 G. Minard1
G. Minard1 F. H. Tran1
F. H. Tran1 Van Tran Van1C. Goubert2C. Bellet3G. Lambert4
Van Tran Van1C. Goubert2C. Bellet3G. Lambert4 Khanh Ly Huynh Kim5
Khanh Ly Huynh Kim5 Trang Huynh Thi Thuy5
Trang Huynh Thi Thuy5 P. Mavingui1,6
P. Mavingui1,6 C. Valiente Moro1*
C. Valiente Moro1*- 1Ecologie Microbienne, UMR Centre National de la Recherche Scientifique 5557, USC INRA 1364, VetAgro Sup, FR41 BioEnvironment and Health, Université Claude Bernard Lyon 1, Villeurbanne, France
- 2Laboratoire de Biométrie et Biologie Evolutive, UMR 5558, CNRS, INRIA, VetAgro Sup, Villeurbanne, France
- 3Entente Interdépartementale Rhône-Alpes pour la Démoustication, Chindrieux, France
- 4Entente Interdépartementale de Démoustication du Littoral Méditerranéen, Montpellier, France
- 5Department of Medical Entomology and Zoonotics, Pasteur Institute in Ho Chi Minh City, Vietnam
- 6Université de La Réunion, UMR PIMIT, INSERM U1187, CNRS 9192, IRD 249, Plateforme Technologique CYROI, Saint-Denis, France
The Asian tiger mosquito Aedes albopictus is one of the most significant pathogen vectors of the twenty-first century. Originating from Asia, it has invaded a wide range of eco-climatic regions worldwide. The insect-associated microbiota is now recognized to play a significant role in host biology. While genetic diversity bottlenecks are known to result from biological invasions, the resulting shifts in host-associated microbiota diversity has not been thoroughly investigated. To address this subject, we compared four autochthonous Ae. albopictus populations in Vietnam, the native area of Ae. albopictus, and three populations recently introduced to Metropolitan France, with the aim of documenting whether these populations display differences in host genotype and bacterial microbiota. Population-level genetic diversity (microsatellite markers and COI haplotype) and bacterial diversity (16S rDNA metabarcoding) were compared between field-caught mosquitoes. Bacterial microbiota from the whole insect bodies were largely dominated by Wolbachia pipientis. Targeted analysis of the gut microbiota revealed a greater bacterial diversity in which a fraction was common between French and Vietnamese populations. The genus Dysgonomonas was the most prevalent and abundant across all studied populations. Overall genetic diversities of both hosts and bacterial microbiota were significantly reduced in recently established populations of France compared to the autochthonous populations of Vietnam. These results open up many important avenues of investigation in order to link the process of geographical invasion to shifts in commensal and symbiotic microbiome communities, as such shifts may have dramatic impacts on the biology and/or vector competence of invading hematophagous insects.
Introduction
Mosquitoes are considered by the World Health Organization to be the most medically important disease vectors. The Asian tiger mosquito (Aedes albopictus) is of major concern as it is known to be able to carry 26 arboviruses including Dengue and Chikungunya (Paupy et al., 2009). Furthermore, Ae. albopictus is considered as one of the most geographically invasive species. It has rapidly spread from its native area of South and East Asia to reach various eco-climatic regions in America, Africa, Oceania and Europe (Bonizzoni et al., 2013). The worldwide trades in secondhand tires and lucky bamboo, both of which often contain standing water making them ideal places for mosquito eggs and larvae, have been key factors in Ae. albopictus transportation. Once established in a new region, the tiger mosquito easily adapts and persists in a wide range of habitats, even in temperate climates mainly due to its aptitude to enter into a state of dormancy or “diapause” (Urbanski et al., 2010). Undoubtedly, the intrinsic capacities of the mosquito populations largely play an important role in their ecological plasticity. However, this assumption remains surprising as according to the “paradox of invasion,” recent introductions often imply a burden for the genetic structure of newly introduced populations (reviewed by Handley et al., 2011).
A comprehensive understanding of insect population genetics now requires an integrative approach considering microorganisms as a key component of the system. According to the hologenome theory, Metazoan organisms should no longer be considered as individuals, but rather as holobionts consisting of the host plus all of its associated microorganisms (Zilber-Rosenberg and Rosenberg, 2008). It is the holobiont and its associated hologenome that can be considered as a unit of selection which is impacted by variation, selection, drift and evolution (reviewed by Rosenberg and Zilber-Rosenberg, 2014). Insect holobionts are also difficult to decipher as they may include a range of host-symbiont relationships ranging from parasitism to mutualism (Toft and Andersson, 2010). Numerous studies have demonstrated the contribution of the microbiota to the biology of the host (Blottière et al., 2013; Douglas, 2014). Some mutualistic symbionts favor ecological adaptations in insects (Douglas, 2011, 2014) by playing key roles in extended phenotypes such as growth, nutrition, reproduction, protection against pathogens and tolerance to environmental stresses (Buchner, 1965; Dillon and Dillon, 2004; Moran et al., 2008; Moya et al., 2008). Moreover, the host genotype may also influence in return symbiont communities (Ochman et al., 2010; Muegge et al., 2011). It is thus, important to consider the genetic basis of bacterial microbiota selection.
It is striking that relatively few studies have focused on bacterial microbiota associated with invasive arthropods. Many invasive species harbor genetic modifications caused by founder effects or genetic drift, but the effect of such changes on their microbiota are only just beginning to be documented (Meusnier et al., 2001; Zurel et al., 2011; Ye et al., 2014). As symbiotic microorganisms can change more rapidly and by more diverse processes than the host organism itself they could influence the adaptation and evolution of the holobiont.
Here, we aimed to document whether populations from two representative areas colonized by the Asian tiger mosquito displayed changes in their host genotypes and associated bacterial microbiota. To that end, we sampled mosquitoes from field populations in Vietnam, a country located in the South East Asia where Ae. albopictus originated indicating ancient colonization, and in Metropolitan France, which was more recently invaded by this species. Mitochondrial and nuclear genotypes of mosquitoes and bacterial diversity were compared within and between populations.
Materials and Methods
Sampling Areas and Mosquito Collection
Ae. albopictus specimens were sampled in Metropolitan France and Vietnam. In addition to their contrasted climate and ecology, these two countries were chosen as Vietnam is located in the South East Asia, the region where Ae. albopictus originated indicating ancient colonization, whereas Metropolitan France is a newly invaded zone. Sampling in Metropolitan France was performed between August and September 2012 at Saint-Priest (SP), Portes-Lès-Valence (PLV), and Nice (NC). NC is one of the first invaded sites in France since 2004 (Medlock et al., 2012), whereas PLV and SP were colonized in 2011 and 2012, respectively (data obtained from French health organization INVS). Mosquito sampling in Vietnam was performed during October 2012 at Hồ Chí Minh City (HCM), Bình Du'o'ng (BD), Vung Tàu City (VT), and Bù Gia Mâp (BGM) (Figure 1). All sites were urban or suburban, except BGM located in a protected forest national park. Sampling sites were at least 18 km from each other to avoid sampling populations originated from the same breeding site. Consequently, we assume that individuals collected from the same sites belong to the same population as they share breeding sites, and a total of seven independent populations (three from Metropolitan France and four from Vietnam) were obtained and analyzed. Live adult females were caught with nets or BG-Traps and then identified using morphological characteristics (Rueda, 2004). For some individuals, identification was confirmed by COI barcode sequencing (see below). To control confounding effects from nutritional factors, only females that could be seen to contain no blood upon magnified observation of the gut contents were retained for analysis. Mosquitoes were stored in 100% ethanol at −80°C until used.
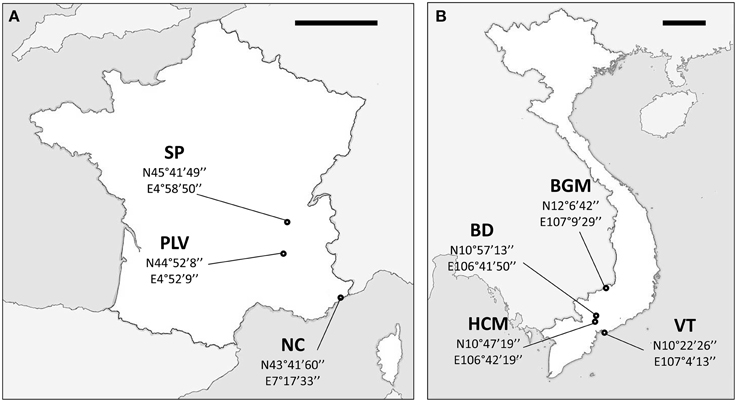
Figure 1. Maps of sampling sites. Sampling sites and GPS coordinates (World Geodetic System 1984) in France (A) and Vietnam (B). NC, Nice; PLV, Porte-lès-Valence; SP, Saint Priest; VT, Vung Tàu City; HCM, Hồ Chí Minh City; BD, Bình Du'o'ng, BGM, Bù Gia Mâp. Scale bars, 200 km.
Quantification of Wolbachia wAlbA and wAlbB Strains
Wolbachia wAlbA and wAlbB were quantified in whole bodies of 10 mosquitoes from each site (Table S1). Their densities were measured in triplicate by qPCR amplification of the Wolbachia wsp gene and normalized with the Ae. albopictus actin gene as described (Zouache et al., 2009). Standard curves were drawn on DNA plasmid pQuantAlb which contains wsp genes of Wolbachia pipientis wAlbA and wAlbB as well as actin gene of Ae. albopictus (Tortosa et al., 2008). Correlations between the two strains were calculated with R software using the Pearson's correlation.
Sample Preparation, Miseq Sequencing, Quality Trimming and Diversity Analysis of Sequences
Previous work (Minard et al., 2014) showed that whole insect body was inappropriate for in-depth analysis of bacterial microbiota of Ae. albopictus by NGS due to the overrepresentation of Wolbachia sequences. Here we used midguts that are known to be a key organ in the metabolism and immunity of mosquitoes as well as the first point of entry for transmitted viruses (Clements, 1992; Jupatanakul et al., 2014; Kenney and Brault, 2014b). In addition this organ was shown to harbor a moderate density of Wolbachia in Ae. albopictus (Zouache et al., 2009).
