucleotides. The water bath was then immediately turned off, and the annealing reaction mixtures were allowed to cool overnight. Double-stranded oligonucleotides were quantified and stored at −20°C (Dosanjh et al., 2007, 2009; West et al., 2012).
Fluorescence Anisotropy Monitored Titrations of HpArsR-P and HpArsR with PureA
A fluorescence anisotropy (FA) assay was used to characterize the interaction of HpArsR-P and HpArsR with the ureA promoter and related mutants. Measurements were taken on an ISS PC-1 spectrofluorimeter configured in the L format with an excitation wavelength of 495 nm and an emission wavelength of 519 nm. The band-pass for excitation was 2 nm and 1 nm for emission. 15 nM of PureA in 50 mM Tris-HCl, 5 mM MgCl2, 100 mM KCl, 5 mM TCEP, pH 7.5 was added to a cuvette that had been pretreated with 5 mM bovine serum albumin (BSA) to prevent adherence of either the protein or DNA to the cuvette walls.
Direct titrations: For direct titrations, either HpArsR-P or HpArsR was titrated into fluorescein labeled ureA (ureA-F) and the change in anisotropy as a function of added protein was measured. The data were analyzed by converting the anisotropy to fraction bound, Fbound (the fraction of HpArsR-P bound to DNA at a given DNA concentration), using the equation (Lakowicz, 1999):
Where rfree is the anisotropy of the fluorescein-labeled oligonucleotide, rbound is the anisotropy of the DNA/protein complex at saturation, and Q, is the quantum yield ratio of the bound to the free form and is calculated from the fluorescence intensity changes that occur (Q = Ibound/Ifree). The typical Q for HpArsR-P DNA binding experiments was Q = 0.87. Fbound was plotted against the protein concentration and fit using a one-site binding model:
Where P is the protein concentration and D is the DNA concentration. Each data point from the FA assay represents the average of 31 readings taken over a time course of 100 s. Each titration was carried out in triplicate.
Competitive Titrations
For competitive titrations, an unlabeled DNA oligomer was titrated into a solution containing 1500 nM HpArsR-P and 5 nM PureA-F and the decrease in anisotropy (r) as the unlabeled DNA oligomer competed with the labeled oligonucleotide was recorded. The resultant anisotropy values were converted to fraction bound. The competition experiments were performed with HpArsR-P concentrations at levels near saturation to minimize the amount of unlabeled DNA required to complete the titrations. Experiments were performed aerobically as no difference in binding was observed between experiments performed anaerobically and aerobically (data not shown).
Binding isotherms were fit using Mathematica (version 8 Wolfram Research) to a model that involved the mass action equations for the three competing equilibria:
Where P is the protein (HpArsR-P), Df is fluorescently labeled DNA, and D represents unlabeled DNA. The value for K1 was determined from the forward titrations and thus used as a known parameter for the fit. Mathematica software was used to combine Equations (1)–(3) and to solve the resulting cubic equation in terms of PDf using non-linear, least squares analysis. All titrations were carried out in triplicate (Dosanjh et al., 2009).
Statistical Analysis
Statistical analysis on qRT-PCR and flow cytometry data was conducted using GraphPad Prism 6.01 (GraphPad Software, La Jolla, CA, USA). A One-Way ANOVA was used to compare basal levels of expression in ΔnikR, ΔarsS, and ΔarsS/nikR to WT. A Two-Way ANOVA with Tukey's correction for multiple comparisons was used to analyze fold differences in expression between WT H. pylori, ΔnikR, ΔarsS, and ΔarsS/nikR. Similarly, mean fluorescent intensity (MFI) values obtained from flow cytometry were assessed using a two-way ANOVA with Tukey's correction for multiple comparisons. A p < 0.05 was considered significant.
Results
Regulation of PureA by HpNikR
HpNikR positively regulates the expression of urease by binding to a partially palindromic sequence located on the ureA promoter (PureA) (Dosanjh et al., 2007, 2009; Evans and Michel, 2012). The DNA sequence that HpNikR recognizes and binds to is AT rich and consists of seven base pairs separated by an eleven base pair linker sequence that is not thought to be directly involved in the protein/DNA recognition event (Figures 1B,C). When either half of the palindrome is mutated to all cytosines, the in vitro affinity of HpNikR for the DNA sequence is diminished by 3 orders of magnitude from a Kd of 8.0 ± 1 nM to a Kd of 1.0 ± 0.09 or 4.9 ± 0.8 μM for the WT/C or C/WT mutants, respectively (Dosanjh et al., 2009; West et al., 2012). When both halves of the palindrome are mutated to all cytosines, DNA binding is fully abrogated (Dosanjh et al., 2009). Thus, both sides of the recognition palindrome appear to be important for HpNikR/DNA binding.
To determine if these in vitro identified DNA sequence requirements are also observed in vivo, a series of Green Fluorescent Protein (GFP) reporter constructs in which the DNA recognition sequence within the ureA promoter was varied, to mirror the modifications that were studied in vitro, were created. These constructs, named WT/WT, WT/C, C/WT, and C/C, were unmodified or modified at each palindrome as indicated in Table 2. To measure the effect of these modifications on the nickel-dependent transcription of urease controlled by endogenous HpNikR, expression of GFP was monitored in H. pylori strain G27 and an isogenic ΔnikR strain grown in the presence of increasing Ni(II) concentrations.
Visual inspection of the strains by fluorescence microscopy showed that fluorescence was only observed in strains carrying constructs in which the ureA promoter was fused to GFP; promoterless controls showed no fluorescence (data not shown). Quantitative analysis of GFP expression was achieved using flow cytometry, as described in the materials and methods. Maximum GFP expression was observed for the WT/WT reporter construct carried in the wildtype H. pylori strain. This was the only reporter construct for which Ni(II) dependence was observed; a statistically significant concentration-dependent increase in GFP expression was observed as the Ni(II) concentration was increased (Figure 2 and Tables 3, 4). In the wildtype H. pylori strain background, GFP expression levels for the WT/C and C/C reporter constructs were similar to the GFP expression levels observed for the WT/WT reporter construct when the media was not supplemented with Ni(II) (Figure 2 and Table 4). In contrast, basal levels of ureA expression in the C/WT background were significantly lower than WT/WT in the wildtype H. pylori strain (p < 0.0001). At high Ni (II) concentrations, the reporter constructs containing mutation of the palindrome half sites (WT/C and C/WT) as well as full site (C/C) exhibited decreased GFP expression levels when compared to the unmodified WT/WT construct (Table 4). En masse, the fact that we observed high basal levels of expression of ureA with some of the mutant constructs was unexpected; our in vitro findings suggest that mutation of both sides of the palindrome completely abrogate HpNikR binding at the ureA promoter (Dosanjh et al., 2009). Given that nickel responsiveness was completely lost for each mutated promoter construct (Figure 2 and Table 4) and because increasing nickel concentrations are known to result in increased HpNikR activity, this finding suggested that another regulator may play a role in ureA expression under the in vivo conditions examined in these experiments.
