 Kateřina Petříčková1
Kateřina Petříčková1 Alica Chroňáková2*
Alica Chroňáková2* Tomáš Zelenka1
Tomáš Zelenka1 Tomáš Chrudimský2Stanislav Pospíšil1Miroslav Petříček1*Václav Krištůfek2
Tomáš Chrudimský2Stanislav Pospíšil1Miroslav Petříček1*Václav Krištůfek2- 1Institute of Microbiology, Czech Academy of Sciences, v. v. i., Prague, Czech Republic
- 2Institute of Soil Biology, Biology Centre, Czech Academy of Sciences, v. v. i., České Budějovice, Czech Republic
A combined approach, comprising PCR screening and genome mining, was used to unravel the diversity and phylogeny of genes encoding 5-aminolevulinic acid synthases (ALASs, hemA gene products) in streptomycetes-related strains. In actinomycetes, these genes were believed to be directly connected with the production of secondary metabolites carrying the C5N unit, 2-amino-3-hydroxycyclopent-2-enone, with biological activities making them attractive for future use in medicine and agriculture. Unlike “classical” primary metabolism ALAS, the C5N unit-forming cyclizing ALAS (cALAS) catalyses intramolecular cyclization of nascent 5-aminolevulinate. Specific amino acid sequence changes can be traced by comparison of “classical” ALASs against cALASs. PCR screening revealed 226 hemA gene-carrying strains from 1,500 tested, with 87% putatively encoding cALAS. Phylogenetic analysis of the hemA homologs revealed strain clustering according to putative type of metabolic product, which could be used to select producers of specific C5N compound classes. Supporting information was acquired through analysis of actinomycete genomic sequence data available in GenBank and further genetic or metabolic characterization of selected strains. Comparison of 16S rRNA taxonomic identification and BOX-PCR profiles provided evidence for numerous horizontal gene transfers of biosynthetic genes or gene clusters within actinomycete populations and even from non-actinomycete organisms. Our results underline the importance of environmental and evolutionary data in the design of efficient techniques for identification of novel producers.
Introduction
5-aminolevulinate (ALA) is a key precursor of a huge family of essential tetrapyrrole compounds. Two distinct ALA biosynthetic pathways have evolved; that for archea, majority of bacteria, and plants where ALA is derived from glutamate via the C5 pathway. Some α-Proteobacteria, fungi, and animals form ALA through condensation of glycine and succinyl-CoA by hemA-encoded aminolevulinate synthase in so called C4 or Shemin pathway. Presence of both pathways in a single cell is extremely rare and their products are always temporally, spatially or functionally separated (Weinstein and Beale, 1983; Mayer and Beale, 1992; Yang and Hoober, 1995). We recently reported one example in actinomycetes, where the C5 pathway is devoted to primary metabolism and the C4 strictly toward feeding biosynthesis of secondary metabolites containing the ALA-derived cyclic moiety, the C5N unit, 2-amino-3-hydroxycyclopent-2-enone (Petříček et al., 2006; Supplementary Figure S1). These secondary metabolites have a range of structures and functions, including linear or aromatic polyketides, glycolipids, macrolides, peptide compounds, and others (Supplementary Figure S2). Many have pharmaceutically attractive biological activities and in some cases, the relevant biosynthetic gene clusters have already been cloned and characterized.
Recently, it has been shown that formation of the C5N unit and its attachment to core structures of antibiotics is directed by three enzymes: cyclizing 5-aminolevulinate synthase (cALAS encoded by a hemA homolog), amide synthase (AMS) and aminolevulinate-CoA ligase (ALL). These always appear to be encoded by three adjacent genes located within a relevant antibiotic biosynthetic gene cluster. We consider the presence of this gene triad to be a specific genetic marker of the C5N unit biosynthetic pathway that “tags” producers of C5N-containing metabolites among others. The key enzyme, cALAS, catalyzes not only condensation of glycine and succinyl-CoA, but also the subsequent cyclization of nascent ALA (Zhang et al., 2010, Supplementary Figure S1). The other two enzymes activate and attach the unit to the metabolite core.
The principal aim of our work was to show the potential of the hemA-targeted genetic screening in identification of novel putative biosynthetic gene clusters encoding C5N-containing metabolites. For the purpose, occurrence of hemA was mapped in a large diverse collection of natural streptomycete isolates and also in the collection of genomic data of actinomycetes in the GenBank. The next aim was to verify the hypothesis, that the specialized, cyclizing type of ALAS, and perhaps the entire gene triad responsible for the C5N biosynthesis, have evolved within individual biosynthetic gene clusters, which in many cases differ diametrically. HemA gene phylogeny should then reflect the diversity of synthesized chemical structures and should provide an important genetic clue in classification of novel putative producers.
Materials and Methods
Genome Scanning
Available actinomycete genomes were scanned for putative ALAS-coding regions using a standard BlastP algorithm with well characterized cALAS as queries (cut off set to 40% identity and 60% similarity). The protein sequences retrieved were inspected for conservation of ALAS-typical regions (Figure 1, Supplementary Table S3).

FIGURE 1. Amino acid sequence alignment of antibiotic-related cyclizing 5-aminolevulinate synthases (ALASs; black) with representatives of standard proteobacterial and human ALAS (gray). Original organisms abbreviated as follows S. – Streptomyces, Sac. – Saccharothrix, K. – Kitasatospora, R. – Rhodobacter, H. – Homo. Amino acid residues directly involved in binding of substrates and the PLP cofactor indicated in bold with arrows (Astner et al., 2005). Residues different from classical ALAS and strictly conserved in cALAS boxed gray and outlined in black when residues are directly connected to the enzyme activity. Conservative amino acid stretches used for the screening PCR method design are indicated.
Isolation of Actinomycete Strains and DNA Extraction
In total, 1,500 streptomycete-like bacterial strains were isolated from 53 environmental samples (Supplementary Table S1) from soils and sediments representing unique habitats around the world (Supplementary Table S2; includes strain isolation sources).
Samples were first homogenized by sieving through a 5 mm sieve then cultured according to Krištůfek et al. (2005). Where necessary, samples were stored for 1–2 days at 4°C. Serially diluted samples were plated onto R2A agar (BD Difco, Franklin Lakes, NJ, USA), McBeth–Scales starch-mineral agar or M2 agar (Shirling and Gottlieb, 1966) for isolation and culturing of streptomycete strains. The plates were then incubated in the dark at 28°C. Bacterial cultures were maintained long-term as spore suspensions in glycerol (15% v/v) at -80°C and/or freeze-dried. One group of strains had been isolated and characterized previously (Chroňáková et al., 2010), consequently we studied one of these in greater detail (S. aureus SOK 1/5-04, BCCO10_5, a new colabomycin E producer; Petříčková et al., 2014).
Several cooperating laboratories (see Acknowledgements, Supplementary Table S1) kindly provided sets of soil/sediment samples or streptomycete strains. Most of these are deposited in the Biology Centre Collection of Organisms, Culture Collection of Soil Actinomycetes in České Budějovice (BCCO1).
Genomic DNA was extracted using the Wizard® Genomic DNA Purification kit (Promega, Madison, WI, USA) with slight modifications. We applied a combination of morphological comparison and genomic fingerprinting (BOX-PCR) to de-replicate strains isolated from the same environment.
Screening for the cALAS Gene
ALAS-specific primers were designed based on the two most conservative amino acid stretches, i.e., VWCSNDYL and DEVHAVGMY (Figure 1), with respect to the strong GC preference on the third codon position typical for streptomycete coding regions. Two degenerative primers were designed: HemA1 (5′-GTS TGG TGY TCS/RGS AAC GAC TAC CT-3′) and HemA3 (5′-GTA CAT SCC SAC SGC GTG SAC CTC GCT-3′). Each sample (25 μL) contained 1x Fast Start PCR Master (Roche Applied Science, Penzberg, Germany), the primers (1.6 μM each) and 200 ng of template DNA. PCR reaction conditions were optimized using chromosomal DNA from previously characterized C5N-compound producers (S. parvulus Tü64, S. bambergiensis ATCC13879, S. nodosus sp. asukaensis ATCC29757) and with S. coelicolor A3(2) and S. lividans TK24 strains used as negative controls. Thermal cycling was performed as follows: Initial denaturation at 95°C for 2 min; followed by 30 cycles of denaturation (94°C/30 s), annealing (60–55°C/30 s; annealing temperature touchdown decreased by 0.5°C per cycle over the first 10 cycles, then kept at 55°C over the following 20 cycles), and extension (72°C/60 s; final extension 72°C for 7 min). The optimized PCR reaction yielded in a single band of 519 bp, with no variation in size. Amplified products were cleaned-up using the MinElute PCR Purification Kit (Qiagen, Hilden, Germany), eluted in 12 μL and sequenced using HemA1 primer. Relatedness of DNA sequences to ALAS genes was verified via a BlastX search of non-redundant protein sequences in GenBank.
Phylogenetic Analysis
Nucleotide datasets were translated into amino acids and aligned in BioEdit 7.0.5.3 (Hall, 1999) using the ClustalW algorithm. All ambiguously aligned and unaligned sites were deleted. Edited datasets were then used in nucleotides to infer phylogeny. Bayesian inference was used to construct phylogenetic trees, computed using MrBayes 3.1.2 (Huelsenbeck and Ronquist, 2005) with five million generations (GTR + I + Γ model with gamma distribution in six categories). After deleting a burn-in, consensual topologies were computed from 45,000 sampled trees.
Analysis of Secondary Metabolite Gene Clusters
Analysis of the putative biosynthetic genes clusters was performed using BlastX2. Prediction of PKS domains specificity was done with the help of AntiSMASH package (Medema et al., 2011; Blin et al., 2013).
Extraction of Secondary Metabolites
Fermentation followed by extraction of secondary metabolites was performed as described by Petříček et al. (2006): After 3–4 days of cultivation (28°C, 200 rpm), mycelia were extracted with acetone and ethyl acetate. The post-cultivation media were extracted with ethyl acetate, too. Both final extracts were combined, dissolved in acetone, and analyzed. UHPLC-DAD-ToF-MS analyses were carried out with a Waters Acquity UPLC System (Waters) using the Acquity UPLC BEH C18 column under the conditions specified in Petříčková et al. (2014).
Genetic Fingerprinting
DNA amplification and PCR fragment separation followed the procedure of Lanoot et al. (2004), using the BOXA1R primer 5′-CTACGGCAAGGCGACGCTGACG-3′ (Versalovic et al., 1994). Fingerprinting was performed as follows: PCR products (20 μL) were separated on 20 cm × 20 cm gels using 130 V, 400 mA for 240 min in 1 × TBE buffer. The gels were stained for 30 min in a 1 × TBE bath supplemented with ethidium bromide (1 mg L-1). A photograph of the gel was stored as TIFF file through a CCD coupled camera by using the Photo-Doc software (Vilber-Lourmat, France). Gels were imported into the GelCompar II software package (Applied Maths, Belgium), normalized and gel track densitometric curve similarity matrices calculated using the Pearson product-moment correlation coefficient. Dendrograms were then constructed using the complete linkage algorithm. Distinct BOX-PCR groups were defined using a 70% similarity limit.
Isolate Identification
Representative ALAS gene-positive isolates were identified through 16S rRNA gene amplification using pA (5′-AGAGTTTGATCCTGGCTCAG-3′) and pH (5′-AAGGAGGTGATCCAGCCGCA-3′) universal bacterial primers and sequencing (pA, pH; Edwards et al., 1989). 16S rDNA amplification was performed using the protocol of Kyselková et al. (2012). Once amplified, the 16S rRNA genes were cleaned-up, sequenced then edited and assembled using Geneious 5.5.6 (Biomatters, Auckland, New Zealand). They were then compared against a type strain database (Ez-Taxon Database3; Chun et al., 2007) to retrieve the most relative species.
Whole Genome Sequencing
Genomic sequence of BCCO10_981 strain was determined on the 454 GS Junior System (Roche). A total of 156, 457 reads comprising 70,475,443 bases were assembled using the Roche GS de novo assembler (version 2.7). The assembly yielded 1,532 contigs (unpublished).
Accession Nos.
GenBank accession numbers for the resultant sequences are listed in Supplementary Tables S1 and S3.
Results
Amino Acid Sequence Identifiers of Cyclizing Form of ALAS (i.e., cALAS)
Using genetic data recently released about a number of evident C5N-compound producers (McAlpine et al., 2005; Ostash et al., 2007; Kalan et al., 2013; Zhang et al., 2013), we were able to align several typical cALASs with “classical” ALAS representatives. This helped us to recognize sequence specificities for “cyclizing” ALAS types (cALAS). The amino acid sequence displayed a number of residues specifically conserved in cALAS and these may be directly connected to the novel cyclization function of the enzymes (Figure 1). Some of the residues, conservatively substituted in cALAS, have previously been shown to be crucial for ALAS enzymatic activity, i.e., T83S, I149L/V, D231T, and T365S, numbered based on the accepted Rhodococcus structure model. Thr83 and Thr365 are localized directly in the catalysis site (Kaufholz et al., 2013). Thr83 is important for positioning of the succinyl-CoA terminal carboxyl, and also directs amino acid specificity of ALAS. However, in cALAS model enzyme from the asukamycin biosynthetic route, its substitution with Ser is crucial for the cyclization function (Petříček et al., 2006). The sequence markers identified can be used to distinguish between ALAS and cALAS.
Identification of hemA Homologs in Genomic Data of Actinomycetes
Sequences of all hemA gene homologs, identified by genome scanning of actinomycete genomic data using BLASTP search with AsuD2 cALAS as a query and listed in Supplementary Table S3, were added to the alignment and a cladogram constructed to illustrate phylogenetic relationships among the genes. Referential genes of the characterized producers (type producer strains) and a few representatives of primary metabolism hemA genes were also included. As expected, the cALAS-encoding representatives were clearly separated from “classical” ALAS. As C5N-carrying compounds display remarkable structural variability and diversity of relevant genetic information, we expected hemA genes evolution to follow that of entire biosynthetic gene clusters. Indeed, the genes show apparent clustering based on the type of C5N-containing compound produced (Figure 2). Two major producer groups were identified: The first, designated as “a purple group,” encompasses producers of simple linear polyketides and reductiomycin, which are characterized by head-to-tail organization of the C5N-encoding gene triplet ams-hemA-all and by direct attachment of the C5N unit to a polyketide chain. The second, “a green group,” represents producers of complex compounds such as sugar-containing polyketides or macrolides. In this case, the hemA gene is located upstream of the ams-all pair in the divergent orientation (Figures 2 and 4). Only two putative hemA loci of Saccharothrix espanaensis show different arrangement of the C5N-encoding genes (discussed later, Figure 6B). Contrary to original expectations, substantial fraction of identified genes (15%) clustered clearly with typical primary metabolism hemA representatives and their products did not show cALAS sequence characteristics.
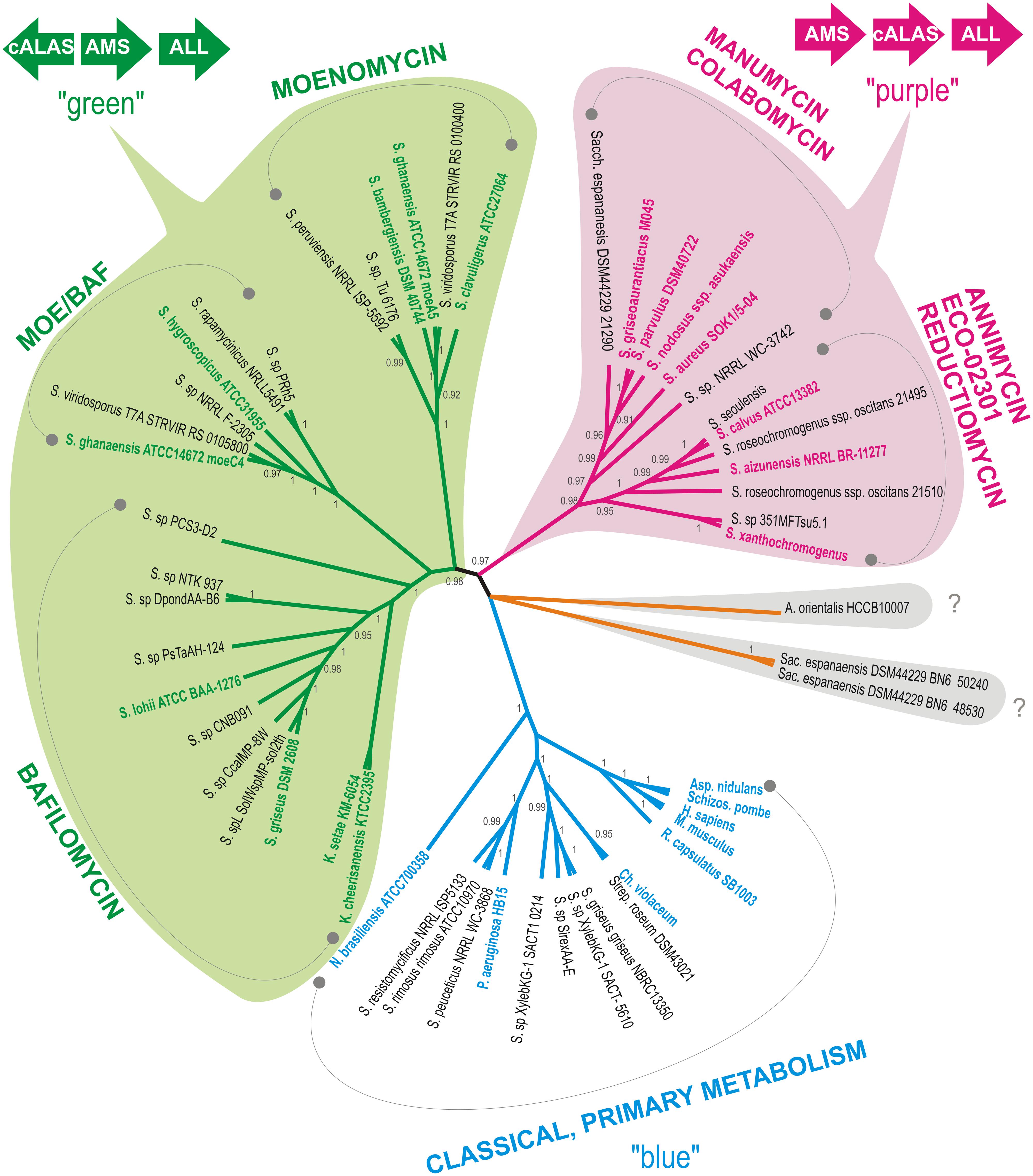
FIGURE 2. Cladogram of hemA was constructed as specified in the Section “Materials and Methods,” posterior probability values are given when higher than 0.90. All branches encompassing genes encoding putative cyclizing type of ALAS (cALAS) are color-shaded, types of secondary metabolites based on characterized producers are indicated in each branch. Reference sequences – genes of primary metabolism ALASs (“blue”) and of secondary metabolite-connected cALASs (“purple” and “green”), are shown in bold. Orange branch covers strains carrying cALAS-encoding hemA genes without a close phylogenetic relationship to any characterized producer strains. Original organisms are abbreviated as follows: S. – Streptomyces, K. – Kitasatospora, A. – Amycolatopsis, N. – Nocardia, Sac. – Saccharothrix, P. – Pseudomonas, Ch. – Chromobacterium, R. – Rhodobacter, Asp. – Aspergillus, Schiz. – Schizosaccharomyces, H. – Homo, M. – Mus. Organization of the C5N biosynthetic gene triad, encoding amide synthase (AMS), cALAS, and aminolevulinate ligase (ALL), conserved within the two major producer groups, designated as “green” and “purple,” is illustrated.
In all identified genomic hemA homologs, the relevant genomic regions were analyzed for presence of putative secondary metabolites biosynthetic gene clusters using BLAST and AntiSMASH searches (see in Materials and Methods).
PCR Screening of Natural Actinomycete Isolates for Presence of ALAS-Encoding Genes
The PCR screening assay, described in the Section “Materials and Methods,” was designed to amplify both ALAS and cALAS-encoding hemA genes. It was applied to 1,500 new streptomycete-like isolates where 14% of strains were found positive, giving a single clear band of invariable size of 519 bp. In some other isolates, slightly smaller fragments were found. These were identified as fragments of putative hemA-related genes encoding 8-amino-7-oxononanoate synthases, which were not subjects of this work. All the hemA-positive isolates were dereplicated using BOX-PCR relatedness survey (see later) to eliminate possible multiple isolates. All 519-bp PCR fragments from 153 dereplicated positives were sequenced and shown to contain part of the hemA-homologous gene. These sequences, and the relevant fragments of the genome-scanning genes, were aligned both with the corresponding regions of cALAS-encoding hemA genes of type producer strains and with examples of classical hemA genes of various origins (Supplementary Table S3, Figure 3). A phylogenetic tree was constructed based on the alignment (Figure 4).
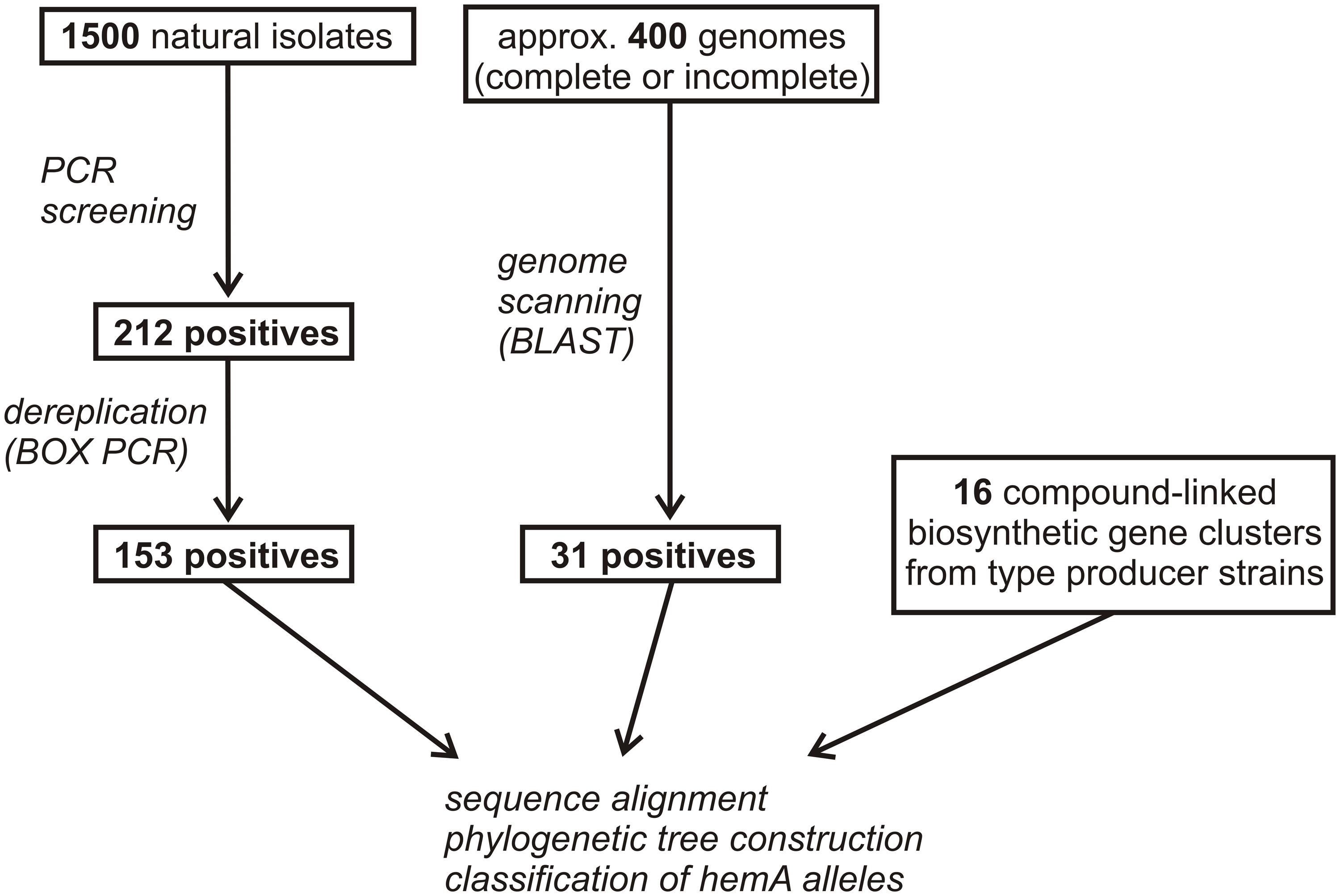
FIGURE 3. Flowchart of hemA positive strains selection. Numbers of screening-derived, genome scanning-derived, and referential producer strains are given.
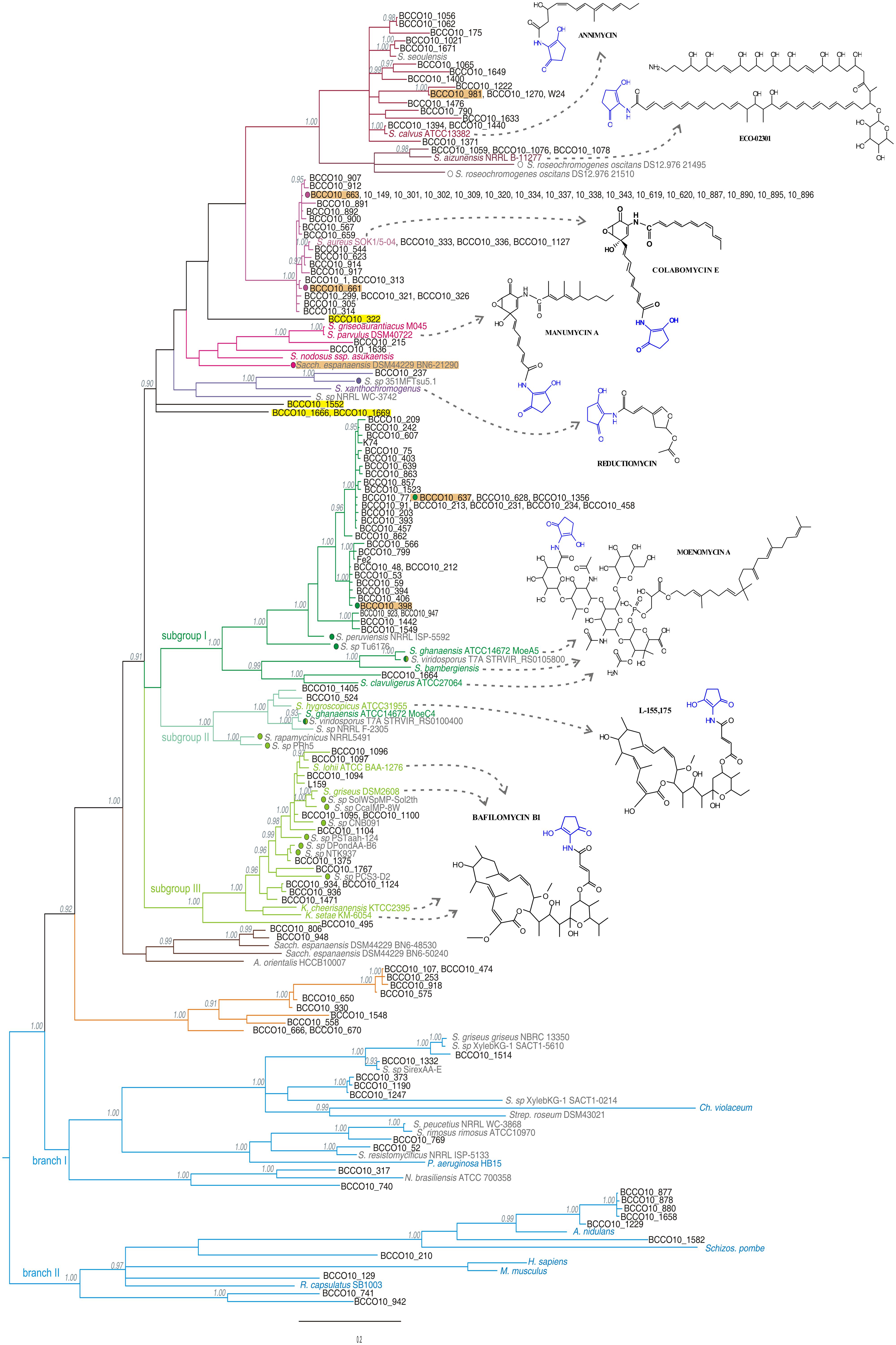
FIGURE 4. Phylogenetic tree designed on the basis of the nucleotide sequence alignment of the hemA gene PCR fragments from the screening, calculated as specified in the Section “Materials and Methods.” Posterior probability values are indicated in gray when higher than 0.90. The individual branches are differentiated by a color code corresponding to the type producers or reference sequences: producers of linear polyketides and reductiomycin in shades of purple, those of moenomycins or bafilomycins in greens, primary metabolism hemA genes in blue, and separate branches with no reference sequences in brown and orange, individually standing isolates are highlighted in yellow. Model structures of produced compounds are indicated for each branch. Sequences from characterized producers of C5N compounds and those of typical animal, fungal and bacterial ALAS genes are shown in colors (corresponds to Figure 2), genome-scanning matches are in gray, screening-based sequences in black letters. Color dots mark the strains with genetic information indicating production of similar compounds as a type strain of the branch. Isolates more closely characterized are highlighted in orange.
The majority of PCR sequences and genomic hits were tightly clustered within two major branches of known C5N compounds producers (green or purple, Figures 2 and 4). Few strains, which also carried genes with characteristics of cALAS-encoding hemA, formed two independent branches (orange and brown, Figure 4). Nineteen screening-based sequences, together with eight genomic sequences, were situated among typical, primary metabolism hemA genes of proteobacteria, bacilli, or fungi (blue groups, Figure 4). Functional and evolutionary aspects of these groups are discussed below.
The “Purple Branch,” the “Head-to-Tail” Operon, the Producers of Linear Polyketides and Reductiomycin
Sixty-six natural isolates were clustered in the linear polyketides branch, together with six uncharacterized genome-based sequences. Thirty eight of the natural isolates formed a separate branch represented by a novel colabomycin E producer, S. aureus SOK1/5-04 (BCCO10_5), which was identified by the genetic screening method described in this work and recently studied in detail by our group (Petříčková et al., 2014). The next two isolates occurred in the manumycin-producer branch (BCCO10_215 and BCCO10_1636; isolated from Sokolov colliery spoil heaps and Mt. Cameroon foggy forest soil, respectively). Twenty-one isolates and three genomic sequences were localized among the PKSI-synthesized polyketide producers, ECO-02301 and annimycin. The reductiomycin branch encompassed one isolate (Amycolatopsis thailandensis BCCO10_237) and two genomic sequences. Four remaining sequences formed individual branches not tightly associated with known producers (Figure 4, in yellow). BCCO10_0322, BCCO10_1552, BCCO10_1666 and BCCO10_1669 may represent producers of either novel compounds or producers of previously characterized C5N-polyketides with genetic data lacking.
To verify the metabolite-based clustering, two strains, BCCO10_661 and BCCO10_663, of the colabomycin group were subjected to UPLC-MS (ultra-performance liquid chromatography tandem mass spectrometry) analysis of secondary metabolites and their putative biosynthetic gene clusters were partially sequenced and compared to that of colabomycin E producer. Both isolates shared the colabomycins production profile of S. aureus SOK1/5-04 and also their gene clusters organization was identical (not shown). Most of the strains of the colabomycin group originated from the same habitat, the Velká Podkrušnohorská spoil heap in the Sokolov brown coal mining area, Czech Republic, though from several distant localities differing mainly in succession age and streptomycete community composition (Chroňáková et al., 2010). Only two were isolated from different habitats: BCCO10_1127 from an agricultural field in Bad Lauchstädt, Germany, and BCCO10_544 from the Domica Cave river sediment, Slovakia (Supplementary Tables S1 and S2). Strains carrying hemA sequences related to colabomycin E branch isolated from similar habitats (H33-35, H37, H39-40, Supplementary Table S2) belonged to five phylogenetic clades according to Labeda et al. (2012) and they differed also by BOX-PCR groups (Supplementary Table S1, Supplementary Figure S3). These findings may support the evidence for lateral transfer of partial or entire biosynthetic gene clusters in streptomycetes in soil recorded previously (Egan et al., 1998; Deng et al., 2011).
Next, S. graminilatus BCCO10_215, clustering tightly with manumycin and asukamycin producers was proved to produce manumycin D and manumycin A in the ratio of 2,5:1, both compounds found also in the characterized producer S. parvulus Tü64 where manumycin A is the most prevalent (data not shown). We have also attempted to isolate annimycin-type compounds from the S. sioyaensis BCCO10_981 originated from the coal-mining recultivated soil in Wyoming, USA, as suggested by the phylogenetic data. No annimycin-type compounds were identified in the metabolic extracts of the strain under various cultivation conditions. Therefore, the genome of the strain was sequenced using 454 pyrosequencing (Roche) and the hemA locus identified (GenBank Accession No. KR364704). The in silico analysis revealed a biosynthetic gene cluster highly similar (more than 90% identity) to that of the annimycin producer, S. calvus ATCC13382. However, one of the ann genes, encoding the antibiotic transporter/resistance protein, is damaged by several deletions and insertions. We believe that this abolishes production of annimycin-type compounds in the S. sioyaensis BCCO10_981.
The “Green Branch,” with “Head-to-Head” Organization of C5N Biosynthetic Genes, the Producers of Isoprenoids and Glycosylated Macrolides
The green branch (Figures 2 and 4) can be divided into three subgroups: the subgroup I comprising 38 isolates and three genomic sequences clustered with three moenomycin producers (Ostash and Walker, 2010); the subgroup II including two isolates, four genomic sequences, and S. hygroscopicus ATCC31955 (Kim et al., 2012) with S. ghanaensis ATCC 14672 (the moeC4 gene), the type producer strains; and the subgroup III of 13 isolates and eight genomic sequences localized around four bafilomycin producers (Ichikawa et al., 2010; Hwang et al., 2013, 2015; Zhang et al., 2013).
Most of the putative moenomycin-type isolates formed a separate branch (subgroup I), with only one strain clustered tightly with the type producer strains. Therefore, two strains S. aurantiacus BCCO10_398 and S. ederensis BCCO10_628 were more closely characterized genetically. Short genomic fragments of their hemA loci were cloned and sequenced. Both showed similar gene organization as moe cluster 1 of the moenomycin producer S. ghanaensis (Ostash et al., 2007), with a short non-homologous region in BCCO10_628 (homologous parts indicated as black rectangles in Figure 5A). We suppose that at least majority of the subgroup I strains produces moenomycin-type compounds.
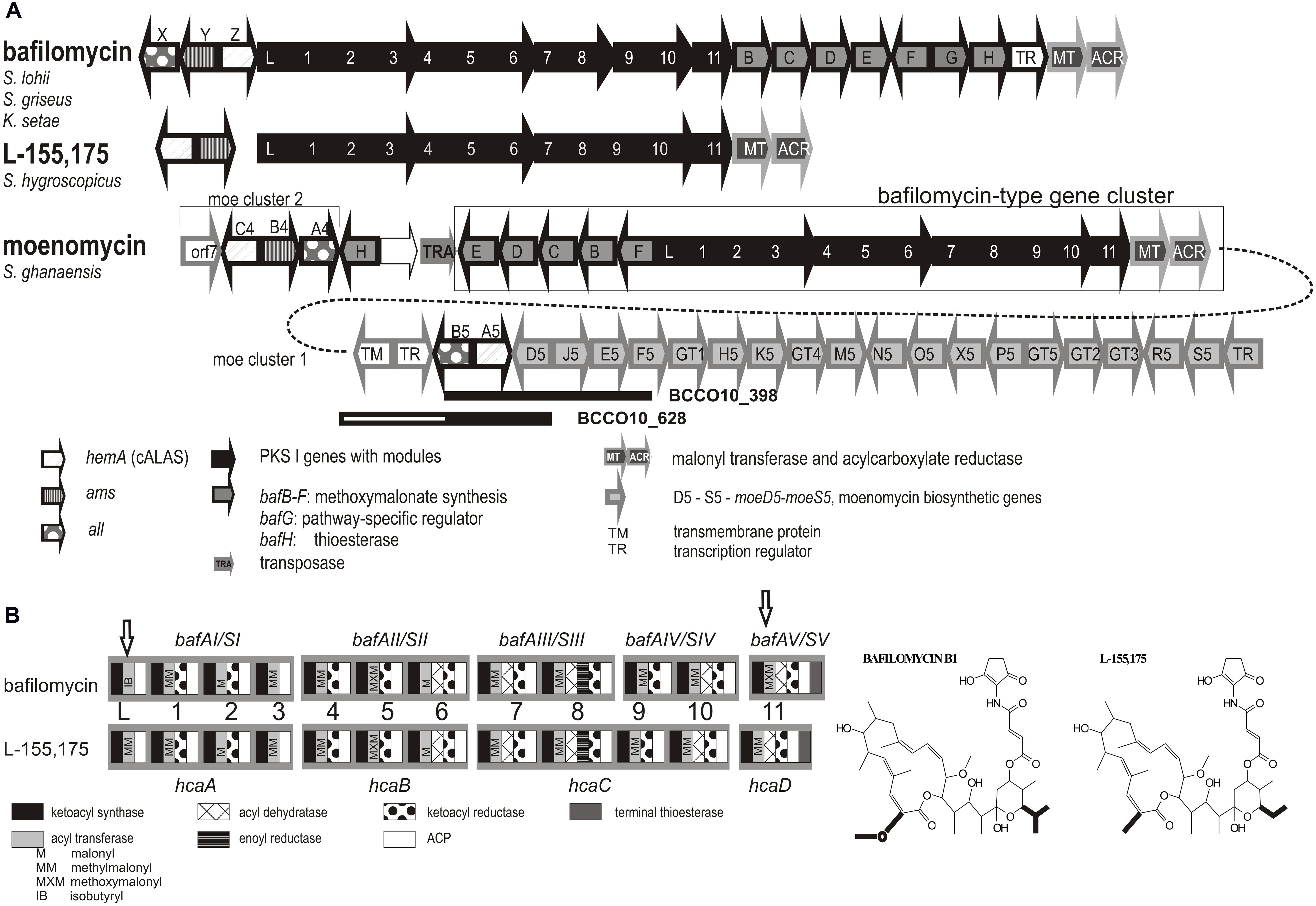
FIGURE 5. Moenomycin and bafilomycin biosynthetic genes clusters. (A) Rearrangement of the moenomycin- and bafilomycin-type clusters in some producers of moenomycin. PKS genes are shown in black, modules indicated by numbers. Bafilomycin biosynthetic genes outlined in black (PKS genes in filled black, tailoring genes filled gray), moenomycin in gray. Homologous regions identified in isolates Nos. BCCO10_398 and BCCO_628 are shown as black rectangles. (B) Domain organization within PKS genes – comparison of closely related clusters encoding biosynthesis of bafilomycin B1 and L-155,175. Specificity of AT domains, corresponding to the differences in the metabolite structures, highlighted in bold, are included (different domains indicated with arrows).
Similarly, the subgroup III seems to include only putative bafilomycin compounds producers clustering tightly with 4 type producers of bafilomycin-type compounds. The hemA sequences in this subgroup are highly similar, with only BCCO10_495 staying a bit separately. Some of the novel isolates (all six ranging between Nos. BCCO10_1094-1104) share a geographical origin with one of the bafilomycin producer S. lohii ATCC BAA-1276, isolated from beach sands in Papua New Guinea (Zhang et al., 2013). In contrast, our strains were isolated from ambrosia beetle galleries and were classified as three different species (Supplementary Tables S1 and S2). The origins of remaining eight positives are diverse. Again, these strains are phylogenetically distant and do not display BOX-PCR genomic similarity (Supplementary Figure S3; Supplementary Table S1). Eight genome-project sequences mostly from insect-associated or marine streptomycetes also occurred in the branch.
Interpretation of the subgroup II was not clear at the first sight: It contained the hca1 (hemA) gene from a producer of a bafilomycin derivative L-155,175 (Kim et al., 2012), but also the moeC4 of moenomycin gene cluster 2 of S. ghanaensis ATCC 14672 (Ostash et al., 2007). This extensively studied strain was reported to contain two physically distant C5N-encoding loci connected with moenomycin production (Figure 5A): The major moe biosynthetic cluster 1 carries impaired genetic information for C5N biosynthesis, with a truncated all, missing ams and non-functional moeA5 (hemA) gene. This is suppressed by moe cluster 2 elsewhere in the genome - moeA4, B4, C4 (Ostash et al., 2007). We found identical situation also in the sequenced genome of S. viridosporus T7A and in the genomic DNA of the moenomycin producer S. bambergiensis, previously analyzed in our laboratory (data not shown). Whereas the hemA genes of moe cluster 1 (i.e., moeA4) fit into the subgroup I, the genes from the cluster 2 (i.e., moeC4) are phylogenetically more related to hca1 of S. hygroscopicus ATCC31955, the L-155,175 producer (Kim et al., 2012). To clarify these findings we inspected the neighborhoods of S. ghanaensis moe cluster 2. Indeed, in the available gapped genomic sequence of S. ghanaensis we identified a putative L-155,175-type biosynthetic gene cluster just next to the moe cluster 2. The type I polyketide synthases encoded there showed approx. 90% amino acid sequence identity with hcaA-D genes products. Also, the PKS module organization and predicted specificities of their acyl transferase domains were the same (Figure 5). Unlike the L-155,175 hca cluster, however, S. ghanaensis moe cluster 2 locus carries all putative methoxymalonyl-CoA biosynthetic genes (bafB-F) as in subgroup III bafilomycin producers, and intact C5N-encoding genes. A putative transposase gene is inserted between the annotated moe cluster 2 and the L-155,175 cluster-like region (Figure 5).
The Orange and Brown Branches – Still Secondary Metabolite Related?
Both the orange and brown branches were clearly separated from any other (Figure 4). The brown branch contained two hemA sequences (BCCO10_806 and BCCO10_948) from natural strain isolates and three from genomic data, while the orange branch included 11 sequences from various biotopes and were assigned to six different streptomycete species (Supplementary Tables S1 and S2). The amino acid sequences encoded by the hemA PCR fragments indicate that the majority of residua typical for cALAS, including the key T83S mutation, are conserved in the brown branch representatives, though some were missing in members of the orange branch (Figure 1, Supplementary Figure S4).
The brown branch contained Amycolatopsis orientalis sequences and two hemA genes from Saccharothrix espanaensis DSM 44229 (ORFs BN6_48530 and BN6_50240). Interestingly, both “brown” natural isolates (BCCO10_948 and BCCO10_806) were also identified as S. espanaensis, though they originate from geographically distant localities (Tennessee re-cultivation area and Sokolov colliery spoil, Czech Republic, respectively). Organization of the C5N-encoding genes in this group was not uniform (Figure 6), but they were always surrounded by putative secondary metabolism genes. In Amycolatopsis and Saccharothrix BN6_48530 loci, PKS I genes were found in the hemA neighborhoods (Figure 6B).
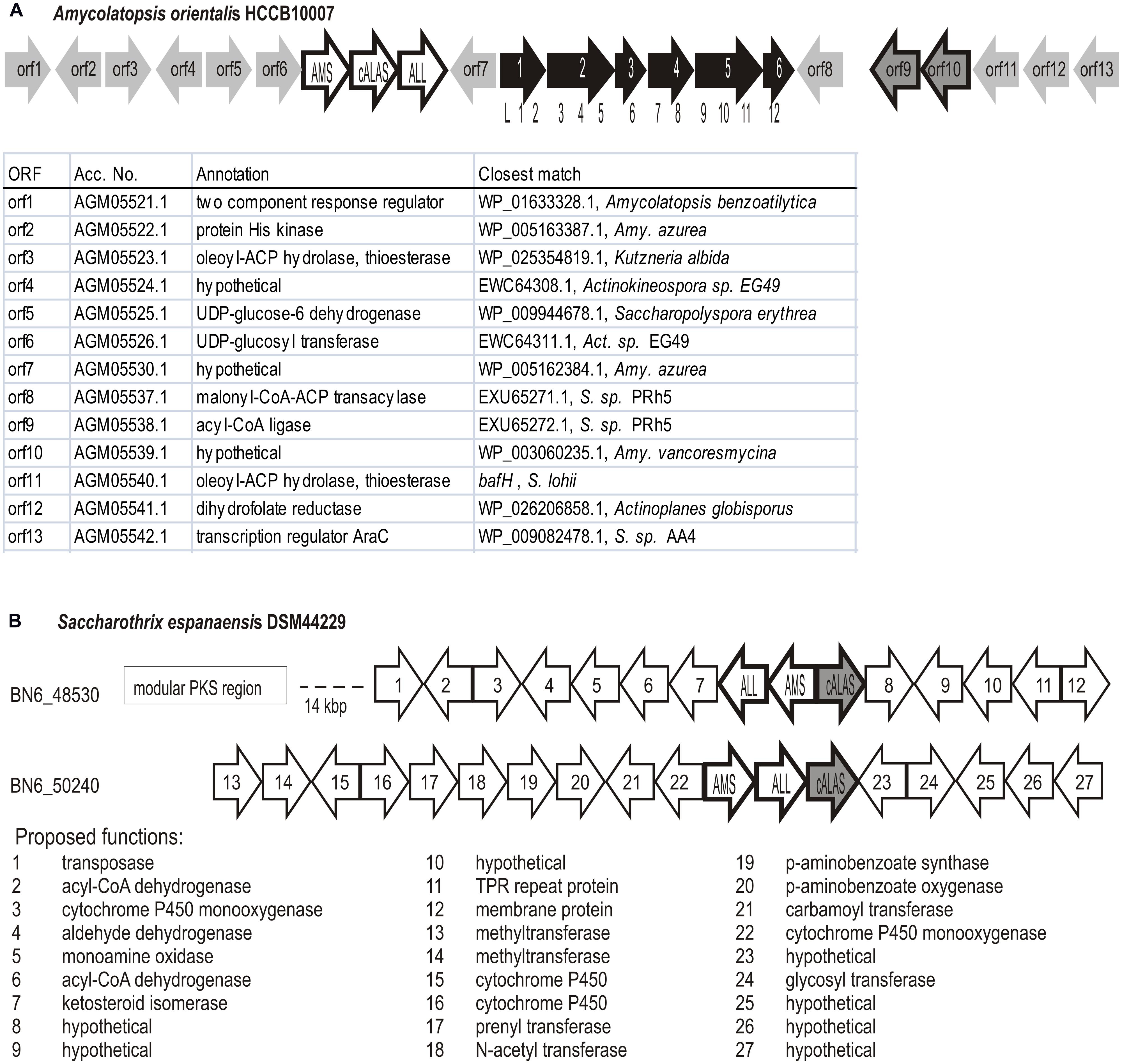
FIGURE 6. Genetic loci containing C5N-like operons in Amycolatopsis and Saccharothrix of the orange branch. The C5N-like operons genes marked AMS, cALAS, ALL. (A) Potential polyketide encoding genetic cluster in Amycolatopsis orientalis HCCB10007. Modular PKS genes shown filled with black, module distribution among the genes indicated by numbers below. The remaining ORF list is shown below the scheme. (B) Saccharothrix espanaensis DSM44229, two non-manumycin-type C5N operons (BN6_48530 and BN6_50240 indicate Gene Bank Accession Nos. of relevant hemA genes) and their genetic context.
General Observations in hemA Genes Putatively Encoding Cyclizing ALAS
All genomic data-derived hemA genes, showing characteristics of the cyclizing type, were routinely found within a triplet of C5N-biosynthetic genes (ams, hemA, all) that was located in a putative secondary metabolite biosynthetic gene cluster. Within the major groups of strains, defined by the hemA phylogeny, these gene clusters typically showed high similarity to the gene clusters of type producer strains (conformity with the type producers indicated in Figure 4 as colored dots in front of the strain names). This applies also to the few natural isolates with more detailed genetic analysis of the hemA loci. In majority of these metabolite-related groups, the homologous biosynthetic gene clusters share the organization of the genes and most of the differences are due to more or less numerous point mutations in individual biosynthetic, regulatory and transport genes. The only exception is the group of manumycin-type metabolites. Here, in most of the known cases, the ordering of individual biosynthetic genes or operons within the clusters differs substantially, though the metabolites share identical core structures. Also, phylogenetic relationships of the corresponding hemA genes are less tight than in other groups.
The Blue Branches – The “Black Box”
Sequences in the blue group were not clustered in any secondary metabolite-related group and were closely related to non-cyclizing primary-metabolism hemA genes. It encompassed eight genome project-derived sequences and 19 screening fits (seven Streptomyces and one Nocardia species; Figure 4; Supplementary Table S1) that formed the two least homogenous branches of all. They do not show typical sequence characteristics of cALAS-encoding genes and have slightly lower GC% contents than usual in streptomycetes. The blue branch I, with the closest match to Chromobacterium violaceum, included the GC-rich sequences, > 62%; whereas the blue branch II more closely matched fungi and included the sequences with GC% < 62. The GC% trend matched those of the closest matching hemA sequences, used as references in each branch (Supplementary Table S3).
ALAS-encoding genetic loci in representatives derived from genome projects indicated no physical association with any C5N-type operon. Their genetic context was not conserved and their secondary metabolism function is speculative. Attempts to identify the closest homologs of the blue group sequences indicate that the blue branch II sequences show stronger match to proteobacterial hemA genes or to those of Bacilli. The branch II sequences are more similar to hemA of Aspergillus. In some cases, we documented several kbp regions surrounding the hemA gene that were identical to their non-actinomycete homologs (see Figure 7 for examples).
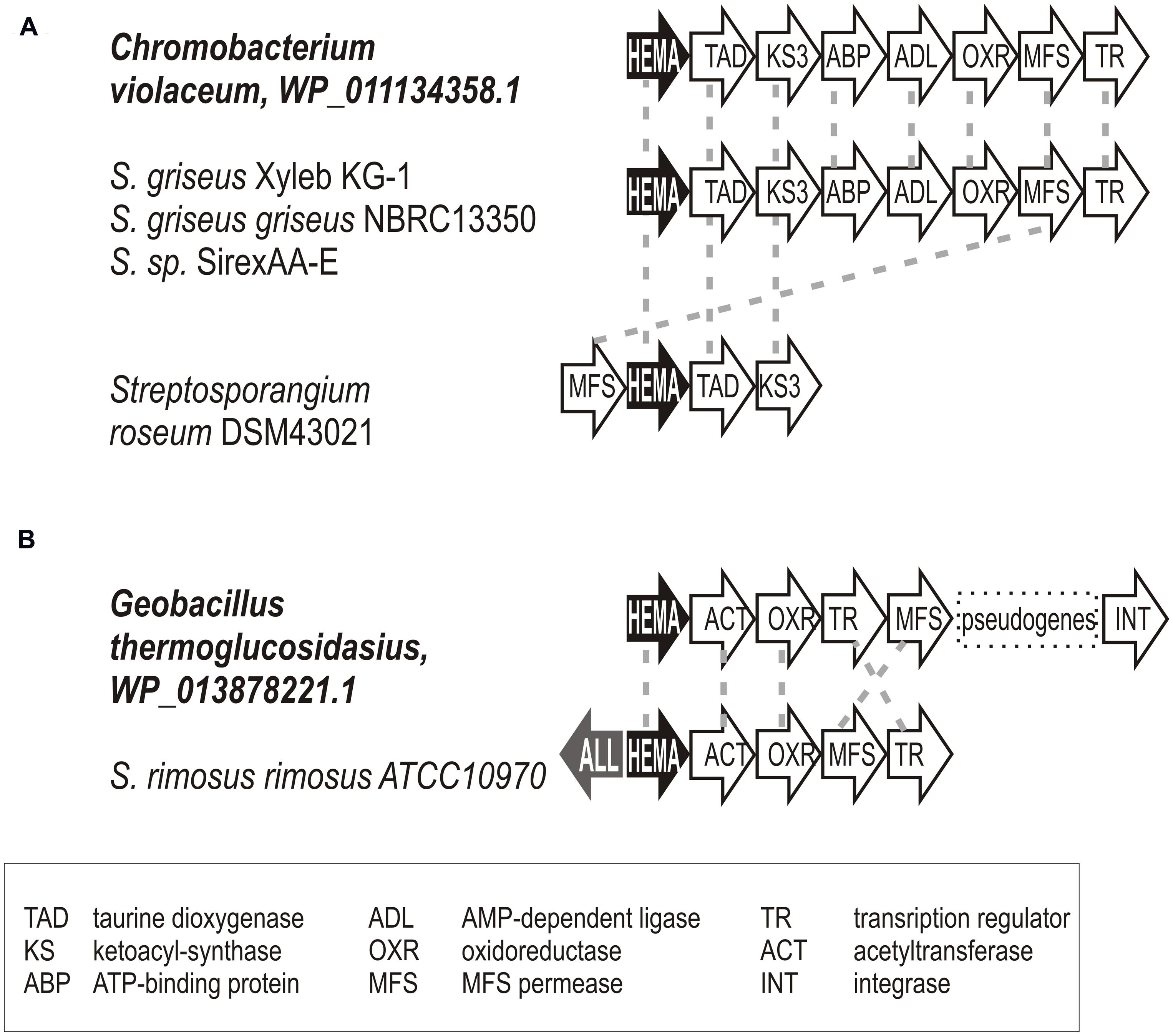
FIGURE 7. Examples documenting the horizontal gene transfer of hemA gene loci between proteobacteria or bacilli and actinomycetes. Relevant genetic loci of putative donors and acceptors are shown; gene homology indicated by dashed gray lines. Gene annotations are given in the figure bottom. (A) Chromobacterium violaceum (B) Geobacillus thermoglucosidasius.
Isolate Identification and Genome Relatedness
To underpin genetic diversity of environmental strains coming into screening procedure, 234 were subjected to a relatedness survey. Genomic fingerprinting indicated high genetic diversity. The BOX-PCR similarity profile divided the environmental isolates into 184 distinct groups, using the Pearson product moment correlation coefficient threshold of 70% (Supplementary Figure S3), of which 147 were unique. The most abundant BOX-PCR groups were clusters B10 and B34, both comprising five strains from two closely related habitats. With the exception of groups B11, B12, B28, and B36, isolates from one BOX-PCR group were usually assigned to the same species or clade (Supplementary Table S1).
The 16S rRNA genes of 131 isolates were sequenced with the aim to characterize most of BOX-PCR groups (at least those comprising of more than one strain). Most environmental isolates had identical or almost identical sequences (98-100% pairwise sequence similarity) to species of genus Streptomyces – 124 strains, A. thailandensis – 1, Kitasatospora atroaurantiaca – 1, K. gansuensis – 1, Lentzea violacea – 1, Nocardia nova – 1 and Saccharothrix espanaensis – 2 (Supplementary Table S1). Forty-nine species have been recognized within the Streptomyces, of which S. aurantiacus, S. aureus, and S. ederensis, along with S. griseus, a group of taxonomically related species, are the most prevalent (comprising 9, 12, 11, and 16 strains, respectively).
Apparently, in four BOX-PCR groups (B11, B12, B28, and B36) a discrepancy between 16S rDNA phylogenetic clade assignment and BOX-PCR grouping was observed. The same BOX-PCR groups contained more or less phylogenetically related species (e.g., S. aureus and S. olivochromogenes in group B11; S. phaeochromogenes and S. olivochromogenes in group B12; S. scabiei and S. lavendulae in group B28; S. prunicolor and S. griseus in group B36). Additionally, species were often represented by several ecotypes (e.g., more than one BOX-PCR group has been assigned to the same species). This was more the rule than the exception, even when strains originated from the same habitat (e.g., S. aurantiacus and S. ederensis from habitat H32). This finding supports the idea that genomic rearrangements and other kinds of genetic flow are occurring in natural strains frequently and should be taken into account.
Discussion
In this work we showed the independent evolution of aminolevulinate synthase genes in actinomycetes, especially of their newly described, cyclizing, secondary metabolism-connected variant encoding cALAS. These represent a key enzyme in the C5N unit formation. Together with accompanying AMS and acyl-CoA ligase they direct biosynthesis and attachment of the functional moiety to different classes of bioactive secondary metabolites of actinomycetes. Thus, the hemA gene may be used as a genetic tag for putative producers of C5N carrying-compounds. This variable group contains secondary metabolites with pharmaceutically attractive potential, such as alternatives for β-lactams, anti-cancer compounds, and immunomodulatory molecules. Definition of metabolite-specific signatures for hemA gene sequences may serve as a useful tool in identifying novel producers of known compounds or their new derivatives, or perhaps in identifying putative producers of novel C5N compound classes. Moreover, our data illustrate the distribution and relative frequency of relevant biosynthetic genes in genomes of actinomycetes. We provide several examples suggesting horizontal gene transfer within actinobacterial communities in particular biotopes, and genetic evidence of gene transfer between bacterial and fungal genera.
Relatively short fragment-sequences of aminolevulinate synthase genes were shown to provide sufficient genetic information to differentiate between putative producers of the individual C5N compounds. The phylogenetic tree of short hemA fragments showed clear clustering of sequences corresponding to compounds. Of the 153 positives analyzed (10% of all screened strains) 88% appear to follow cALAS-specific sequence conservation. Most of these fit into the branches of known type producers (78% of all positives). The remaining 12% of cALAS-like hemA homologs from the screening, together with 9% from genomic matches, were independent of known producer branches (brown and orange branches in Figure 4). These displayed most of cALAS-specific sequence features; hence we can speculate that they form the C5N or similar cyclic units. From the genome data available in the brown group we may also observe, that hemA genes are again accompanied with both all and ams genes here and lay within putative secondary metabolites biosynthetic genes. These represent the hottest candidates for novel types of C5N-containing bioactive compounds producers.
Genomic data suggest that, rarely, streptomycete genomes may carry two putative biosynthetic gene clusters for C5N metabolites of two different classes, as it is in S. ghanaensis ATCC14672 and S. viridosporus T7A. The dysfunctional C5N-encoding locus of one is suppressed by the products of the other, located in a different gene cluster.
Unexpectedly, as high as 12% of screening sequences (and 24% of those from genomic projects) appear to encode non-cyclizing, “classical” ALAS, though this enzyme is not needed in the primary metabolism of actinomycetes as they form 5-aminolevulinate by the different, C5 pathway from glutamate (Petříček et al., 2006). Neither we have observed any obvious connection to secondary metabolism; no conservative, typical secondary metabolism genes were found in the hemA loci. On the other hand, the genetic data provide evidence of intergeneric horizontal gene transfer in soil microorganism communities, even between phylogenetically distant microorganisms, Proteobacteria, Firmicutes, and Actinobacteria, inhabiting the same biotope. This supports recent theories of a horizontal gene transfer between quite distant bacterial phyla (Shintani et al., 2014) or even crossing borders between kingdoms of Bacteria and Fungi, which is much less well understood (Schmitt and Lumbsch, 2009). Whether these “stolen” hemA genes are functional in actinomycetes remains unclear. We can hypothesize, however, that they were used as a starting material for cALAS evolution after their HGT into Streptomyces.
Horizontal gene transfer is also observable in cALAS-encoding hemA genes. For example, groups of hemA-positive isolates fitting to the same metabolite production group (colabomycin, moenomycin, e.g.), which originate from the same biotope, are often classified into different Streptomyces species. This is an important point to take into account when developing screening protocols in the search for novel producers of specific compounds as increased geographic and biotope diversity increases the chances of identifying new producers. Taxonomy data appear to be much less important. Huge parts of chromosomes and giant plasmids are traded within the actinomycete population in individual biotopes (Doroghazi and Buckley, 2010), and this was many times suggested also by our data, e.g., in the set of putative bafilomycin producers. The type strain, S. lohii ATCC BAA-1276, was isolated by a Chinese group from a sandy beach in Papua New Guinea (Zhang et al., 2013). Our own six isolates, isolated from ambrosia beetle galleries in Papua New Guinea, carry highly homologous hemA genes, though taxonomically they are classified as at least three different, non-related species. Of the habitats studied, a number display high genetic variability resulting in diverse secondary-metabolite production, e.g., Sokolov colliery spoil provided strains possessing hemA genes related to manumycin and colabomycin linear polyketides, moenomycin, as well as unknown orange and brown clusters.
Conclusion
Our work proved the hypothesis of correlation between the secondary metabolism-connected hemA gene sequences and a type of the C5N-containing metabolite synthesized. This specifically concerns hemA alleles encoding cyclizing type of aminolevulinate synthase, as a part of the C5N-encoding gene triad. Therefore, phylogenetic analysis of hemA genes may assist in the search for novel producers of bioactive C5N compounds, as in the case of previously characterized producer of novel colabomycin E, S. aureus SOK1/5-04 (Petříčková et al., 2014). The developed PCR screening technique displays high sensitivity and specificity, and is simple, fast, and cheap. Moreover, the presented data imply frequent horizontal transfer of genes and entire clusters within actinomycete populations: Phylogenetically distant streptomycetes of similar geographical origin were shown to carry identical or highly similar hemA genes (bafilomycin producers from Papua New Guinea) and to carry the same biosynthetic gene clusters (colabomycin or moenomycin producers from Sokolov). On the other hand, less frequent hemA genes of non-cyclizing type were not associated with any secondary metabolism genes, showed lower GC contents, and sometimes, obvious homologies with putative donors in hemA chromosomal loci were found. This may suggest more recent horizontal gene transfer event from lower GC donors, which use hemA-encoded enzymes in their primary metabolism. As the gene is not essential for primary metabolism of actinomycetes, we may imagine that a similar event could earlier serve as a starting material for evolution of the novel biosynthetic capability of the acceptor – novel subunit was formed and subsequently attached to different metabolic cores.
Author Contributions
KP planned the experiments, conducted the screening, sequencing and genome scanning, analyzed and interpreted the data, and prepared the manuscript; AC planned and performed phylogenetic analyses, made the whole-genome sequencing, and prepared the phylogenetic parts of the manuscript. TZ isolated genomic DNAs, performed the genetic screening and analyzed the secondary metabolite extracts. TC designed, performed, and interpreted phylogenetic analyses. SP made fermentations and prepared secondary metabolite extracts. MP and VK supervised the experiments, MP also contributed to sequencing and data analysis, and VK collected and isolated all the novel strains.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was financed through Grant LH12191 from the Ministry of Education, Youth and Sports of the Czech Republic. Computational resources were provided by MetaCentrum under Program LM2010005 and CERIT-SC under the CERIT Scientific Cloud Centre Program, OP RDI, CZ.1.05/3.2.00/08.0144. We are indebted to the Slovak Caves Administration in Liptovský Mikuláš (Slovakia) for the sampling and research permit.
We are grateful to Mika Tarkka (Helmholtz Centre for Environmental Research, Halle, Germany), Miroslav Kolařík (Institute of Microbiology, Czech Academy of Sciences, v. v. i., Prague, Czech Republic), Soňa Javoreková (Slovak University of Agriculture in Nitra, Slovakia), Dana Elhottová (Biology Centre, Czech Academy of Sciences, v. v. i. Institute of Soil Biology, České Budějovice, Czech Republic; BC – ISB), and to Fuchao Li (Institute of Oceanology of the Chinese Academy of Science, Qingdao, China) for the gift of strains. We would also like to thank Miloslav Devetter (BC – ISB), Janez Mulec (Karst Research Institute, Research Centre of the Slovenian Academy of Sciences and Arts, Postojna, Slovenia), and Vitalij M. Gryshko (Botanical Garden NAS of Ukraine, Kryvyi Rig, Ukraine) for soil sampling in Cameroon, Georgia, Slovenia, and the Ukraine. Finally, we thank Petra Beníšková, Laura Cibulková, Jiří Petrásek, and Jan Šindelář (BC – ISB) for technical help.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.00814
Footnotes
References
Astner, I., Schulze, J. O., van den Heuvel, J., Jahn, D., Schubert, W. D., and Heinz, D. W. (2005). Crystal structure of 5-aminolevulinate synthase, the first enzyme of heme biosynthesis, and its link to XLSA in humans. EMBO J. 24, 3166–3177. doi: 10.1038/sj.emboj.7600792
Blin, K., Medema, M. H., Kazempour, D., Fischbach, M. A., Breitling, R., Takano, E., et al. (2013). AntiSMASH 2.0-a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 41, 204–212. doi: 10.1093/nar/gkt449
Chroňáková, A., Krištůfek, V., Tichý, M., and Elhottová, D. (2010). Biodiversity of streptomycetes isolated from a succession sequence at a post-mining site and their evidence in Miocene lacustrine sediment. Microbiol. Res. 165, 594–608. doi: 10.1016/j.micres.2009.10.002
Chun, J., Lee, J. H., Jung, Y., Kim, M., Kim, S., Kim, B. K., et al. (2007). EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int. J. Syst. Evol. Microbiol. 57, 2259–2261. doi: 10.1099/ijs.0.64915-0
Deng, M.-R., Guo, J., Li, X., Zhu, C.-H., and Zhu, H.-H. (2011). Granaticins and their biosynthetic gene cluster from Streptomyces vietnamensis: evidence of horizontal gene transfer. Antonie Van Leeuwenhoek 100, 607–617. doi: 10.1007/s10482-011-9615-9
Doroghazi, J. R., and Buckley, D. H. (2010). Widespread homologous recombination within and between Streptomyces species. ISME J. 4, 1136–1143. doi: 10.1038/ismej.2010.45
Edwards, U., Rogall, T., Blocker, H., Emde, M., and Bottger, E. C. (1989). Isolation and direct complete nucleotide determination of entire genes – characterization of a gene coding for 16S-Ribosomal RNA. Nucleic Acids Res. 17, 7843–7853. doi: 10.1093/nar/17.19.7843
Egan, S., Wiener, P., Kallifidas, D., and Wellington, E. M. (1998). Transfer of streptomycin biosynthesis gene clusters within streptomycetes isolated from soil. Appl. Environ. Microbiol. 64, 5061–5063.
Hall, T. A. (1999). Bioedit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98.
Huelsenbeck, J. P., and Ronquist, F. (2005). “Bayesian analysis of molecular evolution using MrBayes,” in Statistical Methods in Molecular Evolution, ed. R. Nielsen (Berlin: Springer), 183–226. doi: 10.1007/0-387-27733-1_7
Hwang, J. Y., Kim, H. S., Kim, S. H., Oh, H. R., and Nam, D. H. (2013). Organization and characterization of a biosynthetic gene cluster for bafilomycin from Streptomyces griseus DSM 2608. AMB Exp. 3:24. doi: 10.1186/2191-0855-3-24
Hwang, J. Y., Kim, S. H., Oh, H. R., Kwon, E., and Nam, D. H. (2015). Analysis of a draft genome sequence of Kitasatospora cheerisanensis KCTC 2395 producing bafilomycin antibiotics. J. Microbiol. 53, 84–89. doi: 10.1007/s12275-015-4340-0
Ichikawa, N., Oguchi, A., Ikeda, H., Ishikawa, J., Kitani, S., Watanabe, Y., et al. (2010). Genome sequence of Kitasatospora setae NBRC 14216(T): an evolutionary snapshot of the family Streptomycetaceae. DNA Res. 17, 393–406. doi: 10.1093/dnares/dsq026
Kalan, L., Gessner, A., Thaker, M. N., Waglechner, N., Zhu, X. M., Szawiola, A., et al. (2013). A cryptic polyene biosynthetic gene cluster in Streptomyces calvus is expressed upon complementation with a aunctional bldA Gene. Chem. Biol. 20, 1214–1224. doi: 10.1016/j.chembiol.2013.09.006
Kaufholz, A. L., Hunter, G. A., Ferreira, G. C., Lendrihas, T., Hering, V., Layer, G., et al. (2013). Aminolaevulinic acid synthase of Rhodobacter capsulatus: high-resolution kinetic investigation of the structural basis for substrate binding and catalysis. Biochem. J. 451, 205–216. doi: 10.1042/BJ20121041
Kim, E. Y., Han, J. W., Lee, J. Y., and Kim, B. S. (2012). Identification of the biosynthetic gene cluster for the antibiotic polyketide L-155,175 in Streptomyces hygroscopicus. Folia Microbiol. 57, 543–550. doi: 10.1007/s12223-012-0173-y
Krištůfek, V., Elhottová, D., Chroňáková, A., Dostálková, I., Picek, T., and Kalčík, J. (2005). Growth strategy of heterotrophic bacterial population along successional sequence on spoil of brown coal colliery substrate. Folia Microbiol. 50, 427–435. doi: 10.1007/BF02931425
Kyselková, M., Chroňáková, A., Volna, L., Němec, J., Ulmann, V., Scharfen, J., et al. (2012). Tetracycline resistance and presence of tetracycline resistance determinants tet(V) and tap in rapidly growing mycobacteria from agricultural soils and clinical isolates. Microbes Environ. 27, 413–422. doi: 10.1264/jsme2.ME12028
Labeda, D. P., Goodfellow, M., Brown, R., Ward, A. C., Lanoot, B., Vanncanneyt, M., et al. (2012). Phylogenetic study of the species within the family Streptomycetaceae. Antonie Van Leeuwenhoek 101, 73–104. doi: 10.1007/s10482-011-9656-0
Lanoot, B., Vancanneyt, M., Dawyndt, P., Cnockaert, M., Zhang, J. L., Huang, Y., et al. (2004). BOX-PCR fingerprinting as a powerful tool to reveal synonymous names in the genus Streptomyces. Emended descriptions are proposed for the species Streptomyces cinereorectus, S. fradiae, S. tricolor, S. colombiensis, S. filamentosus, S. vinaceus and S. phaeopurpureus. Syst. Appl. Microbiol. 27, 84–92. doi: 10.1078/0723-2020-00257
Mayer, S. M., and Beale, S. I. (1992). Succinyl-coenzyme A synthetase and its role in delta-aminolevulinic acid biosynthesis in Euglena gracilis. Plant Physiol. 99, 482–487. doi: 10.1104/pp.99.2.482
McAlpine, J. B., Bachmann, B. O., Piraee, M., Tremblay, S., Alarco, A. M., Zazopoulos, E., et al. (2005). Microbial genomics as a guide to drug discovery and structural elucidation: ECO-02301, a novel antifungal agent, as an example. J. Nat. Prod. 68, 493–496. doi: 10.1021/np0401664
Medema, M. H., Blin, K., Cimermancic, P., de Jager, V., Zakrzewski, P., Fischbach, M. A., et al. (2011). antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 39, W339–W346. doi: 10.1093/nar/gkr466
Ostash, B., Saghatelian, A., and Walker, S. (2007). A streamlined metabolic pathway for the biosynthesis of moenomycin A. Chem. Biol. 14, 257–267. doi: 10.1016/j.chembiol.2007.01.008
Ostash, B., and Walker, S. (2010). Moenomycin family antibiotics: chemical synthesis, biosynthesis, and biological activity. Nat. Prod. Rep. 27, 1594–1617. doi: 10.1039/c001461n
Petříček, M., Petříčková, K., Havlíček, L., and Felsberg, J. (2006). Occurrence of two 5-aminolevulinate biosynthetic pathways in Streptomyces nodosus subsp. asukaensis is linked with the production of asukamycin. J. Bacteriol. 188, 5113–5123. doi: 10.1128/JB.01919-05
Petříčková, K., Pospíšil, S., Kuzma, M., Tylová, T., Jágr, M., Tomek, P., et al. (2014). Biosynthesis of colabomycin E, a new manumycin-family metabolite, involves an unusual chain-length factor. Chembiochem 15, 1334–1345. doi: 10.1002/cbic.201400068
Schmitt, I., and Lumbsch, H. T. (2009). Ancient horizontal gene transfer from bacteria enhances biosynthetic capabilities of Fungi. PLoS ONE 4:e4437. doi: 10.1371/journal.pone.0004437
Shintani, M., Matsui, K., Inoue, J., Hosoyama, A., Ohji, S., Yamazoe, A., et al. (2014). Single-cell analyses revealed transfer ranges of IncP-1, IncP-7, and IncP-9 plasmids in a soil bacterial community. Appl. Environ. Microbiol. 80, 138–145. doi: 10.1128/AEM.02571-13
Shirling, E. B., and Gottlieb, D. (1966). Methods for characterization of streptomycetes. Int. J. Syst. Bacteriol. 16, 313–340. doi: 10.1099/00207713-16-3-313
Versalovic, J., Schneider, M., Bruijn, F. J., and Lupski, J. R. (1994). Genomic fingerprinting of bacteria using repetitive sequence-based polymerase chain reaction. Methods Mol. Cell. Biol. 5, 25–40.
Weinstein, J. D., and Beale, S. I. (1983). Separate physiological roles and subcelullar compartments for two tetrapyrrole biosynthetic pathways in Euglena gracilis. J. Biol. Chem. 72, 6799–6807.
Yang, H. S., and Hoober, J. K. (1995). Divergent pathways for δ-aminolevulinic acid synthesis in 2 Species of Arthrobacter. FEMS Microbiol. Lett. 134, 259–263. doi: 10.1016/0378-1097(95)00417-4
Zhang, W. J., Bolla, M. L., Kahne, D., and Walsh, C. T. (2010). A three enzyme pathway for 2-amino-3-hydroxycyclopent-2-enone formation and incorporation in natural product biosynthesis. J. Am. Chem. Soc. 132, 6402–6411. doi: 10.1021/ja1002845
Keywords: 5-aminolevulinate synthase, C5N unit, Streptomyces, secondary metabolites, gene evolution, genetic screening, horizontal gene transfer
Citation: Petříčková K, Chroňáková A, Zelenka T, Chrudimský T, Pospíšil S, Petříček M and Krištůfek V (2015) Evolution of cyclizing 5-aminolevulinate synthases in the biosynthesis of actinomycete secondary metabolites: outcomes for genetic screening techniques. Front. Microbiol. 6:814. doi: 10.3389/fmicb.2015.00814
Received: 07 May 2015; Accepted: 23 July 2015;
Published: 05 August 2015.
Edited by:
Eric Altermann, AgResearch Ltd, New ZealandReviewed by:
Suleyman Yildirim, Istanbul Medipol University, TurkeyPolpass Arul Jose, Tamil Nadu Agricultural University, India
Virginia Helena Albarracín, PROIMI – CONICET, Argentina
Copyright © 2015 Petříčková, Chroňáková, Zelenka, Chrudimský, Pospíšil, Petříček and Krištůfek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miroslav Petříček, Institute of Microbiology, Czech Academy of Sciences, v. v. i., Vídeňská 1083, 142 20 Prague, Czech Republic,cGV0cmFzQGJpb21lZC5jYXMuY3o=; Alica Chroňáková, Institute of Soil Biology, Biology Centre, Czech Academy of Sciences, v. v. i., Na Sádkách 7, 370 05 České Budějovice, Czech Republic,YWxpY2FjaEB1cGIuY2FzLmN6