Prior to dissection for midgut recovery from 32 individuals (Table S1), female specimens were surface-disinfected with 70% ethanol and rinsed with sterile water as previously described (Minard et al., 2013). All dissection steps were performed under a sterile laminar flow hood in a containment environment. Mosquitoes were dissected in sterile 1 × phosphate buffered saline solution (Life Technologies, NY, USA). For each mosquito, the midgut was separated from the rest of the body. Midguts were then individually crushed with 1-mm diameter beads in ATL lysis buffer (Qiagen, Hilden, Germany) containing 20 mg.ml−1 lysozyme (Euromedex, Strasbourg, France) using a Bioblock Scientific MM 2000 mill (Retsch, Eragny sur Oise, France). DNA was then extracted with Qiagen DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany) following the manufacturer's recommendations for both Gram negative and Gram positive bacteria. Assuming that the remaining mosquito bodies (hereafter referred to as carcasses) should be dominated by Wolbachia, they were pooled per population (Table S1) and DNA extracted as above, and then used as positive controls. Finally, as negative controls to evaluate potential contamination, DNA extraction was carried out without any biological matrix and four independent eluates were concentrated and pooled.
Hypervariable V5-V6 rrs regions were amplified in triplicate for each DNA sample with 30 ng of DNA and modified primers 784F (5′-AGGATTAGATACCCTGGTA-3′) and 1061R (5′-CRRCACGAGCTGACGAC-3′) as described (Andersson et al., 2008) with modifications. Briefly, primers containing a 8-bp multiplex barcode and Illumina adapters were used for PCR amplifications with 1.75 U of Expand High Fidelity Enzyme Mix (Roche, Basel, Switzerland), 1 × Expand High Fidelity Buffer (Roche, Basel, Switzerland), 0.06 mg mL−1 of T4 gene 32 protein (New England Biolabs, Evry, France), 0.06 mg mL−1 of bovine serum albumin (New England Biolabs, Evry, France), 40 μM of dNTP mix, 200 nM of each primer (Life Technologies, Saint Aubin, France). Amplifications were carried out on Biorad C1000 thermal cycler (Biorad, CA, USA) with 5 min at 95°C, followed by 40 cycles at 95°C for 40 s, 54.2°C for 1 min, 72°C for 30 s, with a final extension step of 7 min at 72°C. Forty PCR amplification cycles were necessary to generate an optimal amount of amplicons for Miseq sequencing. The three PCR product replicates from each sample were pooled, purified with Agencourt AMPure XP PCR Purification kit (Beckman Coulter, Villepinte, France), and quantified using the Quant-iT Picogreen dsDNA Assay Kit (Life Technologies, NY, USA).
A total of 40 amplicon libraries were constructed: 32 for individual midguts, 7 for carcasses and 1 for the negative control. Sequencing of each library was performed on the Illumina MiSeq platform (2 × 250-bp paired-end reads) by ProfileXpert—ViroScan 3D (Lyon, France). All FastQ files were deposited at EMBL European Nucleotide Archive (https://www.ebi.ac.uk/ena) under the project accession number PRJEB6896. A total of 9, 222, 165 reads were obtained, paired-end reads were joined with PandaSeq (Masella et al., 2012), trimmed and aligned on the SILVA database release 115 using standard filtering tools in the MOTHUR pipeline (Schloss et al., 2009). Two errors were allowed in primer sequences, read sizes were filtered to be 200–350 bp in length with no ambiguous bases. Chimeras were detected and removed with Perseus implemented in Mothur package. Based on the analysis of clustered sequence rates from 92 to 99% similarity, OTUs were re-adjusted to 97% similarity using a median neighbor algorithm. Sequences were classified according to the SILVA database release 115 at 80% minimum bootstrap using a naïve Bayesian classifier (Wang et al., 2007). OTUs were also kept if there were at least represented by more than one sequence overall samples. Furthermore, OTUs were removed from further analyses if they were detected in the negative control sample and their relative abundance was not at least 10 times greater than that observed in the negative control. This additional quality control criterion allows us to qualify and correct for low concentration contaminants of experimental origin. Richness, α and β diversity indices were calculated using a subsample of the same read number for each sample. Diversity analyses, hierarchical analysis of molecular variance (AMOVA), Non-Metric Multidimensional Scaling ordination and heatmap representations were performed with R software (R Development Core Team, 2009) using ade4 and vegan packages (Dray and Dufour, 2007; Oksanen et al., 2013). To highlight possible country-associated OTUs, extended errors bars were computed and classified according to Welch modified t-test significance (p < 0.05) using STAMP software 2.0.9 (Parks and Beiko, 2010).
Mitochondrial Gene Amplification and Haplotyping
To maximize the quality and quantity of DNA obtained, our previously optimized extraction protocol for individual whole mosquito was used (Minard et al., 2014). A total of 85 individuals were analyzed ranging from 9 to 20 individuals per sampling site (Table S1). The 597-bp region of mtDNA cytochrome c oxidase subunit I (COI) gene was amplified with CI-J-1632 (5′-TGATCAAATTTATAAT-3′) and CI-N-2191 (5′-GGTAAAATTAAAATATAAACTTC-3′) primers using 45 ng of DNA matrix as described (Raharimalala et al., 2012). To analyze haplotypes, COI sequences were aligned with Seaview 4, then diversity and nucleotide composition were calculated with DnaSP (Librado and Rozas, 2009). AMOVA statistical analyses were performed with Arlequin 3.5 × (Excoffier and Lischer, 2010). Sequences of the different COI haplotypes were deposited on Genbank (https://www.ncbi.nlm.nih.gov/genbank) under the accession numbers LM999972-LM999977.
Microsatellite Processing and Genotyping
A total of 199 individuals were genotyped ranging from 22 to 30 individuals per sampling site (Table S1). Amplifications were done with 10 ng of DNA extracted from each individual and master mix from Qiagen Type-it Microsatellite PCR Kit following the manufacturer's recommendations (Qiagen, Hilden, Germany). Amplifications were performed on Biorad C1000 thermal cycler (Biorad, CA, USA) with an optimal protocol for each microsatellite to minimize unspecific artifacts. A set of 11 microsatellite markers previously described (Chambers et al., 1986; Porretta et al., 2006; Beebe et al., 2013) were used; namely AealbA9, AealbB51, AealbB52, AEDC, Alb-di6, Alb-tri3, Alb-tri18, Alb-tri25, Alb-tri41, Alb-tri45, and Alb-tri6 (Table S2). For AeablA9, AealbB51, and AealbB52 microsatellites, the program consisted of 5 min at 94°C, followed by 35 amplification cycles at 94°C for 5 min, 52°C (AealbA9) or 50°C (AealbB51, Aealb0B52) for 30 s and 72°C for 45 s, with a final step of 30 min at 72°C. AEDC microsatellite sequences were amplified with 5 min at 94°C followed by 30 cycles of 45 s at 94°C, 1 min 30 s at 56°C and 45 s at 72°C, with a final step of 30 min at 60°C. Alb-di6, Alb-tri3, Alb-tri18, Alb-tri25, Alb-tri41, Alb-tri45, and Alb-tri6 were amplified as described by Beebe et al. (2013). PCR products were diluted (between 1/60 and 1/100 depending on the relative sensitivity of markers) then 1 μL was mixed with 13.8 μL of ultrapure Hi-Di-formamide TM and 0.2 μL of size marker (MRL 500) and loaded on an ABI Prism 3730XL Genetic Analyzer automated sequencer (Life Technologies, NY, USA). Microsatellites were scored manually with Genemapper 3.0 (LifeTechnologies, NY, USA). Null alleles were evaluated with FreeNA (Chapuis and Estoup, 2007). Diversity indices, linkage disequilibrium, Factorial Correspondence Analysis and hierarchical AMOVA analyses were computed with Genetix 4.05, Fstat 2.9.3.2 and Arlequin 3.5x softwares (Excoffier and Lischer, 2010). The Bayesian structure of populations was evaluated using Structure 2.3.4 (Pritchard et al., 2000) with 100 000 “burn-in” steps followed by 500 000 iterations. Runs from 1 to 8 potential groups (K) were processed with 20 replicates (Figure S1). An admixture model was used with a location prior factor. As recommended for datasets with possible null alleles, a dominant allele option was set in the model. The best fit K-value was chosen with the Evanno method implemented in STRUCTURE HARVESTER (Evanno et al., 2005; Earl and VonHoldt, 2012) and the 20 replicates were averaged with CLUMPP (Jakobsson and Rosenberg, 2007). Finally, a population structure barplot was drawn with DISTRUCT (Rosenberg, 2004). Comparisons between Fst distances and bacterial microbiota Bray-Curtis distances were performed with a Mantel test. Rarefied genetic richness (Ar) and diversity (Hs) were correlated with Shannon bacterial diversity using Spearman's rank correlation. As populations which experienced a recent reduction of their effective size can develop a heterozygosity excess at neutral loci, this parameter was tested using BOTTLENECK software (Cornuet and Luikart, 1996).
Results
Wolbachia wAlbA and wAlbB Strains are Abundant and Positively Correlated with Each Other
Our aim was to study the bacterial microbiota in autochthonous and invasive tiger mosquito populations. In a previous study, we demonstrated that Wolbachia is the predominant bacterial species in Ae. albopictus from Madagascar when using whole body genomic DNA, constituting up to 99% of high throughput sequences recovered (Minard et al., 2014). Here, we first tested whether the two Wolbachia strains wAlbA and wAlbB were also present and dominant in mosquitoes sampled from autochthonous populations at four sites (HCM, BD, VT, BGM) in Vietnam and from invasive populations at three sites (SP, PLV, NC) in France (Figure 1). wAlbA and wAlbB were detected in all individuals of the seven populations. The lowest densities of wAlbA and wAlbB strains detected were 5.25 × 10−5 wsp.actin−1 and 3.09 × 10−3 wsp.actin−1 copies respectively in mosquito samples from BD and the highest densities of 2.44 wsp.actin−1 and 8.53 wsp.actin−1 copies respectively from the BGM population (Figure 2). A significant positive correlation (Pearson's correlation R2 = 0.84, p < 2.2 × 10−16) between wAlbA and wAlbB strains was found among all the populations tested (Figure 2). To check if Wolbachia sequences were overrepresented in bacterial microbiota sequences when applying NGS methods to the whole body, the V5-V6 rrs amplicons were generated from mosquito carcass pools as indicated in material and methods, and sequenced by Miseq technology. Results confirmed a dominance of Wolbachia OTUs (Figure 3) which account for 28% (for HCM) to 91% (for SP) of the sequence dataset, reinforcing the rationale for our choice to avoid using the whole insect body for bacterial community analysis in Ae. albopictus.
