 Thangavel Samikkannu*Deepa RanjithKurapati V. K. RaoVenkata S. R. AtluriEmely PimentelNazira El-HageMadhavan P. N. Nair*
Thangavel Samikkannu*Deepa RanjithKurapati V. K. RaoVenkata S. R. AtluriEmely PimentelNazira El-HageMadhavan P. N. Nair*- Department of Immunology, Institute of NeuroImmune Pharmacology, College of Medicine, Florida International University, Miami, FL, USA
HIV infection and illicit drugs are known to induce oxidative stress and linked with severity of viral replication, disease progression, impaired cell cycle regulation and neurodegeneration. Studies have shown that morphine accelerates HIV infection and disease progression mediated by Reactive oxygen species (ROS). Oxidative stress impact redox balance and ROS production affect cell cycle regulation. However, the role of morphine in HIV associated acceleration of oxidative stress and its link to cell cycle regulation and neurodegeneration has not been elucidated. The aim of present study is to elucidate the mechanism of oxidative stress induced glutathione synthases (GSS), super oxide dismutase (SOD), and glutathione peroxidase (GPx) impact cell cycle regulated protein cyclin-dependent kinase 1, cell division cycle 2 (CDK-1/CDC-2), cyclin B, and cell division cycle 25C (CDC-25C) influencing neuronal dysfunction by morphine co-morbidity with HIV-1 gp120. It was observed that redox imbalance inhibited the GSS, GPx and increased SOD which, subsequently inhibited CDK-1/CDC-2 whereas cyclin B and CDC-25C significantly up regulated in HIV-1 gp120 with morphine compared to either HIV-1 gp120 or morphine treated alone in human microglial cell line. These results suggest that HIV positive morphine users have increased levels of oxidative stress and effect of cell cycle machinery, which may cause the HIV infection and disease progression.
Introduction
HIV infection is a major cause of human death and globally it was estimated that 35 million people are living with HIV (UNAIDS/WHO, 2014). HIV comorbidity with illicit drugs remains a global health problem. Illicit drugs of abuse including opiate is a significant risk factor for HIV infection and AIDS disease progression (Hu et al., 2012; Masvekar et al., 2014; El-Hage et al., 2015). Opiate causes deterioration of neuronal functions in a significant proportion of users resulting in the development of neuronal impairments and HIV associated neurocognitive dysfunction (HAND). Previous studies demonstrate that heroin is deacetylated to morphine, which can affect the central nervous system (CNS), and is very often associated with HIV infection and activated macrophages/microglia, are the major reservoir of viral replication and disease progression (Navia et al., 1986; Kure et al., 1990; Brzezinski et al., 1997). HIV-1 infection is known to cause CNS dysfunction and primarily affects microglial cells and subsequently impact astrocytes and neurons. Clinical observations suggest that selective regions of brain such as the striatum and the hippocampus highly express opioid receptors and have been associated with increased viral titers in HIV-infected patients (Mansour et al., 1987; Mansour and Watson, 1993).
The HIV-1 envelop protein, glycoprotein 120 (gp120) is required for viral entry and it facilitates the viral replication and disease progression mediated by oxidative stress, which subsequently affects the CNS (Brenneman et al., 1994; Galicia et al., 2002). Previous studies have shown that HIV-1 gp120 associated with increased secretion of inflammatory cytokines and chemokines (Holguin et al., 2004), glutamate, arachidonic acid and its metabolites prostaglandin E2 (PGE2) and 5-lipoxygenase (5-LOX; Samikkannu et al., 2011), nitric oxide (NO) and reactive oxygen species (ROS; Ronaldson and Bendayan, 2008; Lisi et al., 2014). In microglia, gp120 induces oxidative stress, ROS, TNF-α, and MCP-1 production and subsequently leading to neuronal cell death (Guo et al., 2013). Also, morphine was found to induce oxidative stress in in vitro as well as in vivo (Kovacic and Cooksy, 2005; Ma et al., 2015). Studies have also shown that morphine impaired cellular function mediated by oxidative stress which affects the CNS (Moron et al., 2009; Beltran-Campos et al., 2015).
Studies have documented that increased level of oxidative stress and ROS is associated with HIV-associated neurocognitive impairment (Shah et al., 2013). Free radicals H2O2 and O2–, induced by oxidative stress are known to generate cellular damage in multiple diseases, including HIV/AIDS (Kashou and Agarwal, 2011). Glutathione (GSH) is a tri peptide and known to be important cell protectant in intracellular anti-oxidant defense mechanisms, and its reduced levels have been correlated with impaired neuronal function (Aoyama et al., 2008). However, oxidative stress and cell cycle reentry seems counterintuitive. Previous report demonstrated that oxidative stress on the cell cycle reveals that increased ROS-induced DNA damage is correlated with cell cycle arrest (Pyo et al., 2013). However, whether ROS-induced stress response undergoes growth arrest or apoptosis may depend in part on where the cell resides in the cell cycle when insulted. Extensive studies have consistently demonstrated that morphine consumption and HIV infection further stimulate the sensitivity and neuronal dysfunction severity that causes HAND (Grigoryan et al., 2010; Masvekar et al., 2014). HIV infected morphine users have synergistically potentiated viral replication and disease progression as compared with HIV positive subjects (Roy et al., 2011; Dutta and Roy, 2012; Masvekar et al., 2014; El-Hage et al., 2015). Despite mounting evidence suggesting morphine use may aggravate HIV infection, mechanistic studies determining the mutual role of morphine and HIV infection on cell cycle regulation and their role in neurodegeneration are yet to be determined. Therefore, we have investigated the mechanism of HIV-1gp120 induced oxidative stress altered cell cycle regulation and morphine co-morbidity accelerated effects in microglial cells.