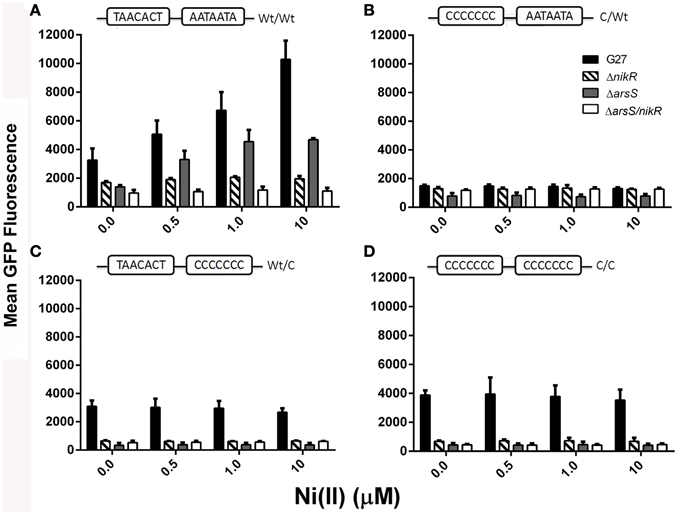
Figure 2. Regulation of PureAby HpNikR and HpArsRS. The mean GFP expression for the PureA WT and mutant constructs expressed in the various parental strains is shown. Each panel corresponds to a different PureA mutant construct that is indicated at the top of each panel: (A)- WT/WT, (B)- C/WT, (C)- WT/C, and (D)- C/C. Shading is as indicated: WT G27 (black bars), ΔnikR (cross bars), ΔarsS (gray bars), and ΔarsS/nikR (white bars) in NiSO4 supplemented (0, 0.5, 1, 10 μM) growth media. Flow cytometry was performed 3–5 times for each strain-reporter plasmid combination. Bars represent the mean GFP fluorescence and the error bars indicate the standard deviation.
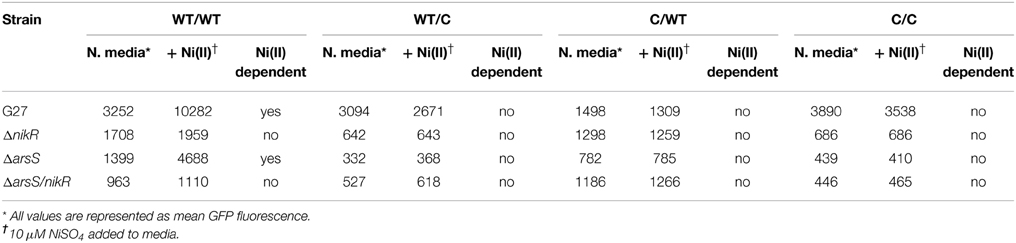
Table 3. Mean GFP fluorescence in normal and 10 μM NiSO4 supplemented media.
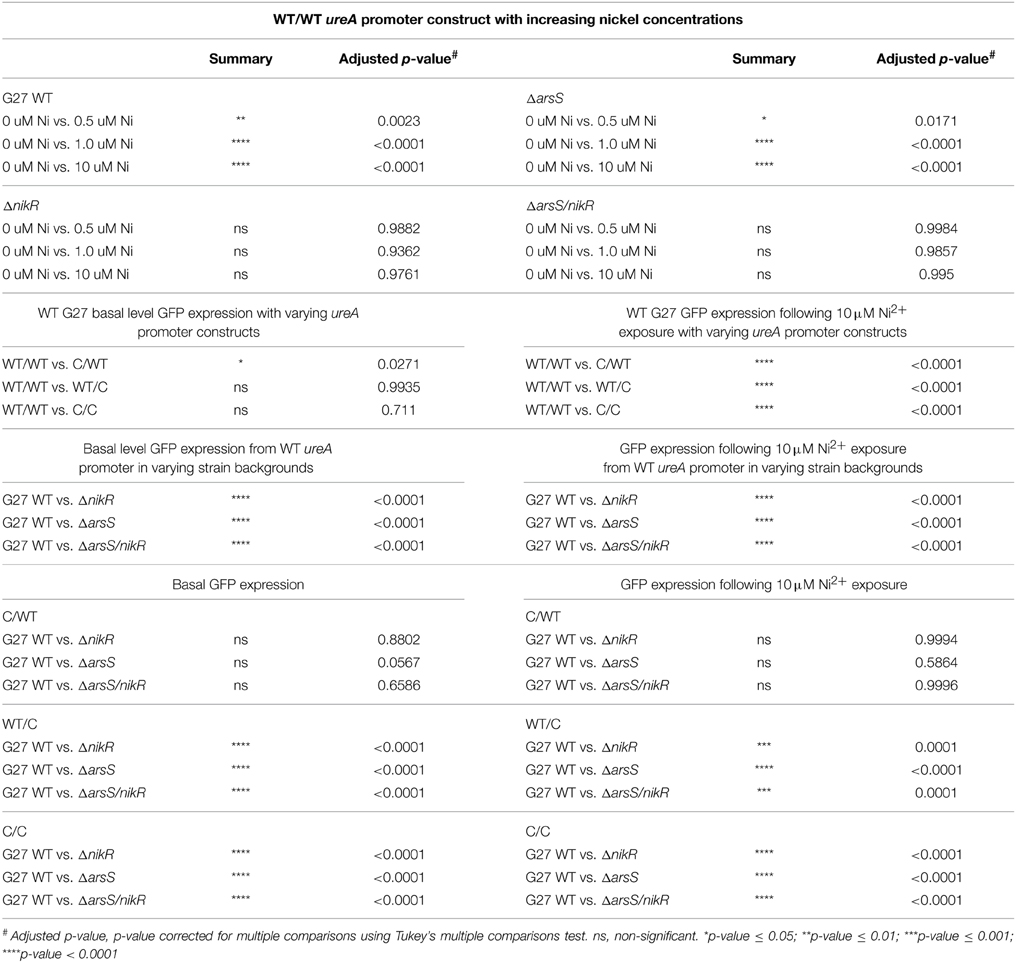
Table 4. Statistical analysis of mean GFP fluorescence.
To specifically examine the contribution of HpNikR to the observed levels of ureA expression, the same four promoter constructs were next examined in an isogenic ΔnikR strain. Interestingly, GFP expression was not entirely abrogated for the WT/WT promoter though expression was not nickel responsive (Figure 2 and Table 4). Once again, this suggests that the GFP expression observed in the wildtype strain is regulated by HpNikR as well as another regulatory factor (Figure 2). Mutating either side of the HpNikR binding sequence in the PureA promoter abrogated the Ni (II) response and resulted in varying effects on basal levels of ureA expression. Mutation of the right half of the recognition sequence (WT/C) resulted in a significant decrease in expression (p = 0.0021) as compared to the WT/C construct in the wildtype strain. Likewise, disruption of both segments of the palindrome (C/C) resulted in a dramatic decrease in ureA expression in the ΔnikR mutant (Figure 2) that was similar to the decrease observed in the WT/C construct (Table 4). Interestingly, mutating the left half (C/WT) of the recognition sequence did not lead to a significant reduction in ureA expression (p = 0.4408) as compared to the C/WT construct in the wildtype strain. When considered together, these findings suggest that an additional regulatory factor is involved in urease transcription. Moreover, given that in vitro studies have clearly shown that HpNikR does not bind to the C/C mutant construct (Dosanjh et al., 2009), the demonstrated decrease in transcription when HpNikR is absent in the strain carrying the C/C mutant construct suggests that HpNikR has a hitherto unidentified indirect role in PureA transcription.
Regulation of PureA by HpArsR
The observation that PureA transcription occurs for the WT/C and C/WT GFP reporter constructs in the presence and absence of HpNikR independent of nickel availability suggested that another factor regulates ureA transcription in these conditions. The two-component system HpArsRS has been shown to mediate pH-responsive urease expression (Pflock et al., 2005, 2006a,b; Wen et al., 2006, 2007). Furthermore, the HpArsR binding site within the ureA promoter partially overlaps the site recognized by HpNikR (Figures 1B,C). To determine if HpArsR was responsible for the observed urease expression, ΔarsS and ΔarsS/nikR deletion strains of H. pylori were constructed and GFP fluorescence was measured for each of the PureA constructs. Of note, this strategy was chosen since HpArsR was previously shown to be essential and cannot be deleted (Beier and Frank, 2000; McDaniel et al., 2001); however, HpArsR regulates urease in its phosphorylated form (HpArsR-P) (Pflock et al., 2005). Therefore, deletion of HpArsS effectively inactivates HpArsR dependent regulation of urease since HpArsS is required for HpArsR phosphorylation (Pflock et al., 2005, 2006a).