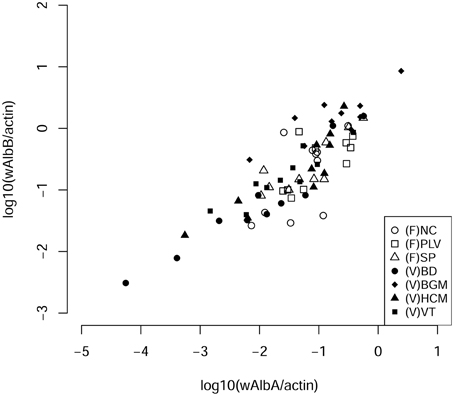
Figure 2. Correlation densities of Wolbachia pipientis. The number of each bacterial strain per cells was evaluated by quantification of wsp genes from each Wolbachia wAlbA and wAlbB strains normalized to the number of host actin gene copies (Pearson's correlation R2 = 0.84, p < 2.2 × 10−16). NC, Nice; PLV, Porte-lès-Valence; SP, Saint Priest; VT, Vung Tàu City; HCM, Hồ Chí Minh City; BD, Bình Du'o'ng, BGM, Bù Gia Mâp.
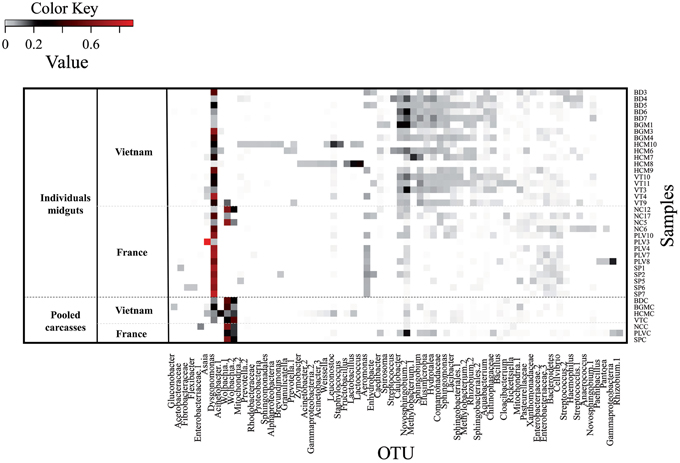
Figure 3. Aedes albopictus microbiota bacterial diversity. The heatmap represents the relative abundance of 3% OTUs in each sample. The Color Key Value refers to the proportion of an OTU in a single sample. The OTUs were represented if their proportion was = 0.01 in at least one sample. They were named by their taxonomic assignment according to Bayesian classification. When more than one OTU get the same assignation, they were differentiated with numbers. Samples are classified according to their own populations. Individual midgut samples from Vietnam were collected in Bình Du'o'ng (BD 3, 4, 5, 6, and 7), Bù Gia Mâp (BGM 1, 3, 4), Hồ Chí Minh City (HCM 6, 7, 8, 9, 10), Vung Tàu City (VT 3, 4, 9, 10, 11). Individual midgut samples from France were collected in Nice (NC 5, 6, 12, 17), Portes-Lès-Valence (PLV 3, 4, 7, 8, 10), and Saint Priest (SP 1, 2, 5, 6, 7). The pooled carcasses of the following individuals were also analyzed for both Vietnamese (BDC, BGMC, HCMC, VTC) and French populations (NCC, PLVC, SPC); the last “C” added to each name indicates carcass.
Midgut Bacterial Community Structure in Vietnamese Autochthonous Populations Compared to French Invasive Ones
The insect gut is a key organ in insect physiology and immunity. Moreover, previous studies have demonstrated that this organ harbored low concentration of Wolbachia in Ae. albopictus adults (Zouache et al., 2009), opening up the possibility to extend the depth of analysis of the gut-associated microbial community. For this purpose, V5-V6 rrs amplicons from 32 individual midgut samples (from 3 to 5 individuals per sampling site) were sequenced with MiSeq technology. Analysis of a negative control showed the presence of bacterial sequences that probably derived from contamination during laboratory sample handling (Table S6). However, the diversity of this control was dissimilar from those of all mosquito samples (Bray-Curtis dissimilarity > 68.6%). For subsequent analysis of sequences associated with mosquito samples, OTUs potentially originating from laboratory contamination were trimmed from the whole dataset. Based on this analysis, a total of 2,088 OTUs were identified in all the midgut samples (between 306 and 1,272 OTUs per sample), with a total of 68 OTUs exceeding 1% in abundance. These OTU numbers were consistent with those previously obtained by high throughput sequencing of midguts from various mosquito species (Osei-Poku et al., 2012). The genus Dysgonomonas was the most prevalent and abundant OTU retrieved from the midgut samples (Figure S3), although its abundance varied from 3% (HCM8) to 72% (SP7) between samples (Figure 3). AMOVA analysis and ordinations were performed to detect the degree of differentiation at various hierarchical levels. No significant variation was observed between sites, indicating a low variability between individuals belonging to a given population. In contrast, variation between countries was found to be significant for the β-diversity measure, explaining a large part of the variation for Bray-Curtis dissimilarities (AMOVA, 22.79%, p < 10−4) (Table 1, Figure 4A). Similar structures were obtained with Unifrac phylogeny based β-diversity distances (Table S3). Consequently, further comparisons of populations were performed at the country level. When compared with the four populations from Vietnam, consisting of a total of 14 individuals, the three populations from France composed of 18 individuals harbored less diverse and more homogeneous bacterial microbiota, indicated by lower values for the Chao 1 richness estimator (Mann–Whitney Wilcoxon, p = 0.002), Shannon-Weather α-diversity (Mann–Whitney Wilcoxon, p < 10−3) and variance of abundance-weighted β-diversity (Figure S2). However, Bray-Curtis (Beta-dispersion, p = 0.33) distances were not significantly different between populations of the two countries.
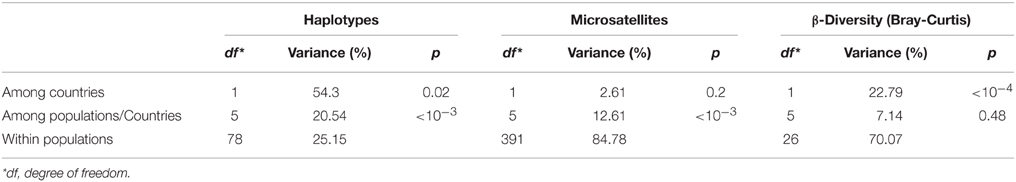
Table 1. AMOVA analysis.
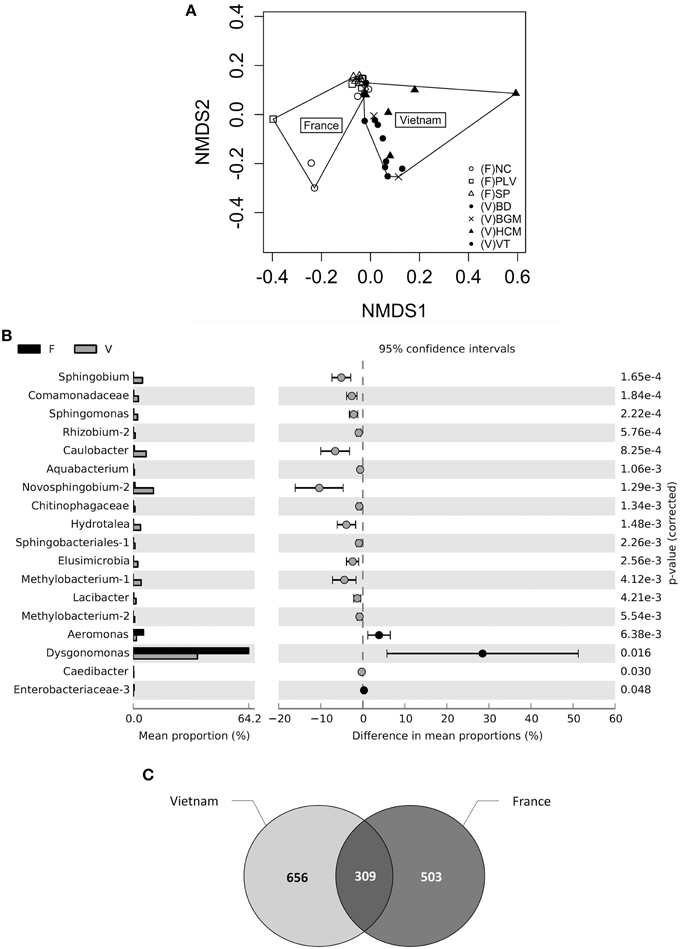
Figure 4. Community structure and OTU-country associations. (A) Non-Metric Multidimentional Scaling plot represents Bray-Curtis β-diversity structure among individuals and populations. The loss function Stress = 0.09 and correlation with true distances R2 = 0.97. NC, Nice; PLV, Porte-lès-Valence; SP, Saint Priest; VT, Vung Tàu City; HCM, Hồ Chí Minh City; BD, Bình Du'o'ng; BGM, Bù Gia Mâp. (B) Difference in OTU abundance between midgut bacterial microbiota of French and Vietnamese Ae. albopictus. Display represents extended error bars of significant fold-changes (P>0.05) between midgut samples from France (F) and Vietnam (V). (C) Venn diagram representing shared OTUs between midgut samples from France and Vietnam. The intersection of both circles represents the number of shared OTUs between France and Vietnam. To avoid size effect, sequences were merged per group, and then subsampled.
Overall, 15 OTUs were shown to be significantly associated with Vietnamese populations (Welch corrected t-test, p < 0.05) (Figure 4B), including various members of Shingomonadaceae family (Sphingobium, Novosphingobium, Sphingomonas). Only three OTUs assigned to Dysgonomonas and Aeromonas genera and Enterobacteriaceae family were shown to be significantly associated with French populations. The OTU overlap was calculated after merging and subsampling sequences at the country level (Figure 4C). The resulting Venn diagram showed 21% of shared OTUs (n = 309), and 34% (n = 503), and 45% (n = 656) specific of France and Vietnam, respectively. Shared OTUs counted for 85% of the overall sequences.
Low Mitochondrial DNA Variation among Ae. albopictus Populations from France and Vietnam
The genetic makeup of Ae. albopictus hosts was evaluated by barcoding based on mitochondrial COI gene sequences. A total of 6 haplotype variants were found in individuals from all populations. The genetic heterogeneity was more pronounced between countries (AMOVA 47.4%, p < 10−3) than between sites (AMOVA 19.79%, p < 10−3) (Table 1). The two major subclades were distinguishable by only one mutation. One subclade included 52 haplotypes (H_1) and the other 28 haplotypes (H_3) (Table S4). H_1 was mostly associated with the populations from Vietnam whereas H_3 was more associated with those from France. However, a mix of both haplotypes was also found in the populations at the PLV site in France (12/20 for H_1 and 7/20 for H_3) and at the BD site in Vietnam (16/17 for H_1 and 1/17 for H_3) (Figure S4).