In this study, we have investigated the effect of HIV-1 gp120 in combination with morphine induced oxidative stress that affect the redox expression in GSS, SOD, GPx, and cell cycle arrest in G0 phase leads CDK-1/CDC-/2, cyclin B, and CDC-25C associated neuropathogenesis. We showed that morphine with HIV-1 gp120 exacerbates the oxidative stress and cell cycle arrest in microglial mediated CNS dysfunction.
Materials and Methods
Cell Culture and Reagents
Cell culture reagents were purchased from Sciencell (Carlsbad, CA, USA). The glutathione synthetase (GSS), super oxide dismutase (SOD), glutathione peroxide (GPx), and CDC25C antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The mouse anti-cyclin B and CDK-1/CDC-2 antibodies were purchased from BD Transduction Laboratories (San Jose, CA, USA). The goat anti-rabbit IgG and goat anti-mouse IgG antibodies were purchased from Santa Cruz Biotechnology. The propidium iodide was purchased from PD bioscience (San Jose, CA, USA). Electrophoresis reagents were purchased from Bio-Rad (Richmond, CA, USA), and nitrocellulose membranes were purchased from Amersham Scientific, Piscataway, NJ, USA. All other reagents were purchased from Sigma–Aldrich (St. Louis, MO, USA).
HIV-1 gp120 Recombinant Proteins
The HIV-1 gp120 (Bal) protein was obtained from the NIH AIDS Research and Reference Reagent Program. The recombinant gp120 proteins were tested >95% purity.
Microglial Cell Line (CHEM-5) Cultures
In this study, we used immortalized microglial CHEM-5 cells. Microglial cells were maintained in Dulbecco’s Modified Eagle Medium supplemented with fetal bovine serum to a final concentration of 10% (catalog # 30- 2020) and 1% antibiotic/antimycotic solution (Sigma-Aldrich, St. Louis, MO, USA).
RNA Extraction and Real time Quantitative PCR (qRT-PCR)
Total RNA from microglia was extracted using the Qiagen RNAeasy mini kit (Invitrogen Life Technologies, Carlsbad, CA, USA) by following the manufacturer’s instructions. The total RNA (3 μg) was used for the synthesis of cDNA. The amplification of cDNA was performed using specific primers for GSS (Assay ID Hs00609286_m1), GPx (Hs01591589_m1), and SOD (Hs00166575_m1); and β-actin (Hs99999903_m1) (Applied Biosystems, Foster City, CA, USA) was used as a housekeeping gene for quantifying real-time PCR. Relative abundance of each mRNA species was assessed using brilliant Q-PCR master mix from Stratagene using Mx3000P instrument that detects and plots the increase in fluorescence versus PCR cycle number to produce a continuous measure of PCR amplification. Relative mRNA species expression was quantitated and the mean fold change in expression of the target gene was calculated using the comparative CT method (Transcript Accumulation Index, TAI = 2_ΔΔCT). All data were controlled for quantity of RNA input by performing measurements on an endogenous reference gene, β-actin. In addition, results on RNA from treated samples were normalized to results obtained on RNA from the control, untreated sample.
Western Blot Analysis
To determine the GSS, SOD, GPx, CDK-1/CDC-2, cyclin B, and CDC25C protein alterations in microglial control, morphine, HIV-1 gp120 and morphine with HIV-1 gp120 treated cells, equal amount of total cellular protein were resolved on a 4–15% gradient polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane and incubated with their respective primary and secondary antibodies. Immunoreactive bands were visualized using a chemiluminescence western blotting system according to the manufacturers’ instructions (Amersham).
Analysis of HIV-1 gp120 and Morphine Impact on Cell Cycle Regulation
Morphine and HIV-1 gp120 induced DNA damage and cell cycle arrest were analyzed by flow cytometry in FACS caliber (BD Bioscience, San Jose, CA). Briefly, Microglial (5 × 105) were separately treated with morphine, HIV-1 gp120, and HIV-1 gp120 with morphine. Cells were harvested and washed twice with PBS. The cells were fixed in 0.5% paraformaldehyde and stained with PI solution after 24 h. The ModFit LT software program (Verity Software House) was used to determine the distribution of cells in subG0/G0 phase of the cell cycle.
Data Analysis
Statistical analysis was performed using GraphPad Prism- version 5. Differences between morphine, HIV-1 gp120 and morphine with HIV-1 gp120 treated and control were calculated using two-way ANOVA method. Values were expressed as mean ± standard error and a significance level of p < 0.05 was used.
Results
HIV-1 gp120 and Morphine Induced Oxidative Stress; and Redox Gene and Protein Expression in Microglial Cells
It is known that HIV infection; HIV-1 gp120 protein and morphine induced ROS production lead to cell death (Ronaldson and Bendayan, 2008; Ma et al., 2015). However, no information is available on HIV-1 gp120 and morphine induced ROS production and redox altered cell cycle arrest and neurodegeneration. Therefore, we determined whether HIV-1 gp120 induce redox expression, cell cycle regulation and neurodegeneration are accelerated by morphine in microglia. Observed results demonstrate that HIV-1 gp120 and morphine significantly increased oxidative stress and subsequently inhibited the redox expression in microglia. The data presented in Figures 1A,B show the dose response in morphine- (0–1000 nm/ml) and gp120—(0–200 ng/ml) treatment for 24 h and its effects on GSS gene expression in microglia, as assessed using quantitative real-time PCR. Microglia treated with morphine significantly down regulated GSS gene expression at 100 nm (p < 0.01), 500 nm (p < 0.001) and 1000 nm (p < 0.0001) compared to control. In HIV-1 gp120 effect on 50 ng (p < 0.01), 100 ng (p < 0.001) and 200 ng (p < 0.0001) compared to control was observed respectively. Figure 2 shows the effects of HIV-1 gp120, morphine and HIV-1 gp120 with morphine impact on redox gene GSS (A), SOD (B), and GPx (C) expression. These studies showed that HIV-1 gp120 and morphine treatment inhibit the levels of GSS and GPx whereas SOD significantly increased, and the combination of HIV-1 gp120 with morphine accelerates these effects compared with control. The present finding suggested that HIV-1 gp120 with morphine effects are accelerated as compared to either HIV-1 gp120 or morphine alone (Figures 2A–C).