To dissect the role of HpArsR (ΔarsS) in ureA expression, transcription was measured for each of the PureA constructs. As expected, because HpNikR is present in the ΔarsS strain background, the WT/WT promoter showed Ni(II) dependent GFP expression. However, the relative amounts of GFP expression were significantly lower in this strain background as compared to the wildtype strain (Table 4): for example, a mean GFP fluorescence of 4688 fluorescence units was measured for the ΔarsS strain in 10 μM nickel as compared to 10,282 fluorescence units for wildtype under the same nickel concentrations (Figure 2 and Table 3). This finding suggests that under non-acidic conditions, HpArsR-P interacts cooperatively with HpNikR to increase expression from the WT urease promoter. In the ΔarsS/nikR background, though a basal level of GFP expression is observed, this expression is nickel independent (Figure 2).
For the WT/C and C/C mutant promoters, the absence of endogenous HpArsS (and thus, HpArsR-P) resulted in lower levels of GFP expression as compared to those observed in the wildtype strains (p < 0.0001 for both). In each case, GFP expression was unaffected by nickel concentration. Similar to what was seen in the ΔnikR background, while the WT/C and C/C promoters resulted in decreased expression, the decrease in ureA expression in the C/WT background was not significant as compared to the same construct carried in the wildtype strain (Table 4). Furthermore, basal levels of ureA expression in ΔnikR and ΔarsS strains were similar regardless of which ureA promoter was present (Table 4). In the ΔarsS/nikR background, the WT/C and C/WT mutant promoters showed levels of expression similar to those observed in the ΔnikR background. As was observed in the ΔnikR single mutant background, none of these promoter constructs was nickel responsive in the double mutant background (Figure 2 and Table 3). In the ΔarsS and ΔarsS/nikR backgrounds, the C/C promoter fusion produced low levels of GFP expression that were slightly less than the levels observed in the ΔnikR background. Significant GFP expression was only observed for this construct in the wildtype strain. Taken en masse, these findings are consistent with cooperative interaction of HpNikR and HpArsR-P at the ureA promoter to achieve maximal regulation of urease. Of note, the role of HpArsR-P in this regulation occurred in the absence of acidic pH, which is considered to be the major environmental signal controlling HpArsR activity (Pflock et al., 2005, 2006a,b; Wen et al., 2006, 2007).
To further investigate the cooperative regulation of ureA by HpNikR and HpArsR-P and to confirm that the use of plasmid based transcriptional fusions was not artificially affecting our results, we next directly assessed ureA expression directly from the chromosome via qPCR analysis on RNA extracted from WT, ΔnikR, ΔarsS, and ΔarsS/nikR H. pylori strains. We assessed ureA expression in strains that were exposed to normal growth media as well as to medias that were 1) supplemented with excess nickel (10 μM), 2) adjusted to acidic pH (pH 5.0), or 3) supplemented with excess nickel and adjusted to acidic pH (10 μM + pH 5.0). Comparison of basal levels of ureA expression between the WT and mutant strains under normal growth conditions showed that there was little to no difference in ureA expression between WT and ΔnikR (p = 0.319); this was expected based on previous data (Ernst et al., 2005). Conversely, a statistically significant decrease in basal ureA expression was seen in the ΔarsS (p = 0.009) and ΔarsS/nikR (p = 0.0116) strains (Figure 3A). As with the data obtained with the GFP fusions (Figure 2), these data suggest that HpArsRS is necessary for maximal expression of ureA under normal growth conditions.
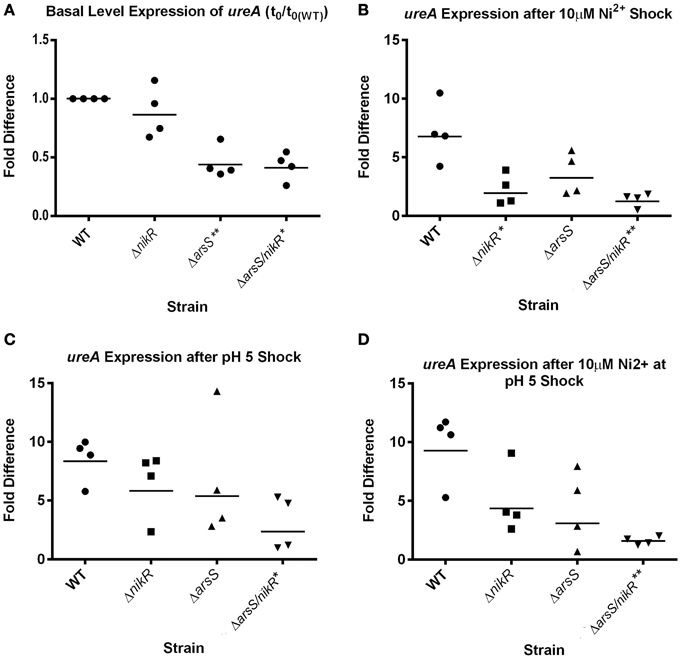
Figure 3. Changes in ureA expression in response to nickel and low pH. qPCR using ureA specific primers was performed on cDNA generated from WT, ΔnikR, ΔarsS, and ΔarsS/nikR strains exposed to 10 μM Ni2+, pH 5.0 or both stress conditions for 90 min following 18 h of growth in normal media. Relative gene expression was calculated using the 2-ΔΔCT method. (A) The basal levels of ureA expression (relative to WT); (B) Changes in ureA expression following shock with 10 μM Ni2+; (C) Changes in ureA expression following shock with pH 5.0; (D) Changes in ureA expression following shock with 10 μM Ni2+ at pH 5.0. Four biologically independent replicates of these experiments were conducted, and each dot represents the fold difference from one replicate with bars representing the geometric mean fold difference. *p < 0.05 compared to WT, **p < 0.01 compared to WT.
Following exposure to 10 μM Ni2+, ureA expression was strongly upregulated in WT H. pylori (approximately six-fold). However, the extent of ureA upregulation was significantly decreased in the ΔnikR strain (p = 0.0159). Although not significant, a decrease in Ni-dependent upregulation of ureA was also observed in the ΔarsS strain background (three-fold increase compared to six-fold in WT) (Figure 3B). Additionally in the ΔarsS/nikR double mutant there was no change in ureA expression upon exposure to nickel (Figure 3B). These data suggest that both HpNikR and HpArsRS are required for maximal expression of ureA upon nickel stress. This point is further supported by the fact that the observed difference in nickel dependent ureA expression between ΔnikR and ΔarsS was not statistically significant (p = 0.5529). Similarly, upon exposure to acidic pH, ureA expression was increased approximately eight-fold in the WT strain, six-fold in ΔnikR, five-fold in ΔarsS but only two-fold in the ΔarsS/nikR double mutant (Figure 3C). A statistically significant difference in ureA expression under low pH was only observed when comparing WT and the ΔarsS/nikR double mutant (p = 0.0467). Given that similar levels of ureA expression were observed in both the ΔnikR and ΔarsS strains, this suggests that both regulatory proteins are necessary for maximal expression in the low pH environment; thus, HpNikR appears to play a previously unknown role in the acid responsive regulation of ureA (Figure 3C). Lastly, changes in ureA expression were monitored following simultaneous exposure to excess nickel and low pH. Again, the largest increase in expression was observed in the WT strain background (nine-fold). Although not statistically significant, ureA expression was only moderately increased in the ΔnikR (four-fold) and ΔarsS (three-fold) strains (Figure 3D). Of note, under these conditions, ΔnikR and ΔarsS were not significantly different from each other (p = 0.8729) or from the ΔarsS/nikR double mutant (p = 0.1722 and p = 0.4847, respectively). However, given that no change in ureA expression was observed for the ΔarsS/nikR double mutant, ureA expression in the ΔarsS/nikR double mutant was significantly different from WT (p = 0.0059) (Figure 3D). En masse, these data support the notion that both HpNikR and HpArsRS are important for regulation of ureA expression under normal conditions as well as in low pH and nickel supplemented environments.
