 Laura M. Coughlan
Laura M. Coughlan Paul D. Cotter
Paul D. Cotter Colin Hill
Colin Hill Avelino Alvarez-Ordóñez
Avelino Alvarez-Ordóñez- 1Teagasc Food Research Centre, Cork, Ireland
- 2Alimentary Pharmabiotic Centre, Cork, Ireland
- 3School of Microbiology, University College Cork, Cork, Ireland
Microorganisms are found throughout nature, thriving in a vast range of environmental conditions. The majority of them are unculturable or difficult to culture by traditional methods. Metagenomics enables the study of all microorganisms, regardless of whether they can be cultured or not, through the analysis of genomic data obtained directly from an environmental sample, providing knowledge of the species present, and allowing the extraction of information regarding the functionality of microbial communities in their natural habitat. Function-based screenings, following the cloning and expression of metagenomic DNA in a heterologous host, can be applied to the discovery of novel proteins of industrial interest encoded by the genes of previously inaccessible microorganisms. Functional metagenomics has considerable potential in the food and pharmaceutical industries, where it can, for instance, aid (i) the identification of enzymes with desirable technological properties, capable of catalyzing novel reactions or replacing existing chemically synthesized catalysts which may be difficult or expensive to produce, and able to work under a wide range of environmental conditions encountered in food and pharmaceutical processing cycles including extreme conditions of temperature, pH, osmolarity, etc; (ii) the discovery of novel bioactives including antimicrobials active against microorganisms of concern both in food and medical settings; (iii) the investigation of industrial and societal issues such as antibiotic resistance development. This review article summarizes the state-of-the-art functional metagenomic methods available and discusses the potential of functional metagenomic approaches to mine as yet unexplored environments to discover novel genes with biotechnological application in the food and pharmaceutical industries.
Introduction
Recent advances in molecular microbiology have revealed that the microbial world extends far beyond what can be revealed by traditional microbiological techniques. Environments once believed to be devoid of life have now been shown to support the growth of microbes. As a consequence, it is now accepted that microorganisms thrive throughout nature, and that at least some microorganisms can be found in almost all known environments. This is due to the fact that microbial life has adjusted to survive under a wide range of harsh or unaccommodating conditions, resulting in a variety of diverse microorganisms adapted to specific niches. This review article explores the molecular methods that can provide access to these specially adapted microbes and, more specifically, their potentially useful genes/molecules and outlines how these approaches can be harnessed by the food and pharmaceutical industries.
Traditional microbiology generally involves obtaining a pure culture as a major step in any study. However, it is estimated that standard laboratory culturing techniques provide information on 1% or less of the bacterial diversity in a given environmental sample (Torsvik et al., 1990). This is most noticeable in what is known as the plate count anomaly, i.e., the discrepancy between the numbers of microorganisms detected by microscopy and the numbers obtained from pure colony counts of cultivated samples (Staley and Konopka, 1985). Although significant advances have been recently made in culturing as-yet-uncultured microbes, e.g., Ling et al. (2015), culture-independent techniques present a more promising effort to access the genetic information contained within the vast number of species in the environment.
Metagenomics presents a molecular tool to study microorganisms via the analysis of their DNA acquired directly from an environmental sample, without the requirement to obtain a pure culture. With this technology, the DNA of microorganisms in a population is analyzed as a whole. Sequencing and analysis of total metagenomic DNA can provide information about several aspects of the sample, allowing one to better characterize the microbial life in a given environment. It can not only reveal the identity of species present but also can provide insight into the metabolic activities and functional roles of the microbes present in a given population (Langille et al., 2013). Expression of the genetic information from an environmental sample in a routinely culturable surrogate host can also overcome in part the barriers faced when dealing with as yet uncultured bacteria. The coupling of this approach with function-based screening of the subsequent colonies to uncover a desired activity that has been conferred onto the host by the inserted environmental DNA in a functional metagenomics approach is a powerful technique for the discovery of novel functional genes from uncultured microorganisms.
In this review article, functional metagenomics is discussed as an emerging molecular technique with potential applications in industrial settings. An overview of the current methodological strategies employed for functional metagenomic analysis of microbial populations, with emphasis on the use of phenotypic-based metagenomic screens for the discovery of novel small molecules, enzymes, and bioactives is provided. The applications of such compounds to the food and pharmaceutical industries are discussed, while highlighting recent successes in this area.
Functional Metagenomics: Methodological Approaches
Sequencing-based Strategies
Metagenomic analyses begin with the isolation of microbial DNA from an environmental sample. The acquired metagenomic DNA specimen should be as pure and of as high quality as possible, and should accurately represent all species present both qualitatively and quantitatively. Direct sequencing of extracted metagenomic DNA, followed by appropriate bioinformatics analyses, can facilitate the elucidation of the functional traits of microorganisms colonizing particular environments (Figure 1).
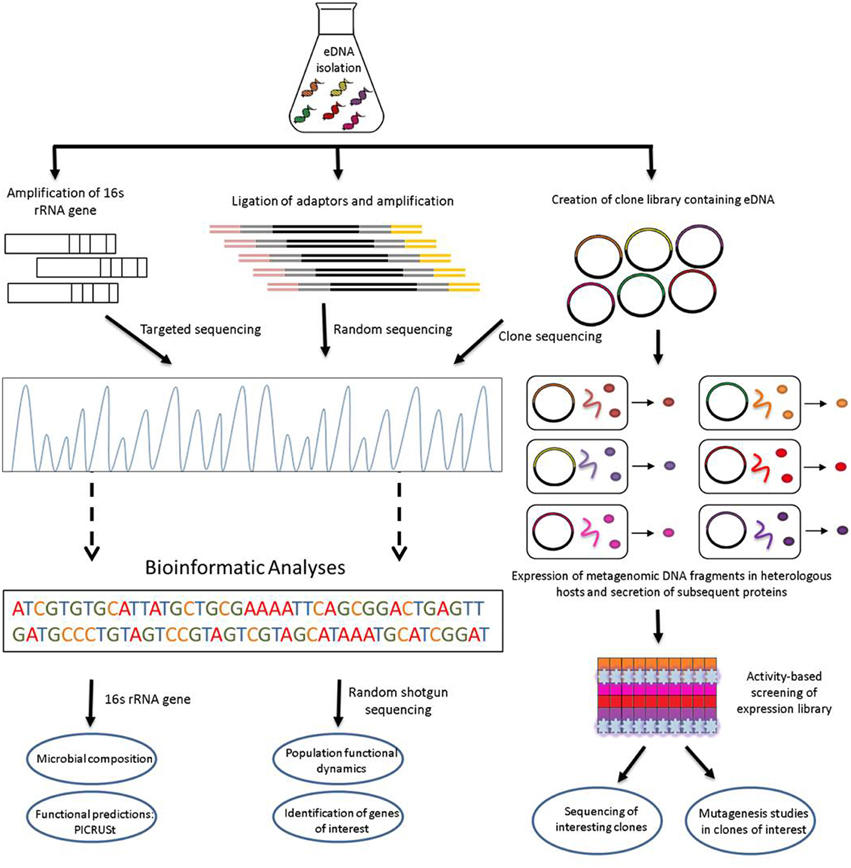
Figure 1. Schematic view of functional metagenomic strategies for the identification of novel biocatalysts and bioactives from environmental DNA.
The initial break from culture-dependent to culture-independent approaches for the microbiological analysis of an environmental sample involved the sequencing of genes encoding microbial ribosomal RNAs (rRNAs). Highly conserved primer binding sites within the bacterial 16S rRNA gene facilitate the amplification and sequencing of hypervariable regions that can provide species-specific signature sequences useful for bacterial identification in an environmental sample (Lane et al., 1985). This technology enables microbiologists to determine phylogenetic relationships between unculturable bacteria and assess and quantify the microbial consistency of a sample. In addition, through 16S rRNA gene sequencing of a metagenomic sample, a functional profile of the bacteria present in a given environment can also be obtained. Information regarding the functional roles of already studied bacterial species is available in database archives, including both cultured bacteria whose functional proteins have been extensively characterized as well as functions assigned to bacterial proteins produced by uncultured bacteria through previous metagenomic studies. Once a member of a previously described bacterial family has been identified in an environmental sample, or an appropriate closest known relative has been appointed, phylogenetic analysis may assign predicted functions to an identified bacterial species by referring to the functional information available regarding that particular taxonomic group. This process can be applied to potentially most, if not all, of the different bacterial species encountered in a sample and therefore community roles can be predicted for the microbes dwelling in the sampled niche without the need for shotgun sequencing (described below). Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) is a computational approach developed by Langille et al. (2013) which can be used to predict the functional properties of microorganisms in a metagenomic sample from characterized relatives in available databases using 16S rRNA sequencing data. By quantifying the individual species abundance in a sample and, in doing so, quantifying the function(s) assigned to that family, PICRUSt can predict the overall functional composition of the community. Keller et al. (2014) also explored this concept through a combinatorial approach of 16S rDNA metabarcoding and single genomics for assessing the compositional and functional diversity of a microbial community. Although these authors were successful in validating their method, this innovative technique requires optimization prior to its introduction into larger and more challenging projects. Microbial eukaryotic communities may also be studied through similar strategies. Eukaryotic-specific primers homologous to the bacterial 16S rRNA can be used to target eukaryotic microbes present in an environmental sample. Bates et al. (2012) used bar-coded pyrosequencing of 18S rRNA to investigate the eukaryotic components of three different lichens, identifying members of the Alveolata, Metazoa, and Rhizaria taxonomic clades. Non-coding DNA located between the small and large subunit eukaryotic rRNA genes, known as the Internal Transcribed Spacer (ITS) regions, are also targeted as a universal DNA marker in Fungi (Schoch et al., 2012). The environmental virome has also been explored through metagenomics by the coupling of sequence-independent amplification of viral nucleic acids with next generation sequencing technologies (Smits and Osterhaus, 2013), particularly in the areas of epidemiology and diagnostics. In addition, genes similar to those of metabolic cells, known as auxiliary metabolic genes (AMGs), have been discovered in viruses (reviewed by Rosario and Breitbart, 2011) and may have potential in the search for industrially relevant enzymes and bioactives.
Environmental DNA random shotgun sequencing, where total metagenomic DNA is sequenced, assembled and annotated, has been shown to be a more useful tool which may be used to analyse at a molecular/species level the metagenome of an environmental sample. In this instance, the functional potential of a microbial population is revealed by directly sequencing the environmental DNA rather than predicting its functional potential based on 16S rRNA data. Some examples of large scale metagenomic studies involving shotgun sequencing are those carried out by Venter et al. (2004), who characterized the microbial population of the Sargasso Sea identifying 1.2 million previously undescribed genes including the first assignment of rhodopsin-like photoreceptors to bacterial species, Warnecke et al. (2007), who analyzed the hindgut paunch microbiota of a Nasutitermes species of wood-feeding termite revealing unprecedented diversity of the microbial community and identifying novel genes involved in cellulose and xylan hydrolysis, Oh et al. (2014), who analyzed the microbial content and subsequent functional capacity of the healthy human skin microbiome through shotgun metagenomic sequencing, and Hess et al. (2011), who deep sequenced 268 gigabases of metagenomic DNA obtained from the microbiota of cow rumen unveiling carbohydrate active genes encoding enzymes capable of degrading biomass, a desirable ability in the development of biofuels as a renewable energy source. Random sequencing of shotgun metagenomic DNA may reveal genes of interest, the probable phylogeny of which can be inferred through searches for homology in non-redundant databases, usually via Basic Local Alignment Search Tool (BLAST) analysis. Thus, random sequencing has the potential to identify the presence of already known genes, with reported beneficial functions, or their homologs in an uncultured microorganism, which can provide additional advantages and improve the functionality of in-use proteins/enzymes/catalysts, e.g., the new variant/homolog may encode a protein that is capable of carrying out a specific catalytic or metabolic function and may also be tolerant to an extreme environment habitually encountered in industrial processes. This approach is also useful for the study of the population dynamics of a community, including genomic evolution (Chandler et al., 2014; Kay et al., 2014) and the distribution and redundancy of functions throughout the community (Mendes et al., 2015).
Nevertheless, the sequence-based approaches to analysing environmental samples are limited to the study and identification of genes and DNA sequences homologous to those that are already known. Consequently, the possibility of using sequence-based methods for the discovery of proteins encoded by novel sequences is restricted. Phenotypic-based screening of constructed metagenomic expression libraries, described in the next section of the manuscript, is better suited to the unearthing of previously undescribed proteins and small molecules.