Evidence for Genetic Reduction in Ae. albopictus Populations Invasive to France
The mosquito nuclear genomic variation was characterized further by genotyping 199 individuals with 11 microsatellite markers. The overall number of alleles per locus varied from 6 (AealbB52) to 30 (Alb-tri 18). Rarefied allele richness of populations from each site was 4.56 for SP, 4.57 for PLV, 5.91 for NC, 6.50 for HCM, 7.47 for VT, 8.25 for BGM, and 8.62 for BD. To test for hypothetical population bottlenecks or expansions, the allelic richness (Ar) and the heterozygosity (He) at each genomic locus were analyzed for each sampling site. For both these analyses of variation, values for populations from France were significantly lower than those from Vietnam (Mann–Whitney, p = 0.0003 and 0.0003, respectively). The bottleneck analysis did not show any significant heterozygosity excess under a two-phase model or a single stepwise model (Table S5). No significant linkage disequilibrium was found between any pair of loci. All populations had a positive inbreeding index (FIS) between 0.12 (for PLV) and 0.196 (for HCM) reflecting an excess of homozygotes (Table 2). Moreover, there were significant scores for the presence of null alleles. However, FST values of the entire mosquito sample were 0.142 (CI95% 0.058–0.269) with ENA correction for null alleles and 0.145 (CI95% 0.061–0.270) without correction. Consequently, the presence of null alleles did not strongly impact the estimation of differentiation. The structure of the populations was evaluated with the Bayesian method of assignment. The optimal number of clusters selected with the second-order change in likelihood method was K = 2 (Figure S1) (Evanno et al., 2005). Populations were clustered in two different genetic groups according to country of origin, except for populations from PLV in France and BD in Vietnam, which harbored a mixture of both genotypes (Figure 5, Figure S5). AMOVA analysis revealed a non-significant variation in the structure among countries (AMOVA 2.6%, p = 0.2) but a moderate variation among sites within a country (AMOVA 13%, p < 10−3) (Table 1).
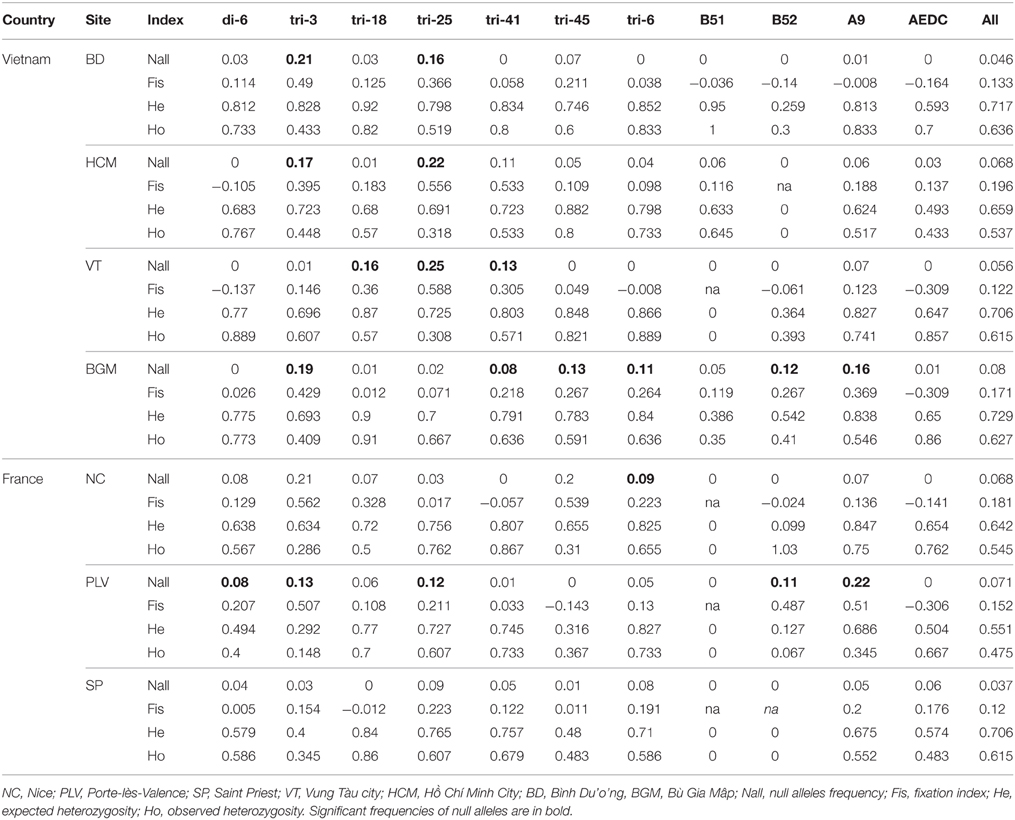
Table 2. Microsatellite characteristics among populations.
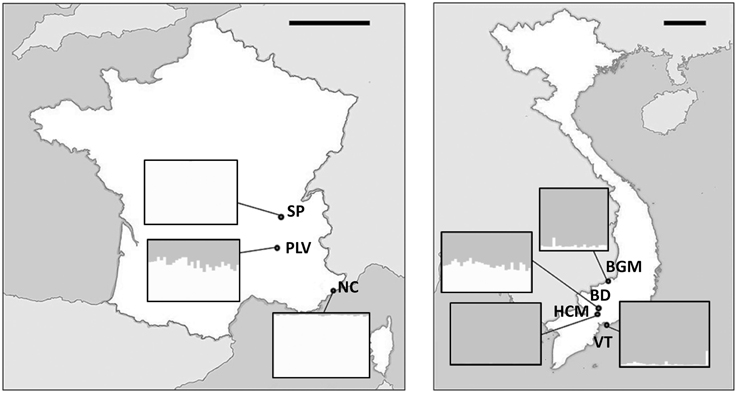
Figure 5. Genetic structures of Aedes albopictus populations. The map of the microsatellite genetic structure (K = 2) for each site. Each bar represents an individual and the grayscale represents the probability that an individual belongs to a population. Scale bar of the maps, 200 km. NC, Nice; PLV, Porte-lès-Valence; SP, Saint Priest; VT, Vung Tàu City; HCM, Hồ Chí Minh City; BD, Bình Du'o'ng; BGM, Bù Gia Mâp.
Positive Correlation between Bacterial and Genetic Diversities of Ae. Albopictus Populations
The populations sampled were compared to assess whether there was any relationship between the bacterial diversity and the genetic diversity of the mosquitoes. Comparative analysis showed a low correlation between bacterial β-diversity (Bray-Curtis dissimilarity distance) and haplotype structure (Mantel, R2 = 0.5, p = 0.02) and no significant correlation with the Cavalli-Sforza Edwards measure of microsatellite genetic distance (Mantel, R2 = 0.20, p = 0.19). However, for all sampling sites, positive correlations were observed between mean bacterial α-diversity (H′) and respectively host Ar (Spearman's rank correlation, ρ = 0.95, p = 8.10−3) (Figure 6A) and host genetic diversity (Hs) (Spearman's rank correlation, ρ = 0.78, p = 0.048) (Figure 6B).
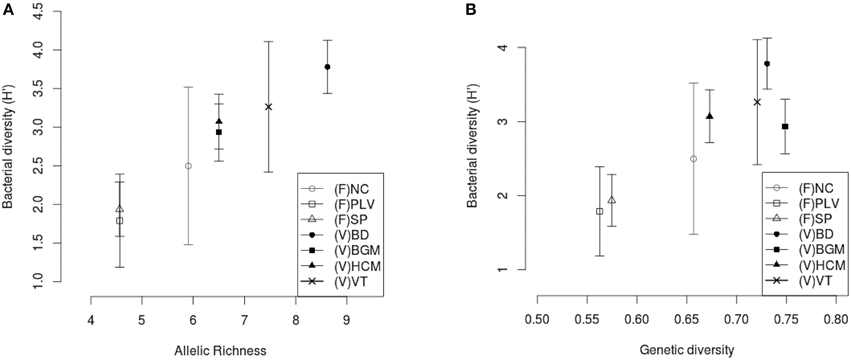
Figure 6. Correlation between host genetic richness, host genetic diversity, and midgut bacterial diversity. The mean bacterial Shannon α-diversity (H′) was correlated with (A) rarefied genetic richness, Ar (Spearman's rank correlation, ρ = 0.95, p = 8.10−3) and (B) diversity, Hs (Spearman's rank correlation, ρ = 0.78, p = 0.048). (F), France; (V), Vietnam; NC, Nice; PLV, Porte-lès-Valence; SP, Saint Priest; VT, Vung Tàu City; HCM, Hồ Chí Minh City; BD, Bình Du'o'ng; BGM, Bù Gia Mâp. Standard deviation of Shannon α-diversity (H′) was represented for each site.
Discussion
Mosquitoes may be regarded as holobiont units in which the host and its microbiota may display symbiotic relationships and multitrophic interactions (Minard et al., 2013). As demonstrated in other insect models such as drosophila and aphids (Engel and Moran, 2013; Lizé et al., 2014), it is envisaged that mosquito-associated bacterial microbiota could influence the ability of the host to respond to biotic and abiotic factors. An essential step forward is to increase our knowledge on the mosquito-associated microbiota. Here, we used a next generation sequencing method and metabarcoding to characterize the composition of bacterial microbiota of invasive and autochthonous populations of Ae. albopictus and to test for correlations with host genotype.
Wolbachia is the most studied bacterium of mosquitoes. Although, some beneficial fitness effects of this endosymbiont have been demonstrated in Ae. albopictus (Dobson et al., 2002), Wolbachia most commonly alters mosquito reproduction by inducing cytoplasmic incompatibility between infected males and uninfected females (Stouthamer et al., 1999). Superinfection with more than one Wolbachia strain makes the system more complex and some models predict that superinfected females have a selective advantage as they should be able to reproduce with every combination of uninfected and strain-infected males (Dobson et al., 2004; Tortosa et al., 2010). Diagnostic PCR on field-caught Ae. albopictus from various countries confirmed this prediction as more than 99% of individuals were superinfected with Wolbachia wAlbA and wAlbB strains (Kittayapong et al., 2002; de Albuquerque et al., 2011; Zouache et al., 2011). Here we showed a 100% prevalence of such double infection in seven Ae. albopictus populations, three from France and four from Vietnam. Other than cytoplasmic incompatibility and the age of the mosquito (Tortosa et al., 2010), factors driving Wolbachia persistence in Ae. albopictus remain largely unknown. Our results demonstrated for the first time a strong correlation between the proportions of the two strains wAlbA and wAlbB in Ae. albopictus natural populations, suggesting a structured mechanism is regulating their co-occurrence. Wolbachia has also been demonstrated to induce selective “sweep” on mitochondria, which drastically reduces the genomic variability of this organelle in Ae. albopictus (Hurst and Jiggins, 2005). Accordingly, the mitochondrial haplotypes of all seven populations showed very low nucleotide diversity in the COI gene (haplotypes differed by only one substitution) and low richness (less than three haplotypes per sampling site after rarefaction). Therefore, mitochondrial markers are highly sensitive to admixture in comparison with nuclear markers such as microsatellites. For this reason, it is not suitable to rely on mitochondrial haplotypes when estimating intraspecific genetic diversity in Ae. albopictus.