Fluorescence Anisotropy to Measure HpArsR-P/PureA Binding in vitro
Based on previous DNase footprinting studies, HpArsR-P binds to two regions of the PureA promoter (Pflock et al., 2005). These two operators are made up of 41 and 57 base pairs, respectively, and the larger operator sequence includes bases recognized by HpNikR (Figures 1B,C). Based upon our in vivo results that suggest co-regulation of the ureA promoter by HpArsR and HpNikR, the in vitro DNA binding properties of HpArsR-P for the PureA sequences recognized by HpNikR were measured using fluorescence anisotropy (FA), which is an approach that has been successfully used to measure HpNikR/PureA binding (Dosanjh et al., 2007, 2009; Evans and Michel, 2012; West et al., 2012). The FA data for HpArsR-P with PureA WT/WT, WT/C, C/WT, and C/C showed comparable binding isotherms (Figure 4), with Kds of 17 ± 2.0, 24 ± 0.5, 20 ± 0.6, and 23 ± 0.7 nM respectively. These findings show that HpArsR-P can bind to the same PureA promoter sequence as HpNikR in vitro, and it can also bind to sequences in which the palindrome recognized by HpNikR is altered. HpArsR did not exhibit any DNA binding (data not shown).
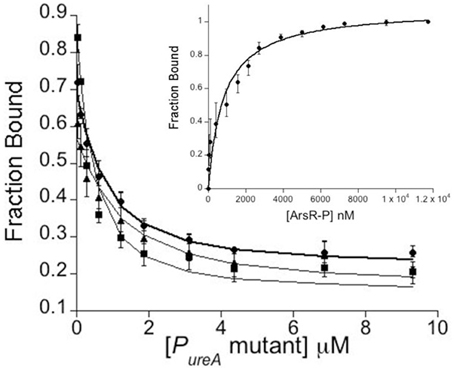
Figure 4. Inset Direct fluorescence anisotropy (FA) titration between HpArsR-P and fluorescein labeled Wt/Wt PureA-F. The data are fit to a 1:1 binding equilibrium. Main Figure: competitive titrations of HpArsR-P with PureA mutants: • PureAWt/C, ■ PureA C/Wt, and ▲ PureA C/C into 5 nM PureA-F and 1.5 μM HpArsR-P. The data are fit to a competitive binding equilibrium. The data shown are the average of three sets of binding data. All FA experiments were performed in 50 mM Tris-HCl, 5 mM MgCl2, 100 mM KCl, 5 mM TCEP, pH 7.5 and 25°C.
Discussion
HpNikR regulates urease production by binding to and activating transcription of PureA (van Vliet et al., 2002, 2004a; Abraham et al., 2006; Dosanjh et al., 2007). The details of the protein/DNA binding interaction have been systematically studied in vitro and a key partially palindromic recognition sequence has been identified (van Vliet et al., 2002, 2004a; Abraham et al., 2006; Dosanjh et al., 2007). When either half-site of the recognition palindrome is modified to all cytosines, the affinity of HpNikR for the DNA is significantly reduced, while mutation of both half sites to all cytosines abrogates binding (Dosanjh et al., 2009). Those findings led us to conclude that these half-sites were the recognition elements required for HpNikR-mediated activation of ureA expression. Herein, we present studies designed to translate our previous in vitro findings to the in vivo conditions present within the H. pylori cell. These in vivo findings revealed that additional factors are involved in the regulation of urease. Specifically, maximal activation of ureA transcription required the HpArsRS two component system in addition to HpNikR.
The initial suggestion that a second factor may regulate ureA transcription came from GFP reporter assays of the PureA WT/C and C/WT half-site mutants. Some level of nickel-independent GFP expression was still observed for all PureA constructs; though, the levels of expression were only nickel dependent in WT/WT (Figure 2 and Table 4). Interestingly, while PureA transcription was significantly decreased for the C/WT half site mutation, WT/C and C/C promoter mutations yielded similar levels of GFP expression to WT/WT in the wildtype H. pylori strain background (Figure 2 and Table 4). These high levels of expression suggested that a second factor was involved in regulating ureA transcription in vivo. This hypothesis was further supported by the levels of ureA expression observed in the ΔnikR strain background. For the PureA half-site mutant, C/WT, the level of ureA transcription was similar to that observed in the wildtype strain while for the WT/C mutant, the level of ureA transcription was approximately five-times less than in the wildtype strain. Additionally, expression of the C/C mutant construct was greatly diminished (approximately six times), indicating that the high level of expression seen for this construct in the wildtype strain background was HpNikR dependent. However, in vitro protein/DNA binding data has clearly demonstrated that HpNikR is unable to physically interact with this altered C/C DNA target. Thus, the role that HpNikR plays in regulating the C/C target appears to be indirect.
Given these data, we sought to identify the other factor required for proper regulation of ureA transcription at neutral pH (pH 7.5). The HpArsRS two-component system is also known to control urease expression in H. pylori. However, this system, which is responsive to pH, is thought to function primarily under acidic conditions (Marcus et al., 2012). The data presented in this study, demonstrate that HpArsRS functions with HpNikR to properly regulate urease expression at neutral pH. Within the HpArsRS two-component system, HpArsS functions as the histidine kinase, and HpArsR functions as the cognate response regulator. Upon sensing acidic pH, HpArsS phosphorylates HpArsR, which results in activation of the regulator (Pflock et al., 2006a; Joseph and Beier, 2007; Gupta et al., 2009; Muller et al., 2009). Interestingly, HpArsS is not essential for H. pylori survival, but HpArsR is essential (Schar et al., 2005). This suggests that the non-phosphorylated form of HpArsR regulates some essential component within the H. pylori cell. HpArsR has been shown to bind some target promoters in its non-phosphorylated form, while it only binds other promoters in its phosphorylated form (Wen et al., 2006). The ureA promoter is known to be bound and regulated by the phosphorylated form of HpArsR (Pflock et al., 2005). Thus, deletion of arsS from G27 results in an inactive HpArsR response regulator in terms of regulation of the ureA promoter. Using this strain we found that although expression of the WT/WT fusion was still nickel-dependent, the expression levels were significantly lower at all Ni (II) concentrations tested than in the G27 background (Figure 2 and Table 4), suggesting a role for HpArsR-P in maximal expression from this promoter. Similarly, the WT/C half-site mutant, showed considerable reduction in PureA::gfp expression in the absence arsS; once again indicating a role for HpArsR-P. Interestingly the C/WT half-site mutant appeared to lock the level of expression of ureA at a basal level regardless of the strain background examined. Conversely, the C/C mutation affected ureA transcription in all of the tested mutant strain backgrounds, suggesting concomitant regulation by both HpNikR and HpArsRS. Additionally, qPCR analysis revealed that basal levels of ureA expression were significantly reduced in the absence of arsS (p = 0.009) but not nikR (p = 0.319) (Figure 3A). Further support for this idea comes from the analysis of ureA expression following exposure to nickel, low pH or both stressors combined. Regardless of the stressor, the highest levels of ureA expression were observed in the WT strain background, with each single deletion showing only moderate levels of ureA and the least amount of expression occurring in the double ΔarsS/nikR strain (Figure 3).