Phenotypic-based Strategies
Functional metagenomic analyses can be carried out on metagenomic libraries via the isolation and purification of DNA from an environmental sample, cloning of the DNA into a suitable vector, heterologous expression of the insert vector containing environmental DNA fragments in a suitable surrogate host (usually Escherichia coli), and analysis of subsequent transformants by either sequencing- or phenotypic-based approaches, or both (Figure 1). Screening of metagenomic libraries through phenotypic-based approaches is carried out to detect the expression of a particular phenotype conferred on the host by inserted DNA. Screening is usually performed on multiple clones simultaneously on a fixed matrix in which the entire group is assayed with an appropriate indicator to reveal the presence of a phenotypically relevant clone. Such assays require the functional protein to be secreted from the host cell to allow for extracellular detection. Metagenomic clones may be grown on specific indicator media, to allow visual identification of an active clone, e.g., hemolytic activity on blood agar (Rondon et al., 2000), lipolytic activity (Henne et al., 2000), etc. In other occasions, the presence of zones of inhibition in soft agar overlay assays using indicator microorganisms can reveal inhibitory or antimicrobial agents produced by an active clone (Tannieres et al., 2013; Iqbal et al., 2014). Libraries may also be screened based on selection approaches. In these circumstances only the clones onto which the activity of interest has been conferred by the metagenomic DNA insert will grow or survive. Selections include for instance the ability to metabolize a given substrate as a clone's sole carbon source (Entcheva et al., 2001), the ability to resist a potent antimicrobial agent (Donato et al., 2010) or the ability to grow in the presence of a lethal concentration of a heavy metal (Staley et al., 2015).
An alternative option for the identification of novel genes, the Substrate-Induced Gene EXpression screening (SIGEX), was developed by Uchiyama et al. (2005). It relies on the principle that catabolic gene expression is generally induced by a specific substrate or metabolite of catabolic enzymes and is controlled by regulatory elements situated close to these genes. With SIGEX, the environmental DNA inserts are fused with a reporter gene encoding green fluorescent protein (gfp) on an operon-trap vector and induced by a target substrate. SIGEX is combined with fluorescent-activated cell sorting (FACS) for the high-throughput selection of GFP-expressing clones. Additionally, the protocol eliminates the incorporation of clones containing self-ligated plasmids and those that are constitutively expressing GFP. Despite some limitations with regard to the applications of this method (reviewed by Yun and Ryu, 2005), SIGEX is an efficient process for the identification of novel catabolic substrate-induced genes. Uchiyama and Miyazaki (2010) went on to expand the capabilities of the SIGEX protocol and developed a reporter assay system for the screening of metagenomic libraries for enzymatic function called Product-Induced Gene EXpression (PIGEX). The system uses a transcriptional activator, which is sensitive to the product of the desired reaction, placed upstream of a gfp gene insert. Should a clone possess the activity of interest, upon exposure to an appropriate substrate, the product of this reaction activates transcription of the chosen transcriptional regulator and in turn gfp causing the clone to fluoresce, allowing easy detection of positive clones. Pooja et al. (2015) identified through PIGEX a periplasmic α-amylase from a cow dung-derived metagenomic library by isolating an active clone that fluoresced in response to a maltose substrate.
Despite the potential usefulness of such systems, phenotypic-based functional metagenomic approaches face a number of complications, to which potential resolutions are currently being devised. To successfully identify a useful gene or protein candidate a series of sequential steps in the cloning and screening process must occur adequately and effectively. Transcription of the entire gene, translation of its mRNA, correct protein folding, and secretion of the active protein from the surrogate host must all be achieved before functional screening even begins. Suitable and efficient screening methods must also be applied to detect the presence of an interesting gene within the metagenomic library. As the probability of identifying a metagenomic clone, among possibly thousands of others, with a specific desired activity is low (Uchiyama and Miyazaki, 2009), high-throughput screening (HTS) protocols may improve the chances of obtaining an active clone, by allowing higher numbers of clones to be screened simultaneously. An obstacle occurring at any of these stages may result in the overlooking of an interesting clone which might have been detected under the correct circumstances.
One aspect of the methodological approach that can be particularly challenging relates to expressing DNA fragments isolated from microorganisms native to diverse and exotic environments in a relatively domesticated host such as E. coli (Banik and Brady, 2010). Even if the foreign DNA is successfully transcribed and translated (perhaps due to the presence of DNA regulatory elements placed on the vector), the correct chaperones required for proper protein folding in the original species may be absent from E. coli. A strategy being explored to overcome host related limitations is the generation of an alternative surrogate expression host that may be more suited to efficiently expressing the environmental DNA at hand. Craig et al. (2009) discovered two novel compounds through functional screening of a soil derived metagenomic library expressed in Ralstonia metallidurans. The library was constructed using E. coli as a heterologous host and then the DNA transferred to R. metallidurans for activity based screening. Two clones showed activity in R. metallidurans, one displaying antimicrobial activity through the expression of a polyketide synthase gene and a second yellow colored clone expressing a carotenoid gene cluster. Clones active in R. metallidurans did not confer the same metabolic abilities onto the E. coli host. This shows the importance of using additional heterologous hosts to identify active clones which may not be expressed in the standard E. coli host. After their initial success, this research group carried out a study to compare six different Proteobacteria as hosts for the same soil derived metagenomic cosmid library (Craig et al., 2010). Each host expressing the library was functionally screened for antimicrobial activity, pigment production and altered colony morphology conferred onto the host by the DNA insert. Bacterial species from common soil-dwelling phyla were chosen as experimental hosts. Five candidate hosts, Agrobacterium tumefaciens, Burkholderia graminis, Caulobacter vibrioides, Pseudomonas putida, and Ralstonia metallidurans, were compared to the standard and most commonly used host, E. coli. Active clones were recovered from the library, having been expressed by different heterologous hosts with minimal overlap between hosts. This study shows the usefulness of Broad-Host Range vectors for overcoming host expression related barriers. Biver et al. (2013a) carried out a study to evaluate the use of an E. coli-Bacillus subtilis shuttle vector to functionally screen a forest soil-derived metagenomic library for antimicrobial activity. Activity based screening identified a novel antimicrobial agent, shown to be proteinaceous in nature though not yet fully characterized, that is active against Bacillus cereus. The DNA fragment responsible for such activity was active in the B. subtilis host alone and no activity was observed when the fragment was expressed in E. coli. Again, the importance of developing multiple host expression systems is highlighted by these findings. Further studies similar to those mentioned above must be carried out to better characterize and therefore more fully understand potential alternative hosts. Another obstacle faced in heterologous expression is the possibility of a DNA fragment being too short to contain a functional gene cluster or operon. The availability of a vector able to accommodate large DNA inserts is also fundamental (Streit and Schmitz, 2004). The use of large insert vectors capable of accommodating biosynthetic gene clusters or operons, and the development of shuttle vectors capable of propagating in more than one heterologous host, are examples of strategies being explored to overcome methodological limitations.
Applications of Interest of Functional Metagenomics in Food and Pharmaceutical Industries
Discovery of Novel Bio-catalysts
Certain microbial enzymes are of particular interest to the food and pharmaceutical industries for the catalysis of reactions which may be difficult or expensive to maintain. This interest stems from the fact that there is often difficulty in synthesizing chemical catalysts that truly mimic the complexity of biological enzymes. Many industrial processes are associated with a large environmental burden. Substituting traditional chemical processes used to produce certain compounds or molecules with enzymatic pathways naturally sourced is a more environmentally friendly approach to large-scale production. As microorganisms can catalyze a vast range of reactions, they are an obvious source of enzymes for industrial applications. Several authors have explored this avenue in the last decade (Table 1).
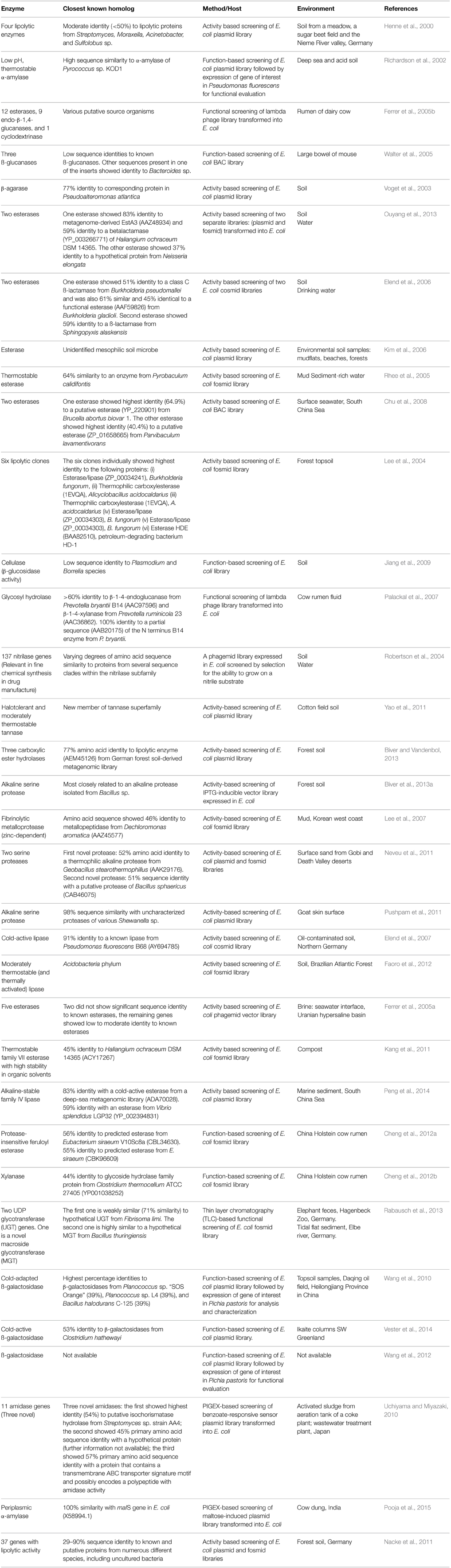
Table 1. Some novel enzymes of industrial interest discovered through functional metagenomics.
Novel enzymes from natural sources are extremely useful in food processing reactions. Many of these relate to reactions that occur in nature to process food for energy but are difficult to mimic on an industrial level, e.g., degradation of starch. In other instances, the search has focused on enzymes that can carry out reactions under extreme conditions, which often prevail in food processing, e.g., high temperatures and extremes of pH. Indeed, microbial enzymes are used for brewing, baking, synthesis of sugar and corn syrups, starch and food processing, texture and flavoring, processing of fruit juices, and production of dairy products and fermented foods, among others, either as recombinant enzymes or by using starter cultures with desirable activities. The following are some examples of industrial food processes which have benefited (and may continue to do so) from access to the diverse repository of enzymes possessed by microorganisms.
In the food industry, starch harvested from sources such as maize, wheat, and potatoes is processed to yield food products such as glucose and fructose syrups, starch hydrolysates, maltodextrins, and cyclodextrins (reviewed by van der Maarel et al., 2002). In recent times, the chemical hydrolysis of starch, which involves acid treatment, is being replaced with enzymatic digestion by starch-hydrolyzing enzymes obtained from natural sources. Starch-modifying enzymes are also added to dough in the baking industry to act as bread anti-staling agents. These starch-converting enzymes usually originate from the α-amylase family or family 13 glycoside hydrolase. Amylases from microbial sources are used in starch processing such as α-amylases from Geobacillus stearothermophilus and Bacillus licheniformis. However, despite the advantages of using enzymatic over chemical hydrolysis (high specificity of enzymes, milder reaction conditions, natural means of processing more acceptable to consumers and to the public), there are limitations with the enzymes currently being used. Starch hydrolysis is carried out at high temperatures, at which α-amylases are usually not active at a pH below 5.9. For the reaction to proceed efficiently, the pH must be raised by the addition of NaOH. As these enzymes also exhibit a Ca2+ dependency, Ca2+ must be added to the reaction in addition to adjusting the pH. Thermostable, Ca2+-independent α-amylases with low pH activity would be ideal for the starch hydrolyzing process. Richardson et al. (2002) identified an α-amylase optimal for the corn wet milling process. They carried out activity based screenings under conditions of temperature and pH similar to those of the corn wet milling process on a large library of metagenomic clones constructed from diverse environmental samples. The clones were also phylogenetically screened for homology to known α-amylases. Three clones were selected which performed well under the given conditions. Phylogenetic analysis revealed that all three enzymes were members of the glycosyl hydrolase family 13. They were expressed in Pseudomonas fluorescens and their activity was compared to the enzymes currently used in industry (from B. licheniformis). One clone was found to have better characteristics for application to the corn wet milling process than the enzyme currently in use. However, further research is needed to improve the low yield of enzyme produced under industrial conditions.