As confirmed herein, analyses of the Ae. albopictus microbiota are systematically dominated by Wolbachia sequences (Minard et al., 2014). In order to investigate other genera we used the midgut tissue, which is recognized to be poorly colonized by this bacterium (Zouache et al., 2009). Moreover, in arthropod vectors, this organ is the main site for multipartite interactions between bacterial microbiota, arboviruses and the host (Jupatanakul et al., 2014; Kenney and Brault, 2014a). Interestingly, 21% of total OTUs richness was found in populations from both France and Vietnam, assuming that some members of the microbiota can be shared among Ae. albopictus from contrasted populations. Despite the relatively low richness of shared OTUs, they accounted for 85.2% of all the sequences. The dominance of the shared microbiota over the transient microbiota is suggestive of positive selection of the interactions that constitute semi-constant gut microbiota (Figure S3), in contrast with the variable microbiota previously described within midguts of field-caught mosquitoes belonging to different species (Osei-Poku et al., 2012). Although, this observation may also be explained by the relatively homogeneity of the habitats of Aedes spp. mosquitoes (low oxygen pressure, a pH comprised between 8 and 10) (Dillon and Dillon, 2004; del Pilar Corena et al., 2005; Saboia-Vahia et al., 2014), and the establishment of habitat-specific bacterial associations. However, some factors such as the type of nutrients ingested and temperature (not regulated in ectotherms) can strongly vary between French and Vietnamese populations. In addition, previous studies highlighted the importance of water of breeding sites in which mosquito larvae and pupae develop. It was shown that mosquitoes acquire a large part of their microbiota from larval stages which themselves depend on the bacterial composition of the water of breeding sites. Moreover, a great variability in diversity of abundance of taxa was shown between stages according to mosquito species (Pumpuni et al., 1996; Minard et al., 2013; Coon et al., 2014; Dada et al., 2014; Gimonneau et al., 2014). Following these observations, the variations observed among the populations studied here could be linked with environmental factors of their habitat.
Among all the midgut samples, a total of 68 dominant OTUs were described and assigned with the name of their most probable phylotype. Dysgonomonas was the most prevalent and abundant one. This genus belongs to the phylum Bacteroidetes, and has already been recently detected in Ae. albopictus from Madagascar (Minard et al., 2014). Prevalent and abundant bacteria belonging to the Bacteroidetes phylum were previously described in Ae. aegypti. In particular, this mosquito species harbors a high relative abundance of Chryseobacterium that is maintained during all different life stages of lab-reared populations (Coon et al., 2014). However, previous studies highlighted that gut mosquito microbiota is largely dominated by Proteobacteria as for Anopheles coluzzi, An. funestus, An. gambiae, An. Stephensi, and Culex tarsalis (Pidiyar et al., 2004; Lindh et al., 2005; Rani et al., 2009; Boissière et al., 2012; Minard et al., 2013; Gimonneau et al., 2014). For these studies one to two major genera were usually found dominant in the midgut, albeit the identity of a given dominant entity changes over individuals or populations (Boissière et al., 2012; Osei-Poku et al., 2012). Therefore, it is surprising that we describe a single prevalent and abundant taxon, Dysgonomonas, in the midgut of all individuals even originated from distantly separated populations, suggesting an evolutionary process that maintains the presence of this particular taxon. Interestingly, Dysgonomonas has also been identified in the gut of different animals such as termites (Coptotermes formosanus, Macrotermes barneyi), house flies (Musca domestica), fruit flies (Drosophila spp.), red palm weevils (Rhynchophorus ferrugineus), and sea bass (Dicentrarchus labrax) (Husseneder et al., 2009; Chandler et al., 2011; Carda-Diéguez et al., 2014; Tagliavia et al., 2014; Yang et al., 2014). This genus has been previously detected in Anopheles stephensi and Culex tarsalis microbiota with a moderate abundance (Rani et al., 2009; Duguma et al., 2013). Its ability to cause lysis of erythrocytes and to synthesize B12 vitamins might be a selective mechanism involved in a mutualistic interaction with female mosquitoes (Hironaga et al., 2008; Husseneder et al., 2009; Lawson et al., 2010; Yang et al., 2014). Some species of Dysgonomonas are both aerobic and facultative anaerobic, which could partly explain how some may adapt to the nearly anoxic insect midgut habitat (Johnson and Barbehenn, 2000; Chouaia et al., 2014). All these observations point to Dysgonomonas having a role in mosquito biology. Finally, certain Dysgonomonas species were also identified as human opportunistic pathogens, raising concern about the possibility of additional mosquito borne diseases and thus highlighting the recent concept of “pathobiome” in arthropod vectors (Hironaga et al., 2008; Lawson et al., 2010; Vayssier-Taussat et al., 2014).
Other bacterial genera detected in Ae. albopictus belong to the Sphingomonadaceae family (Sphingomonas, Sphingobium, Novosphingobium) and were preferentially associated with the autochthonous populations from Vietnam. These bacterial genera are ubiquitous in the environment (Vaz-Moreira et al., 2011; Ashton Acton, 2012) and are also able to colonize a variety of higher organisms (D'Auria et al., 2013; Zhang et al., 2013; Dai et al., 2014). They display various catabolic abilities including the production of hydrolases involved in the degradation of oligosaccharides, and in termite hosts they may participate in the degradation of plant compounds (Aylward et al., 2013). Moreover, these bacteria have already been identified in other plant-feeding insects including mosquitoes Anopheles maculipenis, Anopheles gambiae, Anopheles stephensis, and Aedes aegypti (Dong et al., 2009; Ramírez-Puebla et al., 2010; Dinparast Djadid et al., 2011; Gayatri Priya et al., 2012; Terenius et al., 2012; Koroiva et al., 2013). From these observations, it could be assumed that Sphingomonadaceae are important in making plant sugar available to the mosquito host, by degrading oligosaccharides in the mosquito gut or acquiring them from the environment.
Interestingly, the bacterial communities associated with the invasive mosquito populations in France were less diverse and more homogeneous than those associated with autochthonous populations in Vietnam. The living host environment is an important factor impacting the microbiota of insects (Linnenbrink et al., 2013). In mosquitoes, it is known that feeding behavior may drastically modify the structure of gut microbiota and strongly increase inter-individual variation (Wang et al., 2011; Pernice et al., 2014). To avoid the effects of short-term changes in midgut microbiota, we only studied unfed mosquitoes, but long-term feeding effects cannot be ignored. Indeed, mosquito nutrition is mostly based on nectar and Ae. albopictus is known to have a wide nutritional spectrum (Clements, 1992). Interestingly, previous studies of the microflora diversity of different environments highlighted a significant reduction of species richness within the flowering plants (angiosperms) in temperate compared to tropical ecosystems (Francis and Currie, 2003). Therefore, the diversity and availability of plant nutrient sources could explain the reduction and the homogeneity in bacterial diversity we observed in mosquito populations of France. Populations from France also harbor a lower genetic diversity. Genetic reductions in invasive species have been widely documented. The most probable factor explaining genetic reduction in invasive populations is that recent colonization by a reduced effective population size causes a founder effect bottleneck and genetic drift (Dlugosch and Parker, 2008). However, no evidence for a recent founder effect was detected in the invasive populations in France. The short generation time of Ae. albopictus as well as the history of complex and multiple introductions (evidenced especially in Portes-Lès-Valence populations which harbor low allelic richness and genetic diversity but an admixture structure discovered with both haplotype and microsatellite markers) may have erased signs of a past bottleneck. In addition, genetic diversity reductions in France could also be explained by other patterns (e.g. Landscape fragmentation, lowest effective size…). Interestingly, the most genetic diversified of these populations was in Nice, a invaded site since 2004 (Medlock et al., 2012), whereas Portes-Lès-Valence and Saint-Priest were colonized much later. As already suggested in various models (Sommer, 2005), genetic diversity reduction identified with neutral markers could be linked with diversity reduction of genes involved in immunity. Moreover, immune genes could also be under considerable selective pressure that would affect the composition of mosquito microbiota (Wang et al., 2011; Minard et al., 2013). Indeed, gut microbiota are involved in a strong reciprocal interaction with the host immune system in the mosquito gut (Hillyer, 2010; Cirimotich et al., 2011; Weiss and Aksoy, 2011). However, we did not find any correlations between microbiota and mosquito genetic structure based on neutral microsatellite markers. Interestingly, a genetic study of mice using quantitative markers demonstrated that a core microbiome was regulated by a complex polygenic trait likely to involve pleiotropic effects (Benson et al., 2010). However, as a direct link between genetic and microbial structures cannot be proven following our sampling design, further investigations would be necessary. In particular, development of Ae. albopictus quantitative markers would be helpful in pinpointing which host genetic factors could partly shape the microbiota diversity.
Reduction in diversity for both the mosquito host and its associated bacterial microbiota also raises questions about the possible impact on human pathogen transmission. During transmission cycles, mosquito-vectored pathogens pass through the host midgut epithelial membrane to reach hemolymph and salivary glands. This checkpoint is critical for transmission as the pathogen faces the microbiota barrier and its potentially antagonistic activity (enzymes, toxins, etc.) as well as the host immune system (Cirimotich et al., 2011; Weiss and Aksoy, 2011; Wang et al., 2013). Indeed, the microbiota of mosquito vectors was shown to interfere, positively or negatively, with their susceptibility to pathogen infection and transmission capacity (Dennison et al., 2014). For instance, a recent study on Anopheles gambiae mosquitoes suggested that reduction of gut microbiota diversity following ingestion of antibiotics increases the capacity of females to transmit Plasmodium falciparum (Gendrin et al., 2015). In previous works it was demonstrated that a high proportion of viral particles were able to disseminate beyond the insect midgut barrier in Ae. albopictus populations from Cagne-Sur-Mer (~12 km away from Nice in France) (Vega-Rua et al., 2013) whereas the lowest range of Chikungunya virus dissemination was found in mosquitoes from Vietnam (Zouache et al., 2014). Indeed, the midgut microbiota is the first barrier encountered by viruses during their infection process and its diversity could strongly interferes with virus replication (Jupatanakul et al., 2014; Kenney and Brault, 2014a). However, microbiota is not the only factor that can modulate mosquito competence. Vector spatial genetic structure (gene flow, presence of cryptic species, invasion) has previously been demonstrated to greatly influence its interactions with pathogens (reviewed by McCoy, 2008; Léger et al., 2013). In Ae. albopictus recent studies highlighted that complex genetic-genetic-environment interactions impacted the transmission of Chikungunya virus (Zouache et al., 2014). However, the effect of mosquito genetic diversity on disease transmission remains unclear. Nevertheless, previous empirical evidences and models based on host-pathogens systems predicted that host genetic diversity can negatively affect the prevalence of pathogens (reviewed by King and Lively, 2012). It is conceivable that the reduction and change in the mosquito microbiota and its associated immune response could partly explain the efficient vector competence observed in Ae. albopictus populations from Metropolitan France.