Taken together, these data support a model of ureA transcription that involves “cross-talk” between HpNikR and HpArsRS to maximize induction of urease even under neutral conditions. Prior to this study, these two regulators were believed to function independently as HpNikR is responsive to nickel levels and HpArsRS is responsive to acidic shock (Pflock et al., 2005). Therefore, the observation that ureA transcription is 2–2.5 times lower in the ΔarsS background is particularly compelling since, under these conditions, HpNikR is present and functional. Surprisingly, the data presented here also indicate a role for HpNikR in the response to acid stress; previous reports suggested that the acid-induced increase in ureA expression was independent of NikR (Pflock et al., 2005). Using qualitative primer extension, Pflock et al. showed that there were similar increases in ureA expression upon exposure to pH 5.0 in strains with and without nikR (Pflock et al., 2005). However, in this work, we observed that the increase in ureA expression was less than that of wildtype in the ΔnikR mutant strain. The differences in our data and the previously published work could be due to differences in assay sensitivities (primer extension vs. qPCR) as well as differences in exposure to stress conditions. In the previous work, the bacteria were exposed to low pH for 60 min as compared to our 90 min exposure. Perhaps, a longer observation period following exposure to low pH allows for better measurement of the transcriptional changes in response to the stressor. Our data fully support the model that both HpNikR and HpArsRS are necessary for maximal levels of ureA expression in response to low pH regardless of nickel concentration.
Although, the discovery of cross-talk between HpNikR and HpArsR is unexpected, crosstalk, involving HpNikR, HpArsR, or other H. pylori regulatory proteins in general, is not unique. For example, HpNikR and HpFur co-regulate fur transcription (Delany et al., 2001, 2005) and HpArsS and HpFlgS work in concert to recruit and activate urease (Marcus et al., 2012). In addition, regulatory crosstalk among transcription factors has been observed within E. coli. Specifically, the transcription factors MarA and Rob of E. coli, which are involved in the response to chemical stressors consequently enabling antibiotic resistance, are co-regulated through transcriptional cross-talk with each other (Miller et al., 1994; Martin et al., 1996; Martin and Rosner, 1997; Michan et al., 2002; Schneiders and Levy, 2006; McMurry and Levy, 2010; Warner and Levy, 2010).
Though the in vivo transcriptional assays revealed a role of HpArsRS in regulation of ureA transcription, they did not demonstrate whether this role was a direct protein/DNA binding interaction, or an indirect effect via another, yet to be identified, factor. As previous studies have shown a requirement of phosphorylation for DNA binding at the ureA promoter (Pflock et al., 2005), a direct effect would likely involve HpArsR-P (Dietz et al., 2002). Using FA, we examined whether HpArsR-P directly bound to the four DNA targets (WT/WT, WT/C, C/WT, and C/C) by titrating HpArsR-P with fluorescently tagged DNA targets. A change in FA, indicative of binding, was observed for all four DNA targets when HpArsR-P was titrated. No DNA binding was observed when the control non-phosphorylated HpArsR was studied. These data indicate that HpArsRS directly regulates ureA by binding to a 48-mer promoter sequence.
Together, the in vitro and in vivo data obtained for HpArsR-P provide valuable insight into the role of HpArsR from a biophysical and a biological perspective. The in vitro results that we obtained for HpArsR-P binding to ureA (and related mutants) teach us that HpArsR-P binds to the ureA promoter in a very different way than HpNikR. HpNikR requires a specific sequence (the pseudo-palindrome) for high affinity DNA binding (Dosanjh et al., 2009; Evans and Michel, 2012). In contrast, HpArsR-P does not require this specific pseudo-palindromic sequence for binding (i.e., there is equivalent binding when the pseudo-palindromic sequence is modified).
Two factors are often important when proteins bind to DNA: sequence and shape (Rohs et al., 2010; Parker and Tullius, 2011). For HpNikR evidence indicates that sequence - the pseudo-palindromic sequence found within the ureA promoter - is important for binding; when the sequence is modified, binding is abrogated (Dosanjh et al., 2009; Evans and Michel, 2012). In contrast, for HpArsR-P the data indicate that the pseudo-palindromic sequence is not important; when the sequence is modified, binding is not affected (vide supra). This may mean that HpArsR-P/ureA binding is driven by shape (overall conformation/structure of the DNA), rather than sequence, or that the sequence recognized by HpArsR-P contains additional oligonucleotides than the sequence recognized by HpNikR.
The in vivo data, for which the entire promoter is present (rather than the short stretch of DNA utilized in the in vitro binding studies), revealed that when the ureA sequence was modified to the C/WT sequence and HpArsR-P driven expression was measured, the expression decreased. In contrast, HpArsR-P driven expression for all of the other ureA sequences was not dramatically affected. This finding indicates that there must be another factor (or factors), beyond the direct recognition of HpArsR-P with the 48-mer ureA target sequences in vitro, that is important for HpArsR-P regulation of ureA in vivo.
The combination of in vitro and in vivo data presented here allows us to learn both about (i) the very specific binding event between HpArsR-P and ureA (48 mer), which informs on the fundamental biophysical basis of binding, and (ii) the overall regulation by HpArsR-P at the cellular level, which informs on the biological mechanism. We initiated these studies to determine how the in vitro HpNikR/ureA binding that we had previously measured translated in an in vivo setting. The data indicate that the in vivo regulation is more complex that the in vitro protein/DNA binding. Furthermore, we identified HpArsR-P as another key player in this regulation. By then looking at HpArsR-P both in vivo and in vitro, we can draw the same conclusion regarding HpArsR-P function: that its role in regulation in vivo is more complex than its in vitro protein/DNA binding.
Prior to the work presented here, HpNikR and HpArsRS were thought to function as independent regulators of transcription, with HpNikR involved in regulation of urease in response to intracellular nickel availability and HpArsRS involved in urease regulation in response to intracellular acid shock (Pflock et al., 2005). Strikingly, the studies presented here reveal that both HpNikR and HpArsR-P are necessary for maximum Ni(II) dependent regulation of urease in vivo as well as the maximal response to acid shock. The two proteins are not independent regulators but, instead, work cooperatively to regulate ureA transcription. This is the first time that “cross-talk” between HpNikR and HpArsRS has been demonstrated, and further studies will be required to tease out the interactions that promote this cooperative effect.
Author Contributions
Conceived and designed the experiments: AW, BC, DM, SM. Strain Construction: AW, BC, HG, JG. Performed the experiments: AW, HG, BC, DH, OP, SS. Contributed reagents/materials/analysis tools: MF, DH. Wrote the paper: AW, BC, DM, SM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
SLJM thanks NSF CHE1306208 for support of these studies. We are grateful to the members of the Merrell lab for their invaluable microbiology expertise. Prof. Edwin Pozharski (UMB) for assistance with PyMol, Prof. David Scott and Liz Marcus (UCLA School of Medicine) for the ΔarsS::kan-sacB deletion strain of H. pylori and Mohsin Khan (UMB) for editing Figure 1A. DSM thanks Prof. Andrew Snow and Sasha Larsen for their assistance with flow cytometry technique and analysis. The opinions and assertions contained herein are the private ones of the authors and are not to be construed as official or reflecting the views of the NIH, the NSF, the Department of Defense, or the Uniformed Services University of the Health Sciences.