Lipases and esterases are hydrolytic enzymes which play important roles in the food and pharmaceutical industries. Lipases hydrolyze fats into fatty acids and glycerol at the water lipid interface and reverse the reaction in the non-aqueous phase (Gupta et al., 2004). Lipases are exploited by the dairy industry for the hydrolysis of milk fat, releasing short-chain and long-chain fatty acids, creating such features as richness, creaminess or cheesiness depending on the degree of lipolysis, as reviewed by Hasan et al. (2006). For this reason, it is important to use the correct lipolytic enzyme to achieve the right flavor in the final product. Peng et al. (2014) screened a metagenomic library constructed from a Chinese marine sediment for clones displaying lipolytic activity in an E. coli host. They discovered a novel highly alkaline-stable lipase with high specificity for butter milkfat esters. Treatment of butter with the newly identified lipase produced rich and distinctive flavors through the production of palmitic and myristic acids while maintaining the cheesy flavor of the short-chain fatty acids. As palmitic and myristic acids are added to food for their distinctive flavor, the hydrolysis of palmitate and myristate in the production of lipolysed milkfat (LMF) to flavor dairy products is a safe and economically viable potential application of the novel lipase identified in this study. Other dairy applications of lipases include the acceleration of cheese ripening and the enhancement of cheese flavor through the synthesis of short chain fatty acids (SCFAs) and alcohols. Lipases are also used in vegetable oil modification and preservation of baked goods (Hasan et al., 2006). Although in the past lipases used in the food industry were predominantly obtained from animal sources, the microbial world potentially holds a wide range of diverse lipases that can be used in many different industrial applications (Table 1). Examples of pharmaceutical applications of lipases sourced from microbes include the synthesis of an intermediate for the production of an anti-tumor agent (Zhu and Panek, 2001) and the synthesis of intermediates of antimicrobial agents (Kato et al., 1997). Also, through the screening of a metagenomic library constructed from an oil-contaminated German soil sample, Elend et al. (2007) identified a lipolytic cold-activated clone which showed high selectivity for esters of primary alcohols and (R) enantiomers of non-steroidal anti-inflammatory drugs such as ibuprofen. This enzyme has potential in the pharmaceutical industry for the conversion of such anti-inflammatories into an optically pure form.
Esterases catalyze the hydrolysis of an ester into its alcohol and an acid in aqueous solution. They are distinguished from lipases in that they hydrolyze short-chain over long-chain acylglycerols. In the food industry, esterases are used in fat and oil modification and in the fruit juices and alcoholic beverages industries to produce certain flavors and fragrances, as reviewed by Panda and Gowrishankar (2005). Feruloyl esterases hydrolyze the ester bond between ferulic acid (FA) and polysaccharides present in plant cell wall material. They have a dual usefulness as they not only break down plant biomass (which is useful in industrial waste management) but, in doing so, they de-esterify dietary fibers releasing bioactives with potential beneficial health effects (reviewed by Faulds, 2010). In a study carried out by Cheng et al. (2012a), a metagenomic library constructed from the microbial content of a Chinese Holstein cow rumen was functionally screened for feruloyl esterase activity, identifying a protease-insensitive feruloyl esterase capable of releasing FA from wheat straw. This novel enzyme is of particular industrial interest as it showed high thermal and pH stability and was resistant to several proteases including pepsin. A novel xylanase was isolated from the same metagenomic library (Cheng et al., 2012b) and its ability to work synergistically with the newly discovered feruloyl esterase to release xylooligosaccharides (XOS) and FA from wheat straw was assessed. XOS display prebiotic and gut modulatory activities and have other bioactive properties giving them value as food additives, as reviewed by Moure et al. (2006). The novel xylanase was not only effective in working with the feruloyl esterase, but additionally was capable of improving release of FA from wheat straw at a high dose. Esterases also play a role in the synthesis of chiral drugs including medications to relieve pain and reduce inflammation (Bornscheuer, 2002; Shen et al., 2002; Panda and Gowrishankar, 2005).
ß-galactosidases are widely used in the dairy industry for the hydrolysis of lactose to glucose and galactose. Lactose content in milk is reduced to improve taste (lactose is known to absorb undesirable flavors and odors), to accelerate the ripening of cheeses made from treated milk and for the removal of lactose for the production of lactose-free products for intolerant consumers (reviewed by Panesar et al., 2010). The currently commercially available ß-galactosidase for use in the dairy industry, from Kluyveromyces lactis, has a temperature optimum of 50°C and loses much of its enzymatic activity at temperatures below 20°C. Carrying out industrial reactions at lower temperatures is beneficial as it saves energy (and in turn is more economical), it prevents heat destruction of thermosensitive substances such as food compounds, molecules responsible for flavors, taste and nutritional value, etc., and it reduces contamination risks. Cold-active enzymes work at low temperature and can be easily inactivated by rising the temperature to a moderate condition. From a metagenomic library constructed from the ikaite columns of SW Greenland, Vester et al. (2014) isolated a cold-activated ß-galactosidase which can potentially be applied by the dairy industry. The discovered enzyme has an optimal pH of 6 (the natural pH of milk being pH 6.7–6.8) and a temperature optimum of 37°C, but retains lactose hydrolytic activity at 5°C. These properties make it a good candidate for the hydrolysis of lactose into glucose and galactose in milk for the removal of lactose for production of lactose-free products for lactose-intolerant people. In a similar study by Wang et al. (2010) a cold-adapted ß-galactosidase was identified from a metagenomic library expressed in E. coli. The insert from the active clone (encoding a full-length ß-galactosidase) was expressed in Pichia pastoris to assess its candidacy for use in milk treatment and optimal activity was observed at a temperature of 38°C. The enzyme was active at the natural pH of milk.
Flavonoids are plant secondary metabolites found in numerous dietary fruits and vegetables and whose consumption is beneficial to human health (Ververidis et al., 2007a,b). Flavonoids are difficult to source as they are produced by plants at very low levels. Due to their structural complexity enzymatic modification is preferred over a chemical approach for industrial production. Glycosylation of flavonoids influences their water solubility and bioavailability, making glycosyltransferases that are active on flavonoids of great interest to the food and pharmaceutical industries. Rabausch et al. (2013) developed a novel thin-layer chromatography (TLC) based screening method for the identification of flavonoid-modifying enzymes from a metagenomic library. Two novel flavonoid-modifying enzymes with high activity on flavones, flavonols, flavanones, isoflavones, and stilbenes were discovered in this manner.
Proteases hydrolyze peptide bonds and therefore catalyze the degradation of proteins. They have numerous uses in the food industry, including the tenderizing of meat (Ashie et al., 2002), the coagulation of milk and flavor development in the dairy industry (Huang et al., 2011) and the proteolysis of gluten to achieve gluten-free products in the baking industry (Hamada et al., 2013). Proteases may also be used to release beneficial bioactive peptides from polypeptide chains in certain foods (Hafeez et al., 2014; Mora et al., 2014). Currently, commercial proteases used in the food industry are generally sourced from plants and culturable microorganisms. Proteases from as yet uncultured microbial extremophiles would be of use in the carrying out of proteolysis under unconventional reaction conditions. There have been several novel proteases discovered through functional metagenomic methods. For instance, Biver et al. (2013a) identified an oxidant-stable alkaline serine protease from a forest-soil metagenomic library. An alkaline serine protease was also identified in a metagenomic library constructed from goat skin surface samples by Pushpam et al. (2011). These alkaline proteases are examples of microbial enzymes with potential industrial applications, mainly in the detergent industry.
Tannins are naturally occurring water soluble polyphenols which constitute a large percentage of plant material. Tannases catalyze the hydrolysis of tannins, releasing gallic acid, and glucose. Tannases are used in the food industry as a clarifying agent in the manufacture of beverages such as instant teas, fruit juices, beer, and certain wines (Cantarelli et al., 1989; Boadi and Neufeld, 2001). Tannases are also important to the pharmaceutical industry for catalyzing the release of gallic acid (Sariozlu and Kivanc, 2009) which is used in the production of some antibacterial drugs. Additionally, gallic acid is used in the synthesis of propyl gallate, an antioxidant food additive. Tannases isolated from bacteria have typically been restricted to culturable strains, overlooking the diverse potential of those as yet uncultured. Yao et al. (2011) expressed a metagenomic clone library constructed from cotton field in E. coli and screened the transformants for tannase activity, revealing one active clone. Sequence analysis revealed that the active clone encoded a full length tannase gene, which was not found to be closely related to any currently known tannases. Analysis of tannase activity of the enzyme under various industrially relevant conditions was performed and a moderate thermostability of the identified enzyme, which may be useful for food industrial applications, was shown. The enzyme was also found to have a wide range of substrate specificity, making it suitable for applications in both the food and pharmaceutical industries. In 2014, this novel tannase was investigated by Yao et al. (2014) for its suitability for the removal of tannins from a green tea infusion. The presence of tannins in beverages such as green tea is problematic as the ability of tannins to precipitate proteins leads to the formation of a protein haze that is undesirable in terms of product taste and appearance (Wu and Bird, 2010). The tannase enzyme was recombinantly expressed in E. coli and immobilized to several matrices, identifying Ca-alginate beads as the most appropriate support. The immobilized enzyme was effective in the removal of tannins from green tea infusion and was found to possess properties distinct from those of the free enzyme, such as high operational and storage stabilities and a higher temperature and pH optimum.
Discovery of Novel Bioactives
As with the food industry, the use of microbial enzymes is of particular interest for the biosynthesis of pharmaceutical products previously synthesized via chemical means. Thus, functional metagenomics can be applied to the discovery of genes capable of carrying out reactions of interest for the obtaining of bioactives or the synthesis of intermediate compounds in the pharmaceutical industry. One avenue of interest has been the identification and heterologous expression of a microbial biosynthetic pathway capable of producing biotin for industrial purposes (Entcheva et al., 2001; Streit and Entcheva, 2003). Biotin (Vitamin H) is a human and animal dietary requirement and is currently chemically synthesized through industrial processes for addition to food and feed products, with associated negative environmental impacts. The use of biotin-producing microorganisms in place of chemical synthesis offers a greener alternative to conscientious industries. Other microbial biosynthetic genes of interest to the pharmaceutical industry capable of synthesizing other bioactives important for human health and medicine have been also identified by functional metagenomic strategies (listed in Table 2).
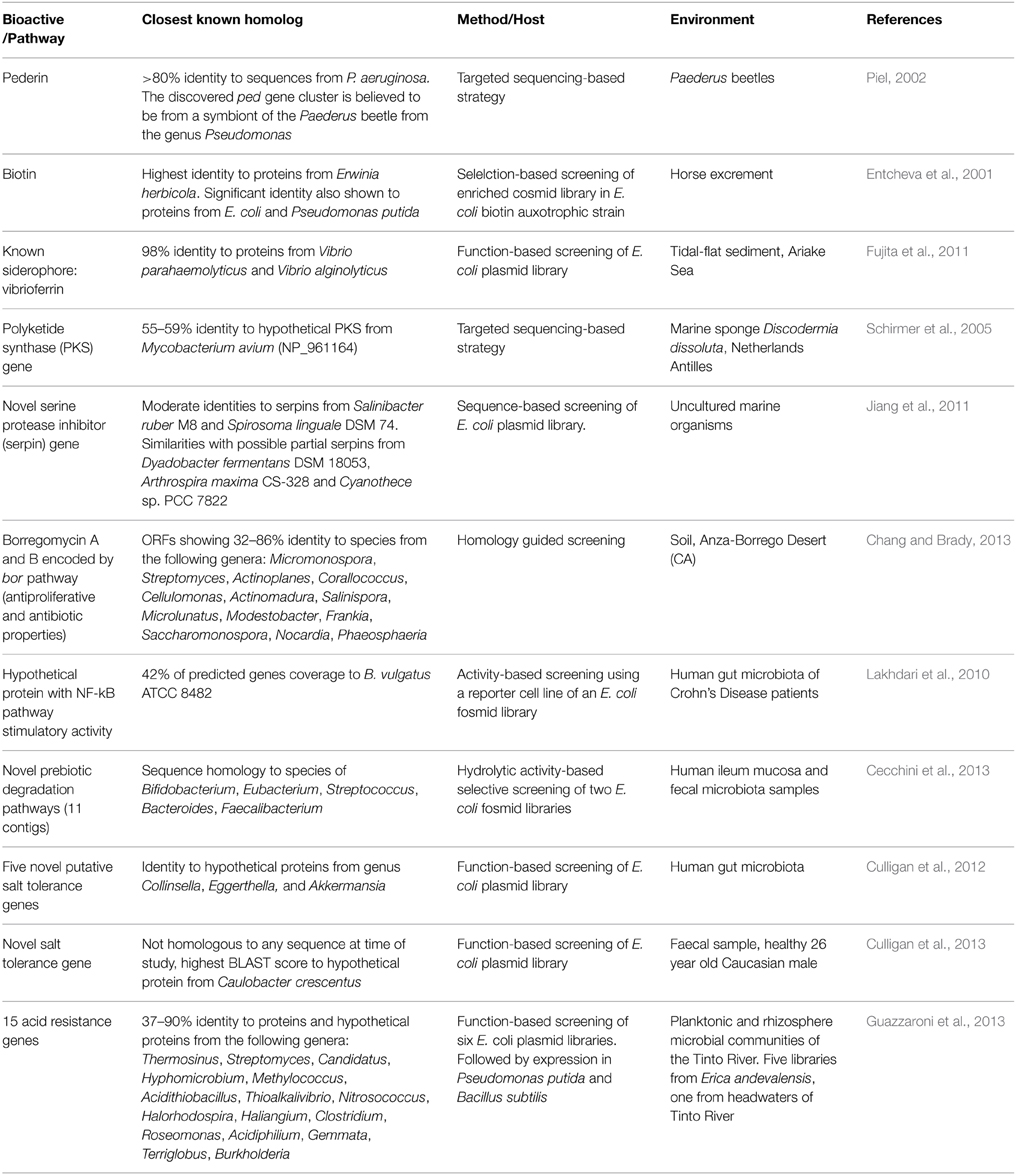
Table 2. Some novel bioactives and biosynthetic pathways of industrial interest discovered through functional metagenomics.