In conclusion, our results suggest a similar pattern in reduction of genetic diversity of Ae. albopictus and bacterial microbiota diversity. This finding provides new insights into the biology of an invasive species and its associated bacterial microbiota. It also highlights the need for further ecological studies to describe how the invasive mosquito population, as well as its hologenome, responds when challenged by new biotic and abiotic factors. Moreover, the dynamics of mosquito-associated eukaryotic and viral microbiota should also be investigated to gain a fully integrated view of the holobiont pathosystem of the Asian tiger mosquito.
Ethical Issues
No ethical issues to be promulgated.
Author Contributions
This work is part of GM's PhD dissertation (supervised by PM and CV) on Aedes albopictus microbiota. GM, PM, and CV conceived the project and sampling design. CB, TH, GL, KL, GM, PM, CV, and VT contributed to collection of specimens. GM, CV, FT, and VT performed all molecular work and genotyping scoring. GM and CG analyzed and interpreted the genotypic data and GM analyzed and interpreted the metabarcoding data. GM wrote the article and all other authors contributed edits and comments.
Funding
Funding for this project was provided by grants from EC2CO CNRS and CMIRA Région Rhône-Alpes. This research was also partially funded by ERA-NET BiodivERsA with the national funders ANR-13-EBID-0007-01, FWF I-1437, and DFG KL 2087/6-1 as part of the 2012-2013 BiodivERsA call for research proposals, and was carried out within the framework of GDRI “Biodiversity and Infectious Diseases in Southeast Asia.” To achieve this work, we used the computing facilities of the PRABI cluster. We also gratefully acknowledge the contribution of the DTAMB platform of the FR41 Bio-Environment and Health (University Lyon 1).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank help by Bernard Kaufmann with population genetics advices, Patricia Luis with map design, Audrey Dubost with bioinformatics analysis, Guillaume Carillo with qPCR experiments, and David Wilkinson for reading the revised version of the manuscript. We also thank the two anonymous reviewers for their helpful comments on the first version of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.00970
Figure S1. Prediction of the best value of K. According to Evanno et al. (2005), the distribution of ΔK (absolute values of the second-order in change of the likelihood distribution divided by the standard deviation of the likelihoods) was plotted for each value of K (number of potential sub-groups) from 2 to 7.
Figure S2. Index comparisons between French and Vietnamese populations according to the 3% OTU richness estimations (Chao1), α-diversity (H') and β -diversity homogeneity (Bray-Curtis Distance to centroid).
Figure S3. Prevalence of Operational Taxonomic Units (OTUs) according to their mean relative abundance in midgut samples. Most abundant OTUs (proportion > 0.01) are named by their assignation according to naïve Bayesian classifier (Bootstrap > 80%). The prevalence and abundance of OTUs were calculated from sequences obtained from midgut samples analyzed in the study.
Figure S4. Map of haplotypes. The six haplotypes proportion (H_1, H_2, H_3, H_4, H_5, H_6) are represented for each site. Scale bar, 200 km. NC, Nice; PLV, Porte-lès-Valence; SP, Saint Priest; VT, Vung Tàu City; HCM, Hồ Chí Minh City; BD, Bình Du'o'ng; BGM, Bù Gia Mâp.
Figure S5. Factorial Correspondence Analysis of mosquitos' genetic structure. Each point represents an individual from a given population. The color indicates different original population of individuals. The ordination is based on multivariate analysis of allelic frequencies of the 11 microsatellites among individuals. NC, Nice; PLV, Porte-lès-Valence; SP, Saint Priest; VT, Vung Tàu City; HCM, Hồ Chí Minh City; BD, Bình Du'o'ng; BGM, Bù Gia Mâp.
Table S1. Sample compositions for the different analyses.
Table S2. Microsatellite primers and information.
Table S3. AMOVA analysis of phylogeny based Unifrac β-diversity.
Table S4. Haplotypes and nucleotide diversity.
Table S5. Bottleneck analysis.
Table S6. Dominant contaminant OTUs found in the negative control.
References
Andersson, A. F., Lindberg, M., Jakobsson, H., Bäckhed, F., Nyrén, P, and Engstrand, L. (2008). Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 3:e2836. doi: 10.1371/journal.pone.0002836
Ashton Acton, Q. (2012). Advances in Sphingomonadaceae Research and Application. Atlanta, GA: ScholarlyEditions.
Aylward, F. O., McDonald, B. R., Adams, S. M., et al. (2013). Comparison of 26 Sphingomonad genomes reveals diverse environmental adaptations and biodegradative capabilities. Appl. Environ. Microbiol. 79, 3724–3733. doi: 10.1128/AEM.00518-13
Beebe, N. W., Ambrose, L., Hill, L. A., Davis, J. B., Hapgood, G., Cooper, R. D., et al. (2013). Tracing the tiger: population genetics provides valuable insights into the Aedes (Stegomyia) albopictus invasion of the Australasian region. PLoS Negl. Trop. Dis. 7:e2361. doi: 10.1371/journal.pntd.0002361
Benson, A. K., Kelly, S. A., Legge, R., Ma, F., Low, S. J., Kim, J., et al. (2010). Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. U.S.A. 107, 18933–18938. doi: 10.1073/pnas.1007028107
Blottière, H. M., de Vos, W. M., Ehrlich, S. D., and Doré, J. (2013). Human intestinal metagenomics: state of the art and future. Curr. Opin. Microbiol. 16, 232–239. doi: 10.1016/j.mib.2013.06.006
Boissière, A., Tchioffo, M. T., Bachar, D., Abate, L., Marie, A., Nsango, S. E., et al. (2012). Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Pathog. 8:e1002742. doi: 10.1371/journal.ppat.1002742
Bonizzoni, M., Gasperi, G., Chen, X., and James, A. A. (2013). The invasive mosquito species Aedes albopictus: current knowledge and future perspectives. Trends Parasitol. 29, 460–468. doi: 10.1016/j.pt.2013.07.003
Buchner, P. (1965). Endosymbiosis of Animals with Plant Microorganisms. Bucks, PA: Interscience Publishers.
Carda-Diéguez, M., Mira, A., and Fouz, B. (2014). Pyrosequencing survey of intestinal microbiota diversity in cultured sea bass (Dicentrarchus labrax) fed functional diets. FEMS Microbiol. Ecol. 87, 451–459. doi: 10.1111/1574-6941.12236
Chambers, D. M., Young, L. F., and Hill, H. S. (1986). Backyard mosquito larval habitat availability and use as influenced by census tract determined resident income levels. J. Am. Mosq. Control Assoc. 2, 539–544.
Chandler, J. A., Lang, J. M., Bhatnagar, S., Eisen, J. A., and Kopp, A. (2011). Bacterial communities of diverse Drosophila species: ecological context of a host-microbe model system. PLoS Genet. 7:e1002272. doi: 10.1371/journal.pgen.1002272
Chapuis, M.-P., and Estoup, A. (2007). Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 24, 621–631. doi: 10.1093/molbev/msl191
Chouaia, B., Gaiarsa, S., Crotti, E., Comandatore, F., Degli Esposti, M., Ricci, I., et al. (2014). Acetic acid bacteria genomes reveal functional traits for adaptation to life in insect guts. Genome Biol. Evol. 6, 912–920. doi: 10.1093/gbe/evu062
Cirimotich, C. M., Ramirez, J. L., and Dimopoulos, G. (2011). Native microbiota shape insect vector competence for human pathogens. Cell Host Microbe 10, 307–310. doi: 10.1016/j.chom.2011.09.006
Clements, A. N. (1992). The Biology of Mosquitoes: Development, Nutrition and Reproduction. London: Chapman & Hall.
Coon, L. K., Vogel, J. K., Brown, M. R., and Strand, M. R. (2014). Mosquitoes rely on their gut microbiota for development. Mol. Ecol. 23, 2727–2739. doi: 10.1111/mec.12771
Cornuet, J. M., and Luikart, G. (1996). Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144, 2001–2014.
D'Auria, G., Peris-Bondia, F., DŽunková, M., Mira, A., Collado, M. C., Latorre, A., et al. (2013). Active and secreted IgA-coated bacterial fractions from the human gut reveal an under-represented microbiota core. Sci. Rep. 3:3515. doi: 10.1038/srep03515
Dada, N., Jumas-Bilak, E., Manguin, S., Seidu, R., Stenström, T. A., and Overgaard, H. J. (2014). Comparative assessment of the bacterial communities associated with Aedes aegypti larvae and water from domestic water storage containers. Parasit. Vectors 7:391. doi: 10.1186/1756-3305-7-391
Dai, J. X., Liu, X. M., and Wang, Y. J. (2014). Diversity of endophytic bacteria in Caragana microphylla grown in the desert grassland of the Ningxia Hui autonomous region of China. Genet. Mol. Res. 13, 2349–2358. doi: 10.4238/2014.April.3.7
de Albuquerque, A. L., Magalhães, T., and Ayres, C. F. J. (2011). High prevalence and lack of diversity of Wolbachia pipientis in Aedes albopictus populations from Northeast Brazil. Memórias do Instituto Oswaldo Cruz 106, 773–776. doi: 10.1590/S0074-02762011000600021
del Pilar Corena, M., VanEkeris, L., Salazar, M. I., Bowers, D., Fiedler, M. M., Silverman, D., et al. (2005). Carbonic anhydrase in the adult mosquito midgut. J. Exp. Biol. 208, 3263–3273. doi: 10.1242/jeb.01739
Dennison, N. J., Jupatanakul, N., and Dimopoulos, G. (2014). The mosquito microbiota influences vector competence for human pathogens. Curr. Opin. Insect Sci. 3, 6–13. doi: 10.1016/j.cois.2014.07.004
Dillon, R. J., and Dillon, V. M. (2004). The gut bacteria of insects: nonpathogenic interactions. Annu. Rev. Entomol. 49, 71–92. doi: 10.1146/annurev.ento.49.061802.123416
Dinparast Djadid, N., Jazayeri, H., Raz, A., Favia, G., Ricci, I., and Zakeri, S. (2011). Identification of the midgut microbiota of An. stephensi and An. maculipennis for their application as a paratransgenic tool against malaria. PloS ONE 6:e28484. doi: 10.1371/journal.pone.0028484
Dlugosch, K. M., and Parker, I. M. (2008). Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol. Ecol. 17, 431–449. doi: 10.1111/j.1365-294X.2007.03538.x
Dobson, S. L., Marsland, E. J., and Rattanadechakul, W. (2002). Mutualistic Wolbachia infection in Aedes albopictus: accelerating cytoplasmic drive. Genetics 160, 1087–1094.