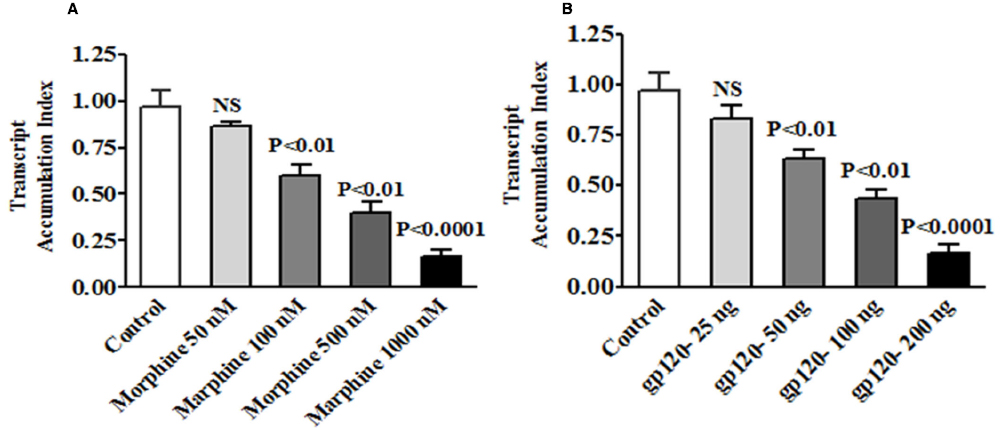
Figure 1. The kinetic studies of Morphine and HIV-1 gp120 protein impact redox dysfunction in GSS gene expression. Microglia (1 × 106 cells/ml) were treated with (A) morphine (0–1000 nM) and (B) HIV-1 gp120 (0–200 ng) for 24 h for dose response studies. RNA was extracted, reverse transcribed, and subjected to quantitative real-time PCR using specific primers for GSS and the housekeeping gene β-actin. Data are expressed as the mean ± SE of the TAI values from three independent experiments.
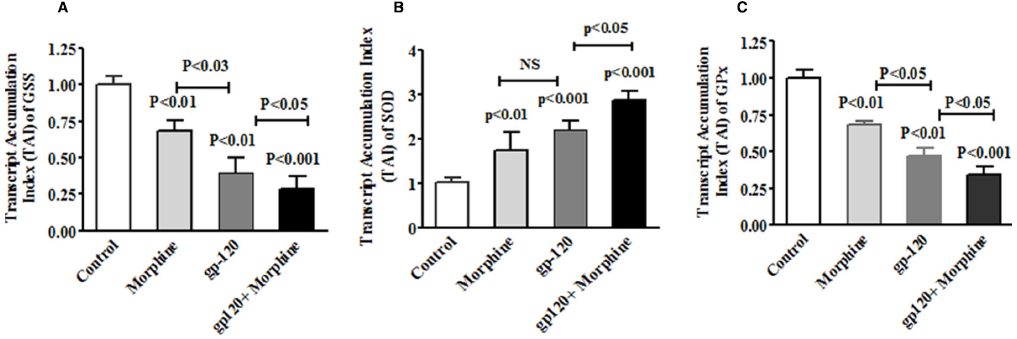
Figure 2. HIV-1 gp120 and morphine effect on redox genes GSS, SOD and GPx expression. Microglia (1 × 106 cells/ml) were treated with HIV-1 gp120 (100 ng), morphine (0.5 μM) and HIV-1 gp120 with morphine. Controls were maintained by drug free medium. The end of the incubation, RNA was extracted and reverse transcribed followed by quantitative real time PCR for GSS (A), SOD (B), GPx (C), and housekeeping β-actin specific primers. Data are expressed as mean ± SE of TAI values of three independent experiments.
Furthermore, we also examined whether HIV-1 gp120 induced oxidative and redox protein regulation is accelerated by morphine. Figure 3 shows the effects of HIV-1 gp120, morphine and HIV-1 gp120 with morphine effects on redox protein GSS (A), SOD (B), and GPx (C). These observed results confirm that HIV-1 gp120 induced redox changes were accelerated by morphine. Taken together, these finding suggested that oxidative stress induced redox changes in HIV infection along with morphine leads to neurodegeneration.