Walter et al. (2005) applied a functional metagenomic method to screen for lichenin-degrading activity in a Bacterial Artificial Chromosome (BAC) library constructed from bacteria obtained from the large-bowel microbiota of mice, identifying three clones with ß-glucanase activity. Glucans cannot be broken down by humans or monogastric animals and so, their hydrolysis relies on bacterial fermentation. As the consumption of glucans is associated with health benefits in humans (Abumweis et al., 2010), glucan hydrolyzing enzymes isolated from bowel-dwelling microbiota may be of interest to pharmaceutical and functional food related industrials. The feed industry may also benefit from the availability of ß-glucanases that improve the digestion of barley-based feed diets by poultry livestock (Von Wettstein et al., 2000).
The development of novel therapeutic strategies relies heavily on gaining a better understanding of human commensals and host-microbe relationships. Lakhdari et al. (2010) established and validated a reporter system capable of detecting immune modulatory activity of metagenomic clones. A metagenomic library constructed from human fecal microbiota of Crohn's Disease (CD) patients was screened for NFkB modulatory activity (whether stimulatory or inhibitory) using an intestinal epithelial cell line transfected with a reporter gene. A clone displaying stimulatory activity of the NF-kB pathway was identified. Although the molecule responsible for the activity is not yet known, two potential candidate loci were determined through transposon mutagenesis: an efflux ABC type transport system and a putative lipoprotein. Phylogenetic analysis showed Bacteroides vulgatus to be the closest known homolog to the source of the insert of interest, an interesting finding as B. vulgatus is a human gut microbe found to be higher in abundance in CD patients than in a control population. This study presents the development of an innovative platform for screening metagenomic libraries and is likely to inspire the creation of other cell-based screening platforms from which a better understanding of human-microbe symbiotic communications can be obtained, advancing the development of novel therapeutic strategies promoting a healthy relationship with the gut microbiota and in turn the entire human microbiome.
Maintaining gut microbiota homeostasis has been shown to contribute to the overall sustaining of human gut health. Probiotics are an oral infusion of high numbers of live beneficial gut microbes formulated into various yogurts and dairy beverage products that, when ingested in adequate amounts, confer a health benefit on the host (Joint, 2001). As an oral formulation, these products face difficulties in efficacy due to insufficient cell numbers reaching the intestine, owing to the necessity of passing through the majority of the GI tract to reach their site of action in the bowel. The harsh pH and osmolarity of the upper GI tract can destroy a large proportion of the ingested cells. Novel acid and salt resistance mechanisms discovered through functional metagenomic studies similar to those of Guazzaroni et al. (2013), who identified an acid resistant metagenomic clone from the Tinto River environment, and Culligan et al. (2013), who discovered a gene conferring salt tolerance onto an E. coli host from a library derived from the human gut microbiota, may be of use in conferring stress resistance to probiotic products. However, this objective faces additional social challenges with respect to consumer acceptance of the use of genetically modified (GM) microorganisms to enhance food products. Although it is generally appreciated by the public that GM cells, organisms and microorganisms are necessary for the production of certain critical biologically active drugs, the thought of everyday food products having been prepared using GM materials is met with a sense of unease, especially in many EU member states. Thus, strict regulations involving the consumption of GM foods and the use of GM organisms in food production and processing have not been made more lenient, as they have in other countries, such as the USA, in recent years. Public transparency and an understanding of the extensive safety and efficacy testing of GM related food products may eventually lead to a change in consumer attitude to bioengineered goods.
Another avenue to maintain human gut health is to promote the growth of beneficial bacteria already present in one's lower GI tract through the use of prebiotics. Prebiotics are non-digestible oligosaccharides (NGOs), usually present in plant material, that are resistant to human digestion in the upper GI tract and are hydrolyzed in the gut by beneficial microbiota to produce SCFAs and organic acids that provide nutritional value to the human host (Gibson and Roberfroid, 1995). Cecchini et al. (2013) used a functional metagenomics approach to investigate the prebiotic hydrolyzing potential of the human gut microbiome by searching for novel prebiotic degradation pathways in a human ileum mucosa and a fecal microbiota derived metagenomic library. They identified high numbers of unknown gut microorganisms capable of hydrolyzing established prebiotics, indicating that the prebiotics tested are not specifically metabolized by their target microorganisms alone. Further investigations must be carried out to determine the effect (if any) of non-specific hydrolysis of prebiotic preparations in the human gut. Galacto-oligosaccharides (GOS) with prebiotic properties can be synthesized through the transgalactosylation activity of ß-galactosidase enzymes on lactose. Wang et al. (2012) validated the ability of a novel ß-galactosidase isolated from a metagenome-derived library for its ability to produce GOS. Carrying out the reaction in an organic-aqueous biphasic media was shown to improve GOS yield. The ß-galactosidase gene discovered in this study is a promising candidate for industrial production of GOS to be used as an additive in various food and dairy products. All of these studies highlight the flexibility of functional metagenomics as a molecular tool not only for identifying new metabolic pathways for biosynthesis of useful/industrially relevant compounds but also for evaluating the efficiency of current therapeutic strategies.
Discovery of Novel Antimicrobials
A major driving force behind the biotechnological applications of functional metagenomics is the search for novel antimicrobials effective in medical settings. Microorganisms produce antibiotic molecules to alleviate competitors in their natural habitat. Natural sources have proved fruitful in the past for providing antibiotic molecules, from the discovery of penicillin produced by Penicillium rubens in 1928 to date. Although most bacterial infections in humans are curable with current antibiotic therapies, the emergent problem of antimicrobial resistance has led to the prevalence of persistent untreatable infections caused by certain pathogens which have developed a resistance to the used antimicrobial therapy. Antibiotic resistance has challenged medical practitioners and researchers and has led to outbreaks of serious untreatable bacterial infections in clinical settings and even community outbreaks have occurred (Alanis, 2005), making antimicrobial resistance a serious threat to human health (World Health Organization, 2014). The rate of antimicrobial drug discovery has declined in recent years, owing in part to a low drug approval rate by governing bodies (Cooper and Shlaes, 2011) and lesser rewards for manufacturers (Fischbach and Walsh, 2009). The exhaustion of products from culturable microorganisms and preferred use of chemical libraries of pure synthetic compounds over natural product exploration (Li and Vederas, 2009) have also contributed. New advances in metagenomics, high throughput screenings (HTS) and metabolic engineering, e.g., Jayasuriya et al. (2007), provide a new lease of life for natural product drug discovery. Functional metagenomic screens can be applied to the identification of novel antimicrobial molecules by screening microbial populations for antimicrobial activity against indicator or clinically relevant microorganisms. So far, this approach has led to the discovery of several novel antimicrobial compounds (Table 3). Gillespie et al. (2002) described the discovery of two novel antimicrobials (turbomycin A and B) exhibiting broad-spectrum activity against both gram-positive and gram-negative bacteria. These antibiotics were identified through activity-based screening of a metagenomic library from soil samples expressed in an E. coli host. Several metagenomic E. coli clones expressing antimicrobial activity were discovered by Macneil et al. (2001) through function-based screening of a BAC library constructed from soil microbial DNA. Metagenomic inserts from active clones were found to be related to the compound indirubin, a cyclin-dependent kinases (CDK) inhibitor used in the treatment of human chronic myelocytic leukemia (Hoessel et al., 1999; Marko et al., 2001). An indirubin compound with antimicrobial activity was also identified through activity-based screening of a forest soil metagenomic library by Lim et al. (2005). More recently, Scanlon et al. (2014) developed a HTS method which enabled them to co-culture recombinant clones from a native staphylococcal-derived metagenomic library with the bacterial pathogen Staphylococcus aureus in hydrogen-in-oil emulsions, with antibiotic activity being rapidly detected using a fluorescent viability assay. Six clones expressing a lysostaphin gene from Staphylococcus simulans with activity against S. aureus were identified in this way. Iqbal et al. (2014) constructed a metagenomic library from Arizona soil hosted by Ralstonia metallidurans. Functional screening for antimicrobial activity against Bacillus subtilis identified six positive clones encoding proteases, a lipase, and enzymes with cell wall lytic activity. These studies highlight the success of applying functional metagenomics to the discovery of novel natural antimicrobials with potential value to the pharmaceutical industry.
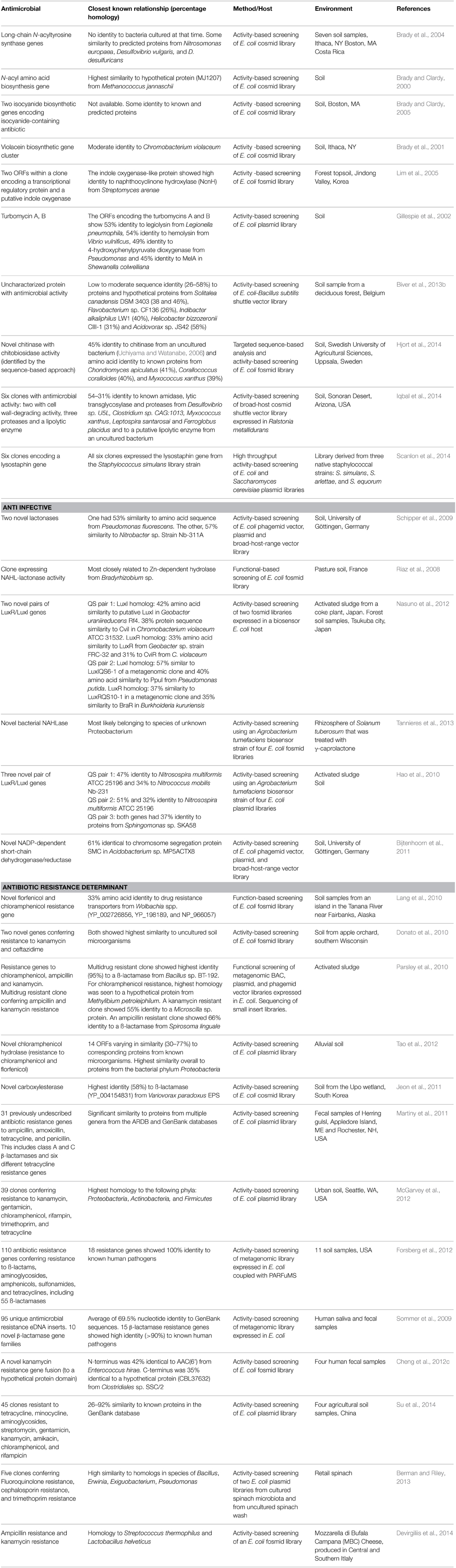
Table 3. Some novel antimicrobials, anti-infectives and antimicrobial resistance genes discovered through functional metagenomics.
