 Gaidi Ren
Gaidi Ren Huayong Zhang1
Huayong Zhang1 Zhongjun Jia
Zhongjun Jia- 1State Key Laboratory of Soil and Sustainable Agriculture, Institute of Soil Science – Chinese Academy of Sciences, Nanjing, China
- 2Key Laboratory of Soil Environment and Pollution Remediation, Institute of Soil Science – Chinese Academy of Sciences, Nanjing, China
Plant endophytic bacteria play an important role in plant growth and health. In the context of climate change, the response of plant endophytic bacterial communities to elevated CO2 at different rice growing stages is poorly understood. Using 454 pyrosequencing, we investigated the response of leaf endophytic bacterial communities to elevated CO2 (eCO2) at the tillering, filling, and maturity stages of the rice plant under different nitrogen fertilization conditions [low nitrogen fertilization (LN) and high nitrogen fertilization (HN)]. The results revealed that the leaf endophytic bacterial community was dominated by Gammaproteobacteria-affiliated families, such as Enterobacteriaceae and Xanthomonadaceae, which represent 28.7–86.8% and 2.14–42.6% of the total sequence reads, respectively, at all tested growth stages. The difference in the bacterial community structure between the different growth stages was greater than the difference resulting from the CO2 and nitrogen fertilization treatments. The eCO2 effect on the bacterial communities differed greatly under different nitrogen application conditions and at different growth stages. Specifically, eCO2 revealed a significant effect on the community structure under both LN and HN levels at the tillering stage; however, the significant effect of eCO2 was only observed under HN, rather than under the LN condition at the filling stage; no significant effect of eCO2 on the community structure at both the LN and HN fertilization levels was found at the maturity stage. These results provide useful insights into the response of leaf endophytic bacterial communities to elevated CO2 across rice growth stages.
Introduction
Just as animals have a complex microbiota, plants are normally colonized by diverse microorganisms rather than existing as axenic organisms (Jackson et al., 2013). It has been stated that a plant completely free of microorganisms does (almost) not exist, neither actually nor in the history of land plants (Partida-Martínez and Heil, 2011). Endophytic bacteria, which colonize the internal tissue of the plant, represent a widespread and ancient symbiosis (Partida-Martínez and Heil, 2011). They can influence host growth and function in many ways. Directly, they can contribute to plant growth by improving the availability of nutrients, such as phosphate (Quecine et al., 2012; Ji et al., 2014), by producing plant hormones, such as indole acetic acid (IAA; Quecine et al., 2012; Khan et al., 2014), and by fixing nitrogen (Knoth et al., 2013; Madhaiyan et al., 2013). Indirectly, they may improve the resistance of host plants to pathogens through increased competition for the same ecological niche (Loaces et al., 2011) or production of antimicrobial substances such as antibiotics (Sessitsch et al., 2004).
The identification of plant endophytic bacteria has largely been based on the cultivation-dependent method (Adhikari et al., 2001; Singh et al., 2006; Muthukumarasamy et al., 2007). However, only less than 1% of the microorganisms in the environment can be cultured because of unknown growth requirements of many microorganisms (Amann et al., 1995) or because some microbial cells are in viable but non-cultivable states (Tholozan et al., 1999). A few culture-independent studies have also been reported for the characterization of plant endophytic bacterial communities via 16S rRNA gene-based molecular techniques such as terminal restriction fragment length polymorphism (T-RFLP; Reiter et al., 2002; Manter et al., 2010; Ding et al., 2013; Thijs et al., 2014), the denaturing gradient gel electrophoresis (DGGE) approach (Araújo et al., 2002; Hardoim et al., 2011) and the clone library construction method (Jin et al., 2014). However, due to the relatively lower resolution of these techniques, the diversity of plant endophytic bacteria is far from being understood. The emergence of next-generation sequencing technology allows sequence efforts that are orders of magnitude greater than ever before and therefore have opened a new frontier in microbial diversity determination (Huse et al., 2008; Hollister et al., 2010). This technique has been used to evaluate the diversity of endophytic bacterial communities from the roots of rice (Zhang et al., 2013) and potato (Manter et al., 2010), and the leaves of rice (Ren et al., 2015) and commercial salad (e.g., baby spinach and lettuce; Jackson et al., 2013), and from the leaves and roots of Arabidopsis thaliana (Bodenhausen et al., 2013). This technique has also been used to determine the whole genome sequence of plant endophytic bacteria isolates (Zhu et al., 2012).
The rise in atmospheric CO2 concentration is projected to have a profound impact on the properties and functioning of the terrestrial ecosystem (Ainsworth and Long, 2005; Rosenzweig et al., 2007). A number of research studies have been conducted to understand the changes in plant physiology (Ainsworth et al., 2002; Bernacchi et al., 2003), plant growth and yield (Idso and Idso, 1994; Ainsworth et al., 2002; Belote et al., 2004), and plant community composition (Leadley et al., 1999; Belote et al., 2004) under the condition of elevated atmospheric CO2. However, within the context of climate changes, the response of the plant endophytic bacterial community, despite acting as a paramount player in plant growth and health and nutrient biogeochemical cycling, are poorly understood. The response of the plant endophytic bacterial community to elevated CO2 may be influenced by other environmental factors. Specifically, elevated CO2 often has been shown to have a stimulation effect on plant growth (Ainsworth and Long, 2005; Reich and Hobbie, 2013). However, the magnitude and sustainability of the CO2-enhanced plant biomass accumulation may be influenced by the nitrogen (N) level in the soil (Reich and Hobbie, 2013). A few studies (Kim et al., 2001; Anten et al., 2004; Reich et al., 2006) have shown that increased nitrogen levels in soil amplified the effect of elevated CO2 on plant productivity. Changes in plant growth may further lead to variations in the plant-associated bacterial community structure. Furthermore, over different growth stages of the host plant, the plant endophytic bacterial community is likely subject to dynamic changes over time.
Rice, a main source of nourishment for over 50% of the world’s population, is by far one of the most important staple food crops in the world (Gyaneshwar et al., 2001). The responses of the leaf endophytic bacterial communities from the rice plant to elevated CO2 under different nitrogen fertilization levels at the tillering, filling, and maturity stages was investigated in this study using the 16S rRNA gene-based 454 pyrosequencing technique. Our aim was to reach a deep understanding of the response of the leaf endophytic bacterial community to elevated CO2 at different nitrogen application levels over different rice growth periods.
Materials and Methods
Experimental Site Description
The FACE (free air CO2 enrichment) facility was set up in 2004 in Yangzhou, Jiangsu Province, China (119°42′0″E, 32°35′5″N). This area is a typical agricultural region for rice-wheat rotation in China. Rice planting has been ongoing for more than 50 years in this area. This region is also an important area for rice production in China. The relevant soil properties are as follows: soil bulk density 1.2 g cm-3, sand (2–0.02 mm) 57.8%, silt (0.02–0.002 mm) 28.5%, clay (<0.002 mm) 13.7%, and pH (H2O) 6.8. The climatic conditions are as follows: (1) mean annual precipitation temperature: 15°C; (2) mean annual precipitation: 900–1,000 mm; (3) average annual sunshine: approximately 2,132 h; and (4) annual frostless time: more than 220 days.
FACE System
Details of the set-up and performance of the China rice-FACE facility have been described previously (Okada et al., 2001; Liu et al., 2002). Briefly, the FACE system consists of six plots distributed in different paddy fields having similar soil characteristics, nutrient levels (total nitrogen 1.45 g kg-1, total phosphorus 0.63 g kg-1, total potassium 14.0 g kg-1, available phosphorus 10.1 mg kg-1, available potassium 70.5 mg kg-1), and agronomic histories. Three plots were randomly allocated for the elevated CO2 treatments (hereinafter called eCO2) and three were maintained under ambient conditions (hereinafter referred to as aCO2). In the eCO2 plots, the rice plants were grown in octagonal “rings” with a diameter of 12 m. The target CO2 concentration in the center of the eCO2 rings was 200 ± 40 μmol mol-1 higher than that of the aCO2. To avoid treatment crossover, the eCO2 rings were separated from aCO2 plots by more than 90 m. Pure CO2 gas was sprayed from peripheral emission tubes, which were set 50–60 cm above the canopy. The CO2 concentration in the eCO2 rings was monitored and controlled by a computer program.
Rice Cultivation
On June 20th, 2010, seedlings of japonica rice cultivar ‘Wuxiangjing 14,’ a major local cultivar with high-yield potential, were manually transplanted at a density of three seedlings per hill and 24 hills m-2 into the plots. Each ring was further partitioned into two identical subplots to test the impact of two nitrogen (N) fertilization levels, which were provided as urea (N, 46%) and a compound fertilizer [(N:P2O5:K2O = 15:15:15, %): low N (LN, 15 g N m-2) and high N (HN, 25 g N m-2)]. For all of the N fertilization levels, the phosphorus (P) and potassium (K) fertilizers were supplied at rates of 7 g P2O5 m-2 and 7 g K2O m-2, respectively. For the HN level, 36% of the N was applied as a basal dressing prior to transplanting (June 19th); 24% of the N was applied as a side dressing at early tillering (July 27th), and 40% of the N was provided at the panicle initiation (August 1st). For the LN fertilization level, 60% of the N was used as a basal dressing; the remainder (40% of the N) was applied at the panicle initiation as supplied in the HN fertilization level. For all of the N levels, P and K were both supplied as a basal dressing. To prevent treatment crossover, a 30-cm polyvinyl chloride (PVC) barrier was pushed 10 cm into the soil between the LN and HN subplots.