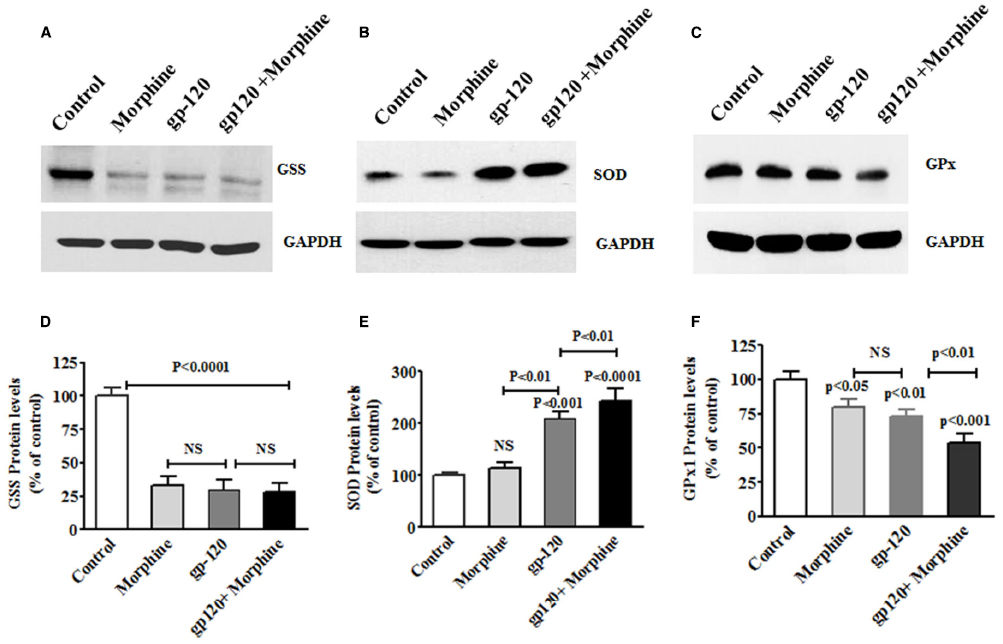
Figure 3. HIV-1 gp120 with morphine effect on redox expression. Microglia (1 × 106 cells/ ml) were treated with HIV-1 gp120 (100 ng), morphine (0.5 μM) and combination of HIV-1 gp120 with morphine. Controls were maintained by drug free medium. The end of the incubation, equal amount of protein lysate were resolved by 4–15% SDS-PAGE and protein expression were analyzed by Western blot showing GSS (A), SOD (B), and GPx (C). (D–F) represented % densitometric values of GSS, SOD, and GPx protein levels (% control). Data are expressed as mean ± SE of three independent experiments.
HIV-1 gp120 and Morphine Effects Cell Cycle Protein in Microglial cells
Oxidative stress is known to induce DNA damage and cell cycle arrest. Therefore, we wanted to test whether exposure to HIV-1 gp120 protein affect cell cycle protein and neurodegeneration. In the present study, HIV-1 gp120 with morphine treated microglial cells showed a significant decrease in CDK-1/CDC-2 whereas cyclin B and CDC25C increased protein expression at 24 h, compared with either HIV-1 gp120 or morphine alone. Figure 4 shows HIV-1 gp120 with morphine effect on CDK1/CDC-2 (A), cyclin B (B), and CDC-25C (C) protein expression as compared to the control. These observations confirmed that morphine accelerates HIV-1 gp120 induced cell cycle effect.
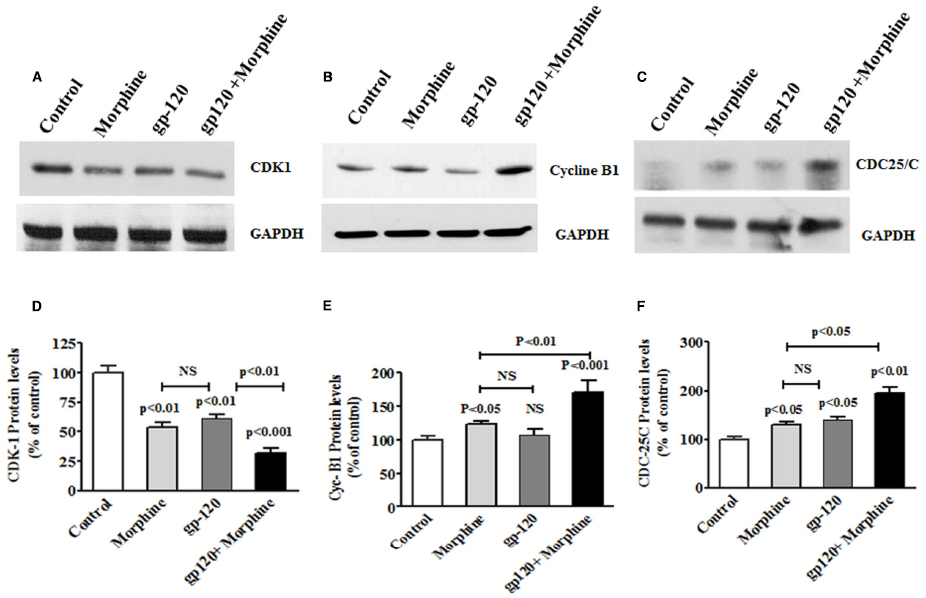
Figure 4. HIV-1 gp120 with morphine effect on cell cycle proteins. Microglia (1 × 106 cells/ml) were treated with HIV-1 gp120 (100 ng), morphine (0.5 μM) and combination of HIV-1 gp120 with morphine. Controls were maintained by drug free medium. The end of the incubation, equal amount of protein lysate were resolved by 4–15% SDS-PAGE and protein expression were analyzed by Western blot showing cycline B1 (A), CDK1 (B), and CDC25C (C). (D–F) represented % densitometric values of Cyc-B1, CDK1, and CDC25C protein levels (% control). Data are expressed as mean ± SE of three independent experiments.
HIV-1 gp120 and Morphine Accelerates Cell Cycle Arrest
HIV infection affects cell cycle regulation and ultimately leads to CNS dysfunctions (Amini et al., 2004; Zhao and Elder, 2005; Liu et al., 2007; Wang et al., 2010). Microglia is one of the major reservoirs of HIV infection and target of illicit drugs in the CNS. Therefore, we examined the effect of HIV-1 gp120 with morphine on cell cycle regulation in microglia. Figure 4 showed that HIV-1 gp120 and morphine treated microglia significantly inhibited cells at subG0 and G0 phase as compared with either HIV-1 gp120 or morphine alone (Figure 5). This study suggested that oxidative stress induced redox inhibition subsequently lead cell cycle arrest and this effect might be accelerated by morphine mediated neurodegeneration.