Dobson, S. L., Rattanadechakul, W., and Marsland, E. J. (2004). Fitness advantage and cytoplasmic incompatibility in Wolbachia single- and superinfected Aedes albopictus. Heredity 93, 135–142. doi: 10.1038/sj.hdy.6800458
Dong, Y., Manfredini, F., and Dimopoulos, G. (2009). Implication of the mosquito midgut microbiota in the defense against malaria parasites. PLoS Pathog. 5:e1000423. doi: 10.1371/journal.ppat.1000423
Douglas, A. E. (2011). Lessons from studying insect symbioses. Cell Host Microbe 10, 359–367. doi: 10.1016/j.chom.2011.09.001
Douglas, A. E. (2014). The molecular basis of bacterial-insect symbiosis. J. Mol. Biol. 426, 3830–3837. doi: 10.1016/j.jmb.2014.04.005
Dray, S., and Dufour, A. B. (2007). The ade4 Package: implementing the duality diagram for ecologists. J. Stat. Softw. 22, 1–20.
Duguma, D., Rugman-Jones, P., Kaufman, M. G., Hall, M. W., Neufeldd, J. D., Southamer, R., et al. (2013). Bacterial communities associated with Culex mosquito larvae and two emergent aquatic plants of bioremediation importance. PLoS ONE 8:e72522. doi: 10.1371/journal.pone.0072522
Earl, D. A., and VonHoldt, B. M. (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361. doi: 10.1007/s12686-011-9548-7
Engel, P., and Moran, N. A. (2013). The gut microbiota of insects – diversity in structure and function. FEMS Microbiol. Rev. 37, 699–735. doi: 10.1111/1574-6976.12025
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Excoffier, L., and Lischer, H. E. L. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Francis, A. P., and Currie, D. J. (2003). A globally consistent richness-climate relationship for angiosperms. Am. Nat. 161, 523–536. doi: 10.1086/368223
Gayatri Priya, N., Ojha, A., Kajla, M. K., Raj, A., and Rajagopal, R. (2012). Host plant induced variation in gut bacteria of Helicoverpa armigera. PLoS ONE 7:e30768. doi: 10.1371/journal.pone.0030768
Gendrin, M., Rodgers, F. H., Yerbanga, R. S., Ouédraogo, J. B., Basáñez, M. G., Cohuet, A., et al. (2015). Antibiotics in ingested human blood affect the mosquito microbiota and capacity to transmit malaria. Nat. Commun. 6:5921. doi: 10.1038/ncomms6921
Gimonneau, G., Tchioffo, M. T., Abate, L., Boissière, A., Awono-Ambéné, P. H., Nsango, S. E., et al. (2014). Composition of Anopheles coluzzii and Anopheles gambiae microbiota from larval to adult stages. Infect. Genet. Evol. 28, 715–724. doi: 10.1016/j.meegid.2014.09.029
Handley, L.-J. L., Estoup, A., Evans, D. M., Thomas, C. E., Lombaert, E., Facon, B., et al. (2011). Ecological genetics of invasive alien species. Biocontrol 56, 409–428. doi: 10.1007/s10526-011-9386-2
Hillyer, J. F. (2010). Mosquito immunity. Adv. Exp. Med. Biol. 708, 218–238. doi: 10.1007/978-1-4419-8059-5_12
Hironaga, M., Yamane, K., Inaba, M., Haga, Y., and Arakawa, Y. (2008). Characterization and antimicrobial susceptibility of Dysgonomonas capnocytophagoides isolated from human blood sample. Jpn. J. Infect. Dis. 61, 212–213.
Hurst, G. D. D., and Jiggins, F. M. (2005). Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc. R. Soc. Lond. B Biol. Sci. 272, 1525–1534. doi: 10.1098/rspb.2005.3056
Husseneder, C., Berestecky, J. M., and Grace, J. K. (2009). Changes in composition of culturable bacteria community in the gut of the Formosan subterranean termite depending on rearing conditions of the host. Ann. Entomol. Soc. Am. 102, 498–507. doi: 10.1603/008.102.0321
Jakobsson, M., and Rosenberg, N. A. (2007). CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806. doi: 10.1093/bioinformatics/btm233
Johnson, K. S., and V Barbehenn, R. (2000). Oxygen levels in the gut lumens of herbivorous insects. J. Insect Physiol. 46, 897–903. doi: 10.1016/S0022-1910(99)00196-1
Jupatanakul, N., Sim, S., and Dimopoulos, G. (2014). The insect microbiome modulates vector competence for arboviruses. Viruses 6, 4294–4313. doi: 10.3390/v6114294
Kenney, J. L., and Brault, A. C. (2014b). The role of environmental, virological and vector interactions in dictating biological transmission of arthropod-borne viruses by mosquitoes. Adv. Virus Res. 89, 39–83. doi: 10.1016/B978-0-12-800172-1.00002-1
King, K. C., and Lively, C. M. (2012). Does genetic diversity limit disease spread in natural host populations? Heredity 109, 199–203. doi: 10.1038/hdy.2012.33
Kittayapong, P., Baisley, K. J., Sharpe, R. G., Baimai, V., and O'Neill, S. L. (2002). Maternal transmission efficiency of Wolbachia superinfections in Aedes albopictus populations in Thailand. Am. J. Trop. Med. Hyg. 66, 103–107.
Koroiva, R., Souza, C. W. O., Toyama, D., Henrique-Silva, F., and Fonseca-Gessner, A. A. (2013). Lignocellulolytic enzymes and bacteria associated with the digestive tracts of Stenochironomus (Diptera: Chironomidae) larvae. Genetic. Mol. Res. 12, 3421–3434. doi: 10.4238/2013.April.2.2
Lawson, P. A., Carlson, P., Wernersson, S., Moore, E. R. B., and Falsen, E. (2010). Dysgonomonas hofstadii sp. nov., isolated from a human clinical source. Anaerobe 16, 161–164. doi: 10.1016/j.anaerobe.2009.06.005
Léger, E., Vourc'h, G., Vial, L., Chevillon, C., and McCoy, D. K. (2013). Changing distributions of ticks: causes and consequences. Exp. Appl. Acarol. 59, 9615. doi: 10.1007/s10493-012-9615-0
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lindh, J. M., Terenius, O., and Faye, I. (2005). 16S rRNA gene-based identification of midgut bacteria from field-caught Anopheles gambiae sensu lato and Anopheles funestus mosquitoes reveals new species related to known insect symbionts. Appl. Environ. Microbiol. 71, 7217–7223. doi: 10.1128/AEM.71.11.7217-7223.2005
Linnenbrink, M., Wang, J., Hardouin, E. A., Künzel, S., Metzler, D., and Baines, J. F. (2013). The role of biogeography in shaping diversity of the intestinal microbiota in house mice. Mol. Ecol. 22, 1904–1916. doi: 10.1111/mec.12206
Lizé, A., McKay, R., and Lewis, Z. (2014). Kin recognition in Drosophila: the importance of ecology and gut microbiota. ISME J. 8, 469–477. doi: 10.1038/ismej.2013.157
Masella, A. P., Bartram, A. K., Truszkowski, J. M., Brown, D. G., and Neufeld, J. D. (2012). PANDAseq: paired-end assembler for Illumina sequences. BMC Bioinformatics 13:31. doi: 10.1186/1471-2105-13-31
McCoy. (2008). The population genetic structure of vectors and our understanding of disease epidemiology. Parasite 15, 444–448. doi: 10.1051/parasite/2008153444
Medlock, J. M., Hansford, K. M., Schaffner, F., Versteirt, V., Hendrickx, G., and Zeller, H., et al. (2012). A review of the invasive mosquitoes in Europe: ecology, public health risks, and control options. Vector Borne Zoonotic Dis. 12, 435–447. doi: 10.1089/vbz.2011.0814
Meusnier, I., Olsen, J. L., Stam, W. T., Destombe, C., and Valero, M. (2001). Phylogenetic analyses of Caulerpa taxifolia (Chlorophyta) and of its associated bacterial microflora provide clues to the origin of the Mediterranean introduction. Mol. Ecol. 10, 931–946. doi: 10.1046/j.1365-294X.2001.01245.x
Minard, G., Mavingui, P., and Moro, C. V. (2013). Diversity and function of bacterial microbiota in the mosquito holobiont. Parasit. Vectors 6:146. doi: 10.1186/1756-3305-6-146
Minard, G., Tran, F.-H., Dubost, A., Tran-Van, V., Mavingui, P., and Moro, C. V. (2014). Pyrosequencing 16S rRNA genes of bacteria associated with wild tiger mosquito Aedes albopictus: a pilot study. Front. Cell. Infect. Microbiol. 4:59. doi: 10.3389/fcimb.2014.00059
Moran, N. A., McCutcheon, J. P., and Nakabachi, A. (2008). Genomics and evolution of heritable bacterial symbionts. Annu. Rev. Genet. 42, 165–190. doi: 10.1146/annurev.genet.41.110306.130119
Moya, A., Peretó, J., Gil, R., and Latorre, A. (2008). Learning how to live together: genomic insights into prokaryote-animal symbioses. Nat. Rev. Genet. 9, 218–229. doi: 10.1038/nrg2319
Muegge, B. D., Kuczynski, J., Knights, D., Clemente, J. C., González, A., and Fontana, L., et al. (2011). Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332, 970–974. doi: 10.1126/science.1198719
Ochman, H., Worobey, M., Kuo, C.-H., Ndjango, J. B. N., Peeters, M., Hahn, B. H., et al. (2010). Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol. 8:e1000546. doi: 10.1371/journal.pbio.1000546
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O'Hara, R. B., et al. (2013). vegan: Community Ecology Package. Available online at: https://cran.r-project.org/web/packages/vegan/index.html
Osei-Poku, J., Mbogo, C. M., Palmer, W. J., and Jiggins, F. M. (2012). Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya. Mol. Ecol. 21, 5138–5150. doi: 10.1111/j.1365-294X.2012.05759.x
Parks, D. H., and Beiko, R. G. (2010). Identifying biologically relevant differences between metagenomic communities. Bioinformatics 26, 715–721. doi: 10.1093/bioinformatics/btq041
Paupy, C., Delatte, H., Bagny, L., Corbel, V., and Fontenille, D. (2009). Aedes albopictus, an arbovirus vector: from the darkness to the light. Microbes Infect. 11, 1177–1185. doi: 10.1016/j.micinf.2009.05.005
Pernice, M., Simpson, S. J., and Ponton, F. (2014). Towards an integrated understanding of gut microbiota using insects as model systems. J. Insect Physiol. 69, 12–20. doi: 10.1016/j.jinsphys.2014.05.016
Pidiyar, V. J., Jangid, K., Patole, M. S., and Shouche, Y. S. (2004). Studies on cultured and uncultured microbiota of wild Culex quinquefasciatus mosquito midgut based on 16s ribosomal RNA gene analysis. Am. J. Trop. Med. Hyg. 70, 597–603.