References
Abraham, L. O., Li, Y., and Zamble, D. B. (2006). The metal- and DNA-binding activities of Helicobacter pylori NikR. J. Inorg. Biochem. 100, 1005–1014. doi: 10.1016/j.jinorgbio.2005.10.014
Bahlawane, C., Dian, C., Muller, C., Round, A., Fauquant, C., Schauer, K., et al. (2010). Structural and mechanistic insights into Helicobacter pylori NikR activation. Nucleic Acids Res. 38, 3106–3118. doi: 10.1093/nar/gkp1216
Baltrus, D. A., Amieva, M. R., Covacci, A., Lowe, T. M., Merrell, D. S., Ottemann, K. M., et al. (2009). The complete genome sequence of Helicobacter pylori strain G27. J. Bacteriol. 191, 447–448. doi: 10.1128/JB.01416-08
Beier, D., and Frank, R. (2000). Molecular characterization of two-component systems of Helicobacter pylori. J. Bacteriol. 182, 2068–2076. doi: 10.1128/JB.182.8.2068-2076.2000
Benanti, E. L., and Chivers, P. T. (2007). The N-terminal arm of the Helicobacter pylori Ni2+-dependent transcription factor NikR is required for specific DNA binding. J. Biol. Chem. 282, 20365–20375. doi: 10.1074/jbc.M702982200
Benanti, E. L., and Chivers, P. T. (2011). Helicobacter pylori NikR protein exhibits distinct conformations when bound to different promoters. J. Biol. Chem. 286, 15728–15737. doi: 10.1074/jbc.M110.196055
Benini, S., Cianci, M., and Ciurli, S. (2011). Holo-Ni2+ Helicobacter pylori NikR contains four square-planar nickel-binding sites at physiological pH. Dalton Trans. 40, 7831–7833. doi: 10.1039/c1dt11107h
Blaser, M. J. (1990). Helicobacter pylori and the pathogenesis of gastroduodenal inflammation. J. Infect. Dis. 161, 626–633. doi: 10.1093/infdis/161.4.626
Borin, B. N., Tang, W., and Krezel, A. M. (2014). Helicobacter pylori RNA polymerase alpha-subunit C-terminal domain shows features unique to varepsilon-proteobacteria and binds NikR/DNA complexes. Protein Sci. 23, 454–463. doi: 10.1002/pro.2427
Carpenter, B. M., McDaniel, T. K., Whitmire, J. M., Gancz, H., Guidotti, S., Censini, S., et al. (2007). Expanding the Helicobacter pylori genetic toolbox: modification of an endogenous plasmid for use as a transcriptional reporter and complementation vector. Appl. Environ. Microbiol. 73, 7506–7514. doi: 10.1128/AEM.01084-07
Carter, E. L., Flugga, N., Boer, J. L., Mulrooney, S. B., and Hausinger, R. P. (2009). Interplay of metal ions and urease. Metallomics 1, 207–221. doi: 10.1039/b903311d
Chivers, P. T., and Sauer, R. T. (1999). NikR is a ribbon-helix-helix DNA-binding protein. Protein Sci. 8, 2494–2500. doi: 10.1110/ps.8.11.2494
Chivers, P. T., and Sauer, R. T. (2000). Regulation of high affinity nickel uptake in bacteria. Ni2+-Dependent interaction of NikR with wild-type and mutant operator sites. J. Biol. Chem. 275, 19735–19741. doi: 10.1074/jbc.M002232200
Chivers, P. T., and Sauer, R. T. (2002). NikR repressor: high-affinity nickel binding to the C-terminal domain regulates binding to operator DNA. Chem. Biol. 9, 1141–1148. doi: 10.1016/S1074-5521(02)00241-7
Chivers, P. T., and Tahirov, T. H. (2005). Structure of Pyrococcus horikoshii NikR: nickel sensing and implications for the regulation of DNA recognition. J. Mol. Biol. 348, 597–607. doi: 10.1016/j.jmb.2005.03.017
Contreras, M., Thiberge, J. M., Mandrand-Berthelot, M. A., and Labigne, A. (2003). Characterization of the roles of NikR, a nickel-responsive pleiotropic autoregulator of Helicobacter pylori. Mol. Microbiol. 49, 947–963. doi: 10.1046/j.1365-2958.2003.03621.x
Copass, M., Grandi, G., and Rappuoli, R. (1997). Introduction of unmarked mutations in the Helicobacter pylori vacA gene with a sucrose sensitivity marker. Infect. Immun. 65, 1949–1952.
Covacci, A., Censini, S., Bugnoli, M., Petracca, R., Burroni, D., Macchia, G., et al. (1993). Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. U.S.A. 90, 5791–5795. doi: 10.1073/pnas.90.12.5791
Cover, T. L., and Blaser, M. J. (1992). Helicobacter pylori and gastroduodenal disease. Annu. Rev. Med. 43, 135–145. doi: 10.1146/annurev.me.43.020192.001031
Danielli, A., Romagnoli, S., Roncarati, D., Costantino, L., Delany, I., and Scarlato, V. (2009). Growth phase and metal-dependent transcriptional regulation of the fecA genes in Helicobacter pylori. J. Bacteriol. 191, 3717–3725. doi: 10.1128/JB.01741-08
Davis, G. S., Flannery, E. L., and Mobley, H. L. (2006). Helicobacter pylori HP1512 is a nickel-responsive NikR-regulated outer membrane protein. Infect. Immun. 74, 6811–6820. doi: 10.1128/IAI.01188-06
Delany, I., Ieva, R., Soragni, A., Hilleringmann, M., Rappuoli, R., and Scarlato, V. (2005). In vitro analysis of protein-operator interactions of the NikR and Fur metal-responsive regulators of coregulated genes in Helicobacter pylori. J. Bacteriol. 187, 7703–7715. doi: 10.1128/JB.187.22.7703-7715.2005
Delany, I., Spohn, G., Rappuoli, R., and Scarlato, V. (2001). The Fur repressor controls transcription of iron-activated and -repressed genes in Helicobacter pylori. Mol. Microbiol. 42, 1297–1309. doi: 10.1046/j.1365-2958.2001.02696.x
Dietz, P., Gerlach, G., and Beier, D. (2002). Identification of target genes regulated by the two-component system HP166-HP165 of Helicobacter pylori. J. Bacteriol. 184, 350–362. doi: 10.1128/JB.184.2.350-362.2002
Dosanjh, N. S., Hammerbacher, N. A., and Michel, S. L. J. (2007). Characterization of the Helicobacter pylori NikR-P(ureA) DNA interaction: metal ion requirements and sequence specificity. Biochemistry 46, 2520–2529. doi: 10.1021/bi062092w
Dosanjh, N. S., and Michel, S. L. J. (2006). Microbial nickel metalloregulation: NikRs for nickel ions. Curr. Opin. Chem. Biol. 10, 123–130. doi: 10.1016/j.cbpa.2006.02.011
Dosanjh, N. S., West, A. L., and Michel, S. L. J. (2009). Helicobacter pylori NikR's interaction with DNA: a two-tiered mode of recognition. Biochemistry 48, 527–536. doi: 10.1021/bi801481j
Dunn, B. E., Cohen, H., and Blaser, M. J. (1997). Helicobacter pylori. Clin. Microbiol. Rev. 10, 720–741.