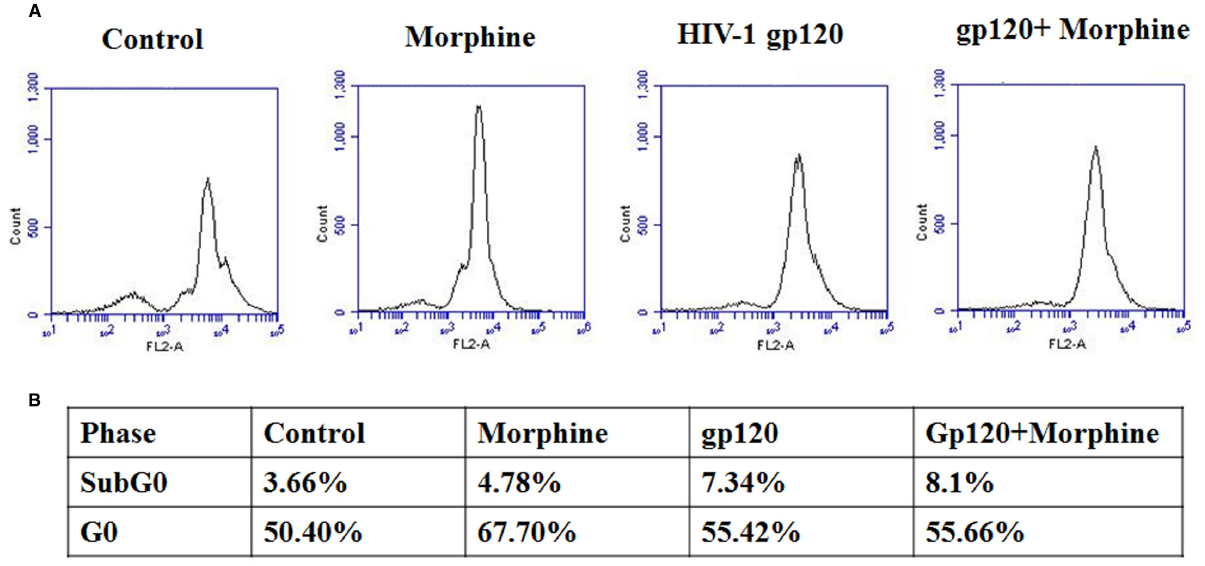
Figure 5. HIV-1 gp120 with morphine effect on cell cycle progression. Microglia (1 × 106 cells/ml) were treated with HIV-1 gp120 (100 ng), morphine (0.5 μM) and combination of HIV-1 gp120 with morphine and its effect on cell cycle transitions were analyzed by flow cytometry (A) and cell cycle phase are expressed as % (B).
Discussion
Studies have shown that oxidative stress plays a wide role in cellular function including immune and neuronal dysfunction, behavioral impairments in HIV-infection and substance abuse (Grigoryan et al., 2010; Masvekar et al., 2014); El-Hage et al., 2015). The oxidative stress induced ROS production is associated with HAD or HAND. Glutathione is an important player in the regulation of redox functions in cellular homeostasis (Schafer and Buettner, 2001). Studies have shown that HIV infection induced oxidative stress and ROS production subsequently affect redox proteins such as GSS, SOD and GPx in HIV infected patients and morphine users (Herzenberg et al., 1997; Koutsilieri et al., 1997; Bhaskar et al., 2015). However, overstimulation of ROS leads to inhibition in redox status GSH/GSSG (Koutsilieri et al., 1997), and subsequently impact the cell cycle machinery in cyclin B, CDK-1/CDC-2 and CDC-25C, which may play a vital role in neuronal dysfunction and disease progression in HAD patients (Klein and Ackerman, 2003; Morris et al., 2012). The CNS microglia cells are the major reservoirs of HIV infection and disease progression; they are modulated by cell cycle arrest induced by CDK-1/CDC-2, cyclin B and CDC-25C that lead to neurodegeneration (Cernak et al., 2005; Wang et al., 2008). However, there are no reports on the effects of HIV-1 gp120 and morphine induced redox imbalance that initiate cell cycle arrest and cellular mechanisms of neurodegeneration. The observed results provide new insights into the functional role of redox expression, which subsequently affects cell cycle machinery in HIV-1 gp120 with morphine.
In the present study, we have demonstrated for the first time that morphine, HIV-1 gp120 and HIV-1 gp120 with morphine showed increased oxidative stress and inhibit redox levels of GSS and GPx and, subsequently increased SOD mRNA and protein expression (Figures 1–3), associated with increased ROS production as compared to the control. It is known that redox dysfunction and GSH/GSSG ratio are the major players in maintaining cellular homeostasis. These studies suggest that morphine with HIV–1 gp120 may have an enhanced role of on oxidative stress as compared to the control. This is consistent with earlier reports of gp120 and morphine induced oxidative stress in microglia and macrophages, respectively where activation of the redox pathway has been observed (Bhat et al., 2004; Ronaldson and Bendayan, 2008; Lisi et al., 2014). Also, studies have shown that oxidative stress and redox impairments are associated with cell cycle arrest and DNA damage in the cerebral cortex brain regions (Klein and Ackerman, 2003). These studies further confirm that the cell cycle process in sub G0 and G0 stage are affected by morphine with HIV-1 gp120 and leading to neurodegeneration.