Certain cell-to-cell communication or quorum sensing molecules and agents with quorum sensing inhibitory (QSI) activities have been also discovered through function-based screening of metagenomic libraries (Table 3). An interesting study by Nasuno et al. (2012) identified two novel sets of quorum sensing (QS) genes from the LuxI family N-acyl-L-homoserine lactone (AHL) synthases and their paired LuxR family transcriptional regulators. These authors constructed metagenomic libraries from an activated sludge from a coke plant and forest soil samples and functionally screened them for the presence of QS genes using a modified E. coli host. This biosensor strain contained a gfp plasmid which produced unstable GFP in response to low levels of five different AHLs, enabling the detection of QS-regulated activity. Other studies which have applied metagenomics for the exploration of QS regulation are reviewed by Kimura (2014). When it comes to treating individuals infected with, or curbing outbreaks of, antimicrobial-resistant pathogens, in some cases quorum sensing inhibitors as an anti-virulence strategy may be a useful course of action. The concept of using quorum sensing inhibitors would also be of benefit to the food industry in the control of undesirable microorganisms in food preparations or food processing environments. Schipper et al. (2009) screened a soil metagenomic library, expressed in E. coli, for QSI activity using an A. tumefaciens based bioassay. The positive clones were expressed in Pseudomonas aeruginosa and were found to be most likely responsible for the reduced motility and biofilm formation observed in the P. aeruginosa host cells expressing the proteins of interest. Of the three active clones isolated, one was found to be similar to a known lactonase, and the remaining two clones were determined to encode novel lactonases.
Certain antimicrobial strategies used in clinical settings could also be applied to the control of bacterial persistence in food development and manufacturing processes. In industrial settings contamination of food products occurs at various stages throughout the food processing cycle. The raw food itself is usually a source of initial contamination. Food can also become contaminated or re-contaminated during its processing, e.g., re-contamination of milk post-pasteurization, resulting in an unsafe or spoiled product. The removal of harmful or spoilage microorganisms from food products and the prevention of microorganisms entering or persisting in food processing is highly desirable. This needs to occur without damaging the structure, texture, taste, and overall quality of food. A potentially powerful application of functional metagenomics with respect to the food industry is screening natural sources for bioactive molecules that function as antimicrobials or inhibitory compounds for use in food safety maintenance strategies. Once the compounds have been identified and mass produced, the ultimate goal is for them to be formulated into safe sanitization products that will not influence the quality of the food product. As microorganisms are widely used and often beneficial to the food industry (e.g., cheese manufacture, brewing), the aim would be to eliminate only those microorganisms which pose a threat to food safety and quality. Screening is performed in a targeted manner to identify isolates producing compounds that inhibit or eliminate the presence of a given problematic microorganism present in the food product or processing equipment. Due to their specificity, bioactives isolated from microorganisms may be used in combination with existing sanitization products. Extremophiles are of particular interest as these could target undesirable microorganisms in extreme environments, which are often present in food processing.
Functional metagenomics can be used to combat antimicrobial resistance via two strategies; through the discovery of novel antibiotics and anti-infectives (as mentioned above) and through the identification of resistance genes in microbial populations. As resistance is transferable, horizontal gene transfer (HGT) being the most common method by which resistance is acquired by previously susceptible strains, resistant genes possessed by environmental bacteria may be acquired by human pathogens. Functional metagenomics can be used to identify novel resistance mechanisms used by bacteria in nature which may not have manifested in the clinical setting yet and so can allow one to predict possible routes via which resistance to current antibiotic therapies could emerge. The studies discussed below provide insight into the diversity of antimicrobial resistance mechanisms, proposing new avenues of research for tackling antibiotic resistance. They also show the value of functional metagenomics as a tool for the investigation of antimicrobial resistance, as reviewed by Mullany (2014). Donato et al. (2010) screened a metagenomic apple orchard soil library for DNA fragments that conferred antibiotic resistance to their E. coli host. Clones were screened for resistance to a selection of 10 antibiotics. The group reported the discovery of two novel enzymes. In one case, a metagenomic clone encoding an aminoglycoside acetyltransferase domain fused to a second acetyltransferase domain displayed resistance to kanamycin. Interestingly, sequence analysis of this clone did not predict antimicrobial resistance. The second interesting clone encoded a bifunctional protein containing a natural fusion of a ß-lactamase and a sigma factor conferring onto the host resistance to ceftazidime. Additional potential chloramphenicol resistance was predicted by sequencing this particular clone, which may evoke further analysis. Tao et al. (2012) used a TLC-based method to screen an alluvial soil-derived metagenomic library for chloramphenicol resistance. They identified a resistant clone harboring a hydrolysate which conferred to the host resistance to chloramphenicol and florfenicol, a synthetic form of chloramphenicol that was employed as a safe antibiotic treatment for use in farming. The enzyme was capable of hydrolyzing both chloramphenicol and florfenicol, with greater efficiency at hydrolyzing florfenicol. Various metagenomic studies have been carried out to identify antimicrobial resistance genes in certain foods. Antibiotic therapies for the treatment of bacterial infections in farm animals select for resistant microbes in food production chains (Hawkey, 2008). Although most microorganisms in foodstuffs are usually not pathogenic, resistant bacteria that survive on products for human consumption may transfer their resistance to opportunistic human pathogens or to the human microbiota. Certain foods (e.g., foods eaten raw) and the human gut microbiota itself may then potentially become a reservoir for antibiotic resistance genes. Retail spinach is commonly eaten raw and thus, has been linked to outbreaks of bacterial infections (Lynch et al., 2009; Wendel et al., 2009). Berman and Riley (2013) functionally screened two spinach-derived metagenomic libraries for resistance to 16 different antimicrobial agents, identifying numerous novel genes conferring resistance to ampicillin, aztreonam, ciprofloxacin, trimethoprim, and trimethoprim-sulfamethoxazole from five different active clones. Their study suggests that microorganisms in close contact with fresh food products, such as plant commensals and saprophytes, may serve as a reservoir of antimicrobial resistance genes. In a study with a similar objective, Devirgiliis et al. (2014) isolated clones displaying ampicillin and kanamycin resistance from a metagenomic library constructed from Mozzarella di Bufala Campana Italian cheese. These studies ultimately show that food products can potentially harbor bacterial species possessing clinically relevant antimicrobial resistance which may be horizontally transferred to pathogens, either directly or by an indirect route through the human microbiota.
Unusual or unexpected antimicrobial resistance mechanisms can be found in nature. Some studies investigating the resistome of uncultured bacteria have explored areas and environments which have not been previously exposed to clinical antibiotics and where endogenous microorganisms have therefore not faced selective pressure to develop antibiotic resistance. A recent study by Fouhy et al. (2014) examined the resistome of the naïve infant gut. A metagenomic library constructed from fecal samples of 22 six-month old infants who had not previously been exposed to antibiotics was screened for resistance to aminoglycoside and β-lactam antibiotics, identifying gentamicin and ampicillin resistant clones. PCR analyses were also carried out to detect DNA sequences encoding aminoglycoside and β-lactam resistance genes not successfully cloned and expressed in the library. One hundred ampicillin resistant clones were identified in their functional screen, conferring resistance via several β-lactamase genes. Aminoglycoside resistant clones were also identified, whose resistance was conferred by acetylation, adenylation, and phosphorylation genes. This study uncovered resistance to clinically relevant antibiotics in a naïve environment. Other studies assessing the resistome of microbial samples from remote areas where little or no antibiotic therapy has been practiced have also identified unexpected resistance (Pallecchi et al., 2008; Bartoloni et al., 2009). More recently, Clemente et al. (2015) examined the bacterial microbiome (from fecal, oral, and skin samples) of 34 Yanomami individuals from an isolated Amerindian village in South America. Among huge microbial diversity observed through 16S rRNA gene sequencing of DNA from the obtained samples, activity-based and culture-independent screening of functional and shotgun metagenomic libraries also revealed resistance genes to clinically relevant antibiotics. These studies further emphasize the diversity of the as yet uncultured microbial world by establishing that genes conveying resistance to current antibiotic therapies can be found in environments void of selective pressure.
Conclusions and Future Prospects
Metagenomics grants access to the huge diversity of the microbial world and has led to significant progress among research communities and in industrial settings with respect to understanding and benefitting from unculturable microbes. Functional metagenomics is a powerful tool for the discovery of novel enzymes and bioactives sourced from as yet uncultured microorganisms. As a relatively new technology, functional metagenomics faces challenges that have yet to be overcome. However, the promise of a technique that has already proven to be fruitful even in its early years suggests that there can be significant rewards if appropriate solutions and further optimization takes place. The development of new screening and selection techniques along with faster and cheaper sequencing technologies will allow for the expansion of a very promising field in microbiology, genetics and the food and pharmaceutical industries.
This article discusses the potential of functional metagenomics to facilitate the development of novel industrial products sourced from as yet uncultured microorganisms. Nonetheless, following the identification of useful proteins and bioactives, challenges ensue in another area, that being the development of a consumer friendly and commercially viable product that can be manufactured in industrially relevant quantities, retains its activity when scaled up (for example when present in high amounts in a large industrial reaction vessel), can be purified and formulated appropriately into a finished product and maintains its stability during shipping and storage. The product also needs to be reasonably easy to use and must be applicable to current industrial demands, i.e., the product must perform efficiently under the proposed/outlined conditions to carry out the job it was bought to do. A successful reaction achieved under laboratory conditions may be difficult to reproduce on an industrial scale. Pilot plant studies must be carried out initially to identify any variables or short comings in the reaction that were not evident at the laboratory stages of development. These studies are a stepping stone between discovery of the interesting active agent and its formulation into a final commercial product. Once deficiencies and other problems have been corrected in the pilot plant phase, further studies must be conducted to qualify the agent at an industrial level and guarantee the development of a robust product that is efficient and true to its intended purpose. The acceptability of the novel enzyme or bioactive and its source microorganism to the relevant regulatory authorities must also be considered.
Once all these limitations are overcome, through access to the seemingly infinite diversity of the microbial world, functional metagenomics presents an opportunity to develop novel innovative products that offer something new and useful to industrial processes or even change for the better or make more convenient the way a current process is carried out.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The financial support of Science Foundation Ireland (SFI) under Grant Number 13/SIRG/2157 is acknowledged.