Ernst, F. D., Kuipers, E. J., Heijens, A., Sarwari, R., Stoof, J., Penn, C. W., et al. (2005). The nickel-responsive regulator NikR controls activation and repression of gene transcription in Helicobacter pylori. Infect. Immun. 73, 7252–7258. doi: 10.1128/IAI.73.11.7252-7258.2005
Ernst, F. D., Stoof, J., Horrevoets, W. M., Kuipers, E. J., Kusters, J. G., and van Vliet, A. H. (2006). NikR mediates nickel-responsive transcriptional repression of the Helicobacter pylori outer membrane proteins FecA3 (HP1400) and FrpB4 (HP1512). Infect. Immun. 74, 6821–6828. doi: 10.1128/IAI.01196-06
Evans, S. E., and Michel, S. L. J. (2012). Dissecting the role of DNA sequence in Helicobacter pylori NikR/DNA recognition. Dalton Trans. 41, 7946–7951. doi: 10.1039/c2dt30504f
Gilbreath, J. J., West, A. L., Pich, O. Q., Carpenter, B. M., Michel, S., and Merrell, D. S. (2012). Fur activates expression of the 2-oxoglutarate oxidoreductase genes (oorDABC) in Helicobacter pylori. J. Bacteriol. 194, 6490–6497. doi: 10.1128/JB.01226-12
Goodwin, A. C., Weinberger, D. M., Ford, C. B., Nelson, J. C., Snider, J. D., Hall, J. D., et al. (2008). Expression of the Helicobacter pylori adhesin SabA is controlled via phase variation and the ArsRS signal transduction system. Microbiology 154, 2231–2240. doi: 10.1099/mic.0.2007/016055-0
Gupta, S. S., Borin, B. N., Cover, T. L., and Krezel, A. M. (2009). Structural analysis of the DNA-binding domain of the Helicobacter pylori response regulator ArsR. J. Biol. Chem. 284, 6536–6545. doi: 10.1074/jbc.M804592200
Jones, M. D., Ademi, I., Yin, X., Gong, Y., and Zamble, D. B. (2015). Nickel-responsive regulation of two novel Helicobacter pylori NikR-targeted genes. Metallomics 7, 662–673. doi: 10.1039/c4mt00210e
Joseph, B., and Beier, D. (2007). Global analysis of two-component gene regulation in H. pylori by mutation analysis and transcriptional profiling. Meth. Enzymol. 423, 514–530. doi: 10.1016/s0076-6879(07)23025-3
Kusters, J. G., van Vliet, A. H., and Kuipers, E. J. (2006). Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 19, 449–490. doi: 10.1128/CMR.00054-05
Lakowicz, J. R. (1999). Principles of Fluorescence Spectroscopy. New York, NY: Kluwer Academic/Plenium Publishers.
Li, Y., and Zamble, D. B. (2009). pH-responsive DNA-binding activity of Helicobacter pylori NikR. Biochemistry 48, 2486–2496. doi: 10.1021/bi801742r
Loh, J. T., Gupta, S. S., Friedman, D. B., Krezel, A. M., and Cover, T. L. (2010). Analysis of protein expression regulated by the Helicobacter pylori ArsRS two-component signal transduction system. J. Bacteriol. 192, 2034–2043. doi: 10.1128/JB.01703-08
Loughlin, M. F. (2003). Novel therapeutic targets in Helicobacter pylori. Expert Opin. Ther. Targets 7, 725–735. doi: 10.1517/14728222.7.6.725
Maier, R. J., Benoit, S. L., and Seshadri, S. (2007). Nickel-binding and accessory proteins facilitating Ni-enzyme maturation in Helicobacter pylori. Biometals 20, 655–664. doi: 10.1007/s10534-006-9061-8
Marcus, E. A., Sachs, G., Wen, Y., Feng, J., and Scott, D. R. (2012). The role of the Helicobacter pylori sensor kinase ArsS in protein trafficking and acid acclimation. J. Bacteriol. 194, 5545–5551. doi: 10.1128/JB.01263-12
Marshall, B. J., and Goodwin, C. S. (1987). Revised nomenclature of Campylobacter pyloridis. Int. J. Syst. Bacteriol. 37, 68. doi: 10.1099/00207713-37-1-68
Marshall, B. J., and Warren, J. R. (1984). Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1, 1311–1315. doi: 10.1016/S0140-6736(84)91816-6
Martin, R. G., Jair, K. W., Wolf, R. E. Jr., and Rosner, J. L. (1996). Autoactivation of the marRAB multiple antibiotic resistance operon by the MarA transcriptional activator in Escherichia coli. J. Bacteriol. 178, 2216–2223.
Martin, R. G., and Rosner, J. L. (1997). Fis, an accessorial factor for transcriptional activation of the mar (multiple antibiotic resistance) promoter of Escherichia coli in the presence of the activator MarA, SoxS, or Rob. J. Bacteriol. 179, 7410–7419.
McCleary, W. R., and Stock, J. B. (1994). Acetyl phosphate and the activation of two-component response regulators. J. Biol. Chem. 269, 31567–31572.
McDaniel, T. K., Dewalt, K. C., Salama, N. R., and Falkow, S. (2001). New approaches for validation of lethal phenotypes and genetic reversion in Helicobacter pylori. Helicobacter 6, 15–23. doi: 10.1046/j.1523-5378.2001.00001.x
McMurry, L. M., and Levy, S. B. (2010). Evidence that regulatory protein MarA of Escherichia coli represses rob by steric hindrance. J. Bacteriol. 192, 3977–3982. doi: 10.1128/JB.00103-10
Michan, C., Manchado, M., and Pueyo, C. (2002). SoxRS down-regulation of rob transcription. J. Bacteriol. 184, 4733–4738. doi: 10.1128/JB.184.17.4733-4738.2002
Miller, P. F., Gambino, L. F., Sulavik, M. C., and Gracheck, S. J. (1994). Genetic relationship between soxRS and mar loci in promoting multiple antibiotic resistance in Escherichia coli. Antimicrob. Agents Chemother. 38, 1773–1779. doi: 10.1128/AAC.38.8.1773
Muller, C., Bahlawane, C., Aubert, S., Delay, C. M., Schauer, K., Michaud-Soret, I., et al. (2011). Hierarchical regulation of the NikR-mediated nickel response in Helicobacter pylori. Nucleic Acids Res. 39, 7564–7575. doi: 10.1093/nar/gkr460
Muller, S., Gotz, M., and Beier, D. (2009). Histidine residue 94 is involved in pH sensing by histidine kinase ArsS of Helicobacter pylori. PLoS ONE 4:e6930. doi: 10.1371/journal.pone.0006930
Parker, S. C., and Tullius, T. D. (2011). DNA shape, genetic codes, and evolution. Curr. Opin. Struct. Biol. 21, 342–347. doi: 10.1016/j.sbi.2011.03.002
Pflock, M., Finsterer, N., Joseph, B., Mollenkopf, H., Meyer, T. F., and Beier, D. (2006a). Characterization of the ArsRS regulon of Helicobacter pylori, involved in acid adaptation. J. Bacteriol. 188, 3449–3462. doi: 10.1128/JB.188.10.3449-3462.2006
Pflock, M., Kennard, S., Delany, I., Scarlato, V., and Beier, D. (2005). Acid-induced activation of the urease promoters is mediated directly by the ArsRS two-component system of Helicobacter pylori. Infect. Immun. 73, 6437–6445. doi: 10.1128/IAI.73.10.6437-6445.2005
Pflock, M., Kennard, S., Finsterer, N., and Beier, D. (2006b). Acid-responsive gene regulation in the human pathogen Helicobacter pylori. J. Biotechnol. 126, 52–60. doi: 10.1016/j.jbiotec.2006.03.045