Moreover, our results show that in morphine, HIV-1 gp120, and morphine with HIV-1 gp120 induction of DNA damage and cell cycle arrest (Figures 4 and 5) is associated with inhibition of CDK-1/CDC-2 and subsequently increased cyclin B1 and CDC-25C. The main observation in this report is that HIV-1 gp120, and morphine with HIV-1 gp120 have higher impact on cell cycle arrest in the G0 phase due to ROS production and subsequently reduced the level of redox expression and CDK-1 inhibition. However, the HIV positive morphine users have higher ROS levels and DNA damage. This suggests that there is an interactive role between morphine and HIV that synergistically potentiates and increases co-morbidity when compared to either morphine use or HIV infection. These results confirm the previous report of HIV-1 gp120 induced cell cycle arrest in CD4+ T Cells (Kolesnitchenko et al., 1995).
Previous studies demonstrated that HIV-Vpr and Nef proteins affect cell cycle regulation which may impact cell cycle process (Yang et al., 2002; Wen et al., 2007; Li et al., 2010). In the present study, HIV-1 gp120 with morphine showed inhibition of CDK-1/CDC-2 and subsequent activation of cyclin B1 and CDC-25C protein as compared to the control. Also, cell cycle arrest in the G0 phase inhibited CDK-1/CDC-2 phosphorylation in microglia could lead to an enhanced DNA damages, which alters neuronal function and resulting in neurodegeneration. Furthermore, it is known that Gi/Gq-induced synergistic activation of ERK and subsequently affect between Ca2+ and Src family tyrosine kinases (Chan et al., 2005). However, Morphine and HIV-1 gp120 also known to activate ERK and downstream effect on CREB transcription mediated by Ca2+. Also, HIV-1 gp120 induced cell cycle arrest and these effects are accelerated by morphine in additive manner and this effect at Gi/Gq phase. These results suggest that viral replication in HIV infected microglial play a wide role in neuronal dysfunction, once HIV-1 gp120 or morphine effects altered DNA damages and subsequently increased CDC-25C in cell cycle arrest mediated by cyclin B.
Overall, the data provides evidence of interaction of morphine and HIV-1 gp120 leading to increase in oxidative stress and redox inhibition which is associated with increased levels of DNA damage and subsequently affecting cell cycle process. Based on these results, redox imbalance potentiates the HIV viral replication and disease progression by impairing the neuronal function, which may lead to neurodegeneration. The present study supports some of earlier reports in the dysfunction of cell cycle machinery and increased neurodegeneration in morphine using HIV infected patients as well as in the presence of HIV derived proteins (Krathwohl and Kaiser, 2004; Malik et al., 2014).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
The present study was supported by grants from National Institute of Health (NIH): DA 027049, DA 025576 and MH085259.
References
Amini, S., Khalili, K., and Sawaya, B. E. (2004). Effect of HIV-1 Vpr on cell cycle regulators. DNA Cell Biol. 23, 249–260. doi: 10.1089/104454904773819833
Aoyama, K., Watabe, M., and Nakaki, T. (2008). Regulation of neuronal glutathione synthesis. J. Pharmacol. Sci. 108, 227–238. doi: 10.1254/jphs.08R01CR
Beltran-Campos, V., Silva-Vera, M., Garcia-Campos, M. L., and Diaz-Cintra, S. (2015). Effects of morphine on brain plasticity. Neurologia 30, 176–180. doi: 10.1016/j.nrleng.2014.08.001
Bhaskar, A., Munshi, M., Khan, S. Z., Fatima, S., Arya, R., Jameel, S., et al. (2015). Measuring glutathione redox potential of HIV-1-infected macrophages. J. Biol. Chem. 290, 1020–1038. doi: 10.1074/jbc.M114.588913
Bhat, R. S., Bhaskaran, M., Mongia, A., Hitosugi, N., and Singhal, P. C. (2004). Morphine-induced macrophage apoptosis: oxidative stress and strategies for modulation. J. Leukoc. Biol. 75, 1131–1138. doi: 10.1189/jlb.1203639
Brenneman, D. E., Mccune, S. K., Mervis, R. F., and Hill, J. M. (1994). gp120 as an etiologic agent for NeuroAIDS: neurotoxicity and model systems. Adv. Neuroimmunol. 4, 157–165. doi: 10.1016/S0960-5428(06)80252-4
Brzezinski, M. R., Spink, B. J., Dean, R. A., Berkman, C. E., Cashman, J. R., and Bosron, W. F. (1997). Human liver carboxylesterase hCE-1: binding specificity for cocaine, heroin, and their metabolites and analogs. Drug Metab. Dispos. 25, 1089–1096.
Cernak, I., Stoica, B. A., Byrnes, K. R., Giovanni, S. D., and Faden, A. I. (2005). Role of the cell cycle in the pathobiology of central nervous system trauma. Cell Cycle 4, 1286–1293. doi: 10.4161/cc.4.9.1996
Chan, A. S. L., Yeung, W. W. S., and Wong, Y. H. (2005). Integration of G protein signals by extracellular signal-regulated protein kinases in SK-N-MC neuroepithelioma cells. J. Neurochem. 94, 1457–1470. doi: 10.1111/j.1471-4159.2005.03304.x
Dutta, R., and Roy, S. (2012). Mechanism(s) involved in opioid drug abuse modulation of HAND. Curr. HIV Res. 10, 469–477. doi: 10.2174/157016212802138805
El-Hage, N., Rodriguez, M., Dever, S. M., Masvekar, R. R., Gewirtz, D. A., and Shacka, J. J. (2015). HIV-1 and morphine regulation of autophagy in microglia: limited interactions in the context of HIV-1 infection and opioid Abuse. J. Virol. 89, 1024–1035. doi: 10.1128/jvi.02022-14
Galicia, O., Sanchez-Alavez, M., Mendez Diaz, M., Navarro, L., and Prospero-Garcia, O. (2002). [HIV glycoprotein 120: possible etiological agent of AIDS-associated dementia]. Rev. Invest. Clin. 54, 437–452.