Porretta, D., Gargani, M., Bellini, R., Calvitti, M., and Urbanelli, S. (2006). Isolation of microsatellite markers in the tiger mosquito Aedes albopictus (Skuse). Mol. Ecol. Notes 6, 880–881. doi: 10.1111/j.1471-8286.2006.01384.x
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Pumpuni, C. B., Demaio, J., Kent, M., Davis, J. R., and Beier, J. C. (1996). Bacterial population dynamics in three anopheline species: the impact on Plasmodium sporogonic development. Am. J. Trop. Med. Hyg. 54, 214–218.
Raharimalala, F. N., Ravaomanarivo, L. H., Ravelonandro, P., Rafarasoa, L. S., Zouache, K., Tran-Van, V., et al. (2012). Biogeography of the two major arbovirus mosquito vectors, Aedes aegypti and Aedes albopictus (Diptera, Culicidae), in Madagascar. Parasit. Vectors 5, 56. doi: 10.1186/1756-3305-5-56
Ramírez-Puebla, S. T., Rosenblueth, M., Chávez-Moreno, C. K., de Lyra, M. C. P., Tecante, A., and Martínez-Romero, E. (2010). Molecular phylogeny of the genus Dactylopius (Hemiptera: Dactylopiidae) and identification of the symbiotic bacteria. Environ. Entomol.39, 1178–1183. doi: 10.1603/EN10037
Rani, A., Sharma, A., Rajagopal, R., Adak, T., and Bhatnagar, R. K. (2009). Bacterial diversity analysis of larvae and adult midgut microflora using culture-dependent and culture-independent methods in lab-reared and field-collected Anopheles stephensi-an Asian malarial vector. BMC Microbiol. 9:96. doi: 10.1186/1471-2180-9-96
R Development Core Team. (2009). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available online at: http://www.R-project.org
Rosenberg, E., and Zilber-Rosenberg, I. (2014). The Hologenome Concept: Human, Animal and Plant Microbiota. Berlin Heidelberg: Springer International Publishing.
Rosenberg, N. A. (2004). Distruct: a program for the graphical display of population structure. Mol. Ecol. Notes 4, 137–138. doi: 10.1046/j.1471-8286.2003.00566.x
Rueda, L. M. (2004). Pictorial keys for the identification of mosquitoes (Diptera: Culicidae) associated with dengue virus transmission. Zootaxa 589, 1–60.
Saboia-Vahia, L., Cuervo, P., Borges-Veloso, A., de Souza, N. P., Britto, C., Dias-Lopes, G., et al. (2014). The midgut of Aedes albopictus females expresses active trypsin-like serine peptidases. Parasit. Vectors 7:253. doi: 10.1186/1756-3305-7-253
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Sommer, S. (2005). The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front. Zool. 2:16. doi: 10.1186/1742-9994-2-16
Stouthamer, R., Breeuwer, J. A., and Hurst, G. D. (1999). Wolbachia pipientis: microbial manipulator of arthropod reproduction. Annu. Rev. Microbiol. 53, 71–102. doi: 10.1146/annurev.micro.53.1.71
Tagliavia, M., Messina, E., Manachini, B., Cappello, S., and Quatrini, P. (2014). The gut microbiota of larvae of Rhynchophorus ferrugineus Oliver (Coleoptera: Curculionidae). BMC Microbiol. 14:136. doi: 10.1186/1471-2180-14-136
Terenius, O., Lindh, J. M., Eriksson-Gonzales, K., Bussière, L., Laugen, A. T., Bergquist, H., et al. (2012). Midgut bacterial dynamics in Aedes aegypti. FEMS Microbiol. Ecol. 80, 556–565. doi: 10.1111/j.1574-6941.2012.01317.x
Toft, C., and Andersson, S. G. E. (2010). Evolutionary microbial genomics: insights into bacterial host adaptation. Nat. Rev. Genet. 11, 465–475. doi: 10.1038/nrg2798
Tortosa, P., Charlat, S., Labbé, P., Dehecq, J. S., Barré, H., and Weill, M. (2010). Wolbachia age-sex-specific density in Aedes albopictus: a host evolutionary response to cytoplasmic incompatibility? PLoS ONE 5:e9700. doi: 10.1371/journal.pone.0009700
Tortosa, P., Courtiol, A., Moutailler, S., Failloux, A. B., and Weill, M. (2008). Chikungunya-Wolbachia interplay in Aedes albopictus. Insect Mol. Biol. 17, 677–684. doi: 10.1111/j.1365-2583.2008.00842.x
Urbanski, J. M., Benoit, J. B., Michaud, M. R., Denlinger, D. L., and Armbruster, P. (2010). The molecular physiology of increased egg desiccation resistance during diapause in the invasive mosquito, Aedes albopictus. Proc. R. Soc. Lond. B Biol. Sci. 277, 2683–2692. doi: 10.1098/rspb.2010.0362
Vayssier-Taussat, M., Albina, E., Citti, C., Cosson, J. F., Jacques, M. A., Lebrun, M. H., et al. (2014). Shifting the paradigm from pathogens to pathobiome: new concepts in the light of meta-omics. Front. Cell. Infect. Microbiol. 4:29. doi: 10.3389/fcimb.2014.00029
Vaz-Moreira, I., Nunes, O. C., and Manaia, C. M. (2011). Diversity and Antibiotic Resistance Patterns of Sphingomonadaceae Isolates from Drinking Water. Appl. Environ. Microbiol. 77, 5697–5706. doi: 10.1128/AEM.00579-11
Vega-Rua, A., Zouache, K., Caro, V., Diancourt, L., Delaunay, P., Grandadam, M., et al. (2013). High efficiency of temperate Aedes albopictus to transmit chikungunya and dengue viruses in the Southeast of France. PLoS ONE 8:e59716. doi: 10.1371/journal.pone.0059716
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wang, Y., Gilbreath, T. M., III, Kukutla, P., Yan, G., and Xu, J. (2011). Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS ONE 6:e24767. doi: 10.1371/journal.pone.0024767
Wang, Y., Wang, Y., Zhang, J., Xu, W., Zhang, J., and Huang, F. S. (2013). Ability of TEP1 in intestinal flora to modulate natural resistance of Anopheles dirus. Exp. Parasitol. 134, 460–465. doi: 10.1016/j.exppara.2013.04.003
Weiss, B., and Aksoy, S. (2011). Microbiome influences on insect host vector competence. Trends Parasitol. 27, 514–522. doi: 10.1016/j.pt.2011.05.001
Yang, Y.-J., Zhang, N., Ji, S.-Q., Lan, X., Shen, Y. L., Li, F. L., et al. (2014). Dysgonomonas macrotermitis sp. nov., isolated from the hindgut of a fungus-growing termite. Int. J. Syst. Evol. Microbiol. 64, 2956–2961. doi: 10.1099/ijs.0.061739-0
Ye, L., Amberg, J., Chapman, D., Gaikowski, M., and Liu, W.-T. (2014). Fish gut microbiota analysis differentiates physiology and behavior of invasive Asian carp and indigenous American fish. ISME J. 8, 541–551. doi: 10.1038/ismej.2013.181
Zhang, X., Lin, L., Zhu, Z., Yang, X., Wang, Y., and An, Q. (2013). Colonization and modulation of host growth and metal uptake by endophytic bacteria of Sedum alfredii. Int. J. Phytoremediation 15, 51–64. doi: 10.1080/15226514.2012.670315
Zilber-Rosenberg, I., and Rosenberg, E. (2008). Role of microorganisms in the evolution of animals and plants: the hologenome theory of evolution. FEMS Microbiol. Rev. 32, 723–735. doi: 10.1111/j.1574-6976.2008.00123.x
Zouache, K., Fontaine, A., Vega-Rua, A., Mousson, L., Thiberge, J. M., Lourenco-De-Oliveira, R., et al. (2014). Three-way interactions between mosquito population, viral strain and temperature underlying chikungunya virus transmission potential. Proc. R. Soc. Lond. B Biol. Sci. 281:20141078. doi: 10.1098/rspb.2014.1078
Zouache, K., Raharimalala, F. N., Raquin, V., Tran-Van, V., Raveloson, L. H. R., Ravelonandro, P., et al. (2011). Bacterial diversity of field-caught mosquitoes, Aedes albopictus and Aedes aegypti, from different geographic regions of Madagascar. FEMS Microbiol. Ecol. 75, 377–389. doi: 10.1111/j.1574-6941.2010.01012.x
Zouache, K., Voronin, D., Tran-Van, V., Mousson, L., Failloux, A. B., Mavingui, et al. (2009). Persistent Wolbachia and cultivable bacteria infection in the reproductive and somatic tissues of the mosquito vector Aedes albopictus. PLoS ONE 4:e6388. doi: 10.1371/journal.pone.0006388
Keywords: Aedes albopictus, Dysgonomonas, holobiont, microbiota, microsatellite, phylogeography, Wolbachia
Citation: Minard G, Tran FH, Van VT, Goubert C, Bellet C, Lambert G, Kim KLH, Thuy THT, Mavingui P and Valiente Moro C (2015) French invasive Asian tiger mosquito populations harbor reduced bacterial microbiota and genetic diversity compared to Vietnamese autochthonous relatives. Front. Microbiol. 6:970. doi: 10.3389/fmicb.2015.00970
Received: 24 July 2015; Accepted: 01 September 2015;
Published: 22 September 2015.
Edited by:
Joerg Graf, University of Connecticut, USAReviewed by:
David William Waite, University of Auckland, New ZealandBrian Weiss, Yale University, USA
Copyright © 2015 Minard, Tran, Van, Goubert, Bellet, Lambert, Kim, Thuy, Mavingui and Valiente Moro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: C. Valiente Moro, Ecologie Microbienne, Université Claude Bernard Lyon 1, Bat. André Lwoff, 10 Rue Raphaël Dubois, 69100 Villeurbanne, France,Y2xhaXJlLnZhbGllbnRlLW1vcm9AdW5pdi1seW9uMS5mcg==