References
Abumweis, S. S., Jew, S., and Ames, N. P. (2010). Beta-glucan from barley and its lipid-lowering capacity: a meta-analysis of randomized, controlled trials. Eur. J. Clin. Nutr. 64, 1472–1480. doi: 10.1038/ejcn.2010.178
Alanis, A. J. (2005). Resistance to antibiotics: are we in the post-antibiotic era? Arch. Med. Res. 36, 697–705. doi: 10.1016/j.arcmed.2005.06.009
Ashie, I. N. A., Sorensen, T. L., and Nielsen, P. M. (2002). Effects of papain and a microbial enzyme on meat proteins and beef tenderness. J. Food Sci. 67, 2138–2142. doi: 10.1111/j.1365-2621.2002.tb09516.x
Banik, J. J., and Brady, S. F. (2010). Recent application of metagenomic approaches toward the discovery of antimicrobials and other bioactive small molecules. Curr. Opin. Microbiol. 13, 603–609. doi: 10.1016/j.mib.2010.08.012
Bartoloni, A., Pallecchi, L., Rodriguez, H., Fernandez, C., Mantella, A., Bartalesi, F., et al. (2009). Antibiotic resistance in a very remote Amazonas community. Int. J. Antimicrob. Agents 33, 125–129. doi: 10.1016/j.ijantimicag.2008.07.029
Bates, S. T., Berg-Lyons, D., Lauber, C. L., Walters, W. A., Knight, R., and Fierer, N. (2012). A preliminary survey of lichen associated eukaryotes using pyrosequencing. Lichenologist 44, 137–146. doi: 10.1017/S0024282911000648
Berman, H. F., and Riley, L. W. (2013). Identification of novel antimicrobial resistance genes from microbiota on retail spinach. BMC Microbiol. 13:272. doi: 10.1186/1471-2180-13-272
Bijtenhoorn, P., Mayerhofer, H., Muller-Dieckmann, J., Utpatel, C., Schipper, C., Hornung, C., et al. (2011). A novel metagenomic short-chain dehydrogenase/reductase attenuates Pseudomonas aeruginosa biofilm formation and virulence on Caenorhabditis elegans. PLoS ONE 6:e26278. doi: 10.1371/journal.pone.0026278
Biver, S., Portetelle, D., and Vandenbol, M. (2013a). Characterization of a new oxidant-stable serine protease isolated by functional metagenomics. Springerplus 2:410. doi: 10.1186/2193-1801-2-410
Biver, S., Steels, S., Portetelle, D., and Vandenbol, M. (2013b). Bacillus subtilis as a tool for screening soil metagenomic libraries for antimicrobial activities. J. Microbiol. Biotechnol. 23, 850–855. doi: 10.4014/jmb.1212.12008
Biver, S., and Vandenbol, M. (2013). Characterization of three new carboxylic ester hydrolases isolated by functional screening of a forest soil metagenomic library. J. Ind. Microbiol. Biotechnol. 40, 191–200. doi: 10.1007/s10295-012-1217-7
Boadi, D. K., and Neufeld, R. J. (2001). Encapsulation of tannase for the hydrolysis of tea tannins. Enzyme Microb. Technol. 28, 590–595. doi: 10.1016/S0141-0229(01)00295-2
Bornscheuer, U. T. (2002). Microbial carboxyl esterases: classification, properties and application in biocatalysis. FEMS Microbiol. Rev. 26, 73–81. doi: 10.1111/j.1574-6976.2002.tb00599.x
Brady, S. F., Chao, C. J., and Clardy, J. (2004). Long-chain N-acyltyrosine synthases from environmental DNA. Appl. Environ. Microbiol. 70, 6865–6870. doi: 10.1128/AEM.70.11.6865-6870.2004
Brady, S. F., Chao, C. J., Handelsman, J., and Clardy, J. (2001). Cloning and heterologous expression of a natural product biosynthetic gene cluster from eDNA. Org. Lett. 3, 1981–1984. doi: 10.1021/ol015949k
Brady, S. F., and Clardy, J. (2000). Long-chain N-acyl amino acid antibiotics isolated from heterologously expressed environmental DNA. J. Am. Chem. Soc. 122, 12903–12904. doi: 10.1021/ja002990u
Brady, S. F., and Clardy, J. (2005). Cloning and heterologous expression of isocyanide biosynthetic genes from environmental DNA. Angew. Chem. Int. Ed Engl. 44, 7063–7065. doi: 10.1002/anie.200501941
Cantarelli, C., Brenna, O., Giovanelli, G., and Rossi, M. (1989). Beverage stabilization through enzymatic removal of phenolics. Food Biotechnol. 3, 203–213. doi: 10.1080/08905438909549709
Cecchini, D. A., Laville, E., Laguerre, S., Robe, P., Leclerc, M., Dore, J., et al. (2013). Functional metagenomics reveals novel pathways of prebiotic breakdown by human gut bacteria. PLoS ONE 8:e72766. doi: 10.1371/journal.pone.0072766
Chandler, J. A., Thongsripong, P., Green, A., Kittayapong, P., Wilcox, B. A., Schroth, G. P., et al. (2014). Metagenomic shotgun sequencing of a Bunyavirus in wild-caught Aedes aegypti from Thailand informs the evolutionary and genomic history of the Phleboviruses. Virology 464–465, 312–319. doi: 10.1016/j.virol.2014.06.036
Chang, F. Y., and Brady, S. F. (2013). Discovery of indolotryptoline antiproliferative agents by homology-guided metagenomic screening. Proc. Natl. Acad. Sci. U.S.A. 110, 2478–2483. doi: 10.1073/pnas.1218073110
Cheng, F. S., Sheng, J. P., Cai, T., Jin, J., Liu, W. Z., Lin, Y. M., et al. (2012a). A protease-insensitive feruloyl esterase from China holstein cow rumen metagenomic library: expression, characterization, and utilization in ferulic acid release from wheat straw. J. Agric. Food Chem. 60, 2546–2553. doi: 10.1021/jf204556u
Cheng, F. S., Sheng, J. P., Dong, R. B., Men, Y. J., Gan, L., and Shen, L. (2012b). Novel xylanase from a holstein cattle rumen metagenomic library and its application in xylooligosaccharide and ferulic acid production from wheat straw. J. Agric. Food Chem. 60, 12516–12524. doi: 10.1021/jf302337w
Cheng, G., Hu, Y., Yin, Y., Yang, X., Xiang, C., Wang, B., et al. (2012c). Functional screening of antibiotic resistance genes from human gut microbiota reveals a novel gene fusion. FEMS Microbiol. Lett. 336, 11–16. doi: 10.1111/j.1574-6968.2012.02647.x
Chu, X. M., He, H. Z., Guo, C. Q., and Sun, B. L. (2008). Identification of two novel esterases from a marine metagenomic library derived from South China Sea. Appl. Microbiol. Biotechnol. 80, 615–625. doi: 10.1007/s00253-008-1566-3
Clemente, J. C., Pehrsson, E. C., Blaser, M. J., Sandhu, K., Gao, Z., Wang, B., et al. (2015). The microbiome of uncontacted Amerindians. Sci. Adv. 1:e1500183. doi: 10.1126/sciadv.1500183
Cooper, M. A., and Shlaes, D. (2011). Fix the antibiotics pipeline. Nature 472, 32. doi: 10.1038/472032a
Craig, J. W., Chang, F. Y., and Brady, S. F. (2009). Natural products from environmental DNA hosted in Ralstonia metallidurans. ACS Chem. Biol. 4, 23–28. doi: 10.1021/cb8002754
Craig, J. W., Chang, F. Y., Kim, J. H., Obiajulu, S. C., and Brady, S. F. (2010). Expanding small-molecule functional metagenomics through parallel screening of broad-host-range cosmid environmental DNA libraries in diverse proteobacteria. Appl. Environ. Microbiol. 76, 1633–1641. doi: 10.1128/AEM.02169-09
Culligan, E. P., Sleator, R. D., Marchesi, J. R., and Hill, C. (2012). Functional metagenomics reveals novel salt tolerance loci from the human gut microbiome. ISME J. 6, 1916–1925. doi: 10.1038/ismej.2012.38
Culligan, E. P., Sleator, R. D., Marchesi, J. R., and Hill, C. (2013). Functional environmental screening of a metagenomic library identifies stlA; a unique salt tolerance locus from the human gut microbiome. PLoS ONE 8:e82985. doi: 10.1371/journal.pone.0082985
Devirgiliis, C., Zinno, P., Stirpe, M., Barile, S., and Perozzi, G. (2014). Functional screening of antibiotic resistance genes from a representative metagenomic library of food fermenting microbiota. Biomed Res. Int. 2014:290967. doi: 10.1155/2014/290967
Donato, J. J., Moe, L. A., Converse, B. J., Smart, K. D., Berklein, F. C., McManus, P. S., et al. (2010). Metagenomic analysis of apple orchard soil reveals antibiotic resistance genes encoding predicted bifunctional proteins. Appl. Environ. Microbiol. 76, 4396–4401. doi: 10.1128/AEM.01763-09
Elend, C., Schmeisser, C., Hoebenreich, H., Steele, H. L., and Streit, W. R. (2007). Isolation and characterization of a metagenome-derived and cold-active lipase with high stereospecificity for (R)-ibuprofen esters. J. Biotechnol. 130, 370–377. doi: 10.1016/j.jbiotec.2007.05.015
Elend, C., Schmeisser, C., Leggewie, C., Babiak, P., Carballeira, J. D., Steele, H. L., et al. (2006). Isolation and biochemical characterization of two novel metagenome-derived esterases. Appl. Environ. Microbiol. 72, 3637–3645. doi: 10.1128/AEM.72.5.3637-3645.2006
Entcheva, P., Liebl, W., Johann, A., Hartsch, T., and Streit, W. R. (2001). Direct cloning from enrichment cultures, a reliable strategy for isolation of complete operons and genes from microbial consortia. Appl. Environ. Microbiol. 67, 89–99. doi: 10.1128/AEM.67.1.89-99.2001
Faoro, H., Glogauer, A., Couto, G. H., de Souza, E. M., Rigo, L. U., Cruz, L. M., et al. (2012). Characterization of a new Acidobacteria-derived moderately thermostable lipase from a Brazilian Atlantic Forest soil metagenome. FEMS Microbiol. Ecol. 81, 386–394. doi: 10.1111/j.1574-6941.2012.01361.x
Faulds, C. B. (2010). What can feruloyl esterases do for us? Phytochem. Rev. 9, 121–132. doi: 10.1007/s11101-009-9156-2
Ferrer, M., Golyshina, O. V., Chernikova, T. N., Khachane, A. N., Martins Dos Santos, V. A., Yakimov, M. M., et al. (2005a). Microbial enzymes mined from the Urania deep-sea hypersaline anoxic basin. Chem. Biol. 12, 895–904. doi: 10.1016/j.chembiol.2005.05.020
Ferrer, M., Golyshina, O. V., Chernikova, T. N., Khachane, A. N., Reyes-Duarte, D., Dos Santos, V., et al. (2005b). Novel hydrolase diversity retrieved from a metagenome library of bovine rumen microflora. Environ. Microbiol. 7, 1996–2010. doi: 10.1111/j.1462-2920.2005.00920.x
Fischbach, M. A., and Walsh, C. T. (2009). Antibiotics for emerging pathogens. Science 325, 1089–1093. doi: 10.1126/science.1176667
Forsberg, K. J., Reyes, A., Wang, B., Selleck, E. M., Sommer, M. O., and Dantas, G. (2012). The shared antibiotic resistome of soil bacteria and human pathogens. Science 337, 1107–1111. doi: 10.1126/science.1220761
Fouhy, F., Ogilvie, L. A., Jones, B. V., Ross, R. P., Ryan, A. C., Dempsey, E. M., et al. (2014). Identification of aminoglycoside and beta-lactam resistance genes from within an infant gut functional metagenomic library. PLoS ONE 9:e0108016. doi: 10.1371/journal.pone.0108016
Fujita, M. J., Kimura, N., Sakai, A., Ichikawa, Y., Hanyu, T., and Otsuka, M. (2011). Cloning and heterologous expression of the vibrioferrin biosynthetic gene cluster from a marine metagenomic library. Biosci. Biotechnol. Biochem. 75, 2283–2287. doi: 10.1271/bbb.110379
Gibson, G. R., and Roberfroid, M. B. (1995). Dietary modulation of the human colonic microbiota: introducing the concept of prebiotics. J. Nutr. 125, 1401–1412.
Gillespie, D. E., Brady, S. F., Bettermann, A. D., Cianciotto, N. P., Liles, M. R., Rondon, M. R., et al. (2002). Isolation of antibiotics turbomycin a and B from a metagenomic library of soil microbial DNA. Appl. Environ. Microbiol. 68, 4301–4306. doi: 10.1128/AEM.68.9.4301-4306.2002
Guazzaroni, M. E., Morgante, V., Mirete, S., and Gonzalez-Pastor, J. E. (2013). Novel acid resistance genes from the metagenome of the Tinto River, an extremely acidic environment. Environ. Microbiol. 15, 1088–1102. doi: 10.1111/1462-2920.12021
Gupta, R., Gupta, N., and Rathi, P. (2004). Bacterial lipases: an overview of production, purification and biochemical properties. Appl. Microbiol. Biotechnol. 64, 763–781. doi: 10.1007/s00253-004-1568-8
Hafeez, Z., Cakir-Kiefer, C., Roux, E., Perrin, C., Mido, L., and Dary-Mourot, A. (2014). Strategies of producing bioactive peptides from milk proteins to functionalize fermented milk products. Food Res. Int. 63, 71–80. doi: 10.1016/j.foodres.2014.06.002
Hamada, S., Suzuki, K., Aoki, N., and Suzuki, Y. (2013). Improvements in the qualities of gluten-free bread after using a protease obtained from Aspergillus oryzae. J. Cereal Sci. 57, 91–97. doi: 10.1016/j.jcs.2012.10.008
Hao, Y., Winans, S. C., Glick, B. R., and Charles, T. C. (2010). Identification and characterization of new LuxR/LuxI-type quorum sensing systems from metagenomic libraries. Environ. Microbiol. 12, 105–117. doi: 10.1111/j.1462-2920.2009.02049.x
Hasan, F., Shah, A. A., and Hameed, A. (2006). Industrial applications of microbial lipases. Enzyme Microb. Technol. 39, 235–251. doi: 10.1016/j.enzmictec.2005.10.016
Hawkey, P. M. (2008). The growing burden of antimicrobial resistance. J. Antimicrob. Chemother. 62, I1–I9. doi: 10.1093/jac/dkn241
Henne, A., Schmitz, R. A., Bomeke, M., Gottschalk, G., and Daniel, R. (2000). Screening of environmental DNA libraries for the presence of genes conferring lipolytic activity on Escherichia coli. Appl. Environ. Microbiol. 66, 3113–3116. doi: 10.1128/AEM.66.7.3113-3116.2000
Hess, M., Sczyrba, A., Egan, R., Kim, T. W., Chokhawala, H., Schroth, G., et al. (2011). Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331, 463–467. doi: 10.1126/science.1200387
Hjort, K., Presti, I., Elvang, A., Marinelli, F., and Sjoling, S. (2014). Bacterial chitinase with phytopathogen control capacity from suppressive soil revealed by functional metagenomics. Appl. Microbiol. Biotechnol. 98, 2819–2828. doi: 10.1007/s00253-013-5287-x
Hoessel, R., Leclerc, S., Endicott, J. A., Nobel, M. E. M., Lawrie, A., Tunnah, P., et al. (1999). Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol. 1, 60–67.