Grigoryan, A., Shouse, R. L., Durant, T., Mastro, T. D., Espinoza, L., Chen, M., et al. (2010). HIV infection among injection-drug users-34 states, 2004–2007. JAMA 303, 126–128.
Guo, L., Xing, Y., Pan, R., Jiang, M., Gong, Z., Lin, L., et al. (2013). Curcumin protects microglia and primary rat cortical neurons against HIV-1 gp120-mediated inflammation and apoptosis. PLoS ONE 8:e70565. doi: 10.1371/journal.pone.0070565
Herzenberg, L. A., De Rosa, S. C., Dubs, J. G., Roederer, M., Anderson, M. T., Ela, S. W., et al. (1997). Glutathione deficiency is associated with impaired survival in HIV disease. Proc. Natl. Acad. Sci. U.S.A. 94, 1967–1972. doi: 10.1073/pnas.94.5.1967
Holguin, A., O’connor, K. A., Biedenkapp, J., Campisi, J., Wieseler-Frank, J., Milligan, E. D., et al. (2004). HIV-1 gp120 stimulates proinflammatory cytokine-mediated pain facilitation via activation of nitric oxide synthase-I (nNOS). Pain 110, 517–530. doi: 10.1016/j.pain.2004.02.018
Hu, G., Yao, H., Chaudhuri, A. D., Duan, M., Yelamanchili, S. V., Wen, H., et al. (2012). Exosome-mediated shuttling of microRNA-29 regulates HIV Tat and morphine-mediated Neuronal dysfunction. Cell Death Dis. 3, e381. doi: 10.1038/cddis.2012.114
Kashou, A., and Agarwal, A. (2011). Oxidants and antioxidants in the pathogenesis of HIV/AIDS. Open Reprod. Sci. J. 3, 154–161. doi: 10.2174/1874255601103010154
Klein, J. A., and Ackerman, S. L. (2003). Oxidative stress, cell cycle, and neurodegeneration. J. Clin. Invest. 111, 785–793. doi: 10.1172/jci18182
Kolesnitchenko, V., Wahl, L. M., Tian, H., Sunila, I., Tani, Y., Hartmann, D. P., et al. (1995). Human immunodeficiency virus 1 envelope-initiated G2-phase programmed cell death. Proc. Natl. Acad. Sci. U.S.A. 92, 11889–11893. doi: 10.1073/pnas.92.25.11889
Koutsilieri, E., Gotz, M. E., Sopper, S., Sauer, U., Demuth, M., Ter Meulen, V., et al. (1997). Regulation of glutathione and cell toxicity following exposure to neurotropic substances and human immunodeficiency virus-1 in vitro. J. Neurovirol. 3, 342–349. doi: 10.3109/13550289709030748
Kovacic, P., and Cooksy, A. L. (2005). Unifying mechanism for toxicity and addiction by abused drugs: electron transfer and reactive oxygen species. Med. Hypotheses 64, 357–366. doi: 10.1016/j.mehy.2004.07.021
Krathwohl, M. D., and Kaiser, J. L. (2004). HIV-1 promotes quiescence in human neural progenitor cells. J. Infect. Dis. 190, 216–226. doi: 10.1086/422008
Kure, K., Weidenheim, K. M., Lyman, W. D., and Dickson, D. W. (1990). Morphology and distribution of HIV-1 gp41-positive microglia in subacute AIDS encephalitis. Pattern of involvement resembling a multisystem degeneration. Acta Neuropathol. 80, 393–400. doi: 10.1007/BF00307693
Li, G., Park, H. U., Liang, D., and Zhao, R. Y. (2010). Cell cycle G2/M arrest through an S phase-dependent mechanism by HIV-1 viral protein R. Retrovirology 7, 59–59. doi: 10.1186/1742-4690-7-59
Lisi, L., Tramutola, A., Navarra, P., and Dello Russo, C. (2014). Antiretroviral agents increase NO production in gp120/IFN-gamma-stimulated cultures of rat microglia via an arginase-dependent mechanism. J. Neuroimmunol. 266, 24–32. doi: 10.1016/j.jneuroim.2013.10.013
Liu, Y., Wong, T. P., Aarts, M., Rooyakkers, A., Liu, L., Lai, T. W., et al. (2007). NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in Vitro and in Vivo. J. Neurosci. 27, 2846–2857. doi: 10.1523/jneurosci.0116-07.2007
Ma, J., Yuan, X., Qu, H., Zhang, J., Wang, D., Sun, X., et al. (2015). The role of reactive oxygen species in morphine addiction of SH-SY5Y cells. Life Sci. 124, 128–135. doi: 10.1016/j.lfs.2015.01.003
Malik, S., Saha, R., and Seth, P. (2014). Involvement of extracellular signal-regulated kinase (ERK1/2)-p53-p21 axis in mediating neural stem/progenitor cell cycle arrest in co-morbid HIV-drug abuse exposure. J. Neuroimmune Pharmacol. 9, 340–353. doi: 10.1007/s11481-014-9523-7
Mansour, A., Khachaturian, H., Lewis, M. E., Akil, H., and Watson, S. J. (1987). Autoradiographic differentiation of mu, delta, and kappa opioid receptors in the rat forebrain and midbrain. J. Neurosci. 7, 2445–2464.
Mansour, A., and Watson, S. J. (1993). “Anatomical distribution of opioid receptors in mammalian: an overview,” in Opioids I, ed. H. A. Berlin (Berlin: Springer-Verlag), 79–105.
Masvekar, R. R., El-Hage, N., Hauser, K. F., and Knapp, P. E. (2014). Morphine enhances HIV-1(SF162)-mediated neuron death and delays recovery of injured neurites. PLoS ONE 9:e100196. doi: 10.1371/journal.pone.0100196
Moron, J. A., Gullapalli, S., Taylor, C., Gupta, A., Gomes, I., and Devi, L. A. (2009). Modulation of opiate-related signaling molecules in morphine-dependent conditioned behavior: conditioned place preference to morphine induces CREB phosphorylation. Neuropsychopharmacology 35, 955–966. doi: 10.1038/npp.2009.199
Morris, D., Guerra, C., Donohue, C., Oh, H., Khurasany, M., and Venketaraman, V. (2012). Unveiling the mechanisms for decreased glutathione in individuals with HIV infection. Clin. Dev. Immunol. 2012, 734125. doi: 10.1155/2012/734125
Navia, B. A., Jordan, B. D., and Price, R. W. (1986). The AIDS dementia complex: I. Clinical features. Ann. Neurol. 19, 517–524. doi: 10.1002/ana.410190602
Pyo, C.-W., Choi, J. H., Oh, S.-M., and Choi, S.-Y. (2013). Oxidative stress-induced cyclin D1 depletion, and its role in cell cycle processing. Biochim. Biophys. Acta 1830, 5316–5325. doi: 10.1016/j.bbagen.2013.07.030
Ronaldson, P. T., and Bendayan, R. (2008). HIV-1 viral envelope glycoprotein gp120 produces oxidative stress and regulates the functional expression of multidrug resistance protein-1 (Mrp1) in glial cells. J. Neurochem. 106, 1298–1313. doi: 10.1111/j.1471-4159.2008.05479.x
Roy, S., Ninkovic, J., Banerjee, S., Charboneau, R., Das, S., Dutta, R., et al. (2011). Opioid drug abuse and modulation of immune function: consequences in the susceptibility to opportunistic infections. J. Neuroimmune Pharmacol. 6, 442–465. doi: 10.1007/s11481-011-9292-5
Samikkannu, T., Agudelo, M., Gandhi, N., Reddy, P. B., Saiyed, Z., Nwankwo, D., et al. (2011). Human immunodeficiency virus type 1 clade B and C gp120 differentially induce neurotoxin arachidonic acid in human astrocytes: implications for neuroAIDS. J. Neurovirol. 17, 230–238. doi: 10.1007/s13365-011-0026-5
Schafer, F. Q., and Buettner, G. R. (2001). Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 30, 1191–1212. doi: 10.1016/S0891-5849(01)00480-4
Shah, A., Kumar, S., Simon, S. D., Singh, D. P., and Kumar, A. (2013). HIV gp120- and methamphetamine-mediated oxidative stress induces astrocyte apoptosis via cytochrome P450 2E1. Cell Death Dis. 4, e850. doi: 10.1038/cddis.2013.374
UNAIDS/WHO. (2014). AIDS epidemic update. UNAIDS, UNAIDS/JC2552E. Available at: http://www.unaids.org/sites/default/files/media_asset/GARPR_2014_guidelines_en_0.pdf [accessed June 03, 2015].
Wang, T., Gong, N., Liu, J., Kadiu, I., Kraft-Terry, S. D., Schlautman, J. D., et al. (2008). HIV-1 infected astrocytes and the microglial proteome. J. Neuroimmune Pharmacol. 3, 173–186. doi: 10.1007/s11481-008-9110-x
Wang, Y., Shyam, N., Ting, J. H., Akay, C., Lindl, K. A., and Jordan-Sciutto, K. L. (2010). E2F1 localizes predominantly to neuronal cytoplasm and fails to induce expression of its transcriptional targets in Human Immunodeficiency Virus-induced neuronal damage. Neurosci. Lett. 479, 97–101. doi: 10.1016/j.neulet.2010.05.032
Wen, X., Duus, K. M., Friedrich, T. D., and De Noronha, C. M. C. (2007). The HIV1 protein Vpr acts to promote G2 cell cycle arrest by engaging a DDB1 and Cullin4A-containing ubiquitin ligase complex using VprBP/DCAF1 as an adaptor. J. Biol. Chem. 282, 27046–27057. doi: 10.1074/jbc.M703955200
Yang, O. O., Nguyen, P. T., Kalams, S. A., Dorfman, T., Gãttlinger, H. G., Stewart, S., et al. (2002). Nef-mediated resistance of Human Immunodeficiency Virus type 1 to antiviral cytotoxic T lymphocytes. J. Virol. 76, 1626–1631. doi: 10.1128/jvi.76.4.1626-1631.2002
Keywords: HIV-1 gp120, morphine, oxidative stress, cell cycle and microglia
Citation: Samikkannu T, Ranjith D, Rao KVK, Atluri VSR, Pimentel E, El-Hage N and Nair MPN (2015) HIV-1 gp120 and morphine induced oxidative stress: role in cell cycle regulation. Front. Microbiol. 6:614. doi: 10.3389/fmicb.2015.00614
Received: 10 April 2015; Accepted: 03 June 2015;
Published: 23 June 2015.
Edited by:
Akihide Ryo, Yokohama City University, JapanReviewed by:
Hiroaki Takeuchi, Tokyo Medical and Dental University, JapanYukio Sasaki, Yokohama City University, Japan
Copyright © 2015 Samikkannu, Ranjith, Kurapati, Atluri, Pimentel, El-Hage and Nair. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thangavel Samikkannu and Madhavan P.N. Nair, Department of Immunology, Institute of NeuroImmune Pharmacology, College of Medicine, Florida International University, Modesto A. Maidique Campus, 11200 S.W. 8th Street, Miami, FL-33199, USA,c3RoYW5nYXZAZml1LmVkdQ==;bmFpcm1AZml1LmVkdQ==