Rohs, R., Jin, X., West, S. M., Joshi, R., Honig, B., and Mann, R. S. (2010). Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 79, 233–269. doi: 10.1146/annurev-biochem-060408-091030
Romagnoli, S., Agriesti, F., and Scarlato, V. (2011). In vivo recognition of the fecA3 target promoter by Helicobacter pylori NikR. J. Bacteriol. 193, 1131–1141. doi: 10.1128/JB.01153-10
Sachs, G., Kraut, J. A., Wen, Y., Feng, J., and Scott, D. R. (2006). Urea transport in bacteria: acid acclimation by gastric Helicobacter spp. J. Membr. Biol. 212, 71–82. doi: 10.1007/s00232-006-0867-7
Sachs, G., Weeks, D. L., Wen, Y., Marcus, E. A., Scott, D. R., and Melchers, K. (2005). Acid acclimation by Helicobacter pylori. Physiology (Bethesda) 20, 429–438. doi: 10.1152/physiol.00032.2005
Schar, J., Sickmann, A., and Beier, D. (2005). Phosphorylation-independent activity of atypical response regulators of Helicobacter pylori. J. Bacteriol. 187, 3100–3109. doi: 10.1128/JB.187.9.3100-3109.2005
Schneiders, T., and Levy, S. B. (2006). MarA-mediated transcriptional repression of the rob promoter. J. Biol. Chem. 281, 10049–10055. doi: 10.1074/jbc.M512097200
Schreiter, E. R., and Drennan, C. L. (2007). Ribbon-helix-helix transcription factors: variations on a theme. Nat. Rev. Microbiol. 5, 710–720. doi: 10.1038/nrmicro1717
Scott, D. R., Marcus, E. A., Wen, Y., Oh, J., and Sachs, G. (2007). Gene expression in vivo shows that Helicobacter pylori colonizes an acidic niche on the gastric surface. Proc. Natl. Acad. Sci. U.S.A. 104, 7235–7240. doi: 10.1073/pnas.0702300104
Sepulveda, A. R., and Coelho, L. G. (2002). Helicobacter pylori and gastric malignancies. Helicobacter 7(Suppl. 1), 37–42. doi: 10.1046/j.1523-5378.7.s1.6.x
Stoof, J., Kuipers, E. J., and van Vliet, A. H. (2010). Characterization of NikR-responsive promoters of urease and metal transport genes of Helicobacter mustelae. Biometals 23, 145–159. doi: 10.1007/s10534-009-9275-7
Thompson, L. J., Merrell, D. S., Neilan, B. A., Mitchell, H., Lee, A., and Falkow, S. (2003). Gene expression profiling of Helicobacter pylori reveals a growth-phase-dependent switch in virulence gene expression. Infect. Immun. 71, 2643–2655. doi: 10.1128/IAI.71.5.2643-2655.2003
van Vliet, A. H., Ernst, F. D., and Kusters, J. G. (2004a). NikR-mediated regulation of Helicobacter pylori acid adaptation. Trends Microbiol. 12, 489–494. doi: 10.1016/j.tim.2004.09.005
van Vliet, A. H., Kuipers, E. J., Stoof, J., Poppelaars, S. W., and Kusters, J. G. (2004b). Acid-responsive gene induction of ammonia-producing enzymes in Helicobacter pylori is mediated via a metal-responsive repressor cascade. Infect. Immun. 72, 766–773. doi: 10.1128/IAI.72.2.766-773.2004
van Vliet, A. H., Poppelaars, S. W., Davies, B. J., Stoof, J., Bereswill, S., Kist, M., et al. (2002). NikR mediates nickel-responsive transcriptional induction of urease expression in Helicobacter pylori. Infect. Immun. 70, 2846–2852. doi: 10.1128/IAI.70.6.2846-2852.2002
Warner, D. M., and Levy, S. B. (2010). Different effects of transcriptional regulators MarA, SoxS and Rob on susceptibility of Escherichia coli to cationic antimicrobial peptides (CAMPs): Rob-dependent CAMP induction of the marRAB operon. Microbiology 156, 570–578. doi: 10.1099/mic.0.033415-0
Wen, Y., Feng, J., Scott, D. R., Marcus, E. A., and Sachs, G. (2006). Involvement of the HP0165-HP0166 two-component system in expression of some acidic-pH-upregulated genes of Helicobacter pylori. J. Bacteriol. 188, 1750–1761. doi: 10.1128/JB.188.5.1750-1761.2006
Wen, Y., Feng, J., Scott, D. R., Marcus, E. A., and Sachs, G. (2007). The HP0165-HP0166 two-component system (ArsRS) regulates acid-induced expression of HP1186 alpha-carbonic anhydrase in Helicobacter pylori by activating the pH-dependent promoter. J. Bacteriol. 189, 2426–2434. doi: 10.1128/JB.01492-06
Wen, Y., Feng, J., Scott, D. R., Marcus, E. A., and Sachs, G. (2009). The pH-responsive regulon of HP0244 (FlgS), the cytoplasmic histidine kinase of Helicobacter pylori. J. Bacteriol. 191, 449–460. doi: 10.1128/JB.01219-08
Wen, Y., Marcus, E. A., Matrubutham, U., Gleeson, M. A., Scott, D. R., and Sachs, G. (2003). Acid-adaptive genes of Helicobacter pylori. Infect. Immun. 71, 5921–5939. doi: 10.1128/IAI.71.10.5921-5939.2003
West, A. L., Evans, S. E., Gonzalez, J. M., Carter, L. G., Tsuruta, H., Pozharski, E., et al. (2012). Ni(II) coordination to mixed sites modulates DNA binding of HpNikR via a long-range effect. Proc. Natl. Acad. Sci. U.S.A. 109, 5633–5638. doi: 10.1073/pnas.1120283109
West, A. L., St John, F., Lopes, P. E., Mackerell, A. D. Jr., Pozharski, E., and Michel, S. L. J. (2010). Holo-Ni(II)HpNikR is an asymmetric tetramer containing two different nickel-binding sites. J. Am. Chem. Soc. 132, 14447–14456. doi: 10.1021/ja104118r
Wolfram, L., Haas, E., and Bauerfeind, P. (2006). Nickel represses the synthesis of the nickel permease NixA of Helicobacter pylori. J. Bacteriol. 188, 1245–1250. doi: 10.1128/JB.188.4.1245-1250.2006
Keywords: nikR, arsRS, pylori, urease, Helicobacter, regulation, pH
Citation: Carpenter BM, West AL, Gancz H, Servetas SL, Pich OQ, Gilbreath JJ, Hallinger DR, Forsyth MH, Merrell DS and Michel SLJ (2015) Crosstalk between the HpArsRS two-component system and HpNikR is necessary for maximal activation of urease transcription. Front. Microbiol. 6:558. doi: 10.3389/fmicb.2015.00558
Received: 26 March 2015; Accepted: 20 May 2015;
Published: 12 June 2015.
Edited by:
Beiyan Nan, University of California, Berkeley, USAReviewed by:
Deborah Beth Zamble, University of Toronto, CanadaAndrzej M. Krezel, Washington University School of Medicine, USA
Copyright © 2015 Carpenter, West, Gancz, Servetas, Pich, Gilbreath, Hallinger, Forsyth, Merrell and Michel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: D. Scott Merrell, Department of Microbiology and Immunology, Uniformed Services University of the Health Sciences, 4301 Jones Bridge Road, Bethesda, MD 20814, USA,ZG91Z2xhcy5tZXJyZWxsQHVzdWhzLmVkdQ==;
Sarah L. J. Michel, Department of Pharmaceutical Sciences, School of Pharmacy, University of Maryland, 20 Penn Street, Baltimore, MD 21201, USA,c21pY2hlbEByeC51bWFyeWxhbmQuZWR1
†These authors have contributed equally to this work.