Huang, X. W., Chen, L. J., Luo, Y. B., Guo, H. Y., and Ren, F. Z. (2011). Purification, characterization, and milk coagulating properties of ginger proteases. J. Dairy Sci. 94, 2259–2269. doi: 10.3168/jds.2010-4024
Iqbal, H. A., Craig, J. W., and Brady, S. F. (2014). Antibacterial enzymes from the functional screening of metagenomic libraries hosted in Ralstonia metallidurans. FEMS Microbiol. Lett. 354, 19–26. doi: 10.1111/1574-6968.12431
Jayasuriya, H., Herath, K. B., Zhang, C., Zink, D. L., Basilio, A., Genilloud, O., et al. (2007). Isolation and structure of platencin: a FabH and FabF dual inhibitor with potent broad-spectrum antibiotic activity. Angew. Chem. Int. Ed Engl. 46, 4684–4688. doi: 10.1002/anie.200701058
Jeon, J. H., Kim, S. J., Lee, H. S., Cha, S. S., Lee, J. H., Yoon, S. H., et al. (2011). Novel metagenome-derived carboxylesterase that hydrolyzes beta-lactam antibiotics. Appl. Environ. Microbiol. 77, 7830–7836. doi: 10.1128/AEM.05363-11
Jiang, C. J., Hao, Z. Y., Zeng, R., Shen, P. H., Li, J. F., and Wu, B. (2011). Characterization of a novel serine protease inhibitor gene from a marine metagenome. Mar. Drugs 9, 1487–1501. doi: 10.3390/md9091487
Jiang, C. J., Ma, G. F., Li, S. X., Hu, T. T., Che, Z. Q., Shen, P. H., et al. (2009). Characterization of a novel beta-glucosidase-like activity from a soil metagenome. J. Microbiol. 47, 542–548. doi: 10.1007/s12275-009-0024-y
Joint, F. (2001). WHO Expert Consultation on Evaluation of Health and Nutritional Properties of Probiotics in Food Including Powder Milk with Live Lactic Acid Bacteria. Córdoba: Food and Agriculture Organization of the United Nations and the World Health Organization.
Kang, C. H., Oh, K. H., Lee, M. H., Oh, T. K., Kim, B. H., and Yoon, J. (2011). A novel family VII esterase with industrial potential from compost metagenomic library. Microb. Cell Fact. 10:41. doi: 10.1186/1475-2859-10-41
Kato, K., Ono, M., and Akita, H. (1997). New total synthesis of (-)- and (+)-chuangxinmycins. Tetrahedron-Asymmetry 8, 2295–2298. doi: 10.1016/S0957-4166(97)00253-X
Kay, G. L., Sergeant, M. J., Giuffra, V., Bandiera, P., Milanese, M., Bramanti, B., et al. (2014). Recovery of a medieval Brucella melitensis genome using shotgun metagenomics. MBio 5, e01337-14. doi: 10.1128/mBio.01337-14
Keller, A., Horn, H., Forster, F., and Schultz, J. (2014). Computational integration of genomic traits into 16S rDNA microbiota sequencing studies. Gene 549, 186–191. doi: 10.1016/j.gene.2014.07.066
Kim, Y. J., Choi, G. S., Kim, S. B., Yoon, G. S., Kim, Y. S., and Ryu, Y. W. (2006). Screening and characterization of a novel esterase from a metagenomic library. Protein Expr. Purif. 45, 315–323. doi: 10.1016/j.pep.2005.06.008
Kimura, N. (2014). Metagenomic approaches to understanding phylogenetic diversity in quorum sensing. Virulence 5, 433–442. doi: 10.4161/viru.27850
Lakhdari, O., Cultrone, A., Tap, J., Gloux, K., Bernard, F., Ehrlich, S. D., et al. (2010). Functional metagenomics: a high throughput screening method to decipher microbiota-driven NF-kappa B modulation in the human gut. PLoS ONE 5:e13092. doi: 10.1371/journal.pone.0013092
Lane, D. J., Pace, B., Olsen, G. J., Stahl, D. A., Sogin, M. L., and Pace, N. R. (1985). Rapid-determination of 16s Ribosomal-Rna sequences for phylogenetic analyses. Proc. Natl. Acad. Sci. U.S.A. 82, 6955–6959. doi: 10.1073/pnas.82.20.6955
Lang, K. S., Anderson, J. M., Schwarz, S., Williamson, L., Handelsman, J., and Singer, R. S. (2010). Novel florfenicol and chloramphenicol resistance gene discovered in Alaskan soil by using functional metagenomics. Appl. Environ. Microbiol. 76, 5321–5326. doi: 10.1128/AEM.00323-10
Langille, M. G. I., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Lee, D. G., Jeon, J. H., Jang, M. K., Kim, N. Y., Lee, J. H., Lee, J. H., et al. (2007). Screening and characterization of a novel fibrinolytic metalloprotease from a metagenomic library. Biotechnol. Lett. 29, 465–472. doi: 10.1007/s10529-006-9263-8
Lee, S. W., Won, K., Lim, H. K., Kim, J. C., Choi, G. J., and Cho, K. Y. (2004). Screening for novel lipolytic enzymes from uncultured soil microorganisms. Appl. Microbiol. Biotechnol. 65, 720–726. doi: 10.1007/s00253-004-1722-3
Li, J. W., and Vederas, J. C. (2009). Drug discovery and natural products: end of an era or an endless frontier? Science 325, 161–165. doi: 10.1126/science.1168243
Lim, H. K., Chung, E. J., Kim, J. C., Choi, G. J., Jang, K. S., Chung, Y. R., et al. (2005). Characterization of a forest soil metagenome clone that confers indirubin and indigo production on Escherichia coli. Appl. Environ. Microbiol. 71, 7768–7777. doi: 10.1128/AEM.71.12.7768-7777.2005
Ling, L. L., Schneider, T., Peoples, A. J., Spoering, A. L., Engels, I., Conlon, B. P., et al. (2015). A new antibiotic kills pathogens without detectable resistance. Nature 517, 455–459. doi: 10.1038/nature14098
Lynch, M. F., Tauxe, R. V., and Hedberg, C. W. (2009). The growing burden of foodborne outbreaks due to contaminated fresh produce: risks and opportunities. Epidemiol. Infect. 137, 307–315. doi: 10.1017/S0950268808001969
Macneil, I. A., Tiong, C. L., Minor, C., August, P. R., Grossman, T. H., Loiacono, K. A., et al. (2001). Expression and isolation of antimicrobial small molecules from soil DNA libraries. J. Mol. Microbiol. Biotechnol. 3, 301–308.
Marko, D., Schatzle, S., Friedel, A., Genzlinger, A., Zankl, H., Meijer, L., et al. (2001). Inhibition of cyclin-dependent kinase 1 (CDK1) by indirubin derivatives in human tumour cells. Br. J. Cancer 84, 283–289. doi: 10.1054/bjoc.2000.1546
Martiny, A. C., Martiny, J. B. H., Weihe, C., Field, A., and Ellis, J. C. (2011). Functional metagenomics reveals previously unrecognized diversity of antibiotic resistance genes in gulls. Front. Microbiol. 2:238. doi: 10.3389/fmicb.2011.00238
McGarvey, K. M., Queitsch, K., and Fields, S. (2012). Wide variation in antibiotic resistance proteins identified by functional metagenomic screening of a soil DNA library. Appl. Environ. Microbiol. 78, 1708–1714. doi: 10.1128/AEM.06759-11
Mendes, L. W., Tsai, S. M., Navarrete, A. A., de Hollander, M., van Veen, J. A., and Kuramae, E. E. (2015). Soil-borne microbiome: linking diversity to function. Microb. Ecol. 70, 255–265. doi: 10.1007/s00248-014-0559-2
Mora, L., Reig, M., and Toldra, F. (2014). Bioactive peptides generated from meat industry by-products. Food Res. Int. 65, 344–349. doi: 10.1016/j.foodres.2014.09.014
Moure, A., Gullon, P., Dominguez, H., and Parajo, J. C. (2006). Advances in the manufacture, purification and applications of xylo-oligosaccharides as food additives and nutraceuticals. Process Biochem. 41, 1913–1923. doi: 10.1016/j.procbio.2006.05.011
Mullany, P. (2014). Functional metagenomics for the investigation of antibiotic resistance. Virulence 5, 443–447. doi: 10.4161/viru.28196
Nacke, H., Will, C., Herzog, S., Nowka, B., Engelhaupt, M., and Daniel, R. (2011). Identification of novel lipolytic genes and gene families by screening of metagenomic libraries derived from soil samples of the German Biodiversity Exploratories. FEMS Microbiol. Ecol. 78, 188–201. doi: 10.1111/j.1574-6941.2011.01088.x
Nasuno, E., Kimura, N., Fujita, M. J., Nakatsu, C. H., Kamagata, Y., and Hanada, S. (2012). Phylogenetically novel LuxI/LuxR-type quorum sensing systems isolated using a metagenomic approach. Appl. Environ. Microbiol. 78, 8067–8074. doi: 10.1128/AEM.01442-12
Neveu, J., Regeard, C., and Dubow, M. S. (2011). Isolation and characterization of two serine proteases from metagenomic libraries of the Gobi and Death Valley deserts. Appl. Microbiol. Biotechnol. 91, 635–644. doi: 10.1007/s00253-011-3256-9
Oh, J., Byrd, A. L., Deming, C., Conlan, S., Kong, H. H., Segre, J. A., et al. (2014). Biogeography and individuality shape function in the human skin metagenome. Nature 514, 59–64. doi: 10.1038/nature13786
Ouyang, L. M., Liu, J. Y., Qiao, M., and Xu, J. H. (2013). Isolation and biochemical characterization of two novel metagenome-derived esterases. Appl. Biochem. Biotechnol. 169, 15–28. doi: 10.1007/s12010-012-9949-4
Palackal, N., Lyon, C. S., Zaidi, S., Luginbuhl, P., Dupree, P., Goubet, F., et al. (2007). A multifunctional hybrid glycosyl hydrolase discovered in an uncultured microbial consortium from ruminant gut. Appl. Microbiol. Biotechnol. 74, 113–124. doi: 10.1007/s00253-006-0645-6
Pallecchi, L., Bartoloni, A., Paradisi, F., and Rossolini, G. M. (2008). Antibiotic resistance in the absence of antimicrobial use: mechanisms and implications. Expert Rev. Anti Infect. Ther. 6, 725–732. doi: 10.1586/14787210.6.5.725
Panda, T., and Gowrishankar, B. S. (2005). Production and applications of esterases. Appl. Microbiol. Biotechnol. 67, 160–169. doi: 10.1007/s00253-004-1840-y
Panesar, P. S., Kumari, S., and Panesar, R. (2010). Potential applications of immobilized beta-galactosidase in food processing industries. Enzyme Res. 2010:473137. doi: 10.4061/2010/473137
Parsley, L. C., Consuegra, E. J., Kakirde, K. S., Land, A. M., Harper, W. F. Jr., and Liles, M. R. (2010). Identification of diverse antimicrobial resistance determinants carried on bacterial, plasmid, or viral metagenomes from an activated sludge microbial assemblage. Appl. Environ. Microbiol. 76, 3753–3757. doi: 10.1128/AEM.03080-09
Peng, Q., Wang, X., Shang, M., Huang, J., Guan, G., Li, Y., et al. (2014). Isolation of a novel alkaline-stable lipase from a metagenomic library and its specific application for milkfat flavor production. Microb. Cell Fact. 13:1. doi: 10.1186/1475-2859-13-1
Piel, J. (2002). A polyketide synthase-peptide synthetase gene cluster from an uncultured bacterial symbiont of Paederus beetles. Proc. Natl. Acad. Sci. U.S.A. 99, 14002–14007. doi: 10.1073/pnas.222481399
Pooja, S., Pushpanathan, M., Jayashree, S., Gunasekaran, P., and Rajendhran, J. (2015). Identification of periplasmic alpha-amlyase from cow dung metagenome by product induced gene expression profiling (Pigex). Indian J. Microbiol. 55, 57–65. doi: 10.1007/s12088-014-0487-3
Pushpam, P. L., Rajesh, T., and Gunasekaran, P. (2011). Identification and characterization of alkaline serine protease from goat skin surface metagenome. AMB Express 1:3. doi: 10.1186/2191-0855-1-3
Rabausch, U., Juergensen, J., Ilmberger, N., Bohnke, S., Fischer, S., Schubach, B., et al. (2013). Functional screening of metagenome and genome libraries for detection of novel flavonoid-modifying enzymes. Appl. Environ. Microbiol. 79, 4551–4563. doi: 10.1128/AEM.01077-13
Rhee, J. K., Ahn, D. G., Kim, Y. G., and Oh, J. W. (2005). New thermophilic and thermostable esterase with sequence similarity to the hormone-sensitive lipase family, cloned from a metagenomic library. Appl. Environ. Microbiol. 71, 817–825. doi: 10.1128/AEM.71.2.817-825.2005
Riaz, K., Elmerich, C., Moreira, D., Raffoux, A., Dessaux, Y., and Faure, D. (2008). A metagenomic analysis of soil bacteria extends the diversity of quorum-quenching lactonases. Environ. Microbiol. 10, 560–570. doi: 10.1111/j.1462-2920.2007.01475.x
Richardson, T. H., Tan, X. Q., Frey, G., Callen, W., Cabell, M., Lam, D., et al. (2002). A novel, high performance enzyme for starch liquefaction - Discovery and optimization of a low pH, thermostable alpha-amylase. J. Biol. Chem. 277, 26501–26507. doi: 10.1074/jbc.M203183200
Robertson, D. E., Chaplin, J. A., Desantis, G., Podar, M., Madden, M., Chi, E., et al. (2004). Exploring nitrilase sequence space for enantioselective catalysis. Appl. Environ. Microbiol. 70, 2429–2436. doi: 10.1128/AEM.70.4.2429-2436.2004
Rondon, M. R., August, P. R., Bettermann, A. D., Brady, S. F., Grossman, T. H., Liles, M. R., et al. (2000). Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl. Environ. Microbiol. 66, 2541–2547. doi: 10.1128/AEM.66.6.2541-2547.2000
Rosario, K., and Breitbart, M. (2011). Exploring the viral world through metagenomics. Curr. Opin. Virol. 1, 289–297. doi: 10.1016/j.coviro.2011.06.004
Sariozlu, N. Y., and Kivanc, M. (2009). Isolation of gallic acid-producing microorganisms and their use in the production of gallic acid from gall nuts and sumac. Afr. J. Biotechnol. 8, 1110–1115.