Sample Collection
Leaf samples (more than 20 g) were collected on August 4th, September 16th, and October 26th, 2010 when the rice was at the tillering (when the tiller number is continently increasing continuously until it reaches to the point of maximum tillering), filling (when a milky white substance was accumulated in the grains and gradually became the texture of bread dough), and maturity (when the endosperm become hard and opaque, the dry matter of the grain becomes constant, and the grain turns golden brown) stages (Yoshida, 1981), respectively. The collected leaves from each subplot (a total of 12 subplots) were respectively bulked together, placed into sterile bags, put into an incubator (4°C), and brought to the lab. The laboratory analysis (i.e., DNA extraction) was conducted immediately after the sampling was finished at each time point. In total, 12 leaf samples were collected at each stage, and 36 leaf samples were obtained across all of the tested growth stages. The meteorological conditions, including the average daily air temperature, daily solar radiation, and daily precipitation during the sampling period, have been reported previously (Ren et al., 2014). When the tillering-, filling-, and maturity-leaf samples were collected, the average daily temperature was 31.2, 24.1, and 9.1°C, respectively; the mean daily solar radiation was 12.1, 13.7, and 5.14 MJ m-2, respectively; and the daily precipitation at all of the sampling points was 0 mm.
Bacterial Cells Harvest and DNA Extraction
The collection of leaf endosphere bacteria was initiated after washing leaf-surface (i.e., phyllosphere) three times for bacteria, as previously described (Ren et al., 2015). Briefly, the leaves were immersed in sterile TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). Vigorous shaking at 250 rpm was then conducted to dislodge the microbial cells from the leaf surface. The dislodging procedure was repeated three times. Then, the collection of leaf endosphere bacterial cells was performed by submerging ground leaf samples in sterile Tris-EDTA (TE) buffer and washing three times. Similar to the collection method for the leaf-surface (i.e., phyllosphere) bacterial cells, three cycles of washing were performed to collect the leaf endophytic bacterial cells. The cell suspension resulting from each washing process was filtered through sterile glass wool to remove any visible large particles. The resulting three cell suspensions were pooled to maximize the recovery efficiency of the leaf endosphere bacterial cells. The combined cell suspension was then centrifuged, the supernatant was discarded, and the bacterial cell pellets were resuspended in 2 mL of TE buffer for further DNA extraction. Based on our preliminary experiment (Supplementary information), the majority of the phyllosphere bacterial cells were removed from the leaf surface after the three-time removal procedures because the DNA band from the fourth-collection of phyllosphere bacteria was greatly weaker (not visible) compared with that from the initial three-time-collection as revealed by the DNA electrophoresis results. For this reason, the dislodging of the phyllosphere bacterial cells was performed three times in the current study. Our preliminary experiment (Supplementary information) also suggested that the leaf endosphere bacteria would not suffer substantially heavy contamination by the phyllosphere bacteria after three times’ washing procedures for phyllosphere bacteria.
The collected cell suspension of the leaf endosphere bacteria was then used for DNA extraction, as previously reported (Garbeva et al., 2001; Araújo et al., 2002; Ren et al., 2015). In brief, three cycles of freezing with liquid nitrogen for 10 min and thawing at 65°C for 30 min was performed. A DNA-free lysozyme solution (40 μL of 10 mg mL-1) was added to enhance the cell lysis and the resulting solution was incubated at 37°C for 2 h. Then, 200 μL of a 20% sterilized sodium dodecyl sulfate (SDS) solution and 32 μL of a 20 mg mL-1 DNA-free proteinase K (Roche Applied Science) were added and the resulting mixture was incubated at 37°C overnight. Next, 800 μL of 5 mol L-1 sterilized NaCl was added, and the mixture was centrifuged. The supernatant was then purified using an equal volume of chloroform/isoamyl alcohol solution (24:1). The aqueous phase was then transferred. To precipitate the DNA pellets, 0.6 volumes of isopropanol were then added, and the mixture was incubated at 4°C for 30 min and centrifuged at 12,000 rpm for 15 min. Finally, the DNA pellet was washed with 70% pre-cooled ethanol, air dried, and re-dissolved in sterilized TE buffer. The whole cell collection and DNA extraction process was performed aseptically within an aseptic room.
Pyrosequencing and Data Analysis
The 454 pyrosequencing was performed on a Roche 454 GS FLX Titanium pyrosequencing platform. Fusion primers consisting of adaptor A or B, key sequence, barcode and template specific sequences were used in this study. Specifically, the V4–V5 region of the bacterial 16S rRNA gene was amplified with the forward primer 515F [5′-CGTATCGCCTCCCTCGCGCCA+TCAG + (6 bp Barcode) + (GTGCCAGCMGCCGCGG)-3′] and the reverse primer 907R [5′-CTATGCGCCTTGCCAGCCCGC + TCAG + (6 bp Barcode) + (CCGTCAATTCMTTTRAGTTT)-3′]. The 50 μL PCR reaction mixture contained 1 × PCR buffer (Mg2+ plus), 0.2 mM of each deoxynucleoside triphosphate, 0.4 mM of each primer, 1.25 U of TaKaRa Taq HS polymerase (TaKaRa Biotech, Dalian, China), and 1 μL of DNA template. The PCR amplification program included initial denaturation at 94°C for 5 min, followed by 32 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 45 s, and a final extension at 72°C for 5 min. The PCR products were subjected to electrophoresis using a 2.0% agarose gel. The amplicon band with a correct size (475 bp) was excised from the gel and then purified with an agarose gel DNA purification kit (TaKaRa Biotech, Dalian, China). The concentration of the cleaned PCR products was measured using the Qubit hs-DS-DNA kit (Invitrogen, Carlsbad, CA, USA) on a Tecan Infinite F200 Pro plate reader. All of the amplicons were then combined in equimolar amounts and sequenced on a Roche 454 GS FLX Titanium pyrosequencing machine (454 Life Science, Branford, CT, USA).
The pyrosequencing data analysis was processed using the Quantitative Insights into Microbial Ecology (QIIME) pipeline (http://qiime.org/). In brief, low quality sequences, which have lengths of <200 bp, an average quality score of <25, ambiguous nucleotides of >0, homopolymer >6, and primer mismatches were trimmed and the barcodes were determined to assign sequence reads to the proper samples. Then, the chimeras were detected using the UCHIME algorithm based on a database of chimera-free sequences (Edgar et al., 2011). The sequences, which were assigned to a mitochondrial or chloroplast origin were eliminated with the Metaxa software tool (Bengtsson et al., 2011) and the V4–V5 region was extracted with the V-Xtractor software tool (Hartmann et al., 2010). The operational taxonomic units (OTUs) with a 97% sequence similarity were then generated using the uclust clustering method (Edgar, 2010). The phylogenetic affiliation of OTUs was analyzed by the Ribosomal Database Project classifier (Wang et al., 2007). The pyrosequencing data have been deposited in the DNA Data Bank of Japan (DDBJ) under accession numbers DRA001097 and DRA001098.
Due to technical reasons, the sequence depth obtained for each sample differed (Aguirre de Cárcer et al., 2011). In this study, the range of valid sequence depth was 2,865–9,650 sequences for each sample after applying all of the quality filters. To standardize the number of sequences per sample, each community should be rarefied to a common sequence depth (Aguirre de Cárcer et al., 2011; Fierer et al., 2011). In this study, each community was rarefied to 2,800 sequences before further analysis, at which sequence depth the sequence coverage averaged 87.2% determined by the good’s coverage: 1- f1/n, where f1 is the number of singletons (OTUs each represented by one sequence) and n denotes the sequence size (2,800 sequence here; Good, 1953). This observed coverage value suggests that the majority of OTUs had been captured. The rarefaction procedure was repeated 20 times on the sample-by-OTU table. The Bray–Curtis distance, due to its robustness (Faith et al., 1987), was then used to measure the dissimilarity based on the rarefied OTU table. The mean was taken over the 20 distance matrices and used for subsequent analysis. A principal coordinates analysis (PCoA) of the Bray–Curtis distance was performed to determine the change in the community structure. Three different non-parametric analysis methods, including analysis of similarities (ANOSIM), non-parametric multivariate analysis of variance using distance matrices (adonis), and a multi-response permutation procedure (MRPP), was used to examine whether there were significant differences in community structures among the treatments. The Bray–Curtis distance was used for the ANOSIM, adonis, and MRPP analyses. PCoA, ANOSIM, adonis, and MRPP were processed with the “vegan” package in R software version 3.1.2.
Results
Overall Taxonomic Distribution
In total, 210,423 valid sequence reads were recovered across all 36 samples (an average of 5,845 sequences per sample) after applying all of the quality filters (Supplementary Table S1). Given that ≥91.1% of sequence reads could be classified taxonomically at the family or higher taxonomic level, but only 49.4% of sequences could be assigned with a taxonomic identity at the genus level (Supplementary Table S1), the taxonomic distribution of the leaf endophytic bacterial community was then examined at the family level. The results indicated that the leaf endophytic bacterial community was primarily made up of nine families (Figure 1) covering five phyla/classes: Gammaproteobacteria, Firmicutes, Bacteroidetes, Actinobacteria, and Betaproteobacteria. Of the nine families, the most heavily sequenced populations were the Gammaproteobacteria-affiliated families. For example, the relative abundances of two of the Gammaproteobacteria-affiliated families, Enterobacteriaceae and Xanthomonadaceae, reached 28.7–86.8% and 2.14–42.6%, respectively, at all of the tested growth stages (Figure 1); another two Gammaproteobacteria-affiliated families, Moraxellaceae and Pseudomonadaceae, were also abundantly detected at both the tillering and the filling stages, with a relative abundance of 3.20–27.1% for Moraxellaceae and 1.80–11.4% for Pseudomonadaceae (Figure 1). The Firmicutes-affiliated family, Bacillaceae, was abundantly present at the tillering and maturity stages; the Bacteroidetes-affiliated families, Sphingobacteriaceae and Flavobacteriaceae, were abundant at the filling stage; and the Microbacteriaceae, belonging to Actinobacteria, was abundantly found at the maturity stage (Figure 1).
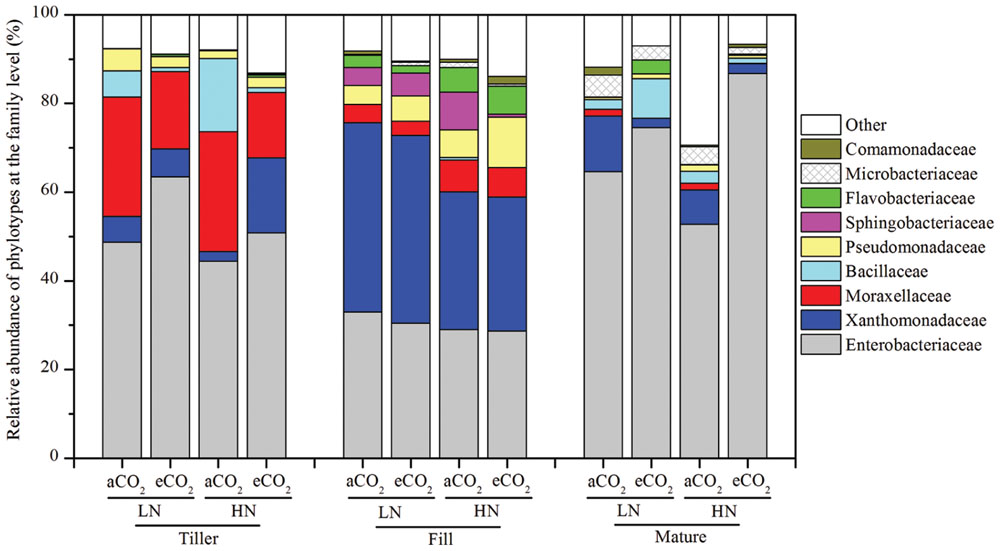
FIGURE 1. The relative abundance of the leaf endophytic bacterial phylotypes at the family level. The designations Tiller, Fill, and Mature indicate that the rice leaves were sampled at the tillering, filling, and maturity stages, respectively. The aCO2 and eCO2 represent the treatments of ambient and elevated CO2, respectively. The LN and HN refer to low and high levels of nitrogen fertilization, respectively. The statistical analysis results are the same as those in Figure 4.
Bacterial Diversity
The observed OTUs (Figure 2A) and the Shannon index (Figure 2B) was used to compare the bacterial richness and diversity, respectively, based on subsampled same number of sequences (2,800 sequences per sample, at which sequence depth the majority of the OTUs have been captured based on the mean sequence coverage (87.2%), which was determined by the good’s coverage). We did not observe consistent changes that could be attributed to eCO2, N fertilization, or different growth stages. For example, eCO2 significantly (P < 0.05) increased the bacterial richness (Figure 2A) and diversity (Figure 2B) under high N fertilization (HN) at the tillering stage, whereas no significant effect was observed at this stage under the low N fertilization (LN) treatment and at other growth stages under the LN and HN treatments. Enhanced N fertilization significantly (P < 0.05) increased the bacterial richness (Figure 2A) and diversity (Figure 2B) under ambient CO2 treatment at the filling stage but it did not show significant influence under eCO2 condition at this stage. In addition, no significant differences between LN and HN were observed at the tillering or maturity stage.
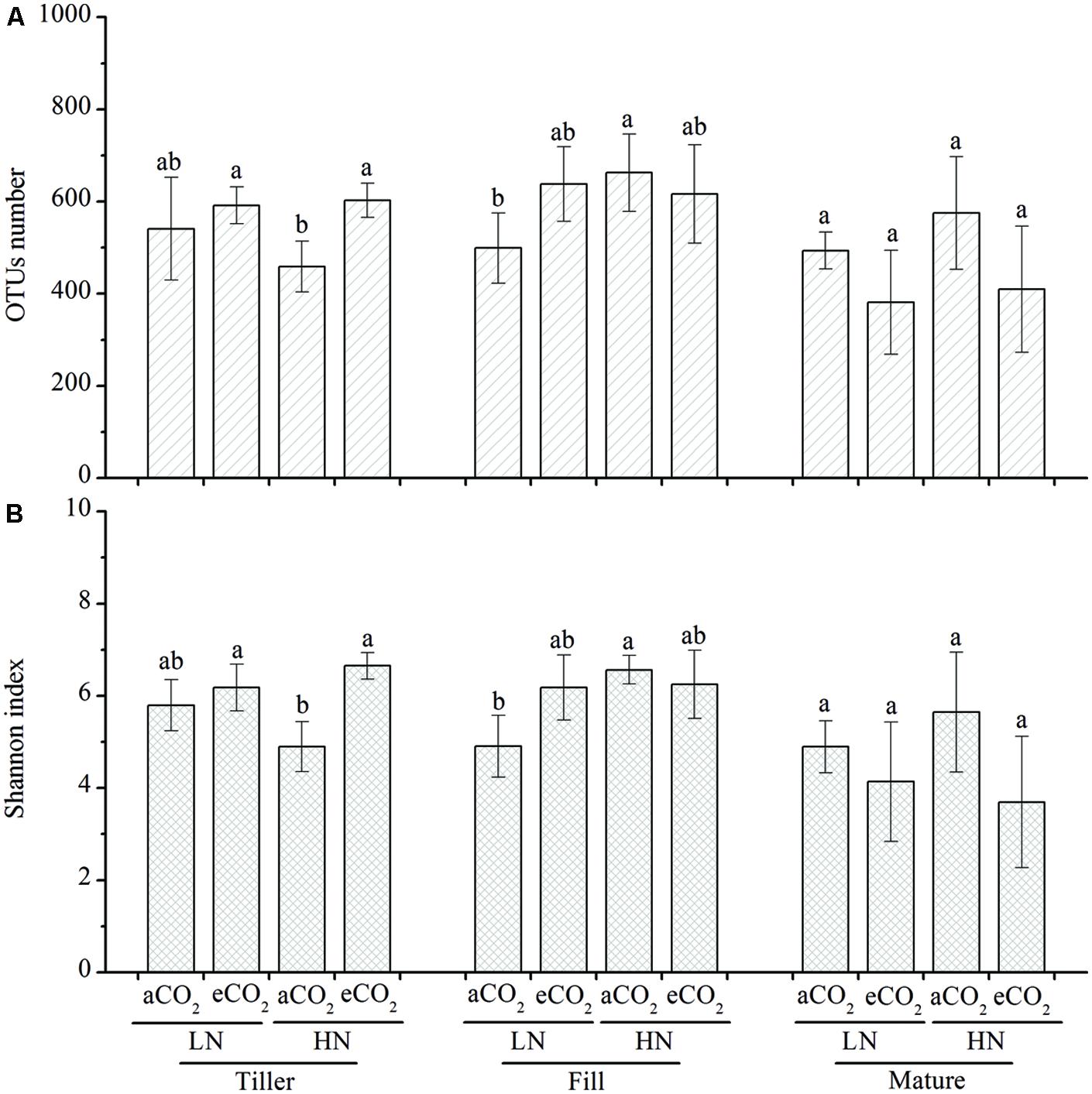
FIGURE 2. The OTUs number (A) and Shannon index (B). The OTUs number and Shannon index were obtained by using 2,800 subsampled sequence reads from each community. The different letters represent significant differences (P < 0.05; Duncan’s multiple range test). All other designations are the same as those in Figure 1.
Bacterial Community Structure
To examine the shift of the leaf endophytic bacterial community structures in response to eCO2 under different N fertilization levels at the tillering, filling, and maturity stages, a PCoA was performed based on the 454 pyrosequencing data (Figure 3). First, to understand how all of the samples were related to each other, a global PCoA was performed on the same ordination plot (Figure 3A). The results revealed that the axis separated the samples based on the growth stage, and the difference in bacterial communities from different treatment (CO2 and N treatments) was less than the difference between the different growth stages. Three non-parametric multivariate analyses (adonis, ANOSIM, and MRPP) also illustrated significant differences (P < 0.05) between the growth stages in the bacterial community structure (Table 1), whereas the significant difference was not always observed when the CO2 and N effects was tested by the three non-parametric multivariate analyses (Table 1). To further understand how bacterial communities from the same growth stage related to one another, the PCoA of the bacterial communities derived from the tillering, filling, and maturity stages were processed on three individual ordination plots (Figures 3B–D). The results showed that the magnitude of the eCO2 effect on the bacterial community structure varied greatly under different N fertilization levels and at different growth stages. The eCO2 showed a significant effect on the community structure under both LN and HN levels at the tillering stage. Specifically, at the tillering stage, the samples from aCO2 and eCO2 samples were distributed in different parts of the PCoA data space under both LN and HN levels, and adonis, ANOSIM, and MRPP further showed significant differences (P < 0.05) between bacterial communities at aCO2 and eCO2 (Table 1). However, a significant effect of eCO2 on the bacterial community structure was only observed under HN, rather than under the LN condition at the filling stage (Table 1). The PCoA result also revealed that the aCO2 and eCO2 samples under the HN level were distributed distantly in the data space at the filling stage, whereas those samples from aCO2 and eCO2 under the LN condition clustered closely (Figure 3C). At the maturity stage, no significant effect of eCO2 on the community structure under both LN and HN fertilization levels were observed (Table 1), although most of aCO2 and eCO2 samples were distributed in a different data space (Figure 3D).
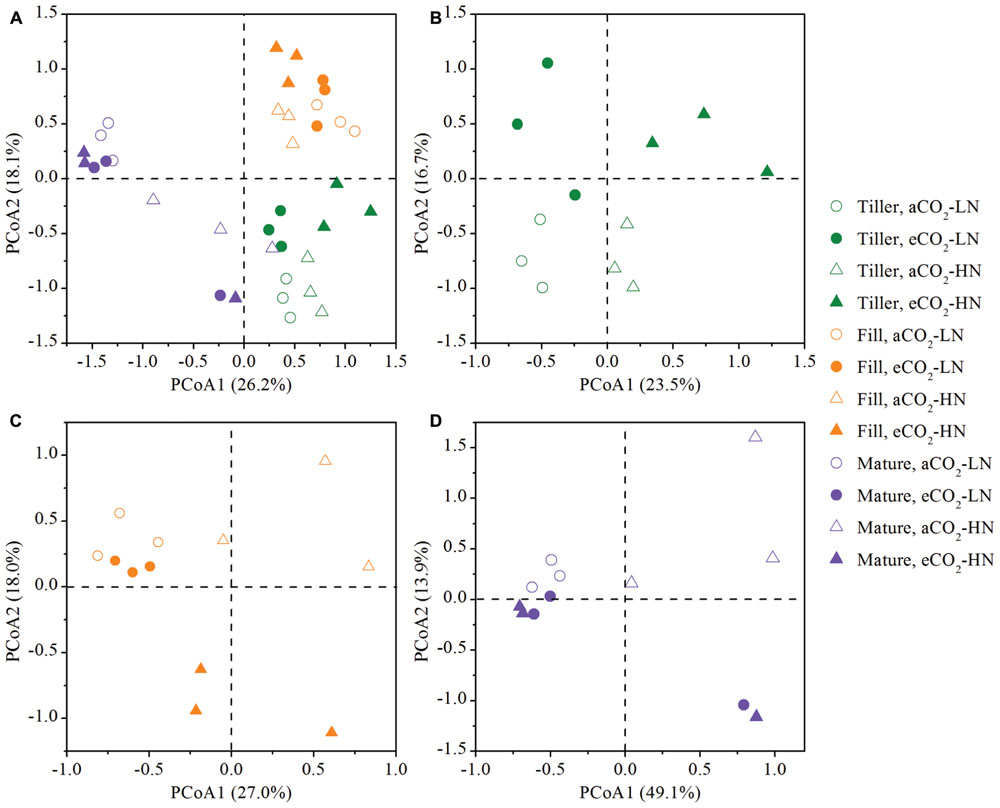
FIGURE 3. Principal coordinates analysis (PCoA) of microbial communities from all samples (A) or those samples collected at the tillering (B), filling (C), and maturity (D) stages, respectively. The percentage in parentheses denotes the proportion of variation explained by each ordination axis. All other designations are the same as those in Figure 1.
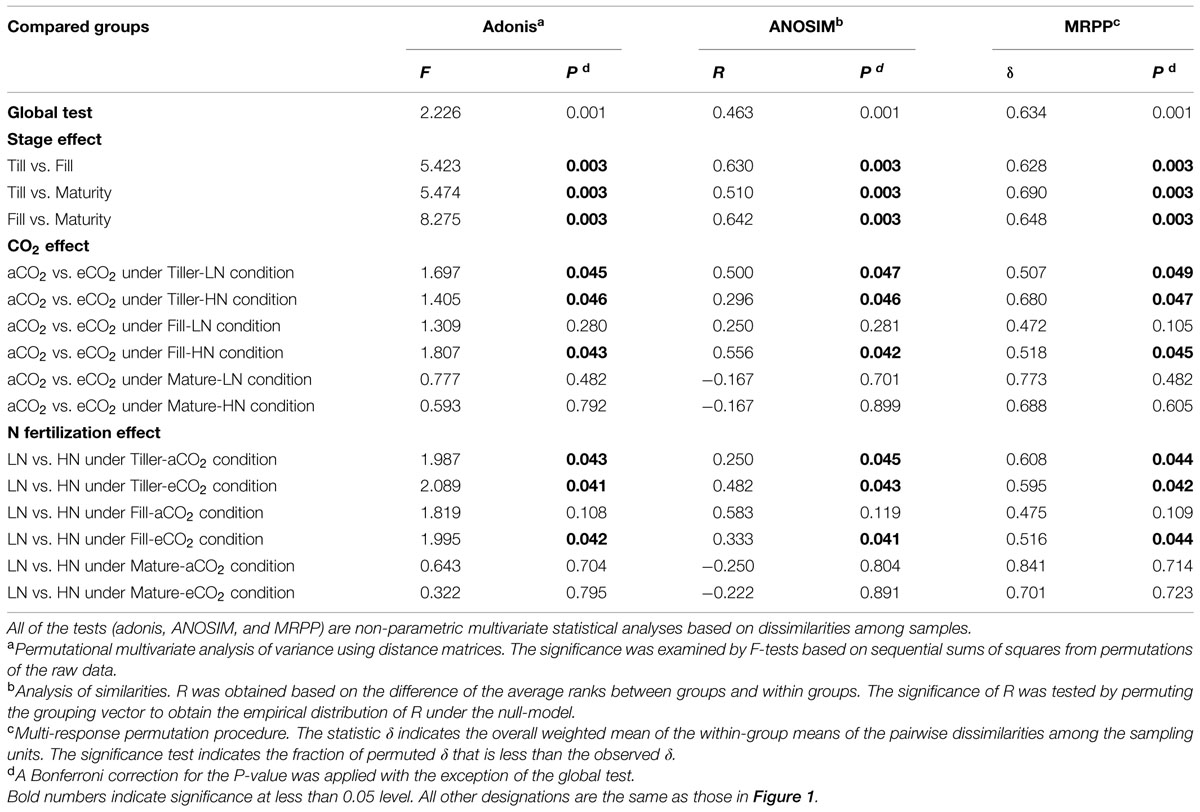
TABLE 1. Significance tests using three statistical approaches to assess the effects of growth stages, CO2, and N fertilization on the overall microbial community structure.
Specific Bacterial Populations in Different Treatments
Given that 91.1% of the sequences could be classified at the family level, but only 49.4% of sequences could be classified at the genus level (Supplementary Table S1), specific bacterial phylotypes were then analyzed at the family level. The relative abundance shifts of the main families in the different treatments are shown in Figure 1. The leaf samples at the same growth stage harbored a similar community composition and the endophytic bacterial communities sampled throughout the growth period were greatly different from one another. For instance, the family Enterobacteriaceae showed a great variations in the relative abundance at different growth stages: they comprised as high as 44.4–63.5% of the entire bacterial community at the tillering stage, and then were reduced to 28.7–33.0% at the filling stage and became dominant again (accounting for 52.5–86.8% of the total sequence reads) at the maturity stage (Figure 1). The family Xanthomonadaceae was abundantly found at the filling stage and comprised 30.2–42.6% of total bacterial community, whereas they had a relatively great lower relative abundance at the tillering and maturity stages, only making up of 2.18–16.8% and 2.14–12.5% of the total sequence reads, respectively (Figure 1). Similarly, the family Moraxellaceae showed a high relative abundance at the tillering stage (14.8–26.9%), whereas only 3.20–6.65% and 0–1.55% were found at the filling and maturity stages, respectively. The family Bacillaceae were abundant at the tillering (0.98–16.5%) and maturity (1.21–8.94%) stages but were rarely detected at the filling stage (0–0.58%). Additionally, the relative abundance of Pseudomonadaceae were exclusively high at the tillering (1.77–4.93%) and filling (4.29–11.4%) stages rather than at the maturity stage (only 0.53–1.42%). Another stage-specific phylotype was the family Sphingobacteriaceae, which was only abundantly found only at the filling stage (0.61–4.07%) rather than at the tillering (0–0.05%) and maturity (0–0.15%) stages. Likewise, the family Flavobacteriaceae was exclusively abundant at the filling and maturity stages and the two families, Microbacteriaceae and Comamonadaceae, were abundantly detected at the filling and maturity stages. These results revealed a stage-specific bacterial community composition of the rice leaves.
The response of the leaf endophytic bacterial taxa to eCO2 under different N fertilization levels was examined. To examine which bacterial taxa responded to eCO2 and how they responded, the net difference in relative abundance of each family between eCO2 and aCO2 was determined to assess the shift of leaf endophytic bacteria in response to eCO2 (Figure 4). Overall, within those nine main bacterial populations, six families, Enterobacteriaceae, Xanthomonadaceae, Moraxellaceae, Sphingobacteriaceae, Microbacteriaceae, and Comamonadaceae, showed significant differences in relative abundance between the aCO2 and eCO2 treatments under the LN or HN condition at one of the tested growth stages (Figure 4). Of them, the family Moraxellaceae was significantly (P < 0.05) decreased in relative abundance due to the eCO2 treatment under the LN condition at the tillering stage (Figure 4A) and under both LN and HN conditions at the maturity stage (Figures 4E,F). The relative abundance of this phylotype also showed a slightly non-significant decrease by eCO2 under both the LN and HN conditions at the filling stage (Figures 4C,D). As for the other five significantly affected families, the changing direction of the relative abundance for a specific phylotype was not consistent (i.e., increase or decrease) in response to eCO2 at different growth stages and under different N fertilization conditions. For example, the most dominant and shared phylotype, Enterobacteriaceae, was significantly increased (P < 0.05) by eCO2 in terms of the relative abundance under the LN condition at the tillering stage (Figure 4A) and under the HN condition at the maturity stage (Figure 4F), whereas a slight decrease was observed at the filling stage (Figures 4C,D). The family Xanthomonadaceae significantly increased in relative abundance under the HN condition at the tillering stage (Figure 4A) whereas a prominent decrease was found under both the LN and HN conditions at the maturity stage (Figures 1 and 4E,F). Similarly, when exposed to eCO2, no consistent changing direction was found for these three families Sphingobacteriaceae, Microbacteriaceae, and Comamonadaceae along the growth stages (Figure 4).
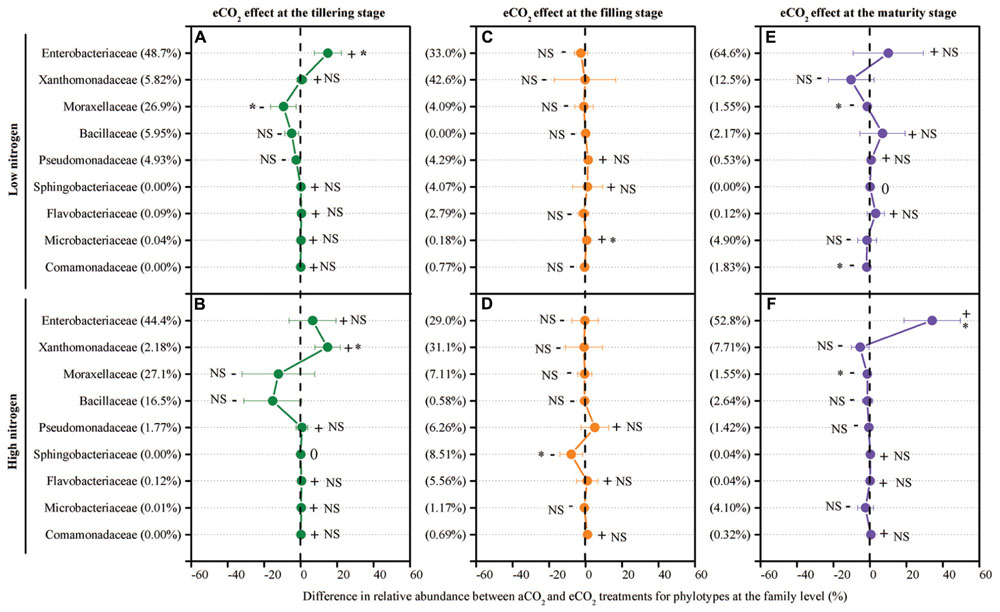
FIGURE 4. The effect of eCO2 on the relative abundance of leaf endophytic bacterial phylotypes at the family level at the tillering (A,B), filling (C,D), and maturity (E,F) stages. The eCO2 effect was examined using the net difference in the relative abundance of bacterial taxa between the eCO2 and aCO2. The net difference in the relative abundance was calculated as the relative abundance under eCO2 minus the relative abundance of the phylotype under aCO2 at each N treatment level. The percentage value in the bracket refers to the relative abundance at aCO2. The error bar denotes the standard error of the mean. The phylotypes are presented in generally descending order based on their relative abundance in the aCO2 control treatment. The symbols “+”, “-”, and “0” indicate that the relative abundance was increased, decreased, or stable compared with the aCO2 control treatment. The symbol “∗” represents significant differences at P < 0.05, and NS represents no significant difference (P > 0.05). All other designations are the same as those in Figure 1.
Discussion
The plant endophytic bacteria from different parts of the rice plant such as roots, stems, leaves, and seeds have been partially characterized by culture-dependent approach, clone library, T-RFLP, and metagenome methods. This study made an in-depth (an average of 5,845 sequences per samples) exploration of the leaf endophytic bacterial community using 454 pyrosequencing. Gammaproteobacteria-affiliated families, members of which have been reported to play a critical role in plant growth (Taghavi et al., 2009; Kim et al., 2012; Witzel et al., 2012), were the most heavily sequenced phylotypes in our study. The predominance of Gammaproteobacteria in the rice leaf endosphere was similar with a reported culture-dependent study showing that members of Gammaproteobacteria, Pantoea sp. and Pseudomonas sp. made up 51% of the entire leaf endophytic bacterial communities in three detected rice varieties (Ferrando et al., 2012). Another study also found the dominance of the Gammaproteobacteria-affiliated genus, Pantoea, in the leaf endosphere of the rice plant (Loaces et al., 2011). In the same paddy field, our previous study has revealed the prevalence of Gammaproteobacteria on the leaf surface (Ren et al., 2014). These two studies indicate that, albeit in different proportions, Gammaproteobacteria commonly and abundantly colonized both the leaf surface and the leaf endosphere of the rice plant. The finding that bacteria taxa were in both the internal and external parts of the rice plant, which was a finding similar to previous studies (Mano et al., 2006, 2007), suggests that the colonizer may come from a similar source. Plant endophytic bacteria have been considered to originate from the external environment and enter the plant through the stomata, lenticles, wounds, areas of emergence of lateral roots and germinating radicles (Mano and Morisaki, 2008). First, the internal bacteria of the leaf in our study may come from rain splashing off the soil since the rice leaves were close to the ground when the rice plant was young. As the plant grows, the bacteria could colonize the expanding leaves. Conversely, the wind and rain, thought to be a source of bacteria residing on the leaf surface (Bodenhausen et al., 2013), could also bring bacteria to the soil and hence to the plant endosphere as the plant grows. Second, the leaf external bacteria could also enter the internal tissue of leaves as endophytes. This may also be an explanation for the common colonization of both the leaf surface and the leaf endosphere. A third explanation is that those bacteria in the seeds or soil may enter the aboveground part including expanding leaves as the plant grows and develops through infecting seeds, the seedling after germination, or the emergence of lateral roots.
In this study, the strongest differences in the bacterial community composition and structure were observed on rice plants of different developmental stages. Conspicuously different microbial community structure in different growth periods was also observed in other plant species such as sugar beets (Beta vulgaris L.; Shi et al., 2014) and weeds (Stellera chamaejasme L.; Jin et al., 2013). Additionally, in our study, the leaves sampled in the same growth period had similar endophytic community compositions, whereas some bacterial populations were only abundantly detected at specific growth stages. Hence, it is hypothesized that the composition and distribution of leaf endophytic bacterial communities across different growth stages were not random, as was the case in leaf epiphytic bacterial communities along the growth period (Redford and Fierer, 2009). The observed temporal variability in the structure and composition of bacterial communities may be explained, in part, by variations in abiotic factors, such as climate conditions, although we cannot state exactly that the shift of a certain bacterial population is caused by specific climate factors. For instance, between the highest and the lowest average daily temperature, the difference was as high as 22.1°C (31.2, 24.1, and 9.1°C at the tillering, filling, and maturity sampling time point, respectively), and the solar radiation also varied greatly between three sampling time points (12.11, 13.86, and 5.14 MJ m-2 day-1, respectively). To some extent, our speculation was supported by previous studies that showed that the composition and fluctuation of endophytic bacterial communities from grapevine leaves was closely related to seasonality (Bulgari et al., 2014). Distinctly seasonal changes of leaf-associated bacterial communities have also been observed on trees (Magnolia grandiflora; Jackson and Denney, 2011). Moreover, the growth stage of plants not only includes changes in climate (water, temperature, and ultraviolet radiation) but also in plant host attributes (photosynthesis, respiration, stomatal conductance, etc.) and hence resource supplies (water, nutrients). Therefore, the temporal variability observed in this study in the structure and composition of leaf endophytic bacterial communities can be the result of a feedback to the host growth stage per se and to season (Peñuelas et al., 2012) or to both. The observation that several unique bacterial taxa were exceptionally abundant at specific stages may be due to the adaptive selection driven by changes in host physiology and by variations in the seasonal environment (Ellis et al., 1999; Hartmann et al., 2009). Further work is needed to identify the driving factors that led to the observed temporal shifts in the stage-specific bacterial populations, to test whether the seasonal pattern was repeatable from year to year, and whether the structure and composition of the leaf endophytic bacterial communities could follow a predictable successional pattern. In particular, to differentiate the contribution of the seasonal effect from the host physiology, it would be valuable to conduct a metabolomics study to investigate rice metabolites at different growth stages and determine their correlation to growth-stage related changes of the endophytes. Alternatively, conducting a controlled parallel experiment (e.g., using a greenhouse to make the plant grow under controlled environmental conditions) to the field experiment would also be helpful to sort out the confounding factors.
The eCO2 could also be an important factor leading to the shift in the leaf endophytic bacterial communities. For example, the significant influence of eCO2 on the community structure was observed under both the LN and HN levels at the tillering stage. At the specific bacterial population level, the relative abundance of the families Enterobacteriaceae, Xanthomonadaceae, Moraxellaceae, Sphingobacteriaceae, Microbacteriaceae, and Comamonadaceae was significantly affected by eCO2 under the LN or HN condition at one of the tested growth stages. We also observed a significant eCO2 effect on some of the abundant phylotypes at a finer taxonomic level, i.e., genus level (Supplementary Figures S1 and S2). Given that the primers are not generally available for each bacteria taxon, the relative abundance (instead of the absolute quantitative data of each bacterial population) was used in this study to elucidate the response patterns of each bacterial phylotype. The importance of eCO2 in structuring the microbial community in soil has been demonstrated in various ecosystems (Montealegre et al., 2000; Drissner et al., 2007; He et al., 2010). Here, we provided evidence that eCO2 could also influence the structure and the composition of plant-associated bacterial communities such as the leaf endophytic bacterial communities.
The eCO2 effect varied greatly under different N fertilization conditions and at the different growth stages. For example, at the tillering stage, we observed a significant influence of eCO2 on the community structure regardless of the N fertilization levels, suggesting a nitrogen-independent response, but a different case was found at the filling stage when the significant impact of eCO2 was observed under enhanced N fertilization rather than under the LN condition. This suggests a nitrogen fertilization-dependent response to eCO2 at the filling stage. Therefore, our results provided evidence for the necessity of incorporating the N fertilization practice and plant growth period when evaluating the biosphere response to elevated CO2 in the context of climate change. Previous studies of the impact of eCO2 on the structure of soil microbial communities have shown that the structure of the soil microbial communities were either stable (Austin et al., 2009; Ge et al., 2010) or changed under the eCO2 condition in various ecosystems (Montealegre et al., 2000; Drissner et al., 2007; He et al., 2010). The influence of eCO2 on microbial communities in soil could depend on various factors, and sampling time and N level should be considered as two of the most important factors in these studies.
A previous rice-FACE study also showed that the N level influenced the eCO2 effect on the leaf-associated bacterial community structure (Ikeda et al., 2015), which was similar to our finding that the response to eCO2 could be nitrogen-dependent at a certain growth stage. In contrast to our results, Ikeda et al. (2015) found that the most prominent effect of eCO2 was observed under a no N fertilization condition rather than under applied N fertilization (8 g m-2). In our study, a significant impact of eCO2 was observed under enhanced N fertilization (25 g m-2) rather than under the LN condition. The difference in growth period may be one of the reasons that lead to these discrepancies (Bulgari et al., 2014). In the Ikeda et al. (2015) study, the research was conducted when the rice plant was at the panicle initiation stage, whereas the significant eCO2 effect was observed at the tilling and filling stages in our study. Additionally, there was a difference in the rice varieties used in the Ikeda et al. (2015; Oryza sativa L. cv. Koshihikari) study and our study (‘Wuxiangjing 14’ japonica rice cultivar), which may also be one of the causal factors (Whipps et al., 2008; Müller et al., 2015).
In summary, culture-independent analysis using 454 pyrosequencing indicated that rice leaves harbored diverse endophytic bacterial phylotypes. The Gammaproteobacteria, which have been reported to play a critical role in plant growth, were found to be the most dominant population in this study. The most evident difference in the community structure and composition were observed on plants of different growth stages. Additionally, eCO2 changed the community structure at certain time points (tillering stage) or under certain N application conditions (HN at the filling stage). The alteration of the composition and structure of leaf endophytic bacterial communities is highly likely to have functional consequences given the versatile functions of plant endophytic bacteria. Further studies should be focused on exploring and testifying the potential roles, such as the functions that are relevant to global N cycling, plant health, and plant growth promotion, of particular phylotypes (e.g., Gammaproteobacteria affiliated families).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Strategic Priority Research Program of the CAS (XDB15040000), the National Science Foundation of China (41090281), and the Distinguished Young Scholar Programme of Jiangsu Province (BK2012048). We gratefully acknowledge Dr. Juan Zhou and Prof. Lianxing Yang from Yangzhou University, and Chen Yan, Jing Xu, and Wanmeng Wang in our lab for sampling assistance. We thank Drs Haoye Tang and Qing Zeng for the meteorological information. We also thank Zhiying Guo in our lab for his help in data analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.00855
References
Adhikari, T. B., Joseph, C. M., Yang, G., Phillips, D. A., and Nelson, L. M. (2001). Evaluation of bacteria isolated from rice for plant growth promotion and biological control of seedling disease of rice. Can. J. Microbiol. 47, 916–924. doi: 10.1139/w01-097
Aguirre de Cárcer, D., Denman, S. E., Mcsweeney, C., and Morrison, M. (2011). Evaluation of subsampling-based normalization strategies for tagged high-throughput sequencing data sets from gut microbiomes. Appl. Environ. Microbiol. 77, 8795–8798. doi: 10.1128/aem.05491-11
Ainsworth, E. A., Davey, P. A., Bernacchi, C. J., Dermody, O. C., Heaton, E. A., Moore, D. J., et al. (2002). A meta-analysis of elevated [CO2] effects on soybean (Glycine max) physiology, growth and yield. Glob. Change Biol. 8, 695–709. doi: 10.1046/j.1365-2486.2002.00498.x
Ainsworth, E. A., and Long, S. P. (2005). What have we learned from 15 years of free-air CO2 enrichment (FACE)? A meta-analytic review of the responses of photosynthesis, canopy properties and plant production to rising CO2. New Phytol. 165, 351–372. doi: 10.1111/j.1469-8137.2004.01224.x
Amann, R. I., Ludwig, W., and Schleifer, K. H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143–169.
Anten, N. P. R., Hirose, T., Onoda, Y., Kinugasa, T., Kim, H. Y., Okada, M., et al. (2004). Elevated CO2 and nitrogen availability have interactive effects on canopy carbon gain in rice. New Phytol. 161, 459–471. doi: 10.1046/j.1469-8137.2003.00943.x
Araújo, W. L., Marcon, J., Maccheroni, W., Van Elsas, J. D., Van Vuurde, J. W. L., and Azevedo, J. L. (2002). Diversity of endophytic bacterial populations and their interaction with Xylella fastidiosa in citrus plants. Appl. Environ. Microbiol. 68, 4906–4914. doi: 10.1128/aem.68.10.4906-4914.2002
Austin, E. E., Castro, H. F., Sides, K. E., Schadt, C. W., and Classen, A. T. (2009). Assessment of 10 years of CO2 fumigation on soil microbial communities and function in a sweetgum plantation. Soil Biol. Biochem. 41, 514–520. doi: 10.1016/j.soilbio.2008.12.010
Belote, R. T., Weltzin, J. F., and Norby, R. J. (2004). Response of an understory plant community to elevated [CO2] depends on differential responses of dominant invasive species and is mediated by soil water availability. New Phytol. 161, 827–835. doi: 10.1111/j.1469-8137.2004.00977.x
Bengtsson, J., Eriksson, K. M., Hartmann, M., Wang, Z., Shenoy, B. D., Grelet, G. A., et al. (2011). Metaxa: a software tool for automated detection and discrimination among ribosomal small subunit (12S/16S/18S) sequences of archaea, bacteria, eukaryotes, mitochondria, and chloroplasts in metagenomes and environmental sequencing datasets. Antonie Van Leeuwenhoek 100, 471–475. doi: 10.1007/s10482-011-9598-6
Bernacchi, C. J., Calfapietra, C., Davey, P. A., Wittig, V. E., Scarascia-Mugnozza, G. E., Raines, C. A., et al. (2003). Photosynthesis and stomatal conductance responses of poplars to free-air CO2 enrichment (PopFACE) during the first growth cycle and immediately following coppice. New Phytol. 159, 609–621. doi: 10.1046/j.1469-8137.2003.00850.x
Bodenhausen, N., Horton, M. W., and Bergelson, J. (2013). Bacterial communities associated with the leaves and the roots of Arabidopsis thaliana. PLoS ONE 8:e56329. doi: 10.1371/journal.pone.0056329
Bulgari, D., Casati, P., Quaglino, F., and Bianco, P. A. (2014). Endophytic bacterial community of grapevine leaves influenced by sampling date and phytoplasma infection process. BMC Microbiol. 14:198. doi: 10.1186/1471-2180-14-198
Ding, T., Palmer, M. W., and Melcher, U. (2013). Community terminal restriction fragment length polymorphisms reveal insights into the diversity and dynamics of leaf endophytic bacteria. BMC Microbiol. 13:1. doi: 10.1186/1471-2180-13-1
Drissner, D., Blum, H., Tscherko, D., and Kandeler, E. (2007). Nine years of enriched CO2 changes the function and structural diversity of soil microorganisms in a grassland. Eur. J. Soil Sci. 58, 260–269. doi: 10.1111/j.1365-2389.2006.00838.x
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Ellis, R. J., Thompson, I. P., and Bailey, M. J. (1999). Temporal fluctuations in the pseudomonad population associated with sugar beet leaves. FEMS Microbiol. Ecol. 28, 345–356. doi: 10.1111/j.1574-6941.1999.tb00589.x
Faith, D. P., Minchin, P. R., and Belbin, L. (1987). Compositional dissimilarity as a robust measure of ecological distance. Vegetatio 69, 57–68. doi: 10.1007/BF00038687
Ferrando, L., Manay, J. F., and Scavino, A. F. (2012). Molecular and culture-dependent analyses revealed similarities in the endophytic bacterial community composition of leaves from three rice (Oryza sativa) varieties. FEMS Microbiol. Ecol. 80, 696–708. doi: 10.1111/j.1574-6941.2012.01339.x
Fierer, N., Mccain, C. M., Meir, P., Zimmermann, M., Rapp, J. M., Silman, M. R., et al. (2011). Microbes do not follow the elevational diversity patterns of plants and animals. Ecology 92, 797–804. doi: 10.1890/10-1170.1
Garbeva, P., Van Overbeek, L., Van Vuurde, J., and Van Elsas, J. (2001). Analysis of endophytic bacterial communities of potato by plating and denaturing gradient gel electrophoresis (DGGE) of 16S rDNA based PCR fragments. Microb. Ecol. 41, 369–383. doi: 10.1007/s002480000096
Ge, Y., Chen, C. R., Xu, Z. H., Oren, R., and He, J. Z. (2010). The spatial factor, rather than elevated CO2, controls the soil bacterial community in a temperate forest ecosystem. Appl. Environ. Microbiol. 76, 7429–7436. doi: 10.1128/aem.00831-10
Good, I. J. (1953). The population frequencies of species and the estimation of population parameters. Biometrika 40, 237–264. doi: 10.1093/biomet/40.3-4.237
Gyaneshwar, P., James, E. K., Mathan, N., Reddy, P. M., Reinhold-Hurek, B., and Ladha, J. K. (2001). Endophytic colonization of rice by a diazotrophic strain of Serratia marcescens. J. Bacteriol. 183, 2634–2645. doi: 10.1128/jb.183.8.2634-2645.2001
Hardoim, P. R., Andreote, F. D., Reinhold-Hurek, B., Sessitsch, A., Van Overbeek, L. S., and Van Elsas, J. D. (2011). Rice root-associated bacteria: insights into community structures across 10 cultivars. FEMS Microbiol. Ecol. 77, 154–164. doi: 10.1111/j.1574-6941.2011.01092.x
Hartmann, A., Schmid, M., Tuinen, D., and Berg, G. (2009). Plant-driven selection of microbes. Plant Soil 321, 235–257. doi: 10.1007/s11104-008-9814-y
Hartmann, M., Howes, C. G., Abarenkov, K., Mohn, W. W., and Nilsson, R. H. (2010). V-Xtractor: an open-source, high-throughput software tool to identify and extract hypervariable regions of small subunit (16 S/18 S) ribosomal RNA gene sequences. J. Microbiol. Methods 83, 250–253. doi: 10.1016/j.mimet.2010.08.008
He, Z. L., Xu, M. Y., Deng, Y., Kang, S., Kellogg, L., Wu, L. Y., et al. (2010). Metagenomic analysis reveals a marked divergence in the structure of belowground microbial communities at elevated CO2. Ecol. Lett. 13, 564–575. doi: 10.1111/j.1461-0248.2010.01453.x
Hollister, E. B., Engledow, A. S., Hammett, A. J. M., Provin, T. L., Wilkinson, H. H., and Gentry, T. J. (2010). Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments. ISME J. 4, 829–838. doi: 10.1038/ismej.2010.3
Huse, S. M., Dethlefsen, L., Huber, J. A., Welch, D. M., Relman, D. A., and Sogin, M. L. (2008). Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 4:e1000255. doi: 10.1371/journal.pgen.1000255
Idso, K. E., and Idso, S. B. (1994). Plant responses to atmospheric CO2 enrichment in the face of environmental constraints: a review of the past 10 years’ research. Agric. For. Meteorol. 69, 153–203. doi: 10.1016/0168-1923(94)90025-6
Ikeda, S., Tokida, T., Nakamura, H., Sakai, H., Usui, Y., Okubo, T., et al. (2015). Characterization of leaf blade- and leaf sheath-associated bacterial communities and assessment of their responses to environmental changes in CO2, temperature, and nitrogen levels under field conditions. Microbes Environ. 30, 51–62. doi: 10.1264/jsme2.ME14117
Jackson, C., and Denney, W. (2011). Annual and seasonal variation in the phyllosphere bacterial community associated with leaves of the Southern Magnolia (Magnolia grandiflora). Microb. Ecol. 61, 113–122. doi: 10.1007/s00248-010-9742-2
Jackson, C. R., Randolph, K. C., Osborn, S. L., and Tyler, H. L. (2013). Culture dependent and independent analysis of bacterial communities associated with commercial salad leaf vegetables. BMC Microbiol. 13:274. doi: 10.1186/1471-2180-13-274
Ji, S. H., Gururani, M. A., and Chun, S. C. (2014). Isolation and characterization of plant growth promoting endophytic diazotrophic bacteria from Korean rice cultivars. Microbiol. Res. 169, 83–98. doi: 10.1016/j.micres.2013.06.003
Jin, H., Yan, Z. Q., Liu, Q., Yang, X. Y., Chen, J. X., and Qin, B. (2013). Diversity and dynamics of fungal endophytes in leaves, stems and roots of Stellera chamaejasme L. in northwestern China. Antonie Van Leeuwenhoek 104, 949–963. doi: 10.1007/s10482-013-0014-2
Jin, H., Yang, X. Y., Yan, Z. Q., Liu, Q., Li, X. Z., Chen, J. X., et al. (2014). Characterization of rhizosphere and endophytic bacterial communities from leaves, stems and roots of medicinal Stellera chamaejasme L. Syst. Appl. Microbiol. 37, 376–385. doi: 10.1016/j.syapm.2014.05.001
Khan, A. L., Waqas, M., Kang, S. M., Al-Harrasi, A., Hussain, J., Al-Rawahi, A., et al. (2014). Bacterial endophyte Sphingomonas sp. LK11 produces gibberellins and IAA and promotes tomato plant growth. J. Microbiol. 52, 689–695. doi: 10.1007/s12275-014-4002-7
Kim, H. J., Lee, J. H., Kang, B. R., Rong, X., Gardener, B. B. M., Ji, H. J., et al. (2012). Draft genome sequence of Pantoea ananatis B1-9, a nonpathogenic plant growth-promoting bacterium. J. Bacteriol. 194, 729. doi: 10.1128/jb.06484-11
Kim, H. Y., Lieffering, M., Miura, S., Kobayashi, K., and Okada, M. (2001). Growth and nitrogen uptake of CO2-enriched rice under field conditions. New Phytol. 150, 223–229. doi: 10.1046/j.1469-8137.2001.00111.x
Knoth, J. L., Kim, S. H., Ettl, G. J., and Doty, S. L. (2013). Effects of cross host species inoculation of nitrogen-fixing endophytes on growth and leaf physiology of maize. GCB Bioenergy 5, 408–418. doi: 10.1111/gcbb.12006
Leadley, P. W., Niklaus, P. A., Stocker, R., and Körner, C. (1999). A field study of the effects of elevated CO2 on plant biomass and community structure in a calcareous grassland. Oecologia 118, 39–49. doi: 10.1007/s004420050701
Liu, G., Han, Y., Zhu, J. G., Okada, M., Nakamura, H., and Yoshimoto, M. (2002). Rice-wheat rotational FACE platform. I. System structure and control. Chin. J. Appl. Ecol. 13, 1253–1258.
Loaces, I., Ferrando, L., and Fernández Scavino, A. (2011). Dynamics, diversity and function of endophytic siderophore-producing bacteria in rice. Microb. Ecol. 61, 606–618. doi: 10.1007/s00248-010-9780-9
Madhaiyan, M., Peng, N., and Ji, L. H. (2013). Complete genome sequence of Enterobacter sp. strain R4-368, an endophytic N-fixing Gammaproteobacterium isolated from surface-sterilized roots of Jatropha curcas L. Genome Announc. 1, e544–e513. doi: 10.1128/genomeA.00544-13
Mano, H., and Morisaki, H. (2008). Endophytic bacteria in the rice plant. Microbes Environ. 23, 109–117. doi: 10.1264/jsme2.23.109
Mano, H., Tanaka, F., Nakamura, C., Kaga, H., and Morisaki, H. (2007). Culturable endophytic bacterial flora of the maturing leaves and roots of rice plants (Oryza sativa) cultivated in a paddy field. Microbes Environ. 22, 175–185. doi: 10.1264/jsme2.22.175
Mano, H., Tanaka, F., Watanabe, A., Kaga, H., Okunishi, S., and Morisaki, H. (2006). Culturable surface and endophytic bacterial flora of the maturing seeds of rice plants (Oryza sativa) cultivated in a paddy field. Microbes Environ. 21, 86–100. doi: 10.1264/jsme2.21.86
Manter, D., Delgado, J., Holm, D., and Stong, R. (2010). Pyrosequencing reveals a highly diverse and cultivar-specific bacterial endophyte community in potato roots. Microb. Ecol. 60, 157–166. doi: 10.1007/s00248-010-9658-x
Montealegre, C. M., Van Kessel, C., Blumenthal, J. M., Hur, H. G., Hartwig, U. A., and Sadowsky, M. J. (2000). Elevated atmospheric CO2 alters microbial population structure in a pasture ecosystem. Glob. Change Biol. 6, 475–482. doi: 10.1046/j.1365-2486.2000.00326.x
Müller, H., Berg, C., Landa, B. B., Auerbach, A., Moissl-Eichinger, C., and Berg, G. (2015). Plant genotype-specific archaeal and bacterial endophytes but similar Bacillus antagonists colonize Mediterranean olive trees. Front. Microbiol. 6:138. doi: 10.3389/fmicb.2015.00138
Muthukumarasamy, R., Kang, U. G., Park, K. D., Jeon, W. T., Park, C. Y., Cho, Y. S., et al. (2007). Enumeration, isolation and identification of diazotrophs from Korean wetland rice varieties grown with long-term application of N and compost and their short-term inoculation effect on rice plants. J. Appl. Microbiol. 102, 981–991. doi: 10.1111/j.1365-2672.2006.03157.x
Okada, M., Lieffering, M., Nakamura, H., Yoshimoto, M., Kim, H. Y., and Kobayashi, K. (2001). Free-air CO2 enrichment (FACE) using pure CO2 injection: system description. New Phytol. 150, 251–260. doi: 10.1046/j.1469-8137.2001.00097.x
Partida-Martínez, L. P., and Heil, M. (2011). The microbe-free plant: fact or artifact ? Front. Plant Sci. 2:100. doi: 10.3389/fpls.2011.00100
Peñuelas, J., Rico, L., Ogaya, R., Jump, A. S., and Terradas, J. (2012). Summer season and long-term drought increase the richness of bacteria and fungi in the foliar phyllosphere of Quercus ilex in a mixed Mediterranean forest. Plant Biol. 14, 565–575. doi: 10.1111/j.1438-8677.2011.00532.x
Quecine, M. C., Araujo, W. L., Rossetto, P. B., Ferreira, A., Tsui, S., Lacava, P. T., et al. (2012). Sugarcane growth promotion by the endophytic bacterium Pantoea agglomerans 33.1. Appl. Environ. Microbiol. 78, 7511–7518. doi: 10.1128/aem.00836-12
Redford, A. J., and Fierer, N. (2009). Bacterial succession on the leaf surface: a novel system for studying successional dynamics. Microb. Ecol. 58, 189–198. doi: 10.1007/s00248-009-9495-y
Reich, P. B., and Hobbie, S. E. (2013). Decade-long soil nitrogen constraint on the CO2 fertilization of plant biomass. Nat. Clim. Chang. 3, 278–282. doi: 10.1038/nclimate1694
Reich, P. B., Hobbie, S. E., Lee, T., Ellsworth, D. S., West, J. B., Tilman, D., et al. (2006). Nitrogen limitation constrains sustainability of ecosystem response to CO2. Nature 440, 922–925. doi: 10.1038/nature04486
Reiter, B., Pfeifer, U., Schwab, H., and Sessitsch, A. (2002). Response of endophytic bacterial communities in potato plants to infection with Erwinia carotovora subsp. atroseptica. Appl. Environ. Microbiol. 68, 2261–2268. doi: 10.1128/aem.68.5.2261-2268.2002
Ren, G. D., Zhang, H. Y., Lin, X. G., Zhu, J. G., and Jia, Z. J. (2014). Response of phyllosphere bacterial communities to elevated CO2 during rice growing season. Appl. Microbiol. Biotechnol. 98, 9459–9471. doi: 10.1007/s00253-014-5915-0
Ren, G. D., Zhu, C. W., Alam, M. S., Tokida, T., Sakai, H., Nakamura, H., et al. (2015). Response of soil, leaf endosphere and phyllosphere bacterial communities to elevated CO2 and soil temperature in a rice paddy. Plant Soil 392, 27–44. doi: 10.1007/s11104-015-2503-8
Rosenzweig, C., Casassa, G., Karoly, D. J., Imeson, A., Liu, C., Menzel, A., et al. (2007). “Assessment of observed changes and responses in natural and managed systems,” in Climate Change 2007: Impacts, Adaptation and Vulnerability. Contribution of Working Group II to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, eds M. L. Parry, O. F. Canziani, J. P. Palutikof, P. J. Van Der Linden, and C. E. Hanson (Cambridge: Cambridge University Press), 79–131.
Sessitsch, A., Reiter, B., and Berg, G. (2004). Endophytic bacterial communities of field-grown potato plants and their plant-growth-promoting and antagonistic abilities. Can. J. Microbiol. 50, 239–249. doi: 10.1139/w03-118
Shi, Y. W., Yang, H. M., Zhang, T., Sun, J., and Lou, K. (2014). Illumina-based analysis of endophytic bacterial diversity and space-time dynamics in sugar beet on the north slope of Tianshan mountain. Appl. Microbiol. Biotechnol. 98, 6375–6385. doi: 10.1007/s00253-014-5720-9
Singh, R., Mishra, R. N., Jaiswal, H., Kumar, V., Pandey, S., Rao, S., et al. (2006). Isolation and identification of natural endophytic rhizobia from rice (Oryza sativa L.) through rDNA PCR-RFLP and sequence analysis. Curr. Microbiol. 52, 345–349. doi: 10.1007/s00284-005-0138-3
Taghavi, S., Garafola, C., Monchy, S., Newman, L., Hoffman, A., Weyens, N., et al. (2009). Genome survey and characterization of endophytic bacteria exhibiting a beneficial effect on growth and development of poplar trees. Appl. Environ. Microbiol. 75, 748–757. doi: 10.1128/aem.02239-08
Thijs, S., Van Dillewijn, P., Sillen, W., Truyens, S., Holtappels, M., D’haen, J., et al. (2014). Exploring the rhizospheric and endophytic bacterial communities of Acer pseudoplatanus growing on a TNT-contaminated soil: towards the development of a rhizocompetent TNT-detoxifying plant growth promoting consortium. Plant Soil 385, 15–36. doi: 10.1007/s11104-014-2260-0
Tholozan, J. L., Cappelier, J. M., Tissier, J. P., Delattre, G., and Federighi, M. (1999). Physiological characterization of viable-but-nonculturable Campylobacter jejuni cells. Appl. Environ. Microbiol. 65, 1110–1116.
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/aem.00062-07
Whipps, J. M., Hand, P., Pink, D., and Bending, G. D. (2008). Phyllosphere microbiology with special reference to diversity and plant genotype. J. Appl. Microbiol. 105, 1744–1755. doi: 10.1111/j.1365-2672.2008.03906.x
Witzel, K., Gwinn-Giglio, M., Nadendla, S., Shefchek, K., and Ruppel, S. (2012). Genome sequence of Enterobacter radicincitans DSM16656T, a plant growth-promoting endophyte. J. Bacteriol. 194, 5469. doi: 10.1128/jb.01193-12
Yoshida, S. (1981). Fundamentals of Rice Crop Science. Los Baños: The International Rice Research Institute.
Zhang, X. X., Gao, J. S., Cao, Y. H., Ma, X. T., and He, J. Z. (2013). Long-term rice and green manure rotation alters the endophytic bacterial communities of the rice root. Microb. Ecol. 66, 917–926. doi: 10.1007/s00248-013-0293-1
Keywords: free-air CO2 enrichment (FACE), growth stage, rice leaves, plant endophyte, bacterial community, 454 pyrosequencing
Citation: Ren G, Zhang H, Lin X, Zhu J and Jia Z (2015) Response of leaf endophytic bacterial community to elevated CO2 at different growth stages of rice plant. Front. Microbiol. 6:855. doi: 10.3389/fmicb.2015.00855
Received: 28 May 2015; Accepted: 06 August 2015;
Published: 31 August 2015.
Edited by:
Gary M. King, Louisiana State University, USAReviewed by:
Yin Chen, University of Warwick, UKChristina Hazard, Ecole Centrale de Lyon – University of Lyon, France
Copyright © 2015 Ren, Zhang, Lin, Zhu and Jia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhongjun Jia, State Key Laboratory of Soil and Sustainable Agriculture, Institute of Soil Science – Chinese Academy of Sciences, East Beijing Road No. 71, Nanjing, Jiangsu 210008, China,amlhQGlzc2FzLmFjLmNu