Scanlon, T. C., Dostal, S. M., and Griswold, K. E. (2014). A high-throughput screen for antibiotic drug discovery. Biotechnol. Bioeng. 111, 232–243. doi: 10.1002/bit.25019
Schipper, C., Hornung, C., Bijtenhoorn, P., Quitschau, M., Grond, S., and Streit, W. R. (2009). Metagenome-derived clones encoding two novel lactonase family proteins involved in biofilm inhibition in Pseudomonas aeruginosa. Appl. Environ. Microbiol. 75, 224–233. doi: 10.1128/AEM.01389-08
Schirmer, A., Gadkari, R., Reeves, C. D., Ibrahim, F., Delong, E. F., and Hutchinson, C. R. (2005). Metagenomic analysis reveals diverse polyketide synthase gene clusters in microorganisms associated with the marine sponge Discodermia dissoluta. Appl. Environ. Microbiol. 71, 4840–4849. doi: 10.1128/AEM.71.8.4840-4849.2005
Schoch, C. L., Seifert, K. A., Huhndorf, S., Robert, V., Spouge, J. L., Levesque, C. A., et al. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. U.S.A. 109, 6241–6246. doi: 10.1073/pnas.1117018109
Shen, D., Xu, J. H., Wu, H. Y., and Liu, Y. Y. (2002). Significantly improved esterase activity of Trichosporon brassicae cells for ketoprofen resolution by 2-propanol treatment. J. Mol. Catal. B Enzym. 18, 219–224. doi: 10.1016/S1381-1177(02)00099-1
Smits, S. L., and Osterhaus, A. D. (2013). Virus discovery: one step beyond. Curr. Opin. Virol. 3, e1–e6. doi: 10.1016/j.coviro.2013.03.007
Sommer, M. O. A., Dantas, G., and Church, G. M. (2009). Functional characterization of the antibiotic resistance reservoir in the human microflora. Science 325, 1128–1131. doi: 10.1126/science.1176950
Staley, C., Johnson, D., Gould, T. J., Wang, P., Phillips, J., Cotner, J. B., et al. (2015). Frequencies of heavy metal resistance are associated with land cover type in the Upper Mississippi River. Sci. Total Environ. 511, 461–468. doi: 10.1016/j.scitotenv.2014.12.069
Staley, J. T., and Konopka, A. (1985). Measurement of insitu activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu. Rev. Microbiol. 39, 321–346. doi: 10.1146/annurev.mi.39.100185.001541
Streit, W. R., and Entcheva, P. (2003). Biotin in microbes, the genes involved in its biosynthesis, its biochemical role and perspectives for biotechnological production. Appl. Microbiol. Biotechnol. 61, 21–31. doi: 10.1007/s00253-002-1186-2
Streit, W. R., and Schmitz, R. A. (2004). Metagenomics - the key to the uncultured microbes. Curr. Opin. Microbiol. 7, 492–498. doi: 10.1016/j.mib.2004.08.002
Su, J. Q., Wei, B., Xu, C. Y., Qiao, M., and Zhu, Y. G. (2014). Functional metagenomic characterization of antibiotic resistance genes in agricultural soils from China. Environ. Int. 65, 9–15. doi: 10.1016/j.envint.2013.12.010
Tannieres, M., Beury-Cirou, A., Vigouroux, A., Mondy, S., Pellissier, F., Dessaux, Y., et al. (2013). A metagenomic study highlights phylogenetic proximity of quorum-quenching and xenobiotic-degrading amidases of the AS-family. PLoS ONE 8:e65473. doi: 10.1371/journal.pone.0065473
Tao, W., Lee, M. H., Wu, J., Kim, N. H., Kim, J. C., Chung, E., et al. (2012). Inactivation of chloramphenicol and florfenicol by a novel chloramphenicol hydrolase. Appl. Environ. Microbiol. 78, 6295–6301. doi: 10.1128/AEM.01154-12
Torsvik, V., Goksoyr, J., and Daae, F. L. (1990). High diversity in DNA of soil bacteria. Appl. Environ. Microbiol. 56, 782–787.
Uchiyama, T., Abe, T., Ikemura, T., and Watanabe, K. (2005). Substrate-induced gene-expression screening of environmental metagenome libraries for isolation of catabolic genes. Nat. Biotechnol. 23, 88–93. doi: 10.1038/nbt1048
Uchiyama, T., and Miyazaki, K. (2009). Functional metagenomics for enzyme discovery: challenges to efficient screening. Curr. Opin. Biotechnol. 20, 616–622. doi: 10.1016/j.copbio.2009.09.010
Uchiyama, T., and Miyazaki, K. (2010). Product-induced gene expression, a product-responsive reporter assay used to screen metagenomic libraries for enzyme-encoding genes. Appl. Environ. Microbiol. 76, 7029–7035. doi: 10.1128/AEM.00464-10
Uchiyama, T., and Watanabe, K. (2006). Improved inverse PCR scheme for metagenome walking. Biotechniques 41, 183–188. doi: 10.2144/000112210
van der Maarel, M. J., van der Veen, B., Uitdehaag, J. C., Leemhuis, H., and Dijkhuizen, L. (2002). Properties and applications of starch-converting enzymes of the alpha-amylase family. J. Biotechnol. 94, 137–155. doi: 10.1016/S0168-1656(01)00407-2
Venter, J. C., Remington, K., Heidelberg, J. F., Halpern, A. L., Rusch, D., Eisen, J. A., et al. (2004). Environmental genome shotgun sequencing of the Sargasso Sea. Science 304, 66–74. doi: 10.1126/science.1093857
Ververidis, F., Trantas, E., Douglas, C., Vollmer, G., Kretzschmar, G., and Panopoulos, N. (2007a). Biotechnology of flavonoids and other phenylpropanoid-derived natural products. Part I: Chemical diversity, impacts on plant biology and human health. Biotechnol. J. 2, 1214–1234. doi: 10.1002/biot.200700084
Ververidis, F., Trantas, E., Douglas, C., Vollmer, G., Kretzschmar, G., and Panopoulos, N. (2007b). Biotechnology of flavonoids and other phenylpropanoid-derived natural products. Part II: Reconstruction of multienzyme pathways in plants and microbes. Biotechnol. J. 2, 1235–1249. doi: 10.1002/biot.200700184
Vester, J. K., Glaring, M. A., and Stougaard, P. (2014). Discovery of novel enzymes with industrial potential from a cold and alkaline environment by a combination of functional metagenomics and culturing. Microb. Cell Fact. 13:72. doi: 10.1186/1475-2859-13-72
Voget, S., Leggewie, C., Uesbeck, A., Raasch, C., Jaeger, K. E., and Streit, W. R. (2003). Prospecting for novel biocatalysts in a soil metagenome. Appl. Environ. Microbiol. 69, 6235–6242. doi: 10.1128/AEM.69.10.6235-6242.2003
Von Wettstein, D., Mikhaylenko, G., Froseth, J. A., and Kannangara, C. G. (2000). Improved barley broiler feed with transgenic malt containing heat-stable (1,3-1,4)-beta-glucanase. Proc. Natl. Acad. Sci. U.S.A. 97, 13512–13517. doi: 10.1073/pnas.97.25.13512
Walter, J., Mangold, M., and Tannock, G. W. (2005). Construction, analysis, and beta-glucanase screening of a bacterial artificial chromosome library from the large-bowel microbiota of mice. Appl. Environ. Microbiol. 71, 2347–2354. doi: 10.1128/AEM.71.5.2347-2354.2005
Wang, K., Li, G., Yu, S. Q., Zhang, C. T., and Liu, Y. H. (2010). A novel metagenome-derived beta-galactosidase: gene cloning, overexpression, purification and characterization. Appl. Microbiol. Biotechnol. 88, 155–165. doi: 10.1007/s00253-010-2744-7
Wang, K., Lu, Y., Liang, W. Q., Wang, S. D., Jiang, Y., Huang, R., et al. (2012). Enzymatic synthesis of galacto-oligosaccharides in an organic-aqueous biphasic system by a novel beta-galactosidase from a metagenomic library. J. Agric. Food Chem. 60, 3940–3946. doi: 10.1021/jf300890d
Warnecke, F., Luginbuhl, P., Ivanova, N., Ghassemian, M., Richardson, T. H., Stege, J. T., et al. (2007). Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450, 560–565. doi: 10.1038/nature06269
Wendel, A. M., Johnson, D. H., Sharapov, U., Grant, J., Archer, J. R., Monson, T., et al. (2009). Multistate Outbreak of Escherichia coli O157:H7 Infection associated with consumption of packaged spinach, August-September 2006: the wisconsin investigation. Clin. Infect. Dis. 48, 1079–1086. doi: 10.1086/597399
World Health Organization. (2014). Antimicrobial Resistance: Global Report On Surveillance. Geneva: World Health Organization.
Wu, D., and Bird, M. R. (2010). The interaction of protein and polyphenol species in ready to drink black tea liquor production. J. Food Process Eng. 33, 481–505. doi: 10.1111/j.1745-4530.2008.00286.x
Yao, J., Chen, Q., Zhong, G., Cao, W., Yu, A., and Liu, Y. (2014). Immobilization and characterization of tannase from a metagenomic library and its use for removal of tannins from green tea infusion. J. Microbiol. Biotechnol. 24, 80–86. doi: 10.4014/jmb.1308.08047
Yao, J., Fan, X. J., Lu, Y., and Liu, Y. H. (2011). Isolation and characterization of a novel tannase from a metagenomic library. J. Agric. Food Chem. 59, 3812–3818. doi: 10.1021/jf104394m
Yun, J., and Ryu, S. (2005). Screening for novel enzymes from metagenome and SIGEX, as a way to improve it. Microb. Cell Fact. 4:8. doi: 10.1186/1475-2859-4-8
Keywords: functional metagenomics, industrial applications, food, pharmacological, catalysts, bioactives, antimicrobials
Citation: Coughlan LM, Cotter PD, Hill C and Alvarez-Ordóñez A (2015) Biotechnological applications of functional metagenomics in the food and pharmaceutical industries. Front. Microbiol. 6:672. doi: 10.3389/fmicb.2015.00672
Received: 26 April 2015; Accepted: 19 June 2015;
Published: 30 June 2015.
Edited by:
Eric Altermann, AgResearch Ltd., New ZealandReviewed by:
William John Kelly, AgResearch Ltd., New ZealandDiego Mora, University of Milan, Italy
Copyright © 2015 Coughlan, Cotter, Hill and Alvarez-Ordóñez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Avelino Alvarez-Ordóñez, Teagasc Food Research Centre, Moorepark, Fermoy, Cork, Ireland,YXZlbGluby5hbHZhcmV6LW9yZG9uZXpAdGVhZ2FzYy5